
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effective electronic tuning of Pt single atoms via heterogeneous atomic coordination of (Co,Ni)(OH)2 for efficient hydrogen evolution†
An
Pei‡
a,
Ruikuan
Xie‡
 c,
Yun
Zhang‡
a,
Yingliang
Feng‡
a,
Weizhen
Wang
d,
Sifan
Zhang
a,
Zinan
Huang
e,
Lihua
Zhu
*ab,
Guoliang
Chai
*cg,
Zhiqing
Yang
*d,
Qingsheng
Gao
e,
Hengqiang
Ye
d,
Congxiao
Shang
b,
Bing Hui
Chen
f and
Zhengxiao
Guo
*b
c,
Yun
Zhang‡
a,
Yingliang
Feng‡
a,
Weizhen
Wang
d,
Sifan
Zhang
a,
Zinan
Huang
e,
Lihua
Zhu
*ab,
Guoliang
Chai
*cg,
Zhiqing
Yang
*d,
Qingsheng
Gao
e,
Hengqiang
Ye
d,
Congxiao
Shang
b,
Bing Hui
Chen
f and
Zhengxiao
Guo
*b
aJiangxi Provincial Key Laboratory of Functional Molecular Materials Chemistry, College of Chemistry and Chemical Engineering, Faculty of Materials Metallurgy and Chemistry, Jiangxi University of Science and Technology, Ganzhou 341000, Jiang Xi, China. E-mail: zhulihua@jxust.edu.cn
bHKU-CAS Joint Laboratory on New Materials and Department of Chemistry, The University of Hong Kong, Hong Kong SAR, China. E-mail: zxguo@hku.hk
cState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences (CAS), Fuzhou, 350002 Fujian, China. E-mail: g.chai@fjirsm.ac.cn
dJi Hua Laboratory, Foshan 528200, China. E-mail: yangzhq@jihualab.ac.cn
eCollege of Chemistry and Materials Science, and Guangdong Provincial Key Laboratory of Functional Supramolecular Coordination Materials and Applications, Jinan University, Guangzhou 510632, China
fCollege of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
gFujian Science & Technology Innovation Laboratory for Optoelectronic Information of China Fuzhou, Fujian 350108, P. R. China
First published on 21st December 2022
Abstract
Hydroxide-supported atomic structures, particularly single atoms, offer a wide scope for active microenvironmental tuning to enhance the catalytic performance, but little has been explored on the electronic synergy between mono- and dual-hydroxides. Here, we propose a way of constructing a Pt1/(Co,Ni)(OH)2/C single-atom catalyst (SAC), with Pt single-atoms, Pt1, stabilized and anchored on the surface of defective (Co,Ni)(OH)2, which is further supported on carbon black. This catalyst exhibits a far superior hydrogen evolution reaction (HER) activity to Pt1/Co(OH)2/C, Pt1/Ni(OH)2/C, Pt1/C, and the commercial 20 wt% Pt/C. Particularly, it shows an almost zero onset overpotential and an outstanding electrocatalytic mass activity for the HER, 29.7 times higher than that of Pt1/C and 115.9 times higher than that of the commercial 20 wt% Pt/C at −0.09 V vs. RHE, respectively. There is negligible attenuation after the chronopotentiometry test at 100 mA cm−2 for 24 h and cyclic voltammetry for 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. Operando Raman spectra clarified that the Volmer step for water decomposition (the H–OH bond breaking) takes place around the defective sites of (Co,Ni)(OH)2. Density functional theory (DFT) calculations confirmed the electronic synergy between the Pt single atoms and bimetallic (Co,Ni)(OH)2, which leads to stable anchoring of Pt and yields an appropriate adsorption energy of *H, leading to rapid H2 generation.
000 cycles. Operando Raman spectra clarified that the Volmer step for water decomposition (the H–OH bond breaking) takes place around the defective sites of (Co,Ni)(OH)2. Density functional theory (DFT) calculations confirmed the electronic synergy between the Pt single atoms and bimetallic (Co,Ni)(OH)2, which leads to stable anchoring of Pt and yields an appropriate adsorption energy of *H, leading to rapid H2 generation.
Broad contextCost-effective electrocatalysts for hydrogen generation, or the hydrogen evolution reaction (HER), from water are crucial for the development of renewable energy technologies to resolve the current energy and environmental crisis. To date, platinum (Pt) is still the most active candidate due to its moderate chemisorption free energy of atomic hydrogen (ΔGH), fitting for the formation and release of H2via the intermediate Pt–*H (*denotes “adsorbed”). However, the HER efficiency depends not only on ΔGH but also on the dissociation energy of water to obtain *H and OH−. The first step towards the dissociation of water on Pt to form Pt–*H is inefficient due to the relatively high water-dissociation barrier. Single atom catalysts (SACs) embedded in a hydrophilic substrate offer the potential to regulate the electronic “micro-environment”, so as to enhance the water dissociation step. Moreover, SACs are also of low cost and have ultra-high atomic efficiency towards diverse electrocatalysis reactions. However, little has been explored on the electronic synergy between mono- and dual-hydroxides. We present a facile strategy for constructing a Pt1/(Co,Ni)(OH)2/C single-atom catalyst (SAC), with Pt single-atoms (Pt1) stabilized and anchored on the surface of (Co,Ni)(OH)2. This catalyst exhibits a far superior hydrogen evolution reaction (HER) activity to Pt1/Co(OH)2/C, Pt1/Ni(OH)2/C, Pt1/C, and the commercial 20 wt% Pt/C, particularly, with an almost zero onset overpotential and an outstanding electrocatalytic mass activity for the HER, 29.7 times higher than that of Pt1/C and 115.9 times higher than that of the commercial 20 wt% Pt/C at −0.09 V vs. RHE, respectively. There is also negligible attenuation after the chronopotentiometry test at 100 mA cm−2 for 24 h and cyclic voltammetry for 20000 cycles. Operando Raman spectra clarified that the Volmer step for water decomposition (the H–OH bond breaking) takes place around (Co,Ni)(OH)2. Density functional theory (DFT) calculations confirmed the electronic synergy between the Pt single atoms and bimetallic (Co,Ni)(OH)2, which leads to stable anchoring of Pt and yields an appropriate adsorption energy of *H, leading to rapid H2 generation. This study opens up a new avenue for developing SACs via effective electronic tuning of Pt single atoms with heterogeneous atomic coordination in hydroxide substrates.
|
1. Introduction
Sustainable clean energy supply is one of the foremost challenges to achieve “net-zero” by 2050.1,2 Hydrogen is a promising clean energy carrier for such purposes.3,4 However, its conventional production by methane reforming is accompanied by the generation of CO2 and also insufficient purity for proton-exchange-membrane fuel cells.5,6 Using renewable power sources, such as solar or wind energy, is environmentally friendly and economical for electrochemical water splitting to produce high-purity H2, which has become increasingly important as the demand for hydrogen is likely to increase by 6–8 fold by 2050.7–10 This will create even more pressure on the already scarce Pt catalyst and its reserves.11,12 Moreover, conventional cathodes show a relatively large overpotential and hence low efficiency (∼60–80%) for H2 evolution, especially under alkaline conditions.11–13 The HER activity is proportional to the coverage of relatively weakly adsorbed H species on a metal (e.g., Pt) catalyst, where a “volcano relationship” exists between the exchange current density and the H-metal bond energy in the HER. The coverage of the adsorbed H species in an alkaline solution is much lower than that in an acidic solution, resulting in the HER efficiency being about two orders of magnitude lower in the former case14,15 On the other hand, the HER under alkaline conditions offers much higher durability and lower dissolution of the cathode catalyst. Thus, it is very desirable but challenging to develop a catalytic cathode through which hydrogen evolution can take place efficiently at a relatively low overpotential under alkaline conditions. Recently, considerable effort has been devoted to various candidate materials, such as transition metal (TM)-based HER catalysts (including Mo,16 Fe,17 Mn,18 Co,19–21 and Ni22–24 catalysts), supported on TM oxides,25 hydroxides,26 phosphides,27 sulfides,28 selenides,29 carbides30 or nitrides.31 However, these non-Pt based catalysts usually show a relatively high overpotential and inferior performance to the commercial 20 wt% Pt/C and are thus unfavorable for industrial applications.32To date, Pt is still the most active candidate due to its moderate free energy for the chemisorption of atomic hydrogen, which is more advantageous than other metals for the formation of H2via the intermediate Pt–*H (* denotes “adsorbed”).33 Unfortunately, the HER efficiency depends not only on ΔGH but also on the dissociation energy of water to obtain *H and *OH. The first step of water dissociation on Pt to form Pt–*H is inefficient due to its sluggish kinetics under alkaline conditions.34 Compared to the Pt nanoparticles and clusters, Pt single-atom catalysts (SACs) show readily regulatable electronic properties and high atom utilization efficiency for various reactions.35–38 In this regard, substrate supports, such as TM oxides,39 hydroxides,40,41 MoS2,42 WOX,43 and MXenes,44 have been recently applied to anchor isolated Pt atoms to enhance the HER performance. Among these, TM hydroxides (e.g., TM = Ni and Co) are characterized by low-cost, high specific surface area, good alkaline stability, and distinctive electronic structures with partially-filled d orbitals to facilitate interactions with charge carriers. Compared with TM oxides, TM hydroxides generally show superior hydrophilicity and optimum binding of *OH to avoid the poisoning of the catalyst during the HER.45–47 On the other hand, the pristine TM hydroxides usually exhibit low efficiency for the HER due to their relatively high hydrogen adsorption energies, typically around 1.71 eV.46 Furthermore, Subbaraman and Tripkovic26 identified that the hydrogen evolution efficiency of 3d-TM hydr(oxy)oxides depends on their affinity for *OH. A strong affinity will lead to *OH accumulation, resulting in poisoning of the active sites and inhibiting subsequent adsorption of H2O. It is found that nickel hydroxide possesses the weakest affinity for *OH, and the 3d-TM (Co, Ni, Fe, and Mn) hydr(oxy)oxides for water dissociation are associated with HO–M2+δ bond strength (0 ≤ δ ≤ 1.5), and the reactivity varies in the order of (Mn < Fe < Co < Ni). In addition, Yu et al.47 demonstrated that β-Ni(OH)2/Pt improves the HER activity compared to α-Ni(OH)2/Pt, due to a larger interlayer spacing which facilitates water adsorption and optimizes hydrogen binding energy with the Pt modified by the electronic effect of β-Ni(OH)2.47 Thus, designing a heterogeneous catalyst with Pt SACs and TM hydroxides is a viable strategy to regulate water dissociation barriers and H2 adsorption energies.
Subbaraman's group first demonstrated the synergistic effect of the edges of the Li+-intercalated Ni(OH)2 clusters that expedited the H–OH bond breaking and the embedded Pt species facilitated the formation of H–H bonds.48 Since then various noble metal/metal hydroxide systems, e.g., Pt NWs/SL/Ni(OH)2,49 Pt/Co(OH)2,50 and Ag/Ni(OH)2,51 have been studied for the alkaline HER. Compared with pure Ni hydroxide, bimetallic hydroxides usually exhibit improved water decomposition due to the existence of electronic synergy to tune the hydrogen binding energy.45,52 For instance, Chen et al. reported the synthesis of Pt single atoms intercalated in Ni3Fe layered double hydroxide (LDH), the interlayer Pt single-atoms greatly improved the electron transfer capability of the Ni3Fe LDH substrate, and the Ni3Fe LDH accelerated the dissociation of water in 1.0 M KOH.45 In addition, the enhanced synergistic effect of Ru/(Fe,Ni)(OH)2 for the HER has also been proposed, where the (Fe,Ni)(OH)2 plays a key role in water dissociation and Ru nanoparticles significantly promoted the formation of H2 molecules.52 Notably, Co is adjacent to Fe and Ni, and all of them show two 4 s electrons in their outermost shell resulting in similar properties. The major difference is the different number of half-filled sub-orbitals (HFOs) on the outermost iso-surface: Co shows three HFOs on the 3d-orbital, while Fe has four and Ni has two HFOs.53 Theoretically, it is easier to tune the electronic structure of Ni with Co than with Fe due to its smaller difference from Ni, and the CoNi double hydroxides were reported to be outstanding for the breakage of the HO–H bonds.46 In addition, these hybrids can take advantage of the high catalytic activities of noble metals and favourable water dissociation ability of metal hydroxides.54 However, the intrinsic functional difference between bimetallic hydroxide and monohydroxide has not been well studied and the bifunctional synergistic catalysis is still unclear. In our recent study, a finely-tuned electronic microenvironment of Pt single atoms and clusters co-located over a (Ni,Co)(OH)2 substrate effectively reduced the energy barrier for hydrogen transfer on (Ni,Co)(OH)2 to enhance hydrogenation reactions.55–58
With the above, we resorted to engineering Pt single atoms and their local environments on defective (Co,Ni)(OH)2, Co(OH)2, and Ni(OH)2 by a facile electrodeposition strategy (see the Methods section for details in the ESI†). The optimized Pt1/(Co,Ni)(OH)2/C nanostructure, with Pt single atoms embedded in [(Co,Ni)(OH)2] nanoparticles (termed Pt1/(Co,Ni)(OH)2) delivered exceptionally high catalytic activity, even with only 1.41 wt% Pt loading. The Pt1/(Co,Ni)(OH)2/C nanostructure exhibited a far superior HER activity (24 mV@10 mA cm−2 with a Tafel slope of 28.7 mV dec−1) to Pt1/Co(OH)2/C (50 mV@10 mA cm−2, 48.7 mV dec−1), Pt1/Ni(OH)2/C (55 mV@10 mA cm−2, 52.1 mV dec−1) and the commercial 20 wt% Pt/C (29 mV@10 mA cm−2, 32.5 mV dec−1) in 1.0 M KOH solution. In particular, Pt1/(Co,Ni)(OH)2/C showed an ultralow overpotential and an outstanding electrocatalytic mass activity for the HER, which was 29.7 times higher than that of Pt1/C and 115.9 times higher than that of the commercial 20 wt% Pt/C at −0.09 V vs. RHE, respectively. More importantly, there was negligible attenuation of Pt1/(Co,Ni)(OH)2/C after chronopotentiometry at a large current density of 100 mA cm−2 for 24 h and cyclic voltammetry for 20000 cycles. The generation of Ni–*OH and Co–*OH was directly observed by operando Raman spectroscopy, indicating the enhancement of (Co,Ni)(OH)2 for water dissociation in an alkaline electrolyte. Density functional theory (DFT) calculations indicated the charge transfer from Pt to O atoms of (Co,Ni)(OH)2, suggesting that the local coordination environment of Pt was altered by (Co,Ni)(OH)2. The adsorbed H2O (*H2O) splits into one *OH staying in the OH defect and one *H on the Pt atom with a downhill free energy of −0.074 eV, which efficiently promoted the H2O dissociation step. In addition, Pt1/(Co,Ni)(OH)2 showed a more appropriate hydrogen adsorption energy (0.145 eV) than pure Pt (−0.260 eV). Therefore, both H2O dissociation (Volmer step) and H2 formation steps (Tafel step) were promoted, making them better HER catalysts than pure Pt and (Co,Ni)(OH)2. However, in contrast with Pt1/(Co,Ni)(OH)2 and Pt1/Ni(OH)2, the water dissociation on Pt1/Co(OH)2 was still endothermic although it was improved over Pt(111) and (Co,Ni)(OH)2. While for Pt1/Ni(OH)2, the adsorption of *H was stronger than on Pt1/(Co,Ni)(OH)2 and Pt1/Co(OH)2, which inhibited the formation of H2. Hence, the Pt1/(Co,Ni)(OH)2 catalyst demonstrated great potential to overcome the bottleneck of low efficiency for hydrogen evolution under alkaline conditions.
2. Results and discussion
The fabrication of single-atom catalysts by electrochemical deposition is facile and universal, and classical cyclic voltammetry (CV) electrochemical deposition helps adjust the load and particle size by controlling the scan number.59 In contrast, the potentiostat deposition was dependent on the deposition time and deposition potential, to a certain extent: the more negative potential results in faster nucleation and easier formation of highly dispersed metallic species into single atoms.60 Herein, the Pt/(Co,Ni)(OH)2/C catalysts were synthesized by a two-step strategy, and (Co,Ni)(OH)2/C was first prepared by a precipitation method. Then, the Pt/(Co,Ni)(OH)2/C samples with different loadings were successfully synthesized by varying the electrodeposition time in a 1.0 mmol L−1 H2PtCl6 solution, and the Pt species were loaded on (Co,Ni)(OH)2 NPs by in situ nucleation and growth (Fig. 1a-Pt1/(Co,Ni)(OH)2/C). Here, the metal loadings of the samples were measured by employing an inductively coupled plasma optical emission spectrometer (ICP-OES) (Table S1, ESI†). As expected, the content of Pt increased with electrodeposition time, from 0.81, 1.41 to 3.67 wt% Pt after deposition for 10, 20 to 30 s, respectively, and the Pt/(Co,Ni)(OH)2/C catalysts were assigned as 0.81 wt% Pt/(Co,Ni)(OH)2/C, Pt1/(Co,Ni)(OH)2/C and 3.67 wt% Pt/(Co,Ni)(OH)2/C, respectively. The Pt1/Co(OH)2/C and Pt1/Ni(OH)2/C, and Pt1/C catalysts were obtained by controlling the same electrodeposition time (20 s) with Pt1/(Co,Ni)(OH)2/C (−0.8 V vs. SCE). Here, we performed HRTEM experiments to show the crystal lattice of (Co,Ni)(OH)2, Fig. S1 (ESI†). The HRTEM image indicates a d-spacing of 0.23 nm for neighboring (101) planes in the hydroxide phase, which is consistent with the SAED result shown in Fig. S2a (ESI†). SAED patterns were recorded from a region with small pieces of the hydroxide and the neighboring amorphous carbon support, respectively. There were diffractions from (101) and (111) lattice planes of (Co,Ni)(OH)2 pieces as shown in Fig. S2a (ESI†) while no diffractions from the amorphous carbon support as shown in Fig. S2b (ESI†). To further confirm the presence of the hydroxide phase of the Pt1/(Co,Ni)(OH)2/C sample, FT-IR analysis was conducted (Fig. S3, ESI†). According to the FT-IR results, the infrared spectra were accurately measured and the existence of hydroxides was confirmed. To determine the existence form of Pt, AC-STEM analysis (Fig. 1b and c) of Pt1/(Co,Ni)(OH)2/C was conducted, confirming the formation of Pt single atoms on (Co,Ni)(OH)2. The density of Pt single atoms on the (Co,Ni)(OH)2 substrate was estimated to be ∼0.61 Pt atoms nm−2 after examination of many AC-STEM images of Pt1/(Co,Ni)(OH)2/C, which is very close to the actual Pt loading (about 0.58 Pt atoms nm−2) determined by ICP-OES and the specific surface area of Pt1 (ESI†), indicating that the Pt single atoms are located on the surface or the subsurface of the (Co,Ni)(OH)2 substrate. In contrast, the density of Pt atoms in commercial 20 wt% Pt/C was calculated to be 163.2 Pt atoms nm−2 which is much higher than that of the Pt1/(Co,Ni)(OH)2/C single atom catalyst. Besides, characterizations using AC-STEM-EDS elemental mapping towards the region of Fig. 1d further reveal the formation of well hybridized Pt and (Co,Ni)(OH)2 NPs (Fig. 1c). Furthermore, the AC-STEM-EDS elemental mapping presents that the signal distribution ranges of O, Co, and Ni elements are coincident and smaller than that of C, indicating that the (Co,Ni)(OH)2 NPs are well distributed over the carbon support (Fig. 1d–g). Elemental mapping demonstrated that Pt single atoms were uniformly distributed on the (Co,Ni)(OH)2 NPs. (Fig. 1h). | ||
| Fig. 1 Fabrication and structural characterizations of Pt1/(Co,Ni)(OH)2/C. (a) Schematic illustration of the fabrication procedures of Pt1/(Co,Ni)(OH)2/C, note: small yellow ellipses on Ni/Co in (a) represent OH defect sites, (b and c) AC-STEM images of Pt1/(Co,Ni)(OH)2/C, and corresponding AC-STEM-EDS maps of Pt1/(Co,Ni)(OH)2/C-(d) C, (e) O, (f) Co, (g) Ni, and (h) Pt. | ||
Powder X-ray diffraction (XRD) patterns of the as-prepared samples were recorded to further determine the crystal form. The apparent diffraction peak at 26.1° is assigned to carbon (002) of carbon paper.61 The crystal phases of 0.81 wt% Pt/(Co,Ni)(OH)2/C, Pt1/(Co,Ni)(OH)2/C, and 3.67 wt% Pt/(Co,Ni)(OH)2/C are identified to be the hybridization of the hexagonal trigonal β-Co(OH)2 (JCPDS-No. 30-0443) and β-Ni(OH)2 (JCPDS-No. 14-0117) structure (Fig. 2A).62,63 Noteworthily, no diffraction peak of Pt species is observed due to Pt being in high dispersion and existing as single atoms. Fig. 2B illustrates the X-ray diffraction (XRD) patterns of Pt1/Co(OH)2/C, Pt1/Ni(OH)2/C, and Pt1/C. The Co(OH)2 and Ni(OH)2 crystal phases are also confirmed to exist in Pt1/Co(OH)2/C and Pt1/Ni(OH)2/C, respectively, whereas Pt1/C only displays the diffraction peak of carbon because the Pt species are highly dispersive. It is of great significance to study the effect of the valence state of the elements and electron transfer between the different elements on the reactivity of the catalysts. Hence, X-ray photoelectron spectroscopy (XPS) was utilized to probe the chemical composition and the chemical state of different species in the catalysts. The binding energies of Pt 4f in Pt1/(Co,Ni)(OH)2/C show a positive shift relative to Pt1/C, uncovering that the Co and Ni atoms in (Co,Ni)(OH)2/C can bring down the electron density of the Pt species (Fig. 2C). Additionally, the peak area and intensity of Pt 4f in Pt1/(Co,Ni)(OH)2/C are larger than those in 0.81 wt% Pt/(Co,Ni)(OH)2/C and lower than those in 3.67 wt% Pt/(Co,Ni)(OH)2/C, which is consistent with the experimental design and ICP-OES results. Noteworthily, the Pt 4f spectra of Pt1/(Co,Ni)(OH)2/C display a positive shift to Pt1/Co(OH)2/C and Pt1/Ni(OH)2/C, suggesting the surface electrons transferred from the Pt species to the Ni and Co species, resulting in an increase in the Pt valence state (Fig. 2C). This effective electronic tuning of Pt single atoms via heterogeneous atomic coordination of (Co,Ni)(OH)2 may be favorable for efficient hydrogen evolution. The XPS spectra of Co 2p, Ni 2p, and C 1s for the as-obtained catalysts are shown in Fig. 2D–F, and it is clear that the intensities of Co and Ni peaks reflect well their surface concentrations (Fig. 2D–F). In addition, the XPS spectra of Pt 4f show that the Pt species are Pt(0) and Ptδ+ due to electron transfer to substrate O species. The value of δ was between 0–2 due to the bonding of Pt–O–M (M = Co and Ni), and Pt was present as Pt(0) and Ptδ+, not ionic Pt (Fig. 2G).42,64,65 In this work, the surface Co species of the Pt1/(Co,Ni)(OH)2/C catalyst is mainly present in an oxidized state as confirmed from the Co 2p XPS spectra (Fig. 2H), such as Co(OH)2.46,50 The XPS spectra of Ni 2p for the Pt1/(Co,Ni)(OH)2/C catalyst show that the Ni species exist in the form of Ni(OH)2, which is in agreement with the XRD results (Fig. 2I). Here, to explore and prove the effect for Pt single atom electronic synergy differences between mono- and dual-hydroxides, it is necessary to combine other characterization and theoretical calculation.
 | ||
| Fig. 2 X-ray diffraction patterns and X-ray photoelectron spectroscopy study of the catalysts. X-ray diffraction (XRD) patterns of (A) (a) 0.81 wt% Pt/(Co,Ni)(OH)2/C, (b) Pt1/(Co,Ni)(OH)2/C, (c) 3.67 wt% Pt/(Co,Ni)(OH)2/C, (B) (a) Pt1/C, (b) Pt1/Co(OH)2/C, (c) Pt1/Ni(OH)2/C, XPS spectra of (C) Pt 4f, (D) Co 2p, (E) Ni 2p, (F) C 1s for the as-obtained catalysts, XPS spectra of (G) Pt 4f, (H) Co 2p, (I) Ni 2p of Pt1/(Co,Ni)(OH)2/C. | ||
After the Pt electrodeposition was accomplished, the evaluation of electrocatalytic activities of the catalysts for the HER was carried out in an N2-saturated 1.0 M KOH solution under ambient conditions using a typical three-electrode configuration (see the Experimental section for details). A series of Pt/(Co,Ni)(OH)2/C samples synthesized with Pt electrodeposition for 10 s, 20 s, 30 s, and (Co,Ni)(OH)2/C without Pt electrodeposition were measured by linear sweep voltammetry (LSV). Pt1/(Co,Ni)(OH)2/C displayed an optimized HER efficiency to 0.81 wt% Pt/(Co,Ni)(OH)2/C and 3.67 wt% Pt/(Co,Ni)(OH)2/C (Fig. 3a). The comparison of the as-prepared catalysts indicated that Pt1/(Co,Ni)(OH)2/C required ultralow overpotentials at 24 and 86 mV to deliver the current densities of 10 and 100 mA cm−2, respectively (Fig. 3a and Fig. S4a, ESI†). The deterioration of the HER performance towards 3.67 wt% Pt/(Co,Ni)(OH)2/C may be due to the excessive time of electrodeposition that results in the agglomeration of Pt species. Moreover, Co(OH)2/C, Ni(OH)2/C, and carbon with Pt electrodeposition for 20 s were also prepared and measured for comparison. Pt1/(Co,Ni)(OH)2/C exhibited much-enhanced activity compared with Pt1/Co(OH)2/C, Pt1/Ni(OH)2/C, (Co,Ni)(OH)2/C, Pt1/C and the commercial 20 wt% Pt/C at different current densities (Fig. 3a–c and Fig. S4, S5, ESI†). The mass activity of the Pt1/(Co,Ni)(OH)2/C catalyst determined by the HER polarization curves at −0.09 V (vs. RHE) was 115.9 times over the commercial 20 wt% Pt/C and 29.7 times over Pt1/C (Fig. 3d–f and Fig. S6, ESI†).32,65,66 The “fluctuation behavior” of the LSV was due to relatively large streams of bubbles being generated at a very large current density, which regularly interrupt the mass transfer process, leading to the evident fluctuation of the polarization curve. Furthermore, an appropriate and widely used frequency range of low (0.01 Hz) to high (100 K Hz) frequency was applied for the HER impedance measurements in this study.67,68 Pt1/(Co,Ni)(OH)2/C demonstrated the smallest charge transfer resistance, which proves the optimized electronic transfer and enhanced intrinsic activity for the HER (Fig. S7 and S8, ESI†). Pt1/(Co,Ni)(OH)2/C demonstrating the smallest charge transfer resistance proves optimized electronic transmission and reaction kinetics account for its enhanced intrinsic activity for the HER (Fig. S7 and S8, ESI†). Moreover, the Tafel slope of Pt1/(Co,Ni)(OH)2/C was 28.7 mV dec−1, lower than that of Pt1/Co(OH)2/C (48.7 mV dec−1) and Pt1/Ni(OH)2/C (52.1 mV dec−1), indicating that bimetallic (Co,Ni)(OH)2/C is more conducive to the HER process than Co(OH)2/C or Ni(OH)2/C. As the Tafel slope of Pt1/(Co,Ni)(OH)2/C (28.7 mV dec−1) was lower than 30 mV dec−1, the HER was likely to follow the Volmer–Tafel mechanism (Fig. 3g).69 However, the HER mechanism of Pt1/C and commercial 20 wt% Pt/C is identified to follow the Volmer–Heyrovsky path due to the Tafel slopes being 56.0 mV dec−1 and 32.5 mV dec−1, respectively (Fig. 3g).69 Rather than Pt/C suffering from sluggish kinetics for breaking the H–OH bonds, the introduction of the (Co,Ni)(OH)2 hybrid in Pt1/(Co,Ni)(OH)2/C has facilitated the water decomposition step (Volmer step), resulting in transformation of the rate-determining step of water decomposition to the formation of the molecular hydrogen step (Tafel step). The linear fitting of the capacitive currents versus CV scanning rates was calculated to determine the double-layer capacitance (CdL) and the electrochemical active area (ECSA) (Fig. S8c, ESI†). Pt1/(Co,Ni)(OH)2/C exhibited the largest CdL, indicating the highest electrochemical active area among 0.81 wt% Pt/(Co,Ni)(OH)2/C, 3.67 wt% Pt/(Co,Ni)(OH)2/C, Pt1/Co(OH)2/C, Pt1/Ni(OH)2/C, and (Co,Ni)(OH)2/C. To further evaluate the intrinsic activity of the catalysts, ECSA normalization was performed for all the as-prepared catalysts (Fig. S10, ESI†) with the CdL provided in the ESI† (Fig. S8, S9 and S14a). Pt1/(Co,Ni)(OH)2/C exhibited the largest current density after being normalized with ECSA, which is 3.5 times over 20 wt% Pt/C and 20.3 times over Pt1/C at a typical potential of −0.09 V. Comparison of Pt1/(Co,Ni)(OH)2/C with Pt1/Co(OH)2/C and Pt1/(Co,Ni)(OH)2/C revealed the effective electronic tuning of Pt single atoms via heterogeneous atomic coordination of (Co,Ni)(OH)2 for efficient hydrogen evolution (Fig. S10, ESI†). For single-atom model catalysts, TOF can be the solid evidence to estimate the intrinsic activity of the catalysts. Therefore, we carried out a TOF analysis (Page 5 and 6 in ESI†) and the curves of TOF against the overpotential were obtained.70 The highest turnover frequency (TOF, s−1) of Pt1/(Co,Ni)(OH)2/C was 5.950 H2 s−1 per active site for the HER which was ∼7.0 times that of Pt1/Co(OH)2/C (0.855 H2 s−1 per active site), 12.3 times that of Pt1/Ni(OH)2/C(0.485 H2 s−1 per active site), and 15.5 times that of Pt1/C (0.385 H2 s−1 per active site) at an overpotential of 0.1 V in 1.0 M KOH solution, also confirming the fastest reaction kinetics among all the as-obtained catalysts (Fig. 3h and Fig. S11, ESI†). The volcano plot of hydrogen adsorption free energy (ΔGH*) of different metal catalysts in the HER is compared in Fig. 3i. Clearly, Pt1/(Co,Ni)(OH)2/C exhibits the highest HER exchange current density and optimized hydrogen binding energy, further confirming its superior HER performance (Fig. S12, ESI†). The summary and comparison of the water dissociation ability of Pt1/(Co,Ni)(OH)2/C and different reported catalysts suggest that Pt1/(Co,Ni)(OH)2/C has a suitable water dissociation energy (Fig. S13, ESI†). Furthermore, Pt1/(Co,Ni)(OH)2/C shows the lowest overpotential and Tafel slope during the alkaline HER activity, compared with those of the state-of-the-art catalysts reported to date (Fig. 3j and Tables S2, S3, ESI†).
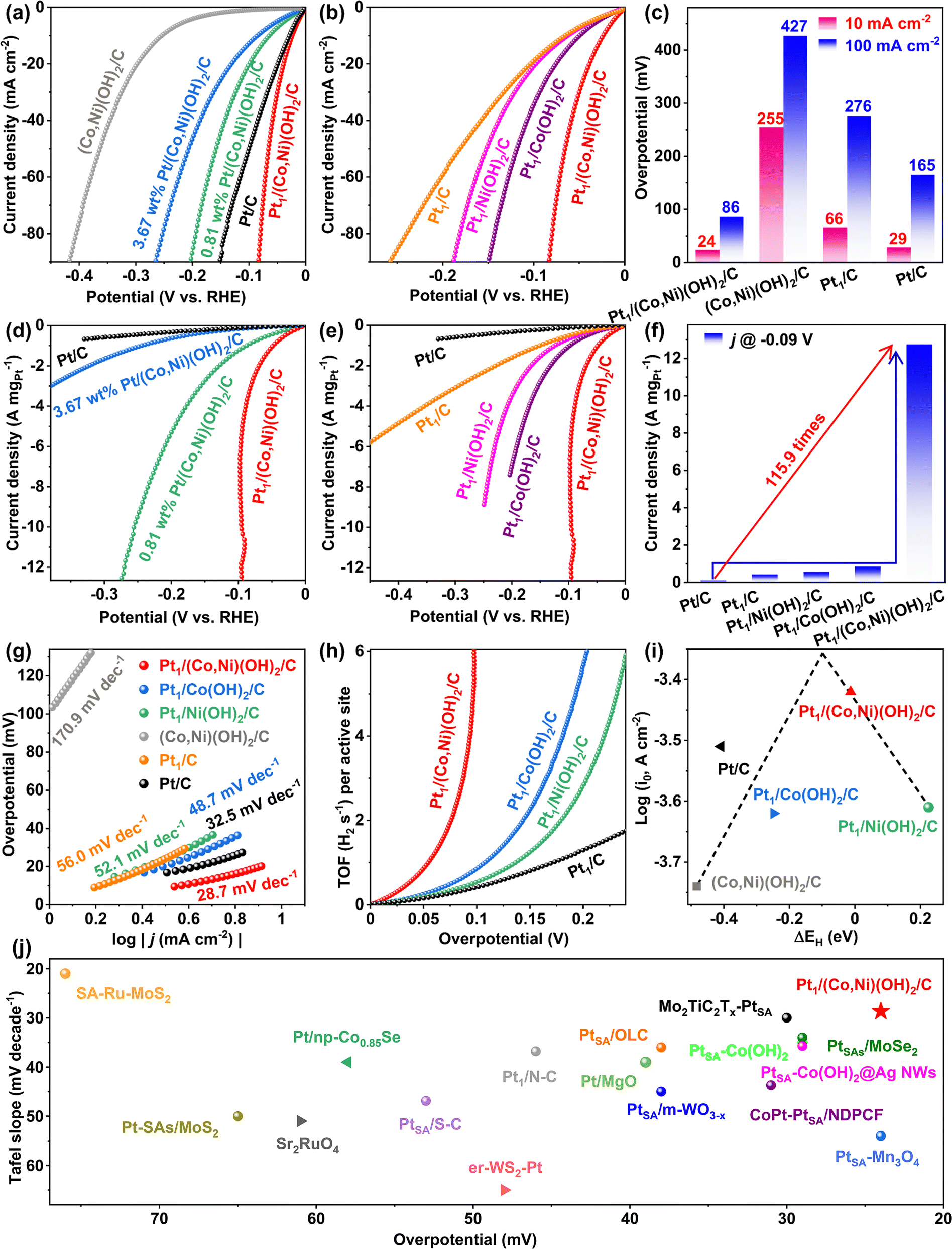 | ||
| Fig. 3 Electrocatalytic performance of Pt1/(Co,Ni)(OH)2/C toward the HER. The HER polarization curves of (a) 0.81 wt% Pt/(Co,Ni)(OH)2/C, Pt1/(Co,Ni)(OH)2/C, 3.67 wt% Pt/(Co,Ni)(OH)2/C, (Co,Ni)(OH)2/C, and commercial 20 wt% Pt/C. (b) Pt1/(Co,Ni)(OH)2/C, Pt1/Co(OH)2/C, Pt1/Ni(OH)2/C, Pt1/C in an 1.0 M KOH solution. Scan rate, 5 mV s−1. (c) Overpotential at 10 mA cm−2, 100 mA cm−2. (d), (e) The HER polarization curves normalized by Pt loading of the as-prepared catalysts for HER. (f) The mass activity of the Pt1/(Co,Ni)(OH)2/C catalyst determined by the HER polarization curves at −0.09 V (vs. RHE). (g) Tafel plots. (h) TOFs of the Pt1/(Co,Ni)(OH)2/C, Pt1/Co(OH)2/C, Pt1/Ni(OH)2/C, and Pt1/C samples. (i) Volcano plot of hydrogen adsorption free energy (ΔGH*) of different metal catalysts in HER. (j) Comparison of alkaline HER activities, in terms of the overpotential at 10 mA cm−2 and the Tafel slope, among the state-of-the-art catalysts reported to date. | ||
Stability is an important factor for practical applications of the HER catalysts. Hence, Pt1/(Co,Ni)(OH)2/C was evaluated by two different long-term stability measurements, along with Pt1/C for comparison. First, chronopotentiometry (CP) tests were conducted for Pt1/C and Pt1/(Co,Ni)(OH)2/C, at a current density of 100 mA cm−2 for 24 h to show the stability on a laboratory scale, as was commonly adopted in the relevant literature for the alkaline HER.71,72 Pt1/C displayed a significant reduction in HER performance after only 5 min at 100 mA cm−2, and the polarization potential demand increased by 181 mV after 24 h of chronopotentiometry to attain a current density of 100 mA cm−2. Even after the electrolyte was renewed, its overpotential (at 10 mA cm−2) still increased by 143 mV while that for the commercial 20 wt% Pt/C increased only 14 mV (Fig. 4(a and b)). In contrast, chronopotentiometry curve of Pt1/(Co,Ni)(OH)2/C showed a slowly increasing overpotential (with the increase of ∼130 mV) lower than that of the commercial 20 wt% Pt/C (with the increase of 205 mV) after 24 h of chronopotentiometry (Fig. 4d). After the renewal of the electrolyte, Pt1/(Co,Ni)(OH)2/C showed a negligible increase (by only 6 mV, Pt/C-14 mV) towards the overpotential of 30 mV at 10 mA cm−2, indicating an outstanding HER stability (Fig. 4(d and e)) Compared with CP at a current density of 100 mA cm−2, CV tests can be conducted over a wider voltage range (0–0.6 V), to attain a current density of 0–400 mA cm−2, which is more convincing than the results of CP at 100 mA cm−2. Hence, to further demonstrate the long-term stability of Pt1/(Co,Ni)(OH)2/C, we made further effort in measuring the stability under cyclic voltammetry via a scan rate of 50 mV s−1 for 20000 cycles for Pt1/(Co,Ni)(OH)2/C and Pt1/C, and 5000 cycles for commercial 20 wt% Pt/C. Noteworthily, the polarization curves of Pt1/(Co,Ni)(OH)2/C showed a much smaller increase in overpotential (21 mV) than Pt1/C (65 mV) commercial 20 wt% Pt/C (36 mV) (Fig. 4(c and f)). Pt1/(Co,Ni)(OH)2/C also shows a smaller attenuation in the electrochemically active surface area (ECSA) than that of Pt1/C, after chronopotentiometry test for 24 h or cyclic voltammetry for 20000 cycles (Fig. S14 and S15, ESI†). More importantly, after 24 h of chronopotentiometry, and after 20000 cycles of long-term stability measurements, the polarization potential of Pt1/(Co,Ni)(OH)2/C (45 mV, 151 mV) at 10 mA cm−2 and 100 mA cm−2 was much lower than that of Pt1/C (131 mV, 337 mV) and commercial 20 wt% Pt/C (65 mV, 327 mV) after 5000 cycles (Fig. 4g). Moreover, a larger scan rate at 200 mV s−1 for another 20000 cycles towards Pt1/(Co,Ni)(OH)2/C was also conducted and in contrast to the scanning rate at 50 mV s−1 it further demonstrated its favorable CV cycle stability. Exactly, there was a negligible decline of the HER activity after tuning the scan rate of 50 to 200 mV s−1, exhibiting improved long-term high current stability of Pt1/(Co,Ni)(OH)2/C, compared with that of Pt1/C and commercial 20 wt% Pt/C (Fig. S16, ESI†). It suggests that the synergistic interaction of highly dispersed Pt anchored on (Co,Ni)(OH)2 nanoparticles and the synergistic effect of Pt1 and (Co,Ni)(OH)2 are conducive to the optimized HER stability. The synergistic effect may be the (Co,Ni)(OH)2 facilitates water decomposition, and the Pt single atoms boost the formation of hydrogen. The specific HER enhancement mechanism of Pt1/(Co,Ni)(OH)2/C required further study by operando Raman spectrometry.
 | ||
| Fig. 4 Electrocatalytic stability measurements of Pt1/C and Pt1/(Co,Ni)(OH)2/C toward the HER. The chronopotentiometry response for (a) Pt1/C, (d) Pt1/(Co,Ni)(OH)2/C at 100 mA·cm−2 for 24 h. Polarization curves of initial (b) Pt1/C, (e) Pt1/(Co,Ni)(OH)2/C, and after chronopotentiometry for 24 h. Polarization curves of initial (c) Pt1/C, (f) Pt1/(Co,Ni)(OH)2/C and after 20000 cycles, initial commercial 20 wt% Pt/C and after 5000 cycles. (g) Overpotential at 10 mA·cm−2, 100 mA·cm−2 for Pt1/(Co,Ni)(OH)2/C, Pt1/C and commercial 20 wt% Pt/C after 24 h chronopotentiometry, and Pt1/(Co,Ni)(OH)2/C, Pt1/C after 20000 cycles, commercial 20 wt% Pt/C after 5000 cycles. | ||
The HER mechanism of Pt1/(Co,Ni)(OH)2/C and Pt1/C was further investigated by employing operando Raman spectrometry. The operando Raman spectra of Pt1/(Co,Ni)(OH)2/C were recorded from −0.8 to −1.3 V (vs. Hg/HgO) with an interval of 0.1 V in 1.0 M KOH. The peak at 475 cm−1 in the operando Raman spectra can be assigned to the M–OH and M–O (M = Ni and Co) vibrations. The increased species of M–OH and M–O were enhanced with applied potentials suggesting that (Co,Ni)(OH)2 were involved in the decomposition of water (Fig. 5a).68,72 Unfortunately, it is hard to distinguish between M–OH and M–O vibrations due to the overlapping of the bands. Since the catalyst itself contains hydroxides, the presence of lattice oxygen may affect the identification of the peak species. To provide solid experimental evidence that the first step for the decomposition of water (the H–OH bond breaking) takes place on the surface of (Co,Ni)(OH)2 rather than the effect of lattice oxygen, the collection of ex situ Raman spectra before the HER, the operando Raman spectra of Pt1/(Co,Ni)(OH)2/C before, during and after the HER was conducted (Fig. 5b). The presence of lattice oxygen was very low in Pt1/(Co,Ni)(OH)2/C, which may be due to the extremely thin (Co,Ni)(OH)2 on the relatively thick carbon support. The low existence of lattice oxygen was mainly due to the presence of (Co,Ni)(OH)2 in Pt1/(Co,Ni)(OH)2/C. The M–OH and M–O intensities during the HER were much stronger than the operando Raman spectra before and after the HER. The comparative observation confirms that the first step for the decomposition of water (the H–OH bond breaking) takes place on the surface of the (Co,Ni)(OH)2 rather than due to the effect of lattice oxygen (Fig. 5b)68,72 The water molecules adsorb on hydrophilic (Co,Ni)(OH)2 and dissociate to generate highly active M–*OH intermediates. After the H–OH bond breaks, the formation of Ni–*OH and Co–*OH intermediates would further expedite the water splitting and induce *H transfer to the single-atom Pt sites to generate Pt–*H. The second step for the decomposition of water is the formation of H2, which is accessible by the interosculation of adjacent Pt–*H species (Tafel step). This process is usually too fast to capture the intermediate of Pt–*H. In contrast, there was no evident peak arising from Pt1/C during the HER, implying that Pt–OH or Pt–O were low in quantity and the dissociation of the H–OH on Pt species was inefficient (Volmer step) (Fig. 5b). Due to weakening of the signal in the solution, the ex situ Raman spectra of both Pt1/(Co,Ni)(OH)2/C and Pt1/C show little sharper peak intensity than those measured at the open-circuit potential in 1.0 M KOH solution (Fig. 5b and c). Similar to the ex situ Raman, the M–OH and M–O vibrations cannot be observed on Pt1/C under open-circuit potential and −1.3 V (vs. Hg/HgO) due to its low HER efficiency. The Pt1/(Co,Ni)(OH)2/C HER indicates a strong synergistic effect between the (Co,Ni)(OH)2 and the Pt single atom: the former, enriched with hydroxyl defect sites, expedites the water decomposition step (step 1, Volmer step), and the latter, with optimized hydrogen adsorption free energy, facilitates the hydrogen formation step (step 2, Tafel step) (Fig. 5d).68,72
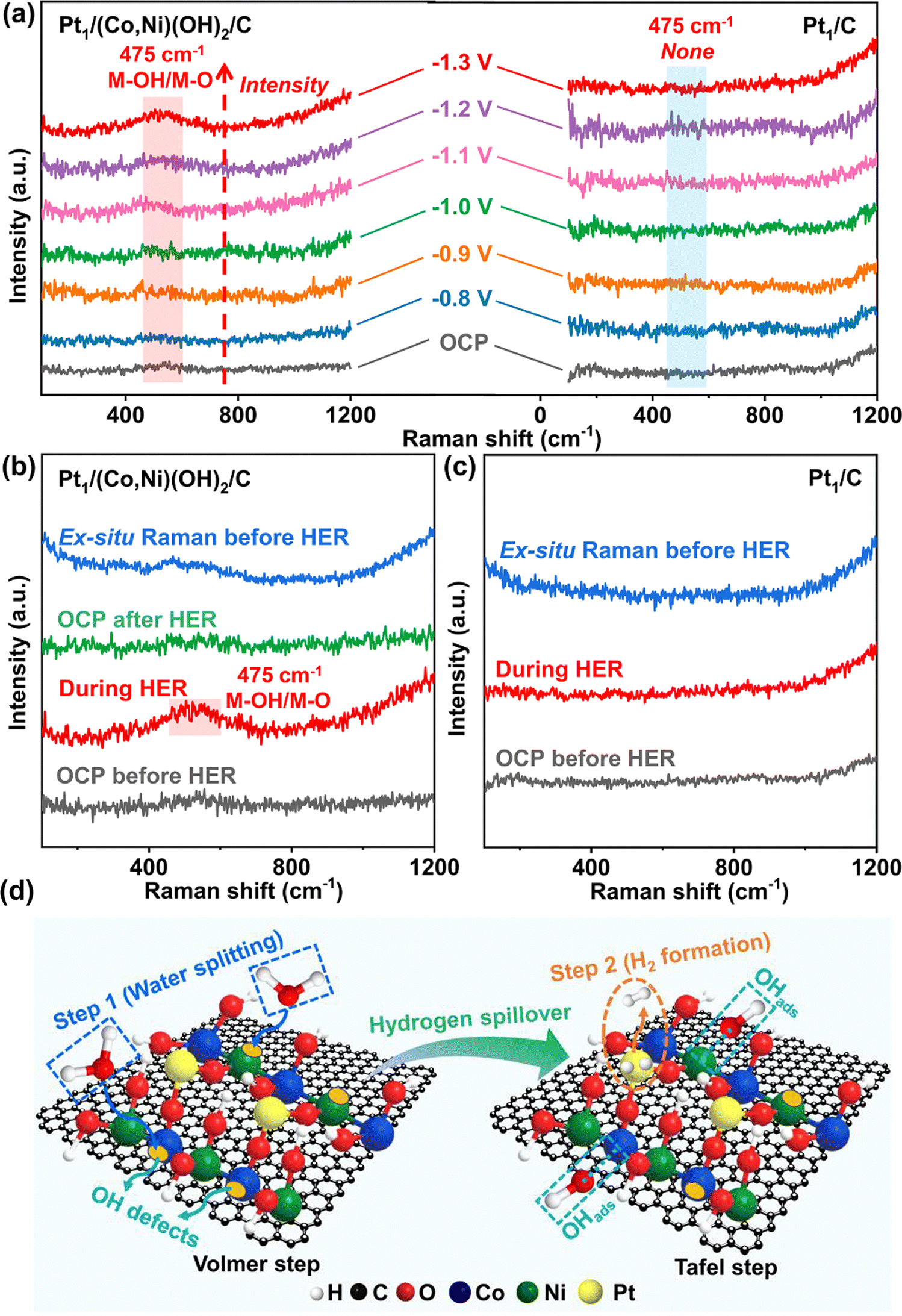 | ||
| Fig. 5 Operando Raman spectra at OCP and different applied potential (vs. Hg/HgO) in 1.0 M KOH for (a) Pt1/(Co,Ni)(OH)2/C and Pt1/C. (b) The ex situ Raman spectra before HER, Operando Raman spectra at OCP before HER, during HER at −1.3 V (vs. Hg/HgO), and OCP after HER for Pt1/(Co,Ni)(OH)2/C, (c) The ex situ Raman spectra before HER, Operando Raman spectra at OCP before HER, and the during HER at −1.3 V (vs. Hg/HgO) for Pt1/C, (d) The Pt1/(Co,Ni)(OH)2/C HER mechanism of synergistic effect among (Co,Ni)(OH)2 surface with hydroxyl defect site expedited the water decomposition step (Step 1, Volmer step) and Pt single atoms with optimized hydrogen adsorption free energy facilitated hydrogen formation step (Step 2, Tafel step). Note: Small yellow ellipses on Ni/Co in (d) represent OH defect sites. | ||
The models of (Co,Ni)(OH)2, (Co,Ni)(OH)2 adsorbed with Pt single atom (Pt1/(Co,Ni)(OH)2) and Pt(111) slabs calculated in this study are shown in Fig. S17–S19 (ESI†). It is well known that the poor performance of Pt for the HER in an alkaline solution is attributed to the sluggish water dissociation step. As shown in Fig. 6d, the dissociation of H2O (*H2O → *H + *OH) is 0.68 eV uphill on pure Pt(111), which is the rate-limiting step. According to previous reports, the catalysts combining Pt and hydroxides show better HER performance than pure Pt in alkaline media.40,45 The improvements are attributed to the abundance of proliferative defects in hydroxides, such as *OH, which are potential active sites for the adsorption and dissociation of H2O. The defected (Co,Ni)(OH)2 with (Pt1/(Ni,Co)(OH)2) and without single Pt adsorption ((Co,Ni)(OH)2) were both considered in this study (see the models in Fig. 6a and b). The dissociation of H2O on (Co,Ni)(OH)2 is 0.93 eV uphill, which leads to a more sluggish HER than that on pure Pt(111). The adsorption of intermediates *H on (Co,Ni)(OH)2 is too strong (−0.32 eV), which inhibits the formation of H2 (*H → *+ 1/2H2). While for Pt1/(Ni,Co)(OH)2, the adsorbed H2O (*H2O) splits into one *OH staying at the OH defect, and one *H on the Pt atom with a downhill free energy of −0.074 eV. Hence, a very efficient H2O dissociation step is realized here. Charge density difference (Fig. 6c) suggests a strong charge transfer between Pt and O atoms of (Co,Ni)(OH)2. The Bader effective charge of Pt on pure Pt is −0.076|e| while 0.27|e| for Pt in Pt1/(Ni,Co)(OH)2, which means that electrons are transferred from the single Pt atom to (Co,Ni)(OH)2. Since the *H and Pt atoms interact covalently (Fig. S20 and S21, ESI†), the more locally negative the charge on the Pt atom, the stronger the *H bonding with Pt. Therefore, *H is relatively weakly adsorbed on Pt1/(Ni,Co)(OH)2 (0.15 eV) compared with pure Pt (−0.26 eV). Pt1/(Co,Ni)(OH)2 shows superior performance for both H2O dissociation and H2 formation steps, compared with pure Pt and (Co,Ni)(OH)2, making it a far more cost-efficient HER catalyst, which is consistent with our experimental findings. The free energy diagrams for Pt1/Ni(OH)2 and Pt1/Co(OH)2 are also presented in Fig. 6d to show the advantage of Pt1/(Ni,Co)(OH)2. Unlike Pt1/(Co,Ni)(OH)2 and Pt1/Ni(OH)2, the water dissociation on Pt1/Co(OH)2 is still endothermic, but with much-improved performance, compared with Pt(111) and (Ni,Co)(OH)2. While for Pt1/Ni(OH)2, the adsorption of *H is stronger than that on Pt1/(Ni,Co)(OH)2 and Pt1/Co(OH)2 which inhibits the formation of H2. In the volcano plot of hydrogen adsorption free energy (ΔGH*) of the as-obtained catalysts in the HER, Pt1/(Co,Ni)(OH)2/C displayed the largest HER exchange current density and the optimized hydrogen binding energy further confirmed its improved HER activity (Fig. 3i). The density of states for 3d states and corresponding d-band centers of Pt atoms in different catalysts is shown in Fig. S22 (ESI†). The d-band center is a good descriptor of metal–adsorbate interactions.73–75 As the d-band center shifts down away from the Fermi level, *H would bind more weakly with the Pt atom (i.e., a lower ΔGH*). It is clear to see that the trend of ΔGH* is consistent with the d-band center as shown in Fig. 6d and Fig. S22 (ESI†). Above analysis suggests that hydroxides can effectively tune the electronic structure of Pt atoms and then ΔGH* on it. Therefore, the advantage of heterogeneous atomic coordination of (Co,Ni)(OH)2 to the effective electronic tuning of Pt single atoms is that the H2O dissociation and H2 formation can be enhanced simultaneously.
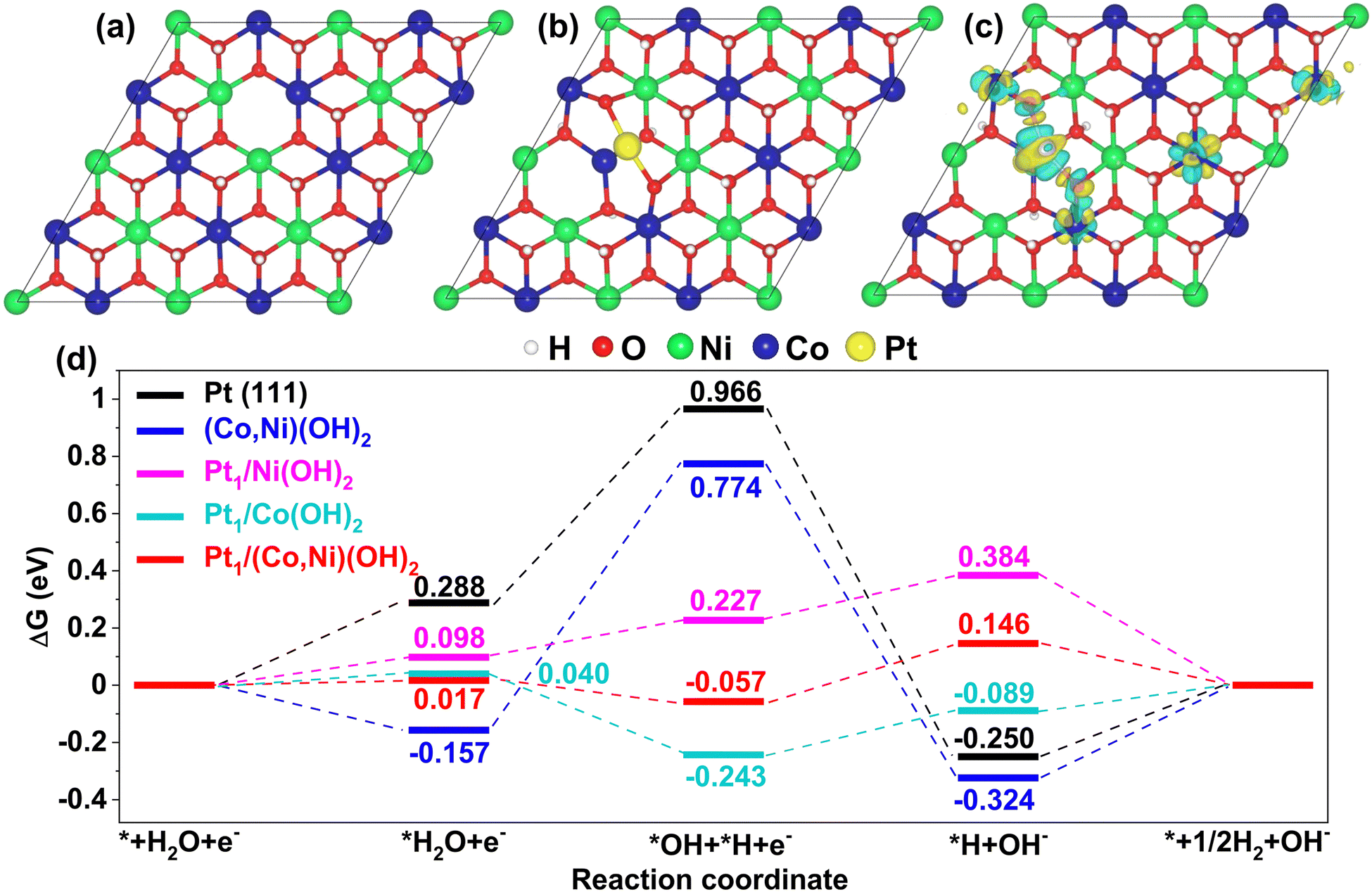 | ||
| Fig. 6 Atomic structures of (a) (Co,Ni)(OH)2 and (b) Pt1/(Co,Ni)(OH)2, (c) charge density difference for the adsorption of single Pt atom on (Co,Ni)(OH)2. The red, green, blue, yellow, and white balls are for O, Co, Ni, Pt, and H atoms, respectively. The charge depletion and accumulation were depicted by blue and yellow isosurfaces, respectively. (d) free energy diagrams for HER on Pt(111), (Co,Ni)(OH)2, Pt1/Ni(OH)2, Pt1/Co(OH)2, and Pt1/(Co,Ni)(OH)2, respectively. | ||
3. Conclusions
A cost-efficient Pt/(Co,Ni)(OH)2/C electrocatalyst for the HER was designed and synthesized to achieve a high density of active single atom Pt sites formed in situ and anchored stably over bimetallic (Co,Ni)(OH)2 nanoparticles, supported further on high-surface-area active carbon. Even with a relatively low Pt loading of 1.41 wt%, Pt1/(Co,Ni)(OH)2/C exhibits a far superior HER activity to Pt1/Co(OH)2/C, Pt1/Ni(OH)2/C, and the commercial Pt/C in 1.0 M KOH solution. Impressively, Pt1/(Co,Ni)(OH)2/C shows an almost zero onset overpotential and an outstanding electrocatalytic mass activity for the HER, 29.7 times greater than that of Pt1/C and 115.9 times higher than that of the commercial 20 wt% Pt/C at −0.09 V vs. RHE, respectively. The synergistic effect between the highly dispersed Pt single atoms and the bimetallic hydroxide (Co,Ni)(OH)2 nanoparticles brings about relatively low energy barriers and/or adequate sorption energies for the two intermediate steps of water-dissociation, to boost the HER activity. The intrinsic activity of defective (Co,Ni)(OH)2 for water decomposition (Volmer step) is enhanced by the Pt species, while (Co,Ni)(OH)2 leads to a charge redistribution of the Pt sites and promotes the formation of H2 (Tafel step). In comparison, combining the heterogeneous atomic coordination of Co(OH)2 and Ni(OH)2 not only accelerates the decomposition of water molecules but also stabilizes and tunes the electronic structure of the Pt single atoms, optimizing the adsorption of *H on Pt simultaneously. This work opens up a new avenue for developing SACs and brings a new understanding of the effective electronic tuning of Pt single atoms by heterogeneous atomic coordination of electronic synergy between mono- and dual-hydroxides for enhancing the catalytic performances of SACs.Author contributions
A. Pei: conceived the idea, designed and performed the experiments, formal analysis, data curation, writing of the original draft, investigation, data curation, and writing-review and editing. R. Xie: performed DFT calculations, data curation, and writing-review and editing. Y. Zhang, Y. Feng and S. Zhang: performed the experiments and data curation. Z. Wang, Z. Huang and C. Shang: investigation. L. Zhu: supervision, funding acquisition, conceptualization, project administration, and writing-review and editing. B. Chen, Q. Gao and H. Ye: supervision and review. G. Chai, Z. Yang and Z. Guo: supervision, conceptualization, and writing-review and editing. The manuscript was written by A. Pei, R. Xie, L. Zhu and Z. Guo. All authors participated in discussion of the research and approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 22162012 and 22202089), the Youth Jinggang Scholars Program in Jiangxi Province ([2019]57), the Thousand Talents Plan of Jiangxi Province (jxsq2019201083), the Natural Science Foundation of Jiangxi Province for Distinguished Young Scholars (Grant No. 20224ACB213005), the Program of Qingjiang Excellent Young Talents, Jiangxi University of Science and Technology (JXUSTQJBJ2019002), the Foundation of State Key Laboratory of Coal Conversion (grant no. J22-23-609), the Jiangxi Provincial Key Laboratory of Functional Molecular Materials Chemistry (20212BCD42018), the Program of Technology Innovation Talents of Ganzhou, the Research Fund Program of Key Laboratory of Fuel Cell Technology of Guangdong Province, the Research Fund Program of Guangdong Provincial Key Lab of Green Chemical Product Technology (GC202102) and the HK Scholars Program, Ji Hua Laboratory [X210141TL210] and Guangdong Province (2021B0301030003). The UGC-TRS (T23-713/22-R) award, the RGC-EU Collaborative Programme initiative (E-HKU704/19), the Environment and Conservation Fund (ECF 2021-152), the “Hong Kong Quantum AI Lab Ltd” funded by the AIR@InnoHK, launched by the Innovation and Technology Commission (ITC), and the URC Platform Technology Fund and the start-up support from the University of Hong Kong are acknowledged.Notes and references
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T. K. Sham, L. M. Liu and G. A. Botton, Nat. Commun., 2016, 7, 13638 CrossRef CAS PubMed.
- S. Ye, F. Luo, Q. Zhang, P. Zhang, T. Xu, Q. Wang, D. He, L. Guo, Y. Zhang, C. He, X. Ouyang, M. Gu, J. Liu and X. Sun, Energy Environ. Sci., 2019, 12, 1000–1007 RSC.
- J. Shan, M. Li, L. F. Allard, S. Lee and M. Flytzani-Stephanopoulos, Nature, 2017, 551, 605–608 CrossRef CAS PubMed.
- I. T. McCrum and M. Koper, Nat. Energy, 2020, 5, 891–899 CrossRef CAS.
- S. T. Wismann, J. S. Engbæk, S. B. Vendelbo, F. B. Bendixen, W. L. Eriksen, K. Aasberg-Petersen, C. Frandsen, I. Chorkendorff and P. M. Mortensen, Science, 2019, 364, 756–759 CrossRef CAS PubMed.
- C. McGlade and P. Ekins, Nature, 2015, 517, 187–190 CrossRef CAS PubMed.
- Z. Sun, G. Wang, S. W. Koh, J. Ge, H. Zhao, W. Hong, J. Fei, Y. Zhao, P. Gao, H. Miao and H. Li, Adv. Funct. Mater., 2020, 30, 2002138 CrossRef CAS.
- R. A. Walters, M. R. Tarbotton and C. E. Hiles, Renewable Energy, 2013, 51, 255–262 CrossRef.
- M. R. Lukatskaya, B. Dunn and Y. Gogotsi, Nat. Commun., 2016, 7, 12647 CrossRef.
- G. Ren, J. Liu, J. Wan, Y. Guo and D. Yu, Appl. Energy, 2017, 204, 47–65 CrossRef.
- C. Li, Z. Chen, H. Yi, Y. Cao, L. Du, Y. Hu, F. Kong, R. Kramer Campen, Y. Gao, C. Du and G. Yin, Angew. Chem., Int. Ed., 2020, 59, 15902–15907 CrossRef CAS PubMed.
- I. Ledezma-Yanez, W. D. Z. Wallace, P. Sebastián-Pascual, V. Climent, J. M. Feliu and M. Koper, Nat. Energy, 2017, 2, 1–7 Search PubMed.
- Y. Zheng, Y. Jiao, A. Vasileff and S. Z. Qiao, Angew. Chem., Int. Ed., 2018, 57, 7568–7579 CrossRef CAS PubMed.
- B. E. Conway and L. Bai, J. Electroanal. Chem., 1986, 198, 149–175 CrossRef CAS.
- B. E. Conway and G. Jerkiewicz, Electrochim. Acta, 2000, 45, 4075–4083 CrossRef CAS.
- N. Dubouis, C. Yang, R. Beer, L. Ries, D. Voiry and A. Grimaud, ACS Catal., 2018, 8, 828–836 CrossRef CAS.
- X. Zou, Y. Wu, Y. Liu, D. Liu, W. Li, L. Gu, H. Liu, P. Wang, L. Sun and Y. Zhang, Chem, 2018, 4, 1139–1152 CAS.
- S. Xu, X. Yu, X. Liu, C. Teng, Y. Du and Q. Wu, J. Colloid Interface Sci., 2020, 577, 379–387 CrossRef CAS PubMed.
- Q. Jin, B. Ren, D. Li, H. Cui and C. Wang, Nano Energy, 2018, 49, 14–22 CrossRef CAS.
- T. Xiong, X. Yao, Z. Zhu, R. Xiao, Y. W. Hu, Y. Huang, S. Zhang and M. S. Balogun, Small, 2022, 18, 2105331 CrossRef CAS.
- Q. Tan, R. Xiao, X. Yao, T. Xiong, J. Li, Y. W. Hu, Y. Huang and M. S. Balogun, Chem. Eng. J., 2022, 433, 133514 CrossRef CAS.
- Q. Zhou, Q. Hao, Y. Li, J. Yu, C. Xu, H. Liu and S. Yan, Nano Energy, 2021, 89, 106402 CrossRef CAS.
- T. Xiong, B. Huang, J. Wei, X. Yao, R. Xiao, Z. Zhu, F. Yang, Y. Huang, H. Yang and M. S. Balogun, J. Energy Chem., 2022, 67, 805–813 CrossRef CAS.
- Y. Huang, L. Hu, R. Liu, Y. Hu, T. Xiong, W. Qiu, M. S. Balogun, A. Pan and Y. Tong, Appl. Catal., B, 2019, 251, 181–194 CrossRef CAS.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Chem., 2014, 5, 5634 CAS.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- Z. Pu, T. Liu, I. S. Amiinu, R. Cheng, P. Wang, C. Zhang, P. Ji, W. Hu, J. Liu and S. Mu, Adv. Funct. Mater., 2020, 30, 2004009 CrossRef CAS.
- Y. Sun, Y. Zang, W. Tian, X. Yu, J. Qi, L. Chen, X. Liu and H. Qiu, Energy Environ. Sci., 2022, 15, 1201–1210 RSC.
- X. Peng, Y. Yan, X. Jin, C. Huang, W. Jin, B. Gao and P. K. Chu, Nano Energy, 2020, 78, 105234 CrossRef CAS.
- S. Li, J. Yang, C. Song, Q. Zhu, D. Xiao and D. Ma, Adv. Mater., 2019, 31, 1901796 CrossRef CAS PubMed.
- J. Lai, B. Huang, Y. Chao, X. Chen and S. Guo, Adv. Mater., 2019, 31, 1805541 CrossRef PubMed.
- K. L. Zhou, C. Wang, Z. Wang, C. B. Han, Q. Zhang, X. Ke, J. Liu and H. Wang, Energy Environ. Sci., 2020, 13, 3082–3092 RSC.
- W. Zhang, J. Zheng, X. Gu, B. Tang, J. Li and X. Wang, Nanoscale, 2019, 11, 9353–9361 RSC.
- Y. Wang, L. Chen, X. Yu, Y. Wang and G. Zheng, Adv. Energy Mater., 2017, 7, 1601390 CrossRef.
- A. Han, Z. Zhang, J. Yang, D. Wang and Y. Li, Small, 2021, 17, 2004500 CrossRef CAS PubMed.
- J. Liu, M. Jiao, L. Lu, H. M. Barkholtz, Y. Li, Y. Wang, L. Jiang, Z. Wu, D. J. Liu, L. Zhuang and C. Ma, Nat. Commun., 2017, 8, 15938 CrossRef CAS PubMed.
- C. Lin, Y. Zhao, H. Zhang, S. Xie, Y. F. Li, X. Li, Z. Jiang and Z. P. Liu, Chem. Sci., 2018, 9, 6803–6812 RSC.
- D. Kunwar, S. Zhou, A. DeLaRiva, H. Xiong, X. I. Pereira-Hernández, S. C. Purdy, R. t. Veen, H. H. Brongersma, J. T. Miller, H. Hashiguchi, L. Kovarik, S. Lin, H. Guo, Y. Wang and A. K. Datye, ACS Catal., 2019, 9, 3978–3990 CrossRef CAS.
- K. L. Zhou, Z. Wang, C. B. Han, X. Ke, C. Wang, Y. Jin, Q. Zhang, J. Liu, H. Wang and H. Yan, Nat. Commun., 2021, 12, 3783 CrossRef CAS PubMed.
- W. Chen, B. Wu, Y. Wang, W. Zhou, Y. Li, T. Liu, C. Xie, L. Xu, S. Du, M. Song, D. Wang, Y. liu, Y. Li, J. Liu, Y. Zou, R. Chen, C. Chen, J. Zheng, Y. Li, J. Chen and S. Wang, Energy Environ. Sci., 2021, 14, 6428–6440 RSC.
- D. Li, X. Chen, Y. Lv, G. Zhang, Y. Huang, W. Liu, Y. Li, R. Chen, C. Nuckolls and H. Ni, Appl. Catal., B, 2020, 269, 118824 CrossRef CAS.
- Y. Shi, Z. R. Ma, Y. Y. Xiao, Y. C. Yin, W. M. Huang, Z. C. Huang, Y. Z. Zheng, F. Y. Mu, R. Huang, G. Y. Shi, Y. Y. Sun, X. H. Xia and W. Chen, Nat. Commun., 2021, 12, 3021 CrossRef.
- J. Park, S. Lee, H. E. Kim, A. Cho, S. Kim, Y. Ye, J. W. Han, H. Lee, J. H. Jang and J. Lee, Angew. Chem., Int. Ed., 2019, 58, 16038–16042 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, G. Xin, C. Chen, C. L. Dong, R. S. Liu, C. P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- W. Chen, B. Wu, Y. Wang, W. Zhou, Y. Li, T. Liu, C. Xie, L. Xu, S. Du, M. Song, D. Wang, Y. liu, Y. Li, J. Liu, Y. Zou, R. Chen, C. Chen, J. Zheng, Y. Li, J. Chen and S. Wang, Energy Environ. Sci., 2021, 14, 6428–6440 RSC.
- Y. Zhang, L. Xue, C. Liang, Y. Chen, J. Liu, C. Shen, Q. Li, Y. Duan, L. Yao, H. Zhang, Y. Cai and Z. Luo, Appl. Surf. Sci., 2021, 561, 150079 CrossRef CAS.
- X. Yu, J. Zhao, L. R. Zheng, Y. Tong, M. Zhang, G. Xu, C. Li, J. Ma and G. Shi, ACS Energy Lett., 2018, 3, 237–244 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- H. Yin, S. Zhao, K. Zhao, A. Muqsit, H. Tang, L. Chang, H. Zhao, Y. Gao and Z. Tang, Nat. Commun., 2015, 6, 6430 CrossRef CAS PubMed.
- Z. Xing, C. Han, D. Wang, Q. Li and X. Yang, ACS Catal., 2017, 7, 7131–7135 CrossRef CAS.
- N. Danilovic, R. Subbaraman, D. Strmcnik, K. C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed., 2012, 124, 12663–12666 CrossRef.
- X. Xiao, X. Wang, X. Jiang, S. Song, D. Huang, L. Yu, Y. Zhang, S. Chen, M. Wang, Y. Shen and Z. Ren, Small Methods, 2020, 4, 1900796 CrossRef CAS.
- L. L. Laurent, C. Barreteau and T. Markussen, Phys. Rev. B, 2019, 100, 174426 CrossRef.
- Y. Zhao, Y. Gao, Z. Chen, Z. Li, T. Ma, Z. Wu and L. Wang, Appl. Catal., B, 2021, 297, 120395 CrossRef CAS.
- L. Zhu, Y. Sun, H. Zhu, G. Chai, Z. Yang, C. Shang, H. Ye, B. H. Chen, A. Kroner and Z. X. Guo, ACS Catal., 2022, 12, 8104–8115 CrossRef CAS.
- Y. Qin, X. Han, S. Gadipelli, J. Guo, S. Wu, L. Kang, J. Callison and Z. X. Guo, J. Mater. Chem. A, 2019, 7, 6543–6551 RSC.
- S. A. Shevlin and Z. X. Guo, Chem. Mater., 2016, 28, 7250–7256 CrossRef CAS.
- J. L. Shi, J. H. Wu, X. J. Zhao, X. L. Xue, Y. F. Gao, Z. X. Guo and S. F. Li, Nanoscale, 2016, 8, 19256–19262 RSC.
- Z. Zhang, C. Feng, C. Liu, M. Zuo, L. Qin, X. Yan, Y. Xing, H. Li, R. Si, S. Zhou and J. Zeng, Nat. Commun., 2020, 11, 1215 CrossRef CAS PubMed.
- Z. L. Liu, R. Huang, Y. J. Deng, D. H. Chen, L. Huang, Y. R. Cai, Q. Wang, S. P. Chen and S. G. Sun, Electrochim. Acta, 2013, 112, 919–926 CrossRef CAS.
- R. Ge, Y. Wang, Z. Li, M. Xu, S. M. Xu, H. Zhou, K. Ji, F. Chen, J. Zhou and H. Duan, Angew. Chem., Int. Ed., 2022, 61, e202200211 CAS.
- F. Wu, X. Guo, G. Hao, Y. Hu and W. Jiang, Adv. Mater. Interfaces, 2019, 6, 1900788 CrossRef CAS.
- Z. Li, X. Zhang, Y. Kang, C. C. Yu, Y. Wen, M. Hu, D. Meng, W. Song and Y. Yang, Adv. Sci., 2021, 8, 2002631 CrossRef CAS PubMed.
- Z. Zhang, J. Liu, J. Wang, Q. Wang, Y. Wang, K. Wang, Z. Wang, M. Gu, Z. Tang, J. Lim and T. Zhao, Nat. Commun., 2021, 12, 5235 CrossRef CAS PubMed.
- J. Wei, K. Xiao, Y. Chen, X. P. Guo, B. Huang and Z. Q. Liu, Energy Environ. Sci., 2022, 15, 4592–4600 RSC.
- K. Jiang, B. Liu, M. Luo, S. Ning, M. Peng, Y. Zhao, Y. R. Lu, T. S. Chan, F. M. de Groot and Y. Tan, Nat. Commun., 2019, 10, 1743 CrossRef PubMed.
- J. Liu, Y. Liu, X. Mu, H. Jang, Z. Lei, S. Jiao, P. Yan, M. G. Kim and R. Cao, Adv. Funct. Mater., 2022, 32, 2204086 CrossRef CAS.
- A. Pei, G. Li, L. Zhu, Z. Huang, J. Ye, Y. C. Chang, S. M. Osman, C. W. Pao, Q. Gao, B. H. Chen and R. Luque, Adv. Funct. Mater., 2022, 2208587 CrossRef CAS.
- X. Tian, P. Zhao and W. Sheng, Adv. Mater., 2019, 31, 180806631 Search PubMed.
- R. Zhang, X. Wang, S. Yu, T. Wen, X. Zhu, F. Yang, X. Sun, X. Wang and W. Hu, Adv. Mater., 2017, 29, 1605502 CrossRef PubMed.
- M. Lu, D. Chen, B. Wang, R. Li, D. Cai, H. Tu, H. Yang, Y. Zhang and W. Han, J. Mater. Chem. A, 2021, 9, 1655–1662 RSC.
- M. Qin, J. Chen, X. Zheng, M. Qi, R. Yang, S. Mao and Y. Wang, Appl. Catal., B, 2022, 316, 121602 CrossRef CAS.
- B. Hammer, Y. Morikawa and J. K. Nørskov, Phys. Rev. Lett., 1996, 76, 2141 CrossRef CAS PubMed.
- A. Ruban, B. Hammer, P. Stoltze, H. L. Skriver and J. K. Nørskov, J. Mol. Catal. A: Chem., 1997, 115, 421–429 CrossRef CAS.
- E. Christoffersen, P. Liu, A. Ruban, H. L. Skriver and J. K. Nørskov, J. Catal., 2001, 199, 123–131 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee02785b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |