
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A high-energy aqueous Zn‖NO2 electrochemical cell: a new strategy for NO2 fixation and electric power generation†
Longtao
Ma
ab,
Shengmei
Chen
 b,
Wenhao
Yan
b,
Guobin
Zhang
b,
Yiran
Ying
c,
Haitao
Huang
c,
Derek
Ho
*b,
Wei
Huang
*a and
Chunyi
Zhi
*bd
b,
Wenhao
Yan
b,
Guobin
Zhang
b,
Yiran
Ying
c,
Haitao
Huang
c,
Derek
Ho
*b,
Wei
Huang
*a and
Chunyi
Zhi
*bd
aFrontiers Science Center for Flexible Electronics, Institute of Flexible Electronics, Northwestern Polytechnical University, Xi'an, 710072, P. R. China. E-mail: iamwhuang@nwpu.edu.cn
bDepartment of Materials Science and Engineering, City University of Hong Kong, 83 Tat Chee Avenue, Kowloon, Hong Kong 999077, P. R. China. E-mail: derekho@cityu.edu.hk; cy.zhi@cityu.edu.hk
cDepartment of Applied Physics and Research Institute for Smart Energy, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong 999077, P. R. China
dCenter for Advanced Nuclear Safety and Sustainable Development, City University of Hong Kong Kowloon, Hong Kong 999077, P. R. China
First published on 24th January 2023
Abstract
Air pollution by nitrogen oxides (NO2) from exhaust gas is a deep-seated problem, thus urgently calling for new capture and abatement technologies. Meanwhile, the electrocatalytic conversion of NO2 to value-added chemicals is a promising strategy for mitigating human-caused imbalances of the global nitrogen cycle. Here, we propose an electrochemical cell based on an aqueous Zn‖NO2 system with a nano-NiO catalyst deposited as the cathode, a metallic Zn foil as the anode and a ZnCl2 aqueous solution as the electrolyte. Importantly, the electrolyte can efficiently capture NO2, then convert it to NO2− and eventually to value-added NH3, while simultaneously producing electric power. As proof of concept, a battery has been fabricated, which exhibits bifunctional activity and stability (>100 h) towards reversible NO2 reduction and evolution reactions. A high cell-level energy density of 553.2 W h kg−1cell/1589.6 W h L−1cell from pouch cells (2.4 Ah) has been achieved. As an additional green feature, the produced NO2− by the Zn‖NO2 cell is subsequently converted to NH3 by a self-powered mechanism, thereby servicing multiple key conversion steps in the nitrogen cycle all within a single device, paving the way to scalable, highly integrated solutions.
Broader contextThe emission of industrial waste gases and vehicle exhaust gases (e.g. NO2, NO, SO2, CO2, CO) from the massive combustion of fossil fuels leads to the need for elimination of the harmful gases and the generation of clean energy. NO2 gas causes great harm to the human body, such as damage to eye, nose, throat, and lung tissues. Therefore, the capture and conversion of harmful NO2 gas to valuable products is of great significance. Metal–gas batteries have been demonstrated to be an effective strategy for efficiently capturing various gases and continuously suppling electrical energy. Zinc metal is an attractive anode material for aqueous batteries, due to its high theoretical capacity (820 mA h g−1), low redox potential (−0.76 V vs. SHE), low cost, abundant reserves, and suitable reactivity. By coupling a Zn anode with a NO2 gas diffusion electrode, the device is capable of performing electrochemical capture and conversion of NO2. Here, we report a 1.8 V, 2.4-Ah-scale aqueous Zn‖NO2 electrochemical system with 3 vol% NO2/air utilized as an energy carrier to store renewable energy, which exhibits an ultrahigh cell level energy density of 553.2 W h kg−1cell and 1589.6 W h L−1cell. The Zn‖NO2 cells are effective for both capturing NO2 and converting NO2 to NO2− species at room temperature. The faradaic efficiency of NO2 → NO2− reaches 96.4%. The produced NO2− can be further converted to NH3 using an electrochemical Haber–Bosch reactor, self-powered by the Zn‖NO2 cells. |
Introduction
The ever-increasing worldwide energy demand and emission of industrial waste gases and vehicle exhaust gases (e.g. NO2, NO, SO2, CO2, CO) from the massive combustion of fossil fuels leads to the need for effective technologies for the capture of exhaust gases and their subsequent conversion into electricity.1–8 Among them, NO2 released into the atmosphere contributes to photochemical smog, stratospheric ozone deterioration and global warming, causing serious environmental and health problems.6,9,10 A strategy is urgently required to convert NO2, which will alleviate NO2 accumulation and simultaneously produce value-added chemicals such as NH3.11–13A variety of metal‖gas electrochemical cells, such as Li‖CO2, Li‖SO2, Al‖CO2, and Zn‖CO2 systems, have been proposed as novel approaches to capture exhaust gas streams while being equipped with the additional benefit of electrical energy production.14–19 For NO2 gas, a Li‖NO2 battery is reported to reduce NO2.20 However, the Li‖NO2 cell produced NO as the final reduzate, which is itself another air pollutant.21,22 Currently, there is no electrochemical method to capture and utilize exhaust NO2 to make value-added chemicals, and to simultaneously produce electrical power.
Zinc (Zn) metal is an attractive anode material for aqueous batteries, due to its high theoretical capacity (820 mA h g−1), low redox potential (−0.76 V vs. SHE), low cost, abundant reserves, and suitable reactivity.7,23–28 By coupling a Zn anode with a NO2 gas diffusion electrode, the device is capable of performing electrochemical capture and conversion of NO2 with an overall reaction as follows:
| Zn + 2 NO2 → Zn(NO2)2E0 = 1.85 V (vs. Zn/Zn2+) | (1) |
The Zn‖NO2 electrochemical capture system may be operated in either secondary (rechargeable) or primary (non-rechargeable) modes. In a secondary cell, reduced NO2 species react with oxidized metal ions to form metal nitrates and electricity during cell discharge (Fig. 1a-i). Ideally, recharging the cell reverses the reaction, consuming electrical energy to release the captured NO2 at the cathode and regenerating the metal anode. Adoption of a secondary electrochemical process would therefore facilitate the separation and concentration of NO2 based on the following electrochemical reaction, as demonstrated in Fig. 1a-ii.
| NO2− − e− → NO2 | (2) |
 | ||
| Fig. 1 Architectures of Zn‖NO2 electrochemistry technology as a capture system. A secondary Zn‖NO2 electrochemical cell, in which (a-i) NO2 is reduced to generate electrical energy and (a-ii) NO2 is concentrated by recharging. (b) A primary Zn‖NO2 in which the captured NO2 is converted to NH3 H2O valuable products. | ||
Another configuration of interest is the primary electrochemical cell, where the metal anode is consumed to produce electrical energy and discharge products, which can be harvested (from the electrode, electrolyte, and other cell components) and converted to valuable chemicals (Fig. 1b) based on the following electrochemical reaction.
| NO2− + 5H+ + 4e− → NH3 + 2H2O | (3) |
Screening catalysts and analysis of catalytic activity
The screening of the electrocatalytic activity for NO2−↔*NO2 conversion (‘*’ denotes the adsorbed state) on various surfaces of metal oxides, such as NiO, CoO, FeO and CuO, is scrutinized and analyzed by density functional theory (DFT) calculations. From density of states (DOS) calculations (Fig. S1, ESI†), the hybridization between the transition metal (Ni, Co, Cu, Fe) 3d and nitrogen/oxygen 2p orbitals at around 2 eV and from −4 to −2.5 eV can be observed, which contributes to the favorable adsorption of NO2. After comparing two different NO2 adsorption configurations (Fig. S2, ESI†), we find that on NiO and FeO, *NO2 prefers configuration 1 in which only the N atom binds to metal site, and on CoO and CuO, *NO2 prefers configuration 2 with both N and O binding to adjacent sites. The charge density difference distribution in Fig. 2a and Fig. S3 (ESI†), shows a clear two-way charge transfer between the metal oxides and NO2, in which charge accumulates in the top/bottom regions of the O atoms and around the metal–N/metal–O bonds, while charge depletion exists around the O atoms. Bader charge analysis suggests that NiO exhibits a relatively smaller value of charge transfer from the surface to *NO2 (0.691 e−), in line with the charge density difference distribution results. Therefore, NO2 is weakly trapped on the surface, which facilitates the desorption and the two-way NO2−↔*NO2 conversion. The Gibbs free energy diagrams in Fig. 2b suggest that the conversion from * + NO2− to *NO2 is energetically favorable, therefore the overall reaction thermodynamics are limited by the NO2 desorption. Results show that both NiO and CoO exhibit a desorption Gibbs free energy change of 2.35 eV, which is much smaller than that for FeO (5.32 eV) and CuO (5.50 eV). These results indicate that NiO has the best performance for catalyzing NO2− ↔*NO2 conversion.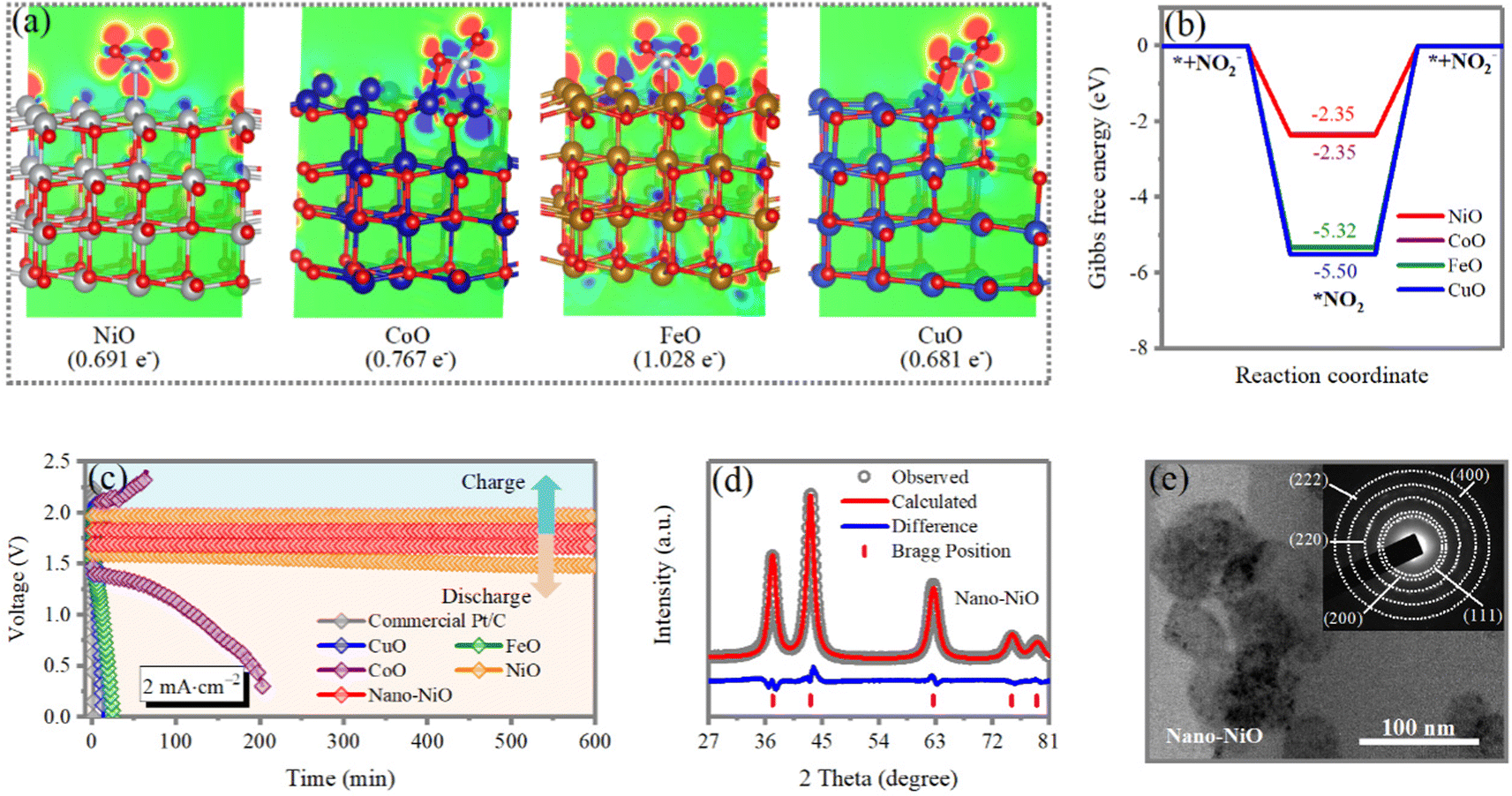 | ||
Fig. 2 (a) 2D contour plot (0.005 e Bohr−1) of the charge density difference distribution (CDDD) of adsorbed *NO2 on NiO, CoO, FeO and CuO with the iso-surface value of 0.005 e Bohr−3. Meanwhile, the number of charge transfers from the metal oxide surface to *NO2 is also denoted. Color code for atoms: Ni: grey; Co: dark blue; Fe: brown; Cu: light blue; O: red; N: cyan. (b) Gibbs free energy diagrams for NO2−↔*NO2 conversion on NiO, CoO, FeO, and CuO surfaces. The purple line represents the Gibbs free energy of CoO, which is covered by that of NiO. (c) The galvanostatic charge/discharge curves of the Zn/NO2 electrochemical cell at a current density of 2 mA cm−2 with commercial Pt/C, CuO, FeO, CoO, NiO and nano-NiO electrocatalysts. (d) XRD pattern of nano-NiO and the corresponding results of the Rietveld refinement. Refined parameters are in space group: R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m; a = 2.97 Å; c = 7.27 Å. (e) TEM image of nano-NiO. The inset is the corresponding SEAD pattern. m; a = 2.97 Å; c = 7.27 Å. (e) TEM image of nano-NiO. The inset is the corresponding SEAD pattern. | ||
To deliver a highly efficient conversion, an electrocatalyst with high activity and selectivity for NO2RR are a prerequisite. The electrocatalytic activities of NO2RR are examined using a three-electrode configuration, in which the catalyst deposited carbon fiber cloth (CFC) is the working electrode, Pt foil is the counter electrode and Ag/AgCl is the reference electrode. Excess NO2 is blown through a gas pump towards the CFC electrode. In general, the NO2RR polarization curve exhibits similar features to those of the hydrogen evolution reaction (HER), including an onset potential and an overpotential. The electrocatalytic performance of nano-NiO (420 mV for overpotential at 10 mA cm−2) remarkably outperforms commercial Pt/C, CuO, FeO, CoO and NiO catalysts in 2M ZnCl2 aqueous solution (Fig. S4a, ESI†). Meanwhile, the nano-NiO catalyst exhibits the lowest Tafel slope (61.1 mV dec−1) (Fig. 4b), suggesting the superiority of nano-NiO for NO2RR. Additionally, the NO2RR behaviors of nano-NiO are examined in different electrolytes of 2M Zn(OTf)2, 2M ZnSO4, 6M KOH and 2M ZnCl2 aqueous solutions. As shown in Fig. S4b and c (ESI†), it shows both the lowest overpotential and Tafel slope in 2M ZnCl2 aqueous solutions, demonstrating the suitability of the ZnCl2 aqueous electrolyte for the NO2RR. Furthermore, the aqueous Zn‖NO2 systems are developed by employing metallic Zn as the anode, deposited electrocatalysts on CFC as the cathode and an aqueous solution of ZnCl2 as the electrolyte. As shown in Fig. 2c, when the batteries based on CuO, FeO and commercial Pt/C electrocatalysts, are charged or discharged, they exhibit a very large overpotential or show a quick voltage drop to zero. This observation indicates the very sluggish electrocatalytic kinetics of Zn‖NO2 batteries using commercial CuO, FeO and Pt/C as catalysts. Although the CoO electrocatalyst enables the Zn‖NO2 battery to operate, the overpotential gradually increases during the charge/discharge processes. Encouragingly, NiO is particularly effective for catalyzing both the reductions of NO2 and the oxidation of NO2−. Nevertheless, the energy efficiency of the Zn‖NO2 cell is only 76.3%, and the output voltage gradually decreases from 1.55 to 1.46 V after 600 min, indicating the gradually reduced electrocatalytic activity of NiO. Therefore, employing the nano-NiO electrocatalyst, the Zn‖NO2 battery can steadily and continuously generate electrical power while the NO2− is efficiently oxidized. The energy efficiency that is calculated by dividing the discharge voltage by the charging voltage, reaches 92.6% when the NiO nanoparticle (nano-NiO) electrocatalyst is used, which is much higher than that of the well-studied Zn–air batteries (<60%).29–33 The enhanced electrocatalytic activity is attributed to sufficient active sites and the rapid transport of electrons and ions with the nanostructuration of the NiO electrocatalyst.
To further enhance the catalytic efficiency of NiO, nano-NiO is fabricated via an ultrasonic crushing method. The morphology and structure are investigated using X-ray diffraction patterns (XRD), scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The fabricated nano-NiO exhibits well-resolved but broader diffraction peaks with high intensity, indicating small crystallite sizes. The crystal structure is successfully refined with the space group of Rm (Fig. 2d), which is similar to that of NiO. The representative SEM image exhibits a uniformly well-defined bean pod-shape with a diameter of ∼100 nm (Fig. S5, ESI†). The TEM image depicts a dense bean pod-like structure, and the corresponding selected area electron diffraction (SAED) pattern confirms further its polycrystalline characteristics (Fig. 2e).
Electrochemical performances of Zn‖NO2 cells
The electrochemical performance of the Zn‖NO2 batteries-based on nano-NiO is investigated. The linear sweep voltammetry (LSV), open circuit voltage (OCV) and galvanostatic charge/discharge curves are conducted following the protocols well developed by Zn–air battery community. As shown in Fig. 3a, the output voltage of the device with the nano-NiO electrocatalyst exhibits a high OCV of 1.83 V under a NO2 atmosphere, close to theoretical value of 1.85 V, which is higher than that based on the commercial NiO electrocatalyst (1.75 V). As shown in Fig. 3b, the peak power densities of the Zn‖NO2 system employing NiO and nano-NiO reach 20.1 and 90.5 mW cm−2, respectively, validating the superior NO2RR kinetics of nano-NiO. The symmetric charge/discharge curves in Fig. 3c indicate the bifunctional electrocatalysis of nano-NiO for NO2RR and NO2− oxidation, surpassing the counterpart values of the NiO-based cells.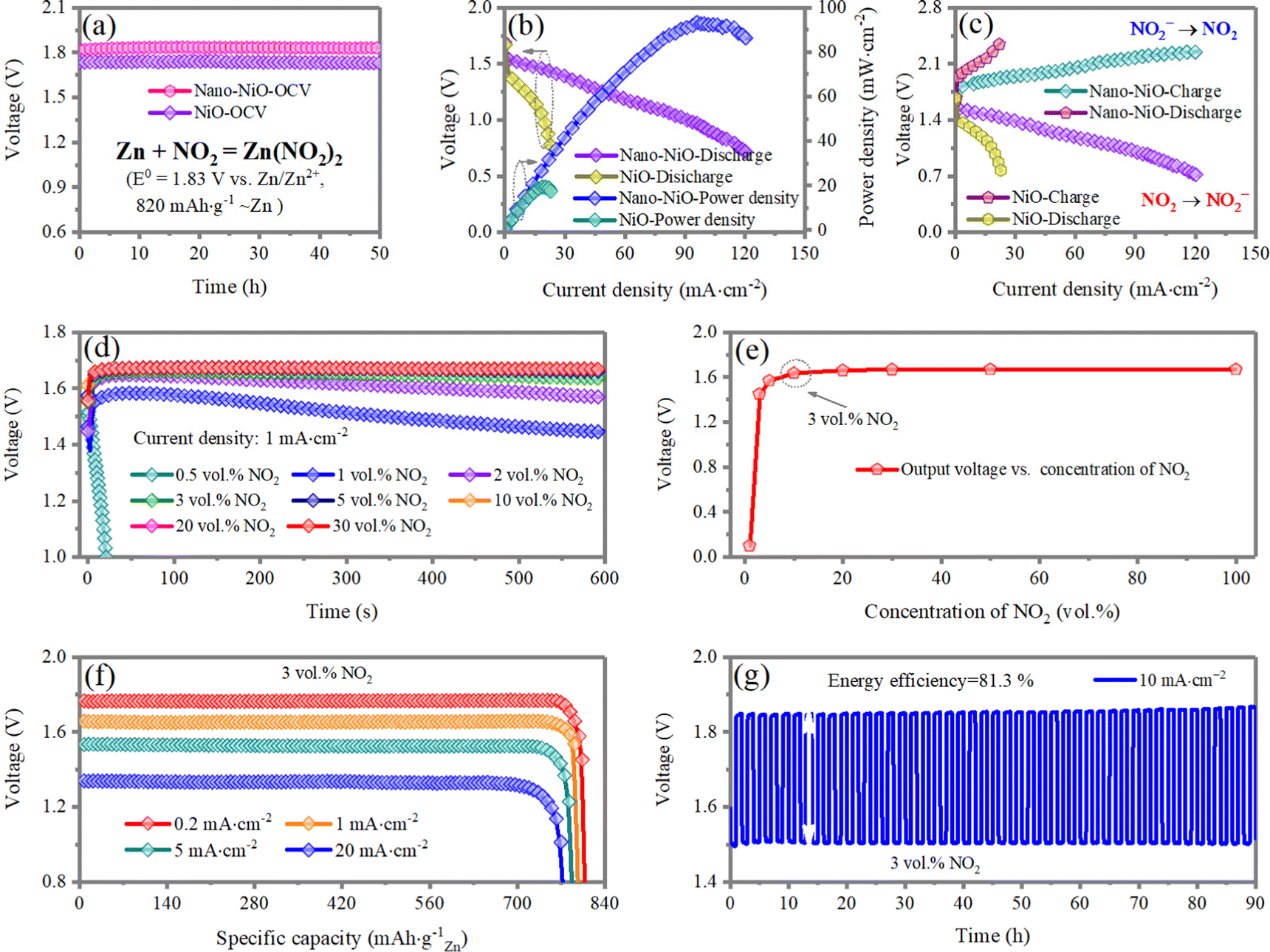 | ||
| Fig. 3 The Zn‖NO2 electrochemical cell with NiO and nano-NiO catalysts. (a) Open circuit voltage over 50 h. (b) The discharged polarization profile and corresponding power density curves at different densities. (c) Charge–discharge polarization profiles at different current densities. (d) Full discharged profiles recorded at different current densities of 0.2, 1, 5, 10 mA cm−2. (e) Cycling performance testing at 2 mA cm−2 using the nano-NiO catalyst. | ||
For the imitation of the practical application of our design, Zn‖NO2 systems employing different concentrations of NO2 gas are studied. It is noted that the mixture of NO2/air is used to simulate the gas contaminated by NO2. As shown in Fig. 3d and e, for a NO2 gas content lower than 3 vol%, the NO2 gas is depleted with the discharge process, while the Zn‖NO2 system can steadily and continuously work with >3 vol% NO2 gas diffusing. Therefore, all subsequent experiments are conducted under the condition of 3 vol% NO2 gas.
The discharge curves of the Zn‖NO2 system at different discharge current densities are shown in Fig. 3f. The battery delivers a high specific capacity of 820 mA h g−1 with an output voltage of 1.79 V at 0.2 mA cm−2. With the current increased up to 20 mA cm−2, the battery can deliver a high capacity of 753 mA h g−1 with 91.8% capacity retention, manifesting an excellent rate capability. The excellent rate capability originates from fast NO2RR kinetics and the high ionic conductivity of the aqueous electrolyte (47.65 mS cm−1) (Fig. S6, ESI†). In order to exclude the participation of air, we investigate the discharge voltage profile of the Zn‖NO2 cell at 1 mA cm−2 with 3 vol% NO2/Ar diffused (Fig. S7, ESI†). It is observed that the electrochemical behavior is almost the same, suggesting that only nitrogen dioxide is involved in the electrochemical reaction. Long-term galvanostatic voltage profiles are collected at an absolute current density of 10 mA cm−2 with a time span of 1 h for each charging and discharging step. In Fig. 3g, there is almost no polarization increase over 90 h and the phase composition of nano-NiO remains unchanged (Fig. S8, ESI†), validating the stable nano-NiO electrocatalyst in the aqueous system and the high reversibility of the gas cathode based on the NO2/NO2− redox reaction. Furthermore, the changes of the pH value are investigated by a home-made operando pH detection configuration (Fig. S9a, ESI†). The pH value near the gas electrode gradually decreases from 3.96 to 3.49 during the discharge process and is followed by a progressive increase to 3.89 at the charged state (Fig. S9b, ESI†), demonstrating a reversible pH change during the discharge/charge process and thus promoting the cyclic stability.
A 553.2 W h kg−1cell, 2.4 Ah-scale pouch-type Zn‖NO2 cell
The Ah-scale Zn‖NO2 cells within a more practical system assembled with a metallic Zn anode are fabricated using a symmetric configuration, wherein the cathodes and electrolytes are positioned on both sides of a metallic Zn anode (Fig. 4a-i and a-ii). The full-cell system can provide an output energy density of 553.2 W h kg−1cell during discharging (calculated based on the mass of total loaded materials on both the electrodes, the electrolyte, and the package, illustrated in the pie chart in Fig. S10, ESI†). The cell-level discharge capacity of 333.3 mA h g−1cell at the current density of 0.2 mA cm−2 is recorded with a nominal cell voltage of 1.78 V using ∼2.4 Ah full cells (Fig. 4b). No obvious potential drop is observed in the galvanostatic discharging at different current densities (0.2 and 5 mA cm−2) until the Zn plate is exhausted or broken, which indicates good stability in the NO2RR test. Under realistic conditions (considering all active and inactive components), the Ah-scale Zn‖NO2 cell achieved ground breaking cell-level specific and volumetric energy densities of 553.2 W h kg−1cell and 1589.6 W h L−1, respectively, based on all active and non-active materials (Fig. S11, ESI†). These are the highest cell energy densities ever reported out of all of commercial Li-ion batteries, Zn-ion batteries and other advanced batteries34–39 (Fig. 4c). Note that the Ah-scale Zn‖NO2 cells demonstrate a cycling stability over 100 h at a current density of 5 mA cm−2 (charge/discharge time of 1 h), and the energy efficiency reaches 80.5%, which is better than all reported Zn–air batteries29–31,33,40,41 (Fig. 4d). The high rechargeability and cyclic stability under practical yet harsh testing conditions indicates exceptional commercialization potential.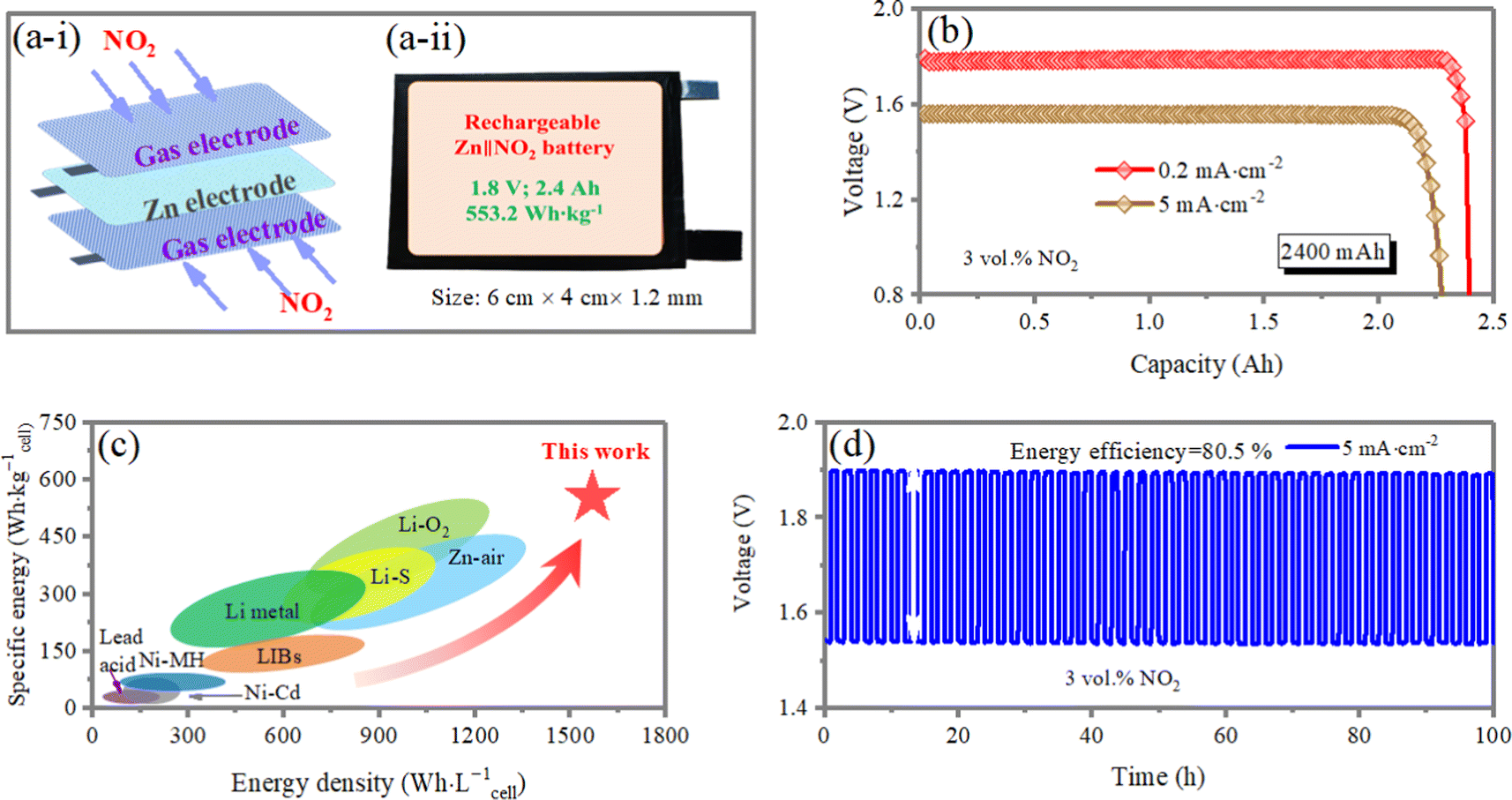 | ||
| Fig. 4 (a-i) Schematic illustration of a rechargeable aqueous Zn‖NO2 pouch cell with a cathode-anode-cathode stack structure. (a-ii) An optical photograph of a rechargeable aqueous Zn‖NO2 pouch cell. (b) The delivered cell capacities of NO2‖Zn‖NO2 cells at different rates of 0.2 and 5 mA cm−2. (c) Ragone plots for projected cell-level specific (W h kg−1(cell)) and volumetric (W h L−1) energy densities with representative commercial and reported batteries. (d) Galvanostatic cyclic charge/discharge performance of a NO2‖Zn‖NO2 cell at a current density of 5 mA cm−2. | ||
Using electrochemical NO2RR to produce value-added chemicals and fuels offers several advantages over traditional chemical engineering synthesis methods, which include operation under mild reaction conditions such as room temperature and ambient pressure, high energy conversion efficiencies, delocalized production and high adaptability and scalability. Another application of the Zn‖NO2 systems is the production of value-added NO2−, which can be further converted to NH3.42,43 Galvanostatic measurements are conducted to electrochemically produce NO2− at different current densities for 1 h (Fig. 5a). The quantity of the produced NO2− is indirectly determined by a chromogenic reaction method coupled with UV-vis absorption, and the standard calibration curve is shown in Fig. S12 (ESI†). The UV-vis spectra of the produced NO2− with different discharge currents are given in Fig. S13a (ESI†), and the corresponding calculated yield of NO2− is displayed in Fig. 5b. The yield of NO2− reaches 3.42 mM cm−2 h−1 and the maximum Faraday efficiency (FE) is up to 96.4%. The yield of NO2− shows an almost linear increase with respect to the discharging current. Similarly, extending the time for NO2− evolution at a current density of 2 mA cm−2, the yield of NO2− also shows a linear increase (Fig. 5c, d and Fig. S13b, ESI†). The above results indicate that the Zn‖NO2 systems can sustain efficient and continuous production of NO2−. The slight reduction of FE can be ascribed to the buildup of NO2− formed on the surface of the cathode, which reduces the number of catalytic active sites for absorbing NO2−. This deficiency can be solved by optimizing the experimental device design, such as adopting a flow cell.
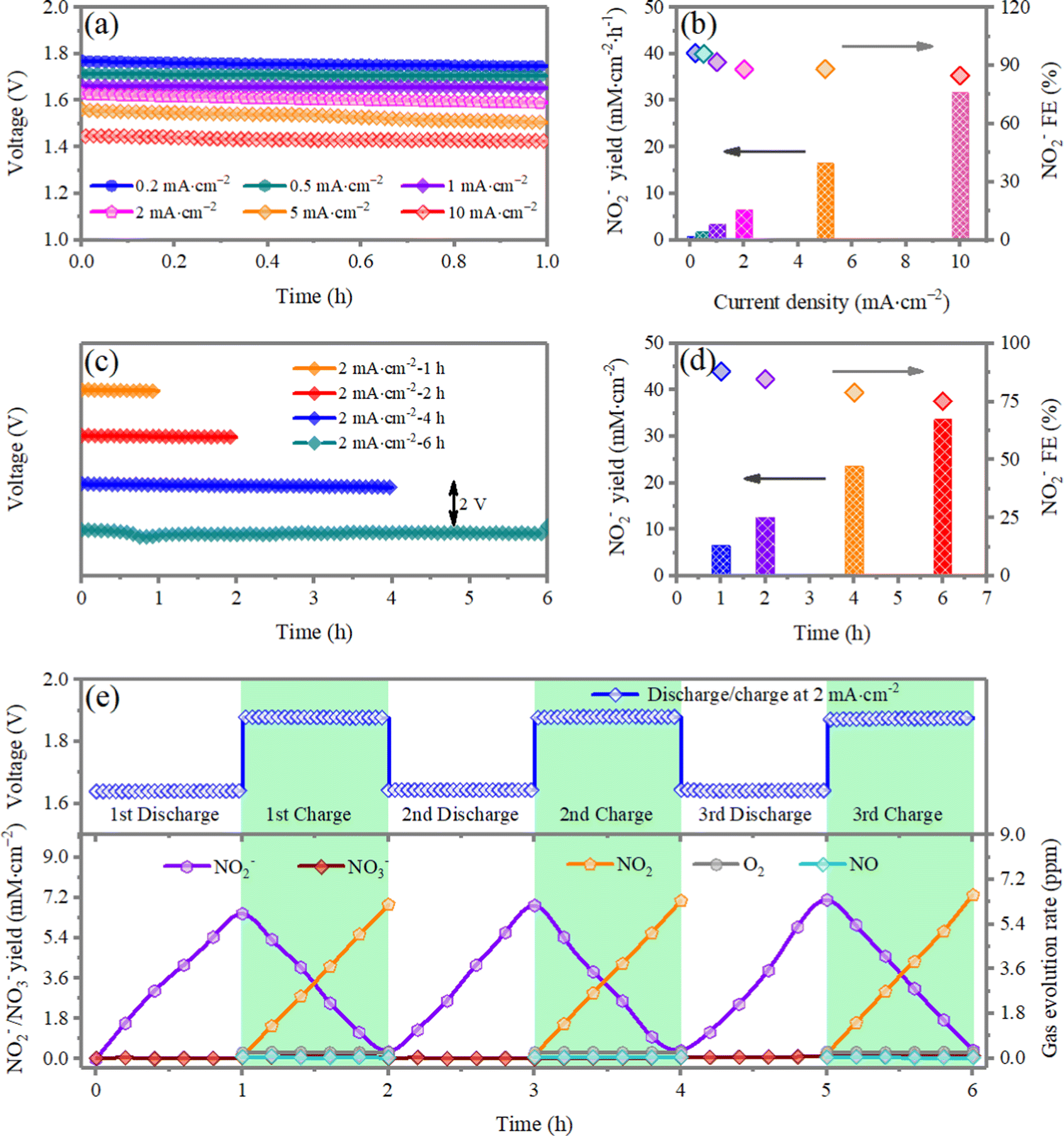 | ||
| Fig. 5 (a) The galvanostatic discharge profile of a Zn‖NO2 electrochemical cell at different current densities of 0.2, 0.5, 1, 2, 5, 10 mA cm−2. The length of the black arrows represents 2 V. (b) Corresponding NO2− species yield rates and faradaic efficiencies (FE). (c) The galvanostatic discharged profile of a Zn‖NO2 electrochemical cell over different discharging times of 1, 2, 4, 6 h at 2 mA cm−2 and (d) corresponding NO2− species yield rates and FE. (e) Discharge–charge curves of first, second and third cycles recorded at 2 mA cm−2 over 2 h for one cycle. (f) Discharged/charged time dependence of the NO2−, NO3−, NO2, NO, O2 yield during cycling. | ||
Characterizations are performed to assess the reversibility of the NO2/NO2− interconversion in a closed environment without oxygen/air, which is assumed for the rechargeable Zn‖NO2 battery system. More accurate quantitative information on both the charge/discharge components and gas evolution have been investigated to further confirm the reaction mechanism and pathway in the Zn‖(3 vol% NO2 in air) system. As shown in Fig. 5e during repeated cycling, the alternated increasing/decreasing and reversible variation trends of NO2 and NO2− clearly indicate the reversible interconversion of NO2 ↔ NO2−. In addition, NO3−, NO and O2 signals cannot be observed, which suggests that there are no NO3−/NO/O2 redox processes during the subsequent cycles. The Gibbs free energy change for the NO2− ↔ *NO2 conversion and the oxygen reduction reaction (ORR) at pH = 5 and 7 on NiO are also compared. As depicted in Fig. S14 (ESI†), the NO2− ↔ *NO2 conversion is more favorable than the ORR at both pH values. Both experimental and theoretical results together validate that, although the mixed NO2/air gas is diffused to the cathode of the Zn‖NO2 electrochemical cell, there is no O2/air involved in the redox reactions during the charge and discharge processes.
Structure evolution of the nano-NiO deposited cathode during the NO2 ↔ NO2− redox process
To unravel the structure evolution of the nano-NiO deposited cathode during the charge/discharge process, ex situ X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and X-ray absorption near-edge spectroscopy (XANES) are performed. As shown in Fig. 6a and b, the discharging process corresponds to Zn0 losing electrons to form Zn2+ ions and the Zn2+ first neutralizes the absorbed NO2− on the surface of the cathode to form Zn(NO2)2. Then Zn(NO2)2 is deposited on the surfaces of the nano-NiO-based gas diffusion electrode. During the charging process, deposited Zn(NO2)2 on the positive electrode is decomposed, which is confirmed by the absence of Zn(NO2)2 in X-ray diffraction (XRD) spectrum.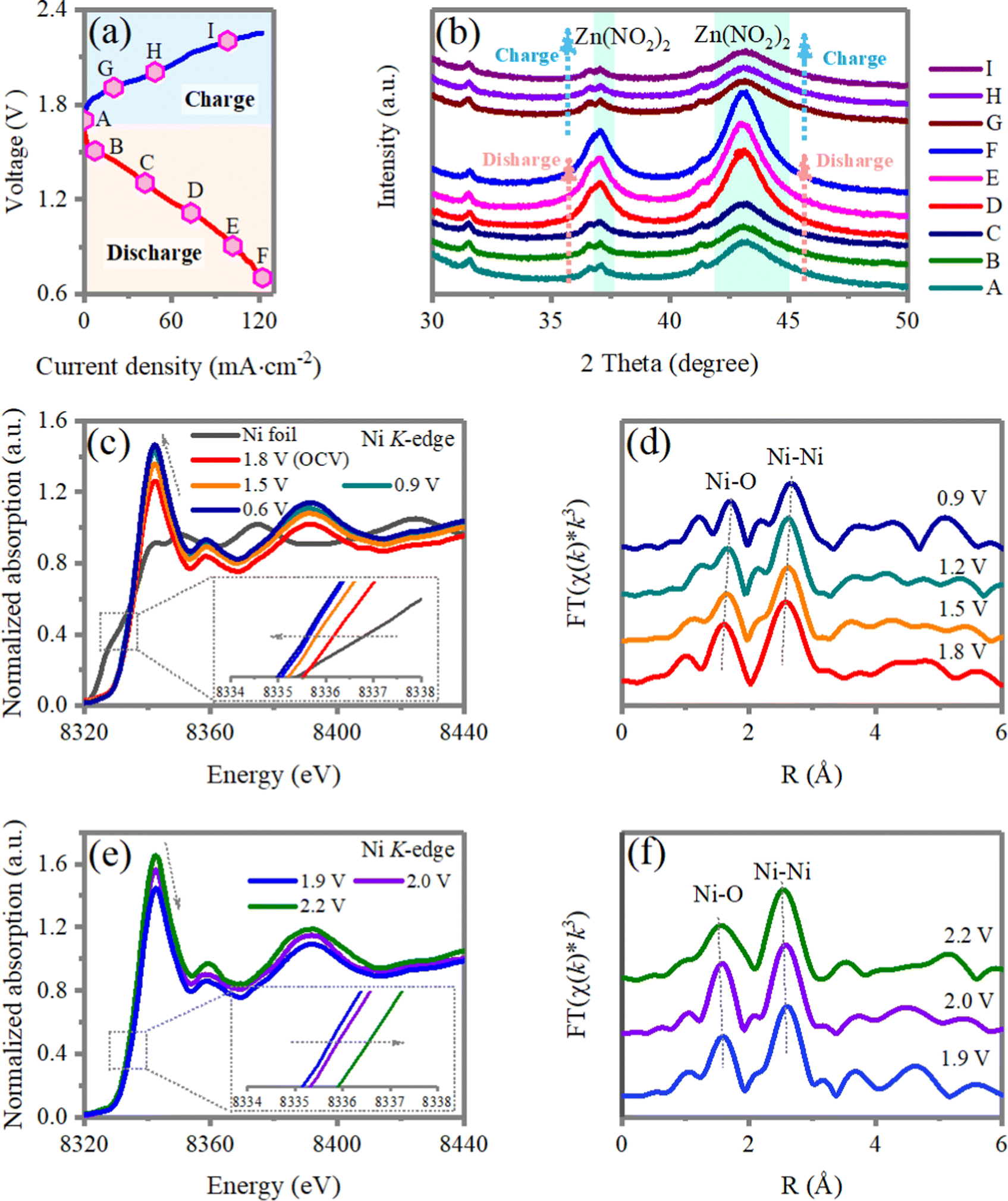 | ||
| Fig. 6 (a) The charge/discharge curves of a Zn‖NO2 electrochemical cell and (b) the corresponding ex situ XRD patterns of the NiO-deposited gas cathode. (c) Ni K-edge XANES spectra and (d) Fourier-transformed EXAFS spectra collected at the Ni K-edge of the nano-NiO composite obtained ex situ at different discharged states. (e) Ni K-edge XANES spectra and (f) Fourier-transformed EXAFS spectra collected at the Ni K-edge of the nano-NiO composite obtained ex situ at different charged states. | ||
Then, the chemistry of the nano-NiO electrocatalysts are studied by ex situ XANES and XPS. Changes in the local electronic and atomic structures of the Ni sites of NiO in the charge/discharge electrocatalytic cycle are monitored by X-ray absorption spectroscopy, including X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) analysis. Fig. 6c–f summarizes the Ni K-edge X-ray absorption spectra during electrochemical cycling, including the discharging process from 1.8 to 0.6 V and charging processes from 1.8 to 2.2 V, respectively. With the discharging process (1.8–0.6 V), H2O is disassociated to Had and OHad by breaking the H–OH bond and the absorbed Had will combine with NO2ad to form HNO2. The Ni K-edge XANES peaks gradually shift to a lower energy compared with the sample at an open-circuit voltage of 1.8 V, demonstrating a gradually decreased average valence state in NiO44,45 (Fig. 6c). The results are in line with the XPS results that the Ni 2p3/2 peaks of NiO after the discharging reaction shift to lower energy, compared with that of pristine NiO (Fig. S15, ESI†). Correspondingly, the Fourier transform of the extended X-ray absorption fine structure (FT-EXAFS) of the interatomic Ni–O distance is enlarged from 1.59 Å to 1.69 Å at an OCV of 0.9 V (Fig. 6d), implying the reduction of the Ni ion with the discharging process. While during the charging process, the H-NO2− is broken and dissociated to Had and NO2ad on the NiO surface. The disassociated Had will occupy the exposed O site of NiO. The Ni K-edge XANES peaks reversibly shift back to higher energy with the charging process (1.9–2.2 V), indicating an increase in the Ni oxidation state46,47 (Fig. 6e). Accordingly, the interatomic Ni–O distance is reduced from 1.69 Å at 1.9 to 1.59 Å at 2.2 V (Fig. 3f). The above results support the reversible transitions of the Ni K-edge, manifesting symmetric Ni2+/x+ (1 < x < 2) redox reactions.
Self-powered Haber–Bosch reactor for NH3 production
Electrochemical nitrate reduction from NO2− into NH3 is promising for the future landscape of NH3 electrosynthesis based on the following six-electron reaction:| NO2− + 7H+ + 6e− → NH3 + 2H2O | (4) |
On the other hand, water electrolysis is a modular alternative H+ source by using a commercially available water splitting setup, based on the following four-electron reaction:
| 2H2O − 4e− → O2 + 4H+ | (5) |
Based on the above reactions of nitrate reduction and water oxidation, we couple the two reactions into an electrochemical Haber–Bosch (eHB) reactor, which produces NH3 from NO2− and H+ at ambient conditions in an overall reaction involving only NO2, H2O, and renewable electrons. For improving the electrocatalytic kinetics, an effective, cheap and low-toxicity catalyst of commercial nano-TiO2 (P25) is employed in the electrocatalytic system for NH3 synthesis. A schematic diagram and an optical photo are depicted in Fig. 7a and Fig. 7b, respectively. The eHB with a dual-compartment electrocatalytic cell, consists of a graphite bipolar plate, a Nafion membrane (NRE-211) and a TiO2/CFC electrode. The eHB reactor can be powered by two Zn‖NO2 cells connected in series to drive the subsequent electrocatalytic reduction for NH3 synthesis. Note that the generated NO2− from the Zn‖NO2 cells can directly flow into the eHB reactor for further NH3 production. Therefore, as long as we keep providing the electrolyte and NO2 gas, the self-powered eHB can keep producing NH3.
 | ||
| Fig. 7 (a) A schematic of two Zn‖NO2 electrochemical cell powered electrochemical Haber–Bosch reactor coupled to a water splitting reactor. (b) The corresponding photograph depicting the model. (c) The voltage and current of the electrochemical Haber–Bosch reactor. (d) The NH3 H2O yield rates and FE. | ||
Fig. 7c shows the voltage and output current of the electrocatalytic cell. With the duration extended, the voltage maintains 3.0 V unchanged and the output current is about 2.1 mA cm−2 over 8 h, demonstrating continuous and stable electrocatalytic reactions for NH3 synthesis. After 1 h of reaction, NH3 is successfully synthesized by the self-powered system. The determination results calculated according to UV-vis absorption (Fig. S16, S17, ESI†), are shown in Fig. 7d. By measuring the solution volume involved in the cathode reaction, the self-powered NH3 yield per hour achieved 4 mM h−1. Therefore, it is feasible to reutilize the residual kinetic energy of the exhaust gas to drive the valuable NH3 synthesis.
Conclusion
This work presents a high-energy rechargeable aqueous Zn‖NO2 electrochemical cell, which demonstrates a new approach for converting NO2 from exhaust gas streams to value-added products while producing large amounts of electrical energy. The aqueous rechargeable Zn‖NO2 system has a 1.8 V discharge voltage based on reversible NO2− ↔ NO2 conversion, and the energy efficiency reaches 81.3% with 3 vol% NO2/air diffused at 10 mA cm−2. The assembled pouch-type 2.4 Ah-scale battery can deliver a high energy density of 553.2 W h kg−1cell and a high volumetric density of 1589.6 W h L−1cell. The system is capable of utilizing NO2 exhaust gas for ammonia synthesis, which is energy-saving and eco-friendly. The yield of the NO2− ions reaches 3.42 mM cm−2 h−1 and the faradaic efficiency is about 96.4%. The produced NO2− can be further converted to NH3 by a self-powered conversion system that consists of two Zn‖NO2 cells connected in series and an electrochemical Haber–Bosch reactor. This work opens a new avenue for NO2 capture/conversion via reversible aqueous metal‖NO2 systems and eco-friendly conversion/storage devices.Author contributions
L. Ma, S. Chen, W. Yan and G. Zhang contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Key R&D Program of China under Project 2019YFA0705104 and the GRF under the project number CityU 11305218. This work was also partially supported by the Fundamental Research Funds for the Central Universities (0515022GH0202253 and 0515022SH0201253), the National Natural Science Foundation of China (52202299) and the Analytical &Testing Center of Northwestern Polytechnical University (2022T006).References
- R. C. Armstrong, C. Wolfram, K. P. de Jong, R. Gross, N. S. Lewis, B. Boardman, A. J. Ragauskas, K. Ehrhardt-Martinez, G. Crabtree and M. V. Ramana, Nat. Energy, 2016, 1(1), 15020 CrossRef.
- M. I. Hoffert, Science, 2010, 329(5997), 1292 CrossRef CAS PubMed.
- R. York, Nat. Clim. Change, 2012, 2(6), 441 CrossRef.
- D. H. Wall, U. N. Nielsen and J. Six, Nature, 2015, 528(7580), 69 CrossRef CAS PubMed.
- X. Zhang, E. A. Davidson, D. L. Mauzerall, T. D. Searchinger, P. Dumas and Y. Shen, Nature, 2015, 528(7580), 51 CrossRef CAS PubMed.
- S. Shiva, X. Wang, L. A. Ringwood, X. Xu, S. Yuditskaya, V. Annavajjhala, H. Miyajima, N. Hogg, Z. L. Harris and M. T. Gladwin, Nat. Chem. Biol., 2006, 2(9), 486 CrossRef CAS PubMed.
- Z. Yang, B. Wang, Y. Chen, W. Zhou, H. Li, R. Zhao, X. Li, T. Zhang, F. Bu, Z. Zhao, W. Li, D. Chao and D. Zhao, Nat. Sci. Rev., 2022, nwac268 CrossRef.
- J. Liu, W. Zhou, R. Zhao, Z. Yang, W. Li, D. Chao, S.-Z. Qiao and D. Zhao, J. Am. Chem. Soc., 2021, 143(38), 15475 CrossRef CAS PubMed.
- P. M. Edwards, S. S. Brown, J. M. Roberts, R. Ahmadov, R. M. Banta, J. A. deGouw, W. P. Dubé, R. A. Field, J. H. Flynn, J. B. Gilman, M. Graus, D. Helmig, A. Koss, A. O. Langford, B. L. Lefer, B. M. Lerner, R. Li, S. M. Li, S. A. McKeen, S. M. Murphy, D. D. Parrish, C. J. Senff, J. Soltis, J. Stutz, C. Sweeney, C. R. Thompson, M. K. Trainer, C. Tsai, P. R. Veres, R. A. Washenfelder, C. Warneke, R. J. Wild, C. J. Young, B. Yuan and R. Zamora, Nature, 2014, 514(7522), 351 CrossRef CAS PubMed.
- J. O. Lundberg, E. Weitzberg and M. T. Gladwin, Nat. Rev. Drug Discovery, 2008, 7(2), 156 CrossRef CAS PubMed.
- C. H. Christensen, T. Johannessen, R. Z. Sørensen and J. K. Nørskov, Catal. Today, 2006, 111(1), 140 CrossRef CAS.
- R. Lan, J. T. S. Irvine and S. Tao, Int. J. Hydrog. Energy, 2012, 37(2), 1482 CrossRef CAS.
- D. Qi, F. Lv, T. Wei, M. Jin, G. Meng, S. Zhang, Q. Liu, W. Liu, D. Ma, M. S. Hamdy, J. Luo and X. Liu, Nano Res. Energy, 2022, 1, e9120022 CrossRef.
- W. I. A. Sadat and L. A. Archer, Sci. Adv., 2016, 2(7), e1600968 CrossRef PubMed.
- H. Park, H. D. Lim, H. K. Lim, W. M. Seong, S. Moon, Y. Ko, B. Lee, Y. Bae, H. Kim and K. Kang, Nat. Commun., 2017, 8(1), 14989 CrossRef PubMed.
- J. Li, L. Wang, Y. Zhao, S. Li, X. Fu, B. Wang and H. Peng, Adv. Funct. Mater., 2020, 30(27), 2001619 CrossRef CAS.
- J. Xie, X. Wang, J. Lv, Y. Huang, M. Wu, Y. Wang and J. Yao, Angew. Chem., Int. Ed., 2018, 57(52), 16996 CrossRef CAS PubMed.
- X. Wang, J. Xie, M. A. Ghausi, J. Lv, Y. Huang, M. Wu, Y. Wang and J. Yao, Adv. Mater., 2019, 31(17), 1807807 CrossRef PubMed.
- T. Ahmad, S. Liu, M. Sajid, K. Li, M. Ali, L. Liu and W. Chen, Nano Res. Energy, 2022, 1, e9120021 CrossRef.
- J. Liang, C. Zhang, D. Liu, T. Wu, Y. Tao, G.-W. Ling, H.-J. Yu, J. Lu and Q.-H. Yang, Room-temperature reduction of NO2 in a Li-NO2 battery: a proof of concept, Sci. Bull., 2020, 65(1), 55 CrossRef CAS PubMed.
- S. W. Bae, S. A. Roh and S. D. Kim, Chemosphere, 2006, 65(1), 170 CrossRef CAS PubMed.
- D. K. Pappas, T. Boningari, P. Boolchand and P. G. Smirniotis, J. Catal., 2016, 334, 1 CrossRef CAS.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17(6), 543 CrossRef CAS PubMed.
- J. Zheng, Q. Zhao, T. Tang, J. Yin, C. D. Quilty, G. D. Renderos, X. Liu, Y. Deng, L. Wang, D. C. Bock, C. Jaye, D. Zhang, E. S. Takeuchi, K. J. Takeuchi, A. C. Marschilok and L. A. Archer, Science, 2019, 366(6465), 645 CrossRef CAS PubMed.
- L. Ma, Q. Li, Y. Ying, F. Ma, S. Chen, Y. Li, H. Huang and C. Zhi, Adv. Mater., 2021, 33(12), 2007406 CrossRef CAS PubMed.
- H. Li, C. Guo, T. Zhang, P. Xue, R. Zhao, W. Zhou, W. Li, A. Elzatahry, D. Zhao and D. Chao, Nano Lett., 2022, 22(10), 4223 CrossRef CAS PubMed.
- X. Wang, X. Li, H. Fan and L. Ma, Nano-Micro Lett., 2022, 14(1), 205 CrossRef CAS PubMed.
- X. Li, X. Wang, L. Ma and W. Huang, Adv. Energy Mater., 2022, 12(37), 2202068 CrossRef CAS.
- L. Ma, S. Chen, Z. Pei, Y. Huang, G. Liang, F. Mo, Q. Yang, J. Su, Y. Gao, J. A. Zapien and C. Zhi, ACS Nano, 2018, 12(2), 1949 CrossRef CAS PubMed.
- S. Chen, L. Ma, S. Wu, S. Wang, Z. Li, A. A. Emmanuel, M. R. Huqe, C. Zhi and J. A. Zapien, Adv. Funct. Mater., 2020, 30(10), 1908945 CrossRef CAS.
- W. Sun, F. Wang, B. Zhang, M. Zhang, V. Küpers, X. Ji, C. Theile, P. Bieker, K. Xu, C. Wang and M. Winter, Science, 2021, 371(6524), 46 CrossRef CAS PubMed.
- J. Pan, Y. Y. Xu, H. Yang, Z. Dong, H. Liu and B. Y. Xia, Adv. Sci., 2018, 5(4), 1700691 CrossRef PubMed.
- S. S. Shinde, J. Y. Jung, N. K. Wagh, C. H. Lee, D. H. Kim, S. H. Kim, S. U. Lee and J. H. Lee, Nat. Energy, 2021, 6(6), 592 CrossRef CAS.
- J. H. Kim, Y. H. Lee, S. J. Cho, J. G. Gwon, H. J. Cho, M. Jang, S. Y. Lee and S. Y. Lee, Energy Environ. Sci., 2019, 12(1), 177 RSC.
- M. Ue, K. Sakaushi and K. Uosaki, Mater. Horiz., 2020, 7(8), 1937 RSC.
- L. Ma, N. Li, C. Long, B. Dong, D. Fang, Z. Liu, Y. Zhao, X. Li, J. Fan, S. Chen, S. Zhang and C. Zhi, Adv. Funct. Mater., 2019, 29(46), 1906142 CrossRef CAS.
- E. Cha, M. Patel, S. Bhoyate, V. Prasad and W. Choi, Nanoscale Horiz., 2020, 5(5), 808–831 RSC.
- X. Yuan, F. Ma, L. Zuo, J. Wang, N. Yu, Y. Chen, Y. Zhu, Q. Huang, R. Holze, Y. Wu and T. van Ree, Electrochem. Energy Rev., 2021, 4(1), 1 CrossRef CAS.
- S. Dühnen, J. Betz, M. Kolek, R. Schmuch, M. Winter and T. Placke, Small Methods, 2020, 4(7), 2000039 CrossRef.
- L. Ma, S. Chen, D. Wang, Q. Yang, F. Mo, G. Liang, N. Li, H. Zhang, J. A. Zapien and C. Zhi, Adv. Energy Mater., 2019, 9(12), 1803046 CrossRef.
- F. Meng, H. Zhong, D. Bao, J. Yan and X. Zhang, J. Am. Chem. Soc., 2016, 138(32), 10226 CrossRef CAS PubMed.
- K. Han, J. Luo, Y. Feng, L. Xu, W. Tang and Z. L. Wang, Energy Environ. Sci., 2020, 13(8), 2450 RSC.
- G. F. Chen, Y. Yuan, H. Jiang, S. Y. Ren, L. X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5(8), 605 CrossRef CAS.
- J. Jiang, F. Sun, S. Zhou, W. Hu, H. Zhang, J. Dong, Z. Jiang, J. Zhao, J. Li, W. Yan and M. Wang, Nat. Commun., 2018, 9(1), 2885 CrossRef PubMed.
- S. Zhao, C. Tan, C. T. He, P. An, F. Xie, S. Jiang, Y. Zhu, K. H. Wu, B. Zhang, H. Li, J. Zhang, Y. Chen, S. Liu, J. Dong and Z. Tang, Nat. Energy, 2020, 5(11), 881 CrossRef CAS.
- G. F. Chen, T. Y. Ma, Z. Q. Liu, N. Li, Y. Z. Su, K. Davey and S. Z. Qiao, Adv. Funct. Mater., 2016, 26(19), 3314 CrossRef CAS.
- H. Doan, I. Kendrick, R. Blanchard, Q. Jia, E. Knecht, A. Freeman, T. Jankins, M. K. Bates and S. Mukerjee, J. Electrochem. Soc., 2021, 168(8), 084501 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee03749a |
| This journal is © The Royal Society of Chemistry 2023 |