
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of an elastic interphase in suppressing gas evolution and promoting uniform electroplating in sodium metal anodes†
Chen
Gong‡
a,
Shengda D.
Pu‡
a,
Shengming
Zhang‡
a,
Yi
Yuan‡
a,
Ziyang
Ning
a,
Sixie
Yang
a,
Xiangwen
Gao
a,
Chloe
Chau
a,
Zixuan
Li
a,
Junliang
Liu
a,
Liquan
Pi
a,
Boyang
Liu
a,
Isaac
Capone
a,
Bingkun
Hu
a,
Dominic L. R.
Melvin
a,
Mauro
Pasta
 a,
Peter G.
Bruce
ab and
Alex W.
Robertson
*c
a,
Peter G.
Bruce
ab and
Alex W.
Robertson
*c
aDepartment of Materials, University of Oxford, Parks Road, Oxford OX1 3PH, UK. E-mail: peter.bruce@materials.ox.ac.uk
bDepartment of Chemistry, University of Oxford, South Parks Road, Oxford OX1 3QZ, UK
cDepartment of Physics, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: alex.w.robertson@warwick.ac.uk
First published on 5th January 2023
Abstract
Ether solvent based electrolytes exhibit excellent performance with sodium battery anodes, outperforming the carbonate electrolytes that are routinely used with the analogous lithium-ion battery. Uncovering the mechanisms that facilitate this high performance for ether electrolytes, and conversely diagnosing the causes of the poor cycling with carbonate electrolytes, is crucial for informing the design of optimized electrolytes that promote fully reversible sodium cycling. An important contributor to the performance difference has been suggested to be the enhanced elasticity of the ether-derived solid–electrolyte interphase (SEI) layer, however experimental demonstration of exactly how this translates to improving the microscopic dynamics of a cycled anode remain less explored. Here, we reveal how this more elastic SEI prevents gas evolution at the interface of the metal anode by employing operando electrochemical transmission electron microscopy (TEM) to image the cycled electrode–electrolyte interface in real time. The high spatial resolution of TEM imaging reveals the rapid formation of gas bubbles at the interface during sodium electrostripping in carbonate electrolyte, a phenomenon not observed for the higher performance ether electrolyte, which impedes complete Na stripping and causes the SEI to delaminate from the electrode. This non-conformal and inflexible SEI must thus continuously reform, leading to increased Na loss to SEI formation, as supported by mass spectrometry measurements. The more elastic ether interphase is better able to maintain conformality with the electrode, preventing gas formation and facilitating flat electroplating. Our work shows why an elastic and flexible interphase is important for achieving high performance sodium anodes.
Broader contextThe development of sodium metal anodes is one of the most promising avenues for attaining high performance sodium–ion rechargeable batteries. However, they suffer from many of the same problems as the lithium metal anode, with endemic low cycling performances. Designing an appropriate electrolyte, and thereby tailoring the form of the anode's solid–electrolyte interphase (SEI), can mitigate this anode degradation. Yet understanding the link between different SEIs and the various complex degradation mechanisms the metal anode undergoes is challenging. We perform operando electrochemical transmission electron microscopy to directly image the cycling of the sodium metal anode in carbonate and ether based electrolytes. The high resolution imaging reveals intensive gas evolution occurs along the electrode interface during electrostripping when the anode is cycled in carbonate electrolyte. The formed gas displaces the electrolyte away from the electrode, preventing ionic transfer. Gas formation is suppressed with ether electrolyte, explaining the much superior cycling performance. We link these observations to the differing mechanical properties of the formed SEI. Our results highlight the crucial importance of a flexible SEI layer in achieving fully reversible Na metal cycling, and the often hidden role gas formation can have in hindering battery performance. |
Introduction
With the demand for low-cost and high-capacity energy storage systems, sodium batteries have attracted significant interest due to promising high energy density chemistries – including Na–sulfur,1–3 Na–oxygen,4–6 and Na–carbon dioxide7 – while being more resource abundant than lithium.8,9 A sodium metal anode, with its high theoretical capacity (1166 mA h g−1) and low electrochemical potential (−2.71 V versus standard hydrogen electrode), is an ideal negative electrode for these future sodium batteries.10,11 Nevertheless, uncontrolled sodium dendrite growth, excessive solid–electrolyte interphase (SEI) buildup, and electrolyte depletion during sodium electroplating and stripping all contribute toward cell fading.12–14 There has been a great deal of study into the similar failure processes that occur with the lithium metal anode,15,16 and as sodium is a fellow monovalent alkali metal it is tempting to conclude that it will exhibit consistent behaviors with lithium. This is not always the case.10,17 Important differences include sodium being more reactive to carbonate solvents,18,19 not showing the same degree of performance improvement with known lithium electrolyte additives,20,21 and the differing mechanical properties of the sodium metal dendrites.19 Dedicated study of the failure mechanisms of the sodium metal anode is necessary to understand these differences and to develop strategies tailored to improving sodium–ion battery performance.The SEI forms on the metal anode due to electrolyte decomposition and electrochemical reactions with the metal. Its structure and composition are centrally important in governing the metal electroplating and stripping performance,22 as it is the interphase that mediates ion diffusion between electrolyte and electrode. An ideal SEI should be uniform and compact, as heterogeneity will favor irregular metal deposition and lead to dendrite growth.10,23 The high instability of the sodium metal SEI, more unstable than that of lithium,24 is a major contributor to the dendrite formation and SEI overgrowth issues that lead to capacity fade in sodium metal batteries. Carbonate ester solvent-based electrolytes, widely used in lithium battery chemistries, have been found to form particularly unstable interphases on sodium metal anodes, yielding much lower coulombic efficiencies than their lithium anode counterparts.18,25 Several strategies have been employed to construct a more stable SEI and improve Na metal anode cycling performance, including adjusting the sodium salt and its concentration,26–28 using different solvents,25,29 and tailoring electrolyte additives.20,30 Employing ether solvent electrolytes has shown particularly promising results, with excellent electroplating and stripping performance; Cui et al. reported that simply changing the electrolyte from a typical carbonate solvent to ether glyme solvents yielded high-reversibility and non-dendritic sodium metal cycling.25
Changing the electrolyte alters the formed SEI, and thus can influence the anode performance. This strategy has been used to good effect in lithium metal anodes, where selecting appropriate electrolytes to tailor for a highly fluorinated interphase has led to excellent reported cycling performances.31–33 In their work, Cui et al. similarly suggested that the improved performance of the glyme based sodium electrolyte was due to it forming a more stable, uniform, and dominantly inorganic SEI.25 To develop an accurate understanding of how manipulating the electrolyte and SEI composition improves the electrochemical performance measured at the cell level, we need to diagnose precisely how it influences and changes the processes that are occurring at the electrode interface. The dynamic nature of these processes, coupled with the fragile nature of the metal–electrolyte interface, strongly favor the use of in situ characterization techniques.34–37In situ optical microscopy has been employed to study the sodium metal deposition structure from carbonate-based electrolyte, with needle-like sodium dendrites and significant levels of gas evolution discovered.30,38 However, finer structural and interfacial dynamics require the higher spatial resolution afforded by in situ liquid–cell transmission electron microscopy (TEM).39
Here, we employ operando electrochemical TEM to reveal the differences in electroplating and stripping behavior when using carbonate or ether solvent sodium electrolytes. The high resolution imaging reveals significant bubble formation with carbonate solvent electrolytes, with particularly intense gas evolution localized at the metal–electrolyte interface during electrostripping, leading to delamination of the SEI from the metal. No bubble formation at the interface was observed when cycling from an ether solvent electrolyte. Atomic force microscopy (AFM) characterization suggests the ether electrolyte forms an SEI that is better able to maintain a conformal coating of the electrode during cycling, limiting opportunities for gas producing side reactions and minimizing sodium loss to SEI reformation.
Results and discussion
1 M NaPF6 dissolved in ethylene carbonate (EC) and dimethyl carbonate (DMC), EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC = 1:1, and 1 M NaPF6 in monoglyme (dimethoxyethane, DME), were employed as representative carbonate and ether electrolytes. In order to better understand the Na degradation mechanisms in carbonate-based electrolyte, the 1 M NaPF6 in EC:DMC was also studied with the commonly used fluoroethylene carbonate (FEC) and vinylene carbonate (VC) additives (10%), as these carbonate additives are known to facilitate better performance30,40,41 and thus represent the best of the candidate carbonate solvent Na electrolytes. The electrochemical performance and impedance behavior were measured in Na‖Cu cells. In the first cycle (Fig. 1a), the carbonate electrolyte cell yielded a poor coulombic efficiency (CE) of 24%. As expected, the inclusion of VC or FEC additives improved the first cycle efficiency significantly, but still only reaching modest CEs of 70% and 74%, respectively. In contrast, the glyme electrolyte cell yielded a CE of 94%, surpassing all the carbonate electrolytes. Following ten cycles (Fig. 1b), the performance of the carbonate electrolyte cells did not change significantly, with measured CEs of 22%, 73%, and 75% for the no additive, VC additive, and FEC additive cells, respectively, suggesting that the low CE of the first cycles were not simply Na loss to initial SEI formation, but instead represent a continued loss mechanism occurring over repeated cycling. After ten cycles the ether electrolyte stabilized at a high CE of over 99.8% (Fig. 1b) and maintained it over 50 cycles (Fig. S1, ESI†), in good agreement with previous findings,25 indicating the lower CE of the first cycle was due to initial interphase formation losses, and that once formed the interphase remained stable over the subsequent cycles. Cells cycled at various rates all exhibited excellent stability with ether electrolyte (Fig. S2, ESI†), and similar resilience to capacity decay was seen in Na3V2(PO4)3/Na full cells (Fig. S3, ESI†). Electrochemical impedance spectroscopy (EIS) measurements (Fig. 1c) showed a significantly improved ionic conductivity for the ether electrolyte. Together, the cycling performance and EIS measurements suggest a critical role for the SEI in discriminating the performances of the two electrolytes.
:DMC = 1:1, and 1 M NaPF6 in monoglyme (dimethoxyethane, DME), were employed as representative carbonate and ether electrolytes. In order to better understand the Na degradation mechanisms in carbonate-based electrolyte, the 1 M NaPF6 in EC:DMC was also studied with the commonly used fluoroethylene carbonate (FEC) and vinylene carbonate (VC) additives (10%), as these carbonate additives are known to facilitate better performance30,40,41 and thus represent the best of the candidate carbonate solvent Na electrolytes. The electrochemical performance and impedance behavior were measured in Na‖Cu cells. In the first cycle (Fig. 1a), the carbonate electrolyte cell yielded a poor coulombic efficiency (CE) of 24%. As expected, the inclusion of VC or FEC additives improved the first cycle efficiency significantly, but still only reaching modest CEs of 70% and 74%, respectively. In contrast, the glyme electrolyte cell yielded a CE of 94%, surpassing all the carbonate electrolytes. Following ten cycles (Fig. 1b), the performance of the carbonate electrolyte cells did not change significantly, with measured CEs of 22%, 73%, and 75% for the no additive, VC additive, and FEC additive cells, respectively, suggesting that the low CE of the first cycles were not simply Na loss to initial SEI formation, but instead represent a continued loss mechanism occurring over repeated cycling. After ten cycles the ether electrolyte stabilized at a high CE of over 99.8% (Fig. 1b) and maintained it over 50 cycles (Fig. S1, ESI†), in good agreement with previous findings,25 indicating the lower CE of the first cycle was due to initial interphase formation losses, and that once formed the interphase remained stable over the subsequent cycles. Cells cycled at various rates all exhibited excellent stability with ether electrolyte (Fig. S2, ESI†), and similar resilience to capacity decay was seen in Na3V2(PO4)3/Na full cells (Fig. S3, ESI†). Electrochemical impedance spectroscopy (EIS) measurements (Fig. 1c) showed a significantly improved ionic conductivity for the ether electrolyte. Together, the cycling performance and EIS measurements suggest a critical role for the SEI in discriminating the performances of the two electrolytes.
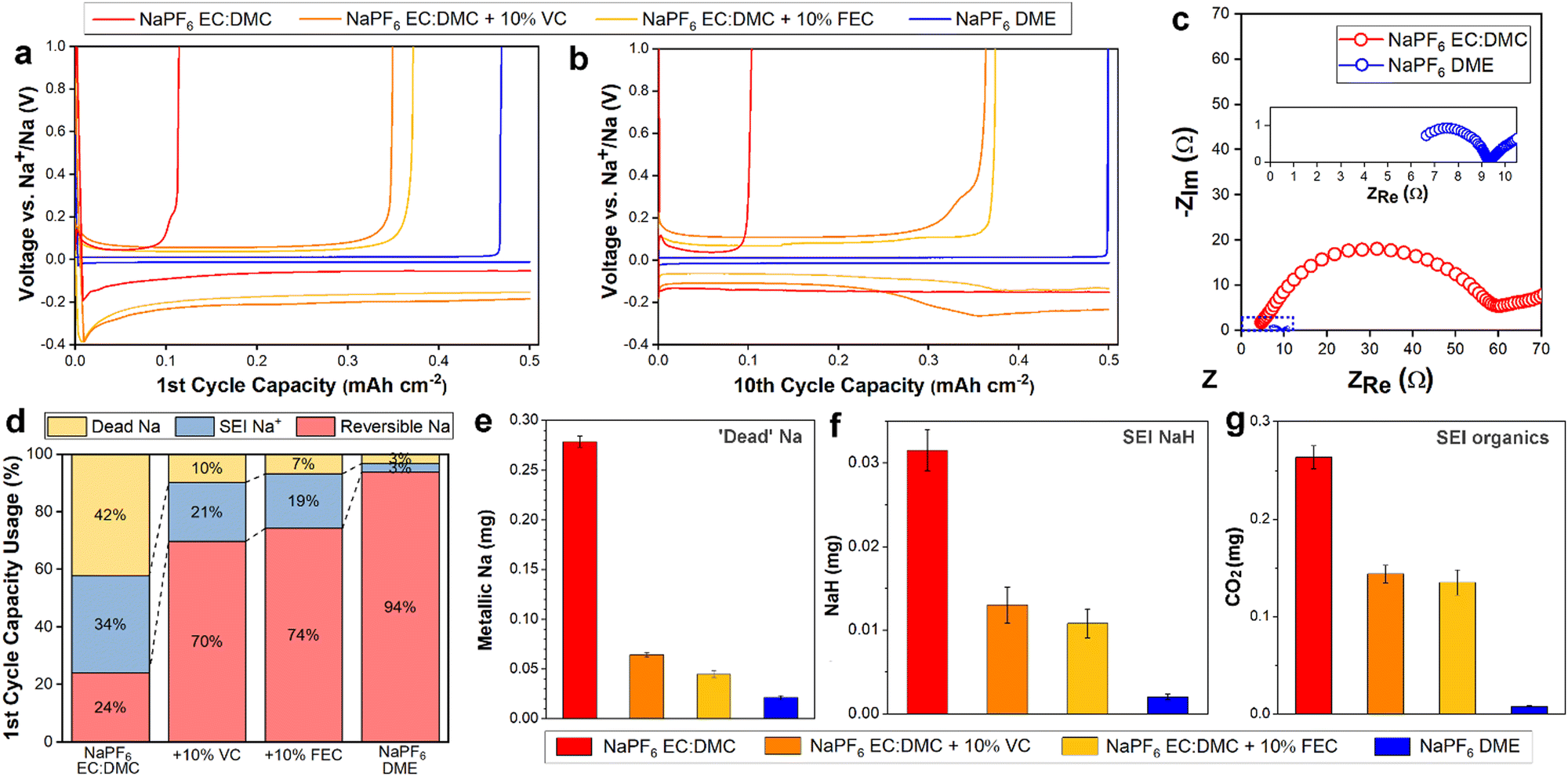 | ||
| Fig. 1 Electrochemical performance comparison of sodium electrolytes with carbonate or ether solvents. Deposition capacity at 0.5 mA cm−2 for 0.5 mA h cm−2 after the (a) first and (b) tenth charge–discharge cycle. (c) EIS characterization of the ether and carbonate Na electrolytes. Magnified view of the EIS of NaPF6 in DME shown in the insert. (d) Relative distribution of Na after the first charge–discharge cycle from titration online MS. Quantitative online MS measurements, after one cycle, of (e) ‘dead’ Na, (f) NaH, and (g) CO2, the signature of organic SEI components such as (CH2OCO2Na)2 and NaOCO2R. | ||
To diagnose the contributions of initial SEI formation to Na loss, and to better quantify the overall size and composition of the SEI, we performed online mass spectrometry (online MS) on the cycled electrolyte cells. The coin cells were plated at 0.5 mA cm−2 for 0.5 mA h cm−2, stripped at 0.5 mA cm−2 to 1 V (vs. Na+/Na) for one cycle, and then transferred to our online MS setup where they were titrated with deuterated water (D2O). By measuring the resultant gas evolution (D2, HD and CO2), the electrically isolated ‘dead’ Na and some SEI components were able to be quantitatively identified (Fig. 1d–g). The quantity of reversible Na is known from the coin cell cycling performance, while the Na consumed by SEI formation is equal to the amount of non-reversible Na minus the quantity of ‘dead’ Na. Following the first cycle, a significant quantity of Na was lost to SEI formation for the carbonate electrolytes (Fig. 1d); 34%, 21%, and 19% for the no additive, VC additive, and FEC additive electrolytes, respectively. Employing an ether solvent electrolyte instead reduced this loss to SEI formation to just 3%, less than a tenth of the relative Na loss compared to the standard carbonate electrolyte. These MS results, along with X-ray photoelectron spectroscopy (XPS) showing a rapid decrease in the intensity of the F 1s peak (indicative of NaF42) with depth for the ether electrolyte electrode (Fig. S4, ESI†), evidence a thinner SEI formed on the electrodes cycled in ether electrolyte. This is supported by the EIS (Fig. 1c), with the carbonate electrolyte resistance measurement roughly seven times higher, as the formation of a thicker SEI layer would be expected to impede transport.
Along with the thinner SEI, the ether electrolyte largely prevented Na loss due to ‘dead’ Na detachment and isolation, limiting it to 3% (Fig. 1d), significantly outperforming even the additive-including electrolytes. ‘Dead’ metal formation is understood to largely arise due to the uneven dissolution of dendritic electroplated structures, leading to metal detachment from the electrode and subsequent electrical isolation.43 Our online MS results suggest the ether electrolyte suppresses dendritic Na deposition, and thus limits possible Na detachment during electrostripping.
Analysis of the products measured, both after one cycle (Fig. 1e–g) and three cycles (Fig. S5, ESI†), allows the quantification of the inorganic SEI product NaH, and organic SEI products (CH2OCO2Na)2 and NaOCO2R (see methods). As per the collated data in Fig. 1d, the relative mass of SEI products was greater in the carbonate solvent electrolyte cells. Interphase NaH forms by reaction of sodium metal with hydrogen gas (or some sodium-based hydrated compounds),44 and has been suggested to hinder Na+ ion transport at the electrode interface.12 The measured organic SEI products can further decompose into inorganic Na2CO3,45 which has been shown to be unstable when exposed to sodium electrolytes.46 Thus the comparatively high quantities of NaH and Na organics found in the carbonate electrolyte SEIs support the poor electrochemical performances measured for them; the NaH will contribute to the insulating character of the SEI (Fig. 1c), and the organics will lead to continued SEI dissolution and replenishment and thus contribute to the sustained low CE (Fig. 1b) and high cumulative SEI product formation (Fig. S5, ESI†). In contrast, the ether electrolyte SEI remains chemically stable, with negligible increase in the quantity of SEI products measured following three cycles compared to one cycle.
The mass spectrometry measurements demonstrated that ether electrolyte limits ‘dead’ Na formation, implying that it inhibits dendritic Na morphologies that are more prone to detachment and isolation. To verify this, we employed operando TEM to directly capture the electrodeposition and dissolution processes from the ether and carbonate electrolytes. Operando electrochemical TEM with a liquid–cell permits the nanoscale visualization of the plating and stripping dynamics from the electrolytes of interest on to a Pt working electrode (WE).35 Galvanostatic electroplating and stripping at 10 mA cm−2 was performed from both DME and EC:DMC electrolytes (Fig. 2). The contrast in TEM is dependent on the relative densities of the imaged structures;47 unfortunately, the density of Na metal is very close to the density of the surrounding solvent (Table S1, ESI†), making the delineation of deposited metal morphologies challenging due to the low contrast, particularly in the case of the ether electrolyte. However, careful examination of the complete electroplating and stripping process from the ether electrolyte reveals a largely uniform deposition and dissolution of sodium (Fig. 2a). This is more clearly seen in the captured real time movie (Movie S1, ESI†). The change in intensity due to sodium plating and stripping is confirmed by box-averaged intensity profile measurements extracted from the same location across the three frames (Fig. 2b). Alongside the largely uniform plating over the WE surface, two micron sized Na structures can be seen to deposit and dissolve, as indicated with arrows in Fig. 2a. The largely flat morphology of the plated Na supports the mass spectrometry measurements, as flat plating and uniform stripping will preclude any ‘dead’ Na detachment. Cycling from the EC:DMC carbonate electrolyte yielded a more disperse deposition morphology (Fig. 2c and Movie S2, ESI†). As opposed to the darker contrast of Na plated from the ether electrolyte, Na metal from the EC:DMC electrolyte appears slightly lighter than the surrounding electrolyte due to the relative density of Na being less than the EC:DMC electrolyte (Table S1, ESI†). To highlight the areas where plating occurred, background subtraction and a false-color look up table were applied to frame 2 (Fig. 2d), revealing the irregular bushy Na deposition morphology. Post-mortem scanning electron microscopy (SEM) imaging of coin-cell electrodes electroplated with Na confirmed the looser and more porous Na morphology for deposition from carbonate solvent electrolyte (Fig. S6, ESI†), with the surface roughness difference of the electrodes also observable by eye (Fig. S7, ESI†).
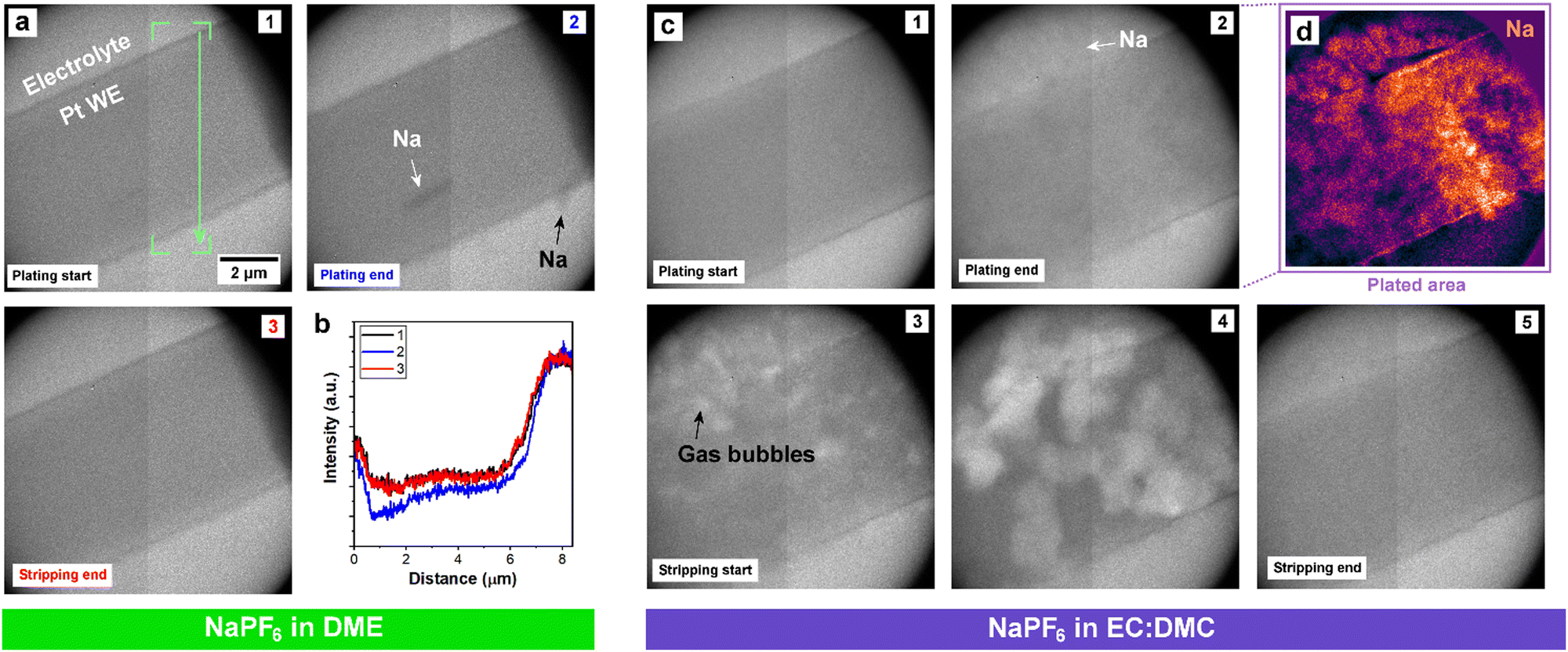 | ||
| Fig. 2 Operando electrochemical liquid–cell TEM imaging. (a) Galvanostatic cycle (10 mA cm−2) of NaPF6 in DME. Annotation in panel 1 denotes where box-averaged intensity profiles were acquired from each panel. (b) Intensity profiles extracted from panels 1 to 3. (c) Galvanostatic cycle (10 mA cm−2) of NaPF6 in EC:DMC. (d) False-color look-up table of panel 2 following background subtraction, highlighting Na deposition. | ||
Cycling from the carbonate electrolyte also revealed many localized bubbles formed across the Na metal deposit (white contrast). Bubble formation started once the bias polarity switched from galvanostatic plating to stripping. These bubbles are not a result of beam damage, as evidenced both by a control experiment (Fig. S8, ESI†), and by their immediate formation upon switching the bias polarity, which instead strongly suggest an electrochemical cause. These small gas bubbles rapidly nucleated on the Na metal surfaces, grew, and eventually dissolved into the electrolyte. Following the completion of the galvanostatic cycle, moving the sample revealed a large bubble (Movie S2, ESI†). This indicates that gas evolution from the cycling process had been sufficient to overcome the saturation limit of the electrolyte in the liquid–cell, leading to degassing/gas accumulation into the large bubble. The excessive bubble formation during stripping, localized directly on the plated Na, while no such effervescence was observed for the ether electrolyte, presents a possible mechanism for the difference in their cycling performances; bubble formation at the stripping interface physically displaces the electrolyte away from the electrode, thus will impede uniform dissolution of the plated Na and lead to the lower CEs exhibited by the carbonate electrolytes.
In order to further explore the localized bubble formation seen in the carbonate electrolyte system, operando electrochemical high angle annular dark field (HAADF) scanning mode TEM (STEM) was performed on cyclic voltammetry cycled NaPF6 in EC:DMC electrolyte with 10% FEC additive (Fig. 3). We studied the carbonate electrolyte with FEC additive as it has been shown to limit gas formation, and so represents the ‘best case’ carbonate electrolyte. HAADF-STEM is a dark field imaging technique, as such the contrast is inverted compared to TEM imaging; i.e., low density features like gas bubbles are dark and high density features like the Pt WE are bright. Faint Na deposits can be initially distinguished in Fig. 3b (white arrows), which increase in contrast with plating time (Fig. 3c). The contrast between metallic Na and the surrounding solvent is low due to the close relative densities, yet the dynamic deposition is observable in the real time movie (Movie S3, ESI†). A bubble can also be seen to have started forming away from the working electrode (Movie S3 and Fig. S9, ESI†), showing that gas formation occurs throughout the cycle and not just during stripping, in agreement with in situ optical microscopy observations. However, once the electrostripping stage of the cyclic voltammetry sweep starts, bubble formation localized at the Na metal surfaces was once more observed (Fig. 3d, yellow arrows). Interestingly, these bubbles are seen to grow toward the working electrode, expanding to fully occupy the spaces enclosed by the SEI shells (Fig. 3g and h). The electrostripping reveals apparent SEI shells left behind, delaminated from the retreating sodium metal, within which the bubbles continue to grow and are contained. It is important to note that it is not possible to reliably distinguish between Na metal and electrolyte inside the SEI shell (darker blue in Fig. 3h) due to their similar contrast, thus liquid electrolyte may be entering the SEI shell following Na dissolution, with the growing gas bubbles subsequently displacing it. These localized bubbles eventually dissipate (Fig. 3e and f, green arrows), presumably through gaps in the SEI shell.
 | ||
| Fig. 3 Operando electrochemical HAADF-STEM imaging of bubble formation from 1 M NaPF6 in EC:DMC with 10% FEC. (a–f) Liquid–cell STEM time-series of cyclic voltammetry cycling showing Na plating and stripping. (g) Magnified views showing bubble formation during stripping. (h) Illustrations corresponding to (g). (i) Cyclic voltammetry profile corresponding to panels (a–f). | ||
That the SEI shells remain protruding from the surface following stripping suggests a SEI with less elasticity forms from carbonate electrolyte. Such mechanical properties mean that the SEI cannot maintain conformality with the retreating metal; the SEI does not follow the electrode's expansion and contraction through the electroplating and stripping cycle, but rather remains at the maximum extent. This may be aided by interceding gas bubbles delaminating the SEI away from the Na metal as it is stripped. The observed detachment would necessitate the SEI to be reformed every cycle, as new Na deposition would be left exposed to the electrolyte, which would explain the sustained capacity loss to SEI formation over repeated cycling experienced by carbonate electrolytes (Fig. 1d).
To further characterize the differences in electroplated morphology, and to evaluate the mechanical properties of the SEI formed, we performed AFM imaging and nanoindentation measurement (Fig. 4 and Fig. S10, S11, ESI†). The morphology of the working electrode electroplated from ether electrolyte was much smoother compared to that plated from carbonate electrolyte (Fig. 4a and c), with a surface roughness of 26 ± 5 nm versus 208 ± 26 nm. After subsequent stripping (Fig. 4b and d), the roughness decreased to 32 ± 8 nm and 6 ± 2 nm for carbonate electrolyte and glyme electrolyte, respectively. The AFM images show that the plating and stripping of Na metal in carbonate electrolyte is highly heterogeneous, forming rough Na islands and pits across the electrode. By contrast, the electrode surface after the first cycle with ether electrolyte exhibited a smooth texture, with an even smaller roughness than that of the pristine Cu foil (roughness: 12.4 ± 2.1 nm, Fig. S12, ESI†), evidencing the excellent flat plating morphology achievable from ether Na electrolyte. This flat morphology will be robust toward detachment, and thus supports the low quantities of measured ‘dead’ Na lost with ether electrolyte (Fig. 1d). AFM indentation measurements reveal the mechanical properties of the SEI formed from the two electrolytes on the Na metal, with stiffer and good elastic performance found for the ether electrolyte SEI and more brittle character for the carbonate electrolyte SEI (Fig. S10 and S11, ESI†), which is in good agreement with similar experiments performed recently on Sn anodes.48 The less elastic SEI formed from the EC:DMC electrolyte may be attributed to the larger amount of carbonate ester (NaOCO2R) and brittle inorganic components (NaH and Na2CO3) formed, as seen in our mass spectrometry measurements (Fig. 1). These measured mechanical properties support our liquid–cell STEM observations, where the non-conformal SEI was seen to rigidly remain following Na electrostripping.
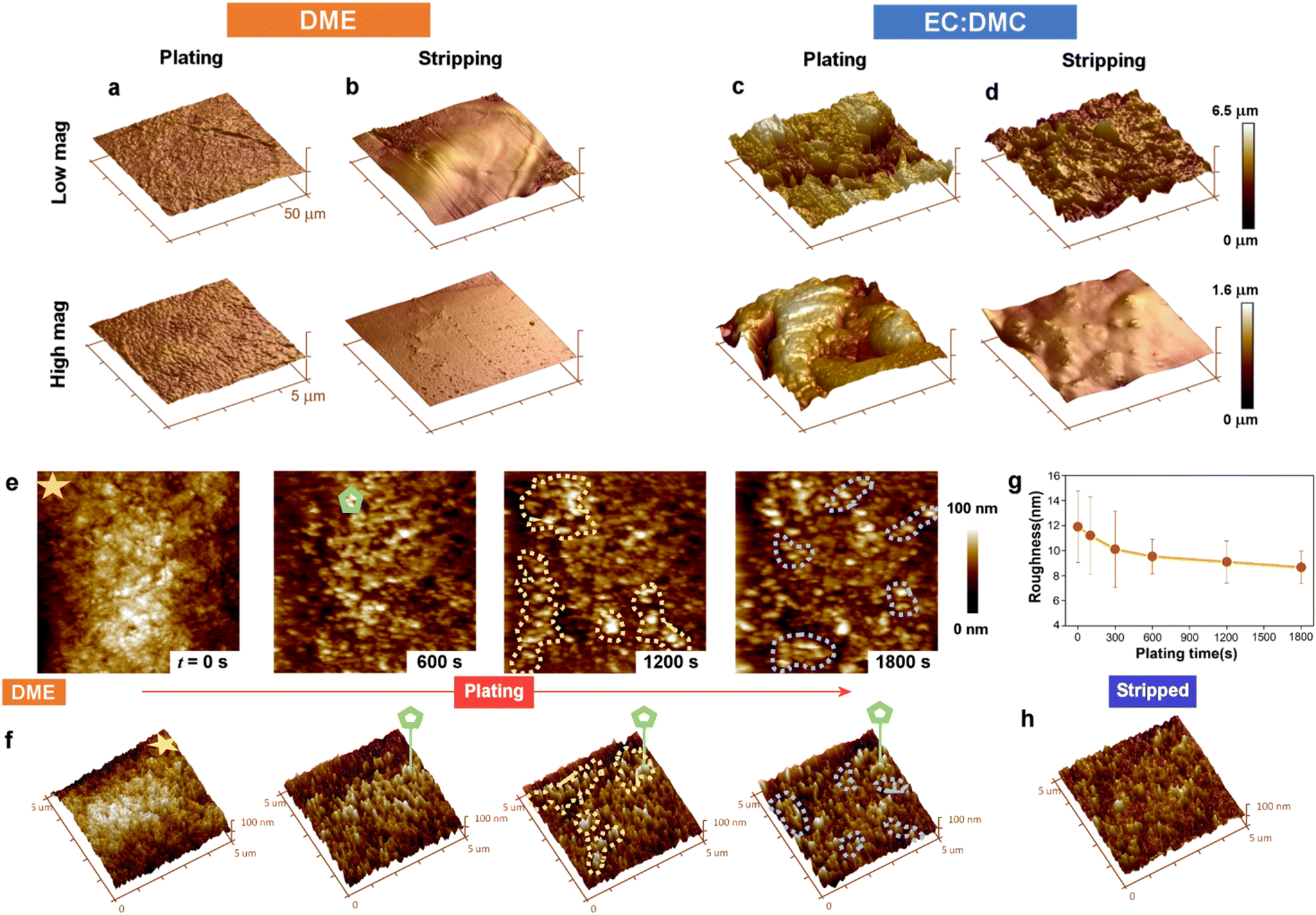 | ||
| Fig. 4 AFM surface profiling of cycled electrodes and in situ AFM imaging of electroplating and stripping. (a–d) AFM images of Cu electrodes galvanostatically cycled (plating at 0.5 mA cm−2 for 0.5 mA h cm−2) in ether and carbonate solvent Na electrolytes, imaged following (a and c) electroplating and (b and d) stripping. The height is indicated by the colour scale. (e) In situ liquid–cell AFM topography time-series of the Cu WE during Na electroplating at 0.5 mA cm−2. (f) Perspective views of the images in (a). (g) Evolution in electrode roughness with electroplating time. (h) The Cu WE after galvanostatic stripping to 1 V (vs. Na+/Na). | ||
In situ liquid–cell AFM imaging captured the roughness evolution over the course of plating, and revealed the dynamics behind the exceptionally flat plating morphology achieved with the ether solvent electrolyte (Fig. 4e–h). After 600 s of plating, Na metal was evenly distributed on the Cu working electrode. With further plating to 1200 s, Na metal was observed to grow at lower sites, highlighted by dashed yellow regions, which were further away from the counter electrode. The same phenomenon was also observed toward the end of plating (1800 s), where the regions labeled by dashed blue curves again demonstrated growth of Na metal at lower sites. The tendency of Na deposition to preferentially occur at lower sites is reflected in the decreasing surface roughness measured over the course of the plating cycle (Fig. 4g), suggesting that the deposition dynamics from ether electrolyte maintains the smoothness of the electrode surface. This electroplating behavior is unexpected, as deposition should typically occur at protrusions rather than recesses due to the higher local electric field, leading to dendrite growth.49,50 The smoothness of the electrode was found to be maintained after stripping (Fig. 4h), with a roughness of 7.1 ± 2.1 nm, confirming the good stripping performance with ether electrolyte.
A possible explanation for this unusual recess deposition mechanism may be the combination of the ether SEI's mechanical properties and its good ionic conductivity. With a higher Young's modulus and a wider elastic region of the SEI formed from the ether electrolyte, a greater compression on Na can be maintained without breakage of the SEI. The compressive stress has been modelled to have a suppression effect on the deposition kinetics by shifting the electrochemical potential at the interface.51 Such suppression of deposition kinetics would be the strongest at the tip of any deposited Na, where the elastic deformation from compressive stress would be the largest, leading to Na deposition at the valleys, as seen in our in situ AFM (Fig. 4e). The SEI layer formed from the ether electrolyte was also significantly thinner, as evidenced by our online-MS, which identified that the Na consumed to form SEI from the EC:DMC electrolyte was ten times more than for the ether solvent in the first cycle. This thinner SEI ensured faster Na+ transport across it compared with that formed from the carbonate electrolyte (Fig. 1c), facilitating a less diffusion-limited process of Na deposition at the interface along with a lower overpotential of Na deposition. This lower overpotential of Na deposition will have aided a denser and more homogeneous growth of Na. The low deposition overpotential from an ionically conductive SEI, together with a strong suppression of deposition kinetics at Na tips, might contribute to a self-regulating Na interface that maintains a flat morphology, as observed in our experiments.
Confirmation by AFM of the enhanced elastic properties of the ether derived SEI formed on Na presents a potential explanation for why interfacial bubble formation was not observed on electrostripping with the ether electrolyte. For the carbonate electrolyte derived SEI, the dissolution of Na metal during stripping led to the rigid SEI delaminating, losing conformal attachment to the anode. This leads to the exposure of previously secluded NaOCO2R and Na2CO3 SEI components to the electrolyte, where they may subsequently react with NaPF6 and release CO2 gas.46 The more elastic SEI derived from the ether electrolyte prevents this, due to it maintaining an intimate conformal contact with the retreating sodium metal during stripping, and it also being composed of fewer NaOCO2R (Fig. 1g) and Na2CO3 components.48 To confirm this mechanism we performed differential electrochemical mass spectrometry (DEMS) on sodium battery cells cycled with ether and carbonate electrolytes, and compared the measured CO2 evolved from the respective cells. The cells were sealed during operation, allowing the evolved gases to accumulate, and then following cycling the gases were released and carried to the MS instrument (see methods). The results show significantly more CO2 evolution following cycling from the carbonate electrolyte cell then from the ether electrolyte (Fig. 5). The greater levels of CO2 evolved from carbonate electrolyte cycled cells support our proposed model of its SEI being more susceptible to CO2 producing side reactions.
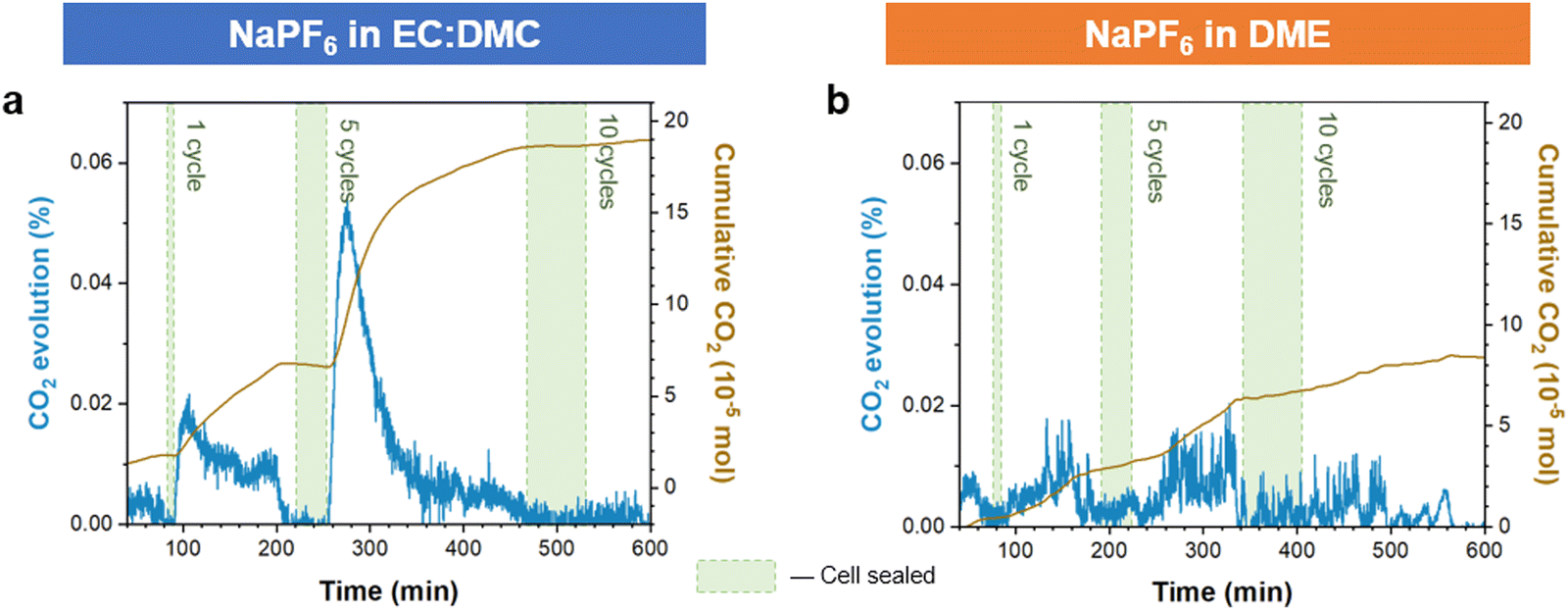 | ||
| Fig. 5 Differential electrochemical mass spectrometry (DEMS) comparison of quantified gas evolution from carbonate and ether electrolyte cells. Measured CO2 evolution following cycling with (a) carbonate (EC:DMC) and (b) ether (DME) solvent electrolytes. Shaded areas are the periods where the cell is sealed and cycled, un-shaded areas are where Ar carrier gas is flowed through the cell to the MS. The cells were cycled cumulatively for 1, 5, and finally 10 cycles at a current density of 0.5 mA cm−2. | ||
We performed further coin cell studies to illustrate how the electrolyte solvent consideration remains relevant for cells beyond that of the metal anode half-cell (i.e., the Na/Cu coin cell). Na cells cycled with hard carbon anodes demonstrate that the use of ether electrolyte drastically improves cell cycle life, with less capacity fade in comparison to an equivalent carbonate electrolyte cell (Fig. S13, ESI†). Recent works using an ether-rather than carbonate-based Na electrolyte with hard carbon anodes have shown this performance improvement as well, and have attributed it to the distinct thin and conformal character of the formed interphase layer.45,52,53 Similar performance improvement with ether solvent electrolyte have been found with Sn anodes. This suggests that the beneficial role of an ether solvent is agnostic to the anode material. The thinner SEI layer formed, with more accommodating mechanical properties that favor conformality during cycling, appears to offer generic utility for any Na anode.
Conclusion
We have explored the mechanisms behind the improved sodium anode performance when cycling in ether rather than carbonate based electrolyte. Operando electrochemical TEM revealed extensive gas bubble formation during stripping along the electrode interface when operated in carbonate electrolyte, while no such interfacial effervescence was observed when cycled with ether electrolyte. This gas formation at the interface will displace the electrolyte, and thus impede complete dissolution of plated sodium. The TEM imaging, alongside AFM, also highlighted the far smoother surface morphology for Na electroplated from ether electrolyte, which in situ AFM showed to be the result of the more elastic and robust SEI. We propose that these favorable mechanical properties also prevent gas formation from SEI reactions that occur during stripping, with the more inflexible non-conformal and brittle SEI derived from carbonate electrolyte more prone to delaminating from the Na and undergoing CO2 producing side reactions. The SEI was imaged by operando TEM to remain fixed in place, losing conformality with the electrode during electrostripping, and thus exposing fresh areas to side reactions with the electrolyte. Our work shows the critical importance of designing electrolytes such that they yield an elastic and robust SEI layer, as these properties promote uniform flat electroplating and inhibit gas producing side reactions. Realizing this beneficial SEI by employing ether electrolytes will still require accommodation for their relatively poor high-voltage stability,54,55 which currently prevent their use with high-voltage cathode materials.56 Continued research into strategies that overcome their limited oxidation stability are ongoing,57–59 and include employing a high salt concentration,60,61 forming a localized high concentration via use of a cosolvent,62 or by inclusion of stabilizing additives.63,64Methods
Electrochemistry
All the electrolyte and coin cell preparation were performed in an argon-filled glove-box (H2O < 0.1 ppm, O2 < 0.1 ppm). The prepared electrolytes were 1 M NaPF6 in EC:DMC = 1:1 (v/v) (battery grade, Kishida Chemical) with and without 10 wt% FEC and VC (anhydrous, ≥99%, Sigma-Aldrich), and 1 M NaPF6 in DME (anhydrous, ≥99.5%, Sigma-Aldrich). The hard carbon and Na3V2(PO4)3 cathode were both purchased from MTI Kejing Corporation (Shenzhen, China), and were used without any further processing. The active mass loading of hard carbon and/or Na3V2(PO4)3 used was around 3 mg cm−2. The water content of prepared electrolytes was measured by Karl Fischer titration (C30S Coulometric KF Titrator, Mettler Toledo) with methanol-free reagents three times, each showing a water content value of <5 ppm. During the electrolyte preparation process, all the tools we used (which include syringes, vials, tweezers, etc.) were dried in vacuum oven for over 12 hours prior to bringing into the glovebox. The coin-cells for online MS were Na‖Cu CR2032 coin cells with two pieces of glass microfiber separators (Whatman GF/D, dried under vacuum oven). They were plated at 0.5 mA cm−2 for 0.5 mA h cm−2 and then stripped at 0.5 mA cm−2 to 1 V. The electrochemical measurements were performed on a Biologic VMP3 system.
Real-time liquid–cell (S)TEM
We used a Protochips Poseidon 510 TEM holder to flow electrolyte into the liquid–cell with a syringe pump, as per our previous work.32 A continually replenished thin layer of electrolyte was thus confined between two Si–SiN chips inside the vacuum of the TEM. The flow rate was 120 μL h−1 during (S)TEM imaging and 240 μL h−1 for electrolyte replenishment after each cycle. Before we introduced the electrolyte we flowed dried DMC or DME for 40 minutes at a 240 μL h−1 flow rate. A Gamry reference 600 was used for cyclic voltammetry and galvanotactic measurements between the reference, working and counter Pt electrodes patterned on the electrochemical chip. The TEM and high angle annular dark field (HAADF) STEM imaging were performed with a JEOL 3000F (300 kV) using a 50-micron condenser aperture. The beam effect is shown in Fig. S6 (ESI†), suggesting the beam dose used was acceptable. The STEM beam current was calibrated by a Faraday cup (10 pA). All of the STEM images were recorded with a pixel dwell time of 3 μs pixel−1 and at 512 × 512 pixels (calculated pixel size of 1.2 × 104 Å2). These imaging conditions correspond to a radiation dose of ∼1.6 × 10−2 e− Å−2. All the electrolyte preparation was performed in an argon glovebox or sealed systems. Cyclic voltammetry used a scan rate of 20 mV s−1.XPS
The Na was deposited from glyme or EC:DMC electrolytes in Na‖Cu coin cells at a current density of 0.5 mA cm−2 for 1 hour. XPS measurements were conducted using a PHI5000 Versa Probe III instrument (Ulvac-PHI, INC.). An Al monochromatic source was used to generate X-rays using a power of 25 W, voltage of 15 kV, and a beam spot size of 100 μm. A pass energy of 55 eV was set for the analyzer. An electron neutralizer gun was used to prevent any surface charge build-up. Depth profiling of samples was done by an Ar ion source at 2 kV and 1.8 μA over an area of 3 × 3 mm2 for 60 seconds and 660 seconds, respectively. The spectra were calibrated according to the signal of adventitious carbon at 284.8 eV. The results were analyzed and fitted via CASAXPS software. Samples were transferred from the glovebox into the chamber via a sealed Ar-filled vessel without exposure to air.Online MS
The procedure for online MS (built in-house) is illustrated in our previous work (Fig. S14, ESI†).32 First, the cycled Na‖Cu CR2032 coin-cells, assembled with two glass microfiber separators (Whatman GF/D, dried under vacuum oven) and plated at 0.5 mA cm−2 for 0.5 mA h cm−2 and then stripped at 0.5 mA cm−2 to 1 V (vs. Na+/Na) for one cycle, were disassembled in an argon glovebox. Then the copper foil and the glass fiber separators on the Cu side were transferred into a well-sealed vial (along with a magnetic stirrer) without further treatment. After the vial was connected to the MS the gas composition inside the vial was analyzed and recorded. When the gas content was stable (checking no D2, O2, CO2 and almost 100% Ar carrier gas), excess degassed D2O (>99.96 atom %D, Sigma-Aldrich) was injected into the vial and then the released D2, HD and CO2 were detected by MS (Prima BT, Thermo Fisher Scientific). Finally, the quantity of metallic Na, NaH and some organic SEI species from the cycling can be calculated from the amount of D2, HD and new CO2 gas detected by MS, according to the following reactions:43| 2Na + 2D2O = 2NaOD + D2↑ | (1) |
| NaH + D2O = NaOD + HD↑; | (2) |
| 2NaOCO2R + D2O = Na2CO3 + 2ROD + CO2↑ | (3) |
| (CH2OCO2Na)2 + D2O = Na2CO3 + (CH2OD)2 + CO2↑ | (4) |
Here, Vm is molar volume of gas (24.79 L mol−1 at 25 °C) and P is the percentage of the target gas in the carrier gas stream. More setup details can be found in our previous work.65,66

We ran a ‘blank’ experiment to control for any potential gas emission from other components of the cell, e.g. the separator, electrodes, etc. For this we prepared an identical coin cell but did not subject it to cycling. This was then disassembled and characterised by online MS as per the standard experiments. The results showed essentially no emission of HD or D2 for the control cell.
DEMS
The sealed vial cell was assembled in a glove box, using Na metal attached to a stainless steel mesh as the working electrode and a Cu foil as the counter electrode. Input and output gas channels were supplied into the vial, and an isolation valve allowed carrier gas to flow to the MS while bypassing the cell. This valve was shut during cycling experiments (of 1 cycle, 5 cycles, and 10 cycles, in that order), with gas produced by the cycling thus sealed inside the vial. Following cycling the valve was opened and the evolved gases, conveyed by the Ar carrier and across a cold-trap to condense any solvent vapor, were detected and recorded by MS. The cell was cycled at a rate of 0.5 mA cm−2, electroplated for 10 min, and electrostripped to a cut-off voltage of +1 V vs. Na.AFM
The Bruker Dimension Icon AFM was used to characterise both ex situ and in situ samples in a glovebox filling with argon (<0.1 ppm H2O, <0.1 ppm O2). All AFM probes used were calibrated according to standard samples (Sapphire and Ti roughness sample from Bruker), interpolating the actual spring constant and the tip radius. The PeakForce QNM model was conducted to capture the topography of cycled electrodes corresponding mechanical data. The 3D topography was reconstructed by NanoScope Analysis 2.0 software with calculated root-mean-square roughness values from at least three different regions. The mechanical nanoindentation experiments were conducted in a 5 μm × 5 μm region with over 100 points evenly disturbed in the field of view. The AFM probes used have a spring constant of around 20 N m−1 with a tip radius of 10 nm. To measure the Young's modulus of the SEI on the cycled electrode without any plastic deformation, the PeakForce setpoint was controlled to 20 nN within the elastic region. The higher 350 nN setpoint was deliberately applied to penetrate and measure the elastic limit of the SEI on the cycled electrode. The force response curves were fitted by Derjaguin–Muller–Toporov (DMT) model to calculate the Young's modulus. All the Young's modulus and elastic region results were statistically summarised to be representative for the samples. This methodology has been widely used to characterise the mechanical of the SEI.48For the in situ AFM study on the DME electrolyte, a closed electrochemical cell with three electrodes was used, as illustrated in Fig. S15 (ESI†). Cu foil was used as the working electrode, a concentric ring of Na metal was used as the counter electrode, and a flake of Na metal on the Cu wire was used as the reference electrode. After the AFM probe approached the cell and immersed into the electrolyte, the cell was closed by a rubber ring around the probe holder. The cell was connected to a Gamry potentiostat to electrochemically plate Na onto the Cu working electrode. The PeakForce QNM in fluid mode was used with fluid-compatible probes with a spring constant around 15 N m−1. After the pristine scan on the Cu surface, the topography of the working electrode was captured during plating from the ether electrolyte at 0.5 mA cm−2 with different capacities of Na plated. The pristine morphology of Cu foil under liquid electrolyte was captured and compared to an identical scan performed in the air, verifying the morphology and capture resolution excluding any side effect from liquid electrolyte (Fig. S12, ESI†).
SEM and FIB
Electroplated Na (plating at 0.5 mA cm−2 for 0.5 mA h cm−2) were characterized by a Carl Zeiss Merlin SEM. The cross-sectioning of plated Na (plating at 0.5 mA cm−2 for 3 mA h cm−2) was examined by Thermo Scientific Helios G4 Plasma FIB DualBeam (or PFIB) system. The cells were disassembled in a glove box, and the sample transfer process was performed by an air-tight holder, making sure the samples were not contaminated by air.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the support and acknowledge the use of the facilities of the DCCEM, at the Materials Department, Oxford (EP/R010145/1). P. G. B. is indebted to the Engineering and Physical Sciences Research Council (EPSRC), including the SUPERGEN Energy Storage Hub (EP/L019469/1), Enabling Next Generation Lithium Batteries (EP/M009521/1), Henry Royce Institute for Advanced Materials, and the Faraday for financial support. A. W. R. thanks the support of the Royal Society.References
- N. Tanibata, H. Tsukasaki, M. Deguchi, S. Mori, A. Hayashi and M. Tatsumisago, Solid State Ionics, 2017, 311, 6–13 CrossRef CAS.
- H. Ye, L. Ma, Y. Zhou, L. Wang, N. Han, F. Zhao, J. Deng, T. Wu, Y. Li and J. Lu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13091–13096 CrossRef CAS PubMed.
- Y. Wang, B. Zhang, W. Lai, Y. Xu, S. Chou, H. Liu and S. Dou, Adv. Energy Mater., 2017, 7, 1602829 CrossRef.
- Q. Sun, Y. Yang and Z.-W. Fu, Electrochem. Commun., 2012, 16, 22–25 CrossRef CAS.
- C. Xia, R. Black, R. Fernandes, B. Adams and L. F. Nazar, Nat. Chem., 2015, 7, 496–501 CrossRef CAS PubMed.
- J. T. Frith, I. Landa-Medrano, I. Ruiz de Larramendi, T. Rojo, J. R. Owen and N. Garcia-Araez, Chem. Commun., 2017, 53, 12008–12011 RSC.
- S. Xu, Y. Lu, H. Wang, H. D. Abruña and L. A. Archer, J. Mater. Chem. A, 2014, 2, 17723–17729 RSC.
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- H. S. Hirsh, Y. Li, D. H. S. Tan, M. Zhang, E. Zhao and Y. S. Meng, Adv. Energy Mater., 2020, 10, 2001274 CrossRef CAS.
- B. Lee, E. Paek, D. Mitlin and S. W. Lee, Chem. Rev., 2019, 119, 5416–5460 CrossRef CAS PubMed.
- X. Zheng, L. Huang, X. Ye, J. Zhang, F. Min, W. Luo and Y. Huang, Chem, 2021, 7, 2312–2346 CAS.
- Y. Xiang, G. Zheng, Z. Liang, Y. Jin, X. Liu, S. Chen, K. Zhou, J. Zhu, M. Lin, H. He, J. Wan, S. Yu, G. Zhong, R. Fu, Y. Li and Y. Yang, Nat. Nanotechnol., 2020, 15, 883–890 CrossRef CAS PubMed.
- A. P. Cohn, N. Muralidharan, R. Carter, K. Share and C. L. Pint, Nano Lett., 2017, 17, 1296–1301 CrossRef CAS PubMed.
- L. Fan and X. Li, Nano Energy, 2018, 53, 630–642 CrossRef CAS.
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J. G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC.
- C. Fang, X. Wang and Y. S. Meng, Trends Chem., 2019, 1, 152–158 CrossRef CAS.
- J. Hu, H. Wang, S. Wang, Y. Lei, L. Qin, X. Li, D. Zhai, B. Li and F. Kang, Energy Storage Mater., 2021, 36, 91–98 CrossRef.
- D. I. Iermakova, R. Dugas, M. R. Palacín and A. Ponrouch, J. Electrochem. Soc., 2015, 162, A7060–A7066 CrossRef CAS.
- Y.-S. Hong, N. Li, H. Chen, P. Wang, W.-L. Song and D. Fang, Energy Storage Mater., 2018, 11, 118–126 CrossRef.
- R. Dugas, A. Ponrouch, G. Gachot, R. David, M. R. Palacin and J. M. Tarascon, J. Electrochem. Soc., 2016, 163, A2333–A2339 CrossRef CAS.
- A. Rudola, D. Aurbach and P. Balaya, Electrochem. Commun., 2014, 46, 56–59 CrossRef CAS.
- E. Peled, J. Electrochem. Soc., 1979, 126, 2047–2051 CrossRef CAS.
- S. Wei, S. Choudhury, J. Xu, P. Nath, Z. Tu and L. A. Archer, Adv. Mater., 2017, 29, 1605512 CrossRef PubMed.
- R. Mogensen, D. Brandell and R. Younesi, ACS Energy Lett., 2016, 1, 1173–1178 CrossRef CAS.
- Z. W. Seh, J. Sun, Y. Sun and Y. Cui, ACS Cent. Sci., 2015, 1, 449–455 CrossRef CAS PubMed.
- I. A. Shkrob, T. W. Marin, Y. Zhu and D. P. Abraham, J. Phys. Chem. C, 2014, 118, 19661–19671 CrossRef CAS.
- L. Schafzahl, I. Hanzu, M. Wilkening and S. A. Freunberger, ChemSusChem, 2017, 10, 401–408 CrossRef CAS PubMed.
- J. Zheng, S. Chen, W. Zhao, J. Song, M. H. Engelhard and J.-G. Zhang, ACS Energy Lett., 2018, 3, 315–321 CrossRef CAS.
- J. Lee, Y. Lee, J. Lee, S.-M. Lee, J.-H. Choi, H. Kim, M.-S. Kwon, K. Kang, K. T. Lee and N.-S. Choi, ACS Appl. Mater. Interfaces, 2017, 9, 3723–3732 CrossRef CAS PubMed.
- R. Rodriguez, K. E. Loeffler, S. S. Nathan, J. K. Sheavly, A. Dolocan, A. Heller and C. B. Mullins, ACS Energy Lett., 2017, 2, 2051–2057 CrossRef CAS.
- C. Cui, C. Yang, N. Eidson, J. Chen, F. Han, L. Chen, C. Luo, P. Wang, X. Fan and C. Wang, Adv. Mater., 2020, 32, 1906427 CrossRef CAS PubMed.
- C. Gong, S. D. Pu, X. Gao, S. Yang, J. Liu, Z. Ning, G. J. Rees, I. Capone, L. Pi, B. Liu, G. O. Hartley, J. Fawdon, J. Luo, M. Pasta, C. R. M. Grovenor, P. G. Bruce and A. W. Robertson, Adv. Energy Mater., 2021, 11, 2003118 CrossRef CAS.
- C. Wang, Y. S. Meng and K. Xu, J. Electrochem. Soc., 2019, 166, A5184–A5186 CrossRef CAS.
- Y. Wu and N. Liu, Chem, 2018, 4, 438–465 CAS.
- S. Pu, C. Gong and A. W. Robertson, R. Soc. Open Sci., 2020, 7, 191204 CrossRef CAS PubMed.
- D. Liu, Z. Shadike, R. Lin, K. Qian, H. Li, K. Li, S. Wang, Q. Yu, M. Liu, S. Ganapathy, X. Qin, Q. Yang, M. Wagemaker, F. Kang, X. Yang and B. Li, Adv. Mater., 2019, 31, 1806620 CrossRef PubMed.
- Y. Yuan, K. Amine, J. Lu and R. Shahbazian-Yassar, Nat. Commun., 2017, 8, 15806 CrossRef CAS.
- Y. Yui, M. Hayashi and J. Nakamura, Sci. Rep., 2016, 6, 1–8 CrossRef PubMed.
- Z. Zeng, P. Barai, S. Y. Lee, J. Yang, X. Zhang, W. Zheng, Y. S. Liu, K. C. Bustillo, P. Ercius, J. Guo, Y. Cui, V. Srinivasan and H. Zheng, Nano Energy, 2020, 72, 104721 CrossRef CAS.
- Y. Lee, J. Lee, J. Lee, K. Kim, A. Cha, S. Kang, T. Wi, S. J. Kang, H.-W. Lee and N.-S. Choi, ACS Appl. Mater. Interfaces, 2018, 10, 15270–15280 CrossRef CAS PubMed.
- M. Han, C. Zhu, T. Ma, Z. Pan, Z. Tao and J. Chen, Chem. Commun., 2018, 54, 2381–2384 RSC.
- X. Zheng, Z. Gu, X. Liu, Z. Wang, J. Wen, X. Wu, W. Luo and Y. Huang, Energy Environ. Sci., 2020, 13, 1788–1798 RSC.
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M. H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed.
- Y. Tian, Y. Sun, D. C. Hannah, Y. Xiao, H. Liu, K. W. Chapman, S.-H. Bo and G. Ceder, Joule, 2019, 3, 1037–1050 CrossRef CAS.
- H. S. Hirsh, B. Sayahpour, A. Shen, W. Li, B. Lu, E. Zhao, M. Zhang and Y. S. Meng, Energy Storage Mater., 2021, 42, 78–87 CrossRef.
- G. G. Eshetu, T. Diemant, M. Hekmatfar, S. Grugeon, R. J. Behm, S. Laruelle, M. Armand and S. Passerini, Nano Energy, 2019, 55, 327–340 CrossRef CAS.
- B. L. Mehdi, J. Qian, E. Nasybulin, C. Park, D. A. Welch, R. Faller, H. Mehta, W. A. Henderson, W. Xu, C. M. Wang, J. E. Evans, J. Liu, J. G. Zhang, K. T. Mueller and N. D. Browning, Nano Lett., 2015, 15, 2168–2173 CrossRef CAS PubMed.
- J. Huang, X. Guo, X. Du, X. Lin, J.-Q. Huang, H. Tan, Y. Zhu and B. Zhang, Energy Environ. Sci., 2019, 12, 1550–1557 RSC.
- Z. Li, J. Huang, B. Yann Liaw, V. Metzler and J. Zhang, J. Power Sources, 2014, 254, 168–182 CrossRef CAS.
- S. D. Pu, C. Gong, X. Gao, Z. Ning, S. Yang, J.-J. Marie, B. Liu, R. A. House, G. O. Hartley, J. Luo, P. G. Bruce and A. W. Robertson, ACS Energy Lett., 2020, 5, 2283–2290 CrossRef CAS.
- E. Wang, Y. Niu, Y.-X. Yin and Y.-G. Guo, ACS Mater. Lett., 2021, 3, 18–41 CrossRef CAS.
- Y. He, P. Bai, S. Gao and Y. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 41380–41388 CrossRef CAS PubMed.
- Y.-E. Zhu, L. Yang, X. Zhou, F. Li, J. Wei and Z. Zhou, J. Mater. Chem. A, 2017, 5, 9528–9532 RSC.
- S. Jiao, X. Ren, R. Cao, M. H. Engelhard, Y. Liu, D. Hu, D. Mei, J. Zheng, W. Zhao, Q. Li, N. Liu, B. D. Adams, C. Ma, J. Liu, J.-G. Zhang and W. Xu, Nat. Energy, 2018, 3, 739–746 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- X. Fan and C. Wang, Chem. Soc. Rev., 2021, 50, 10486–10566 RSC.
- D. Ba, Q. Gui, W. Liu, Z. Wang, Y. Li and J. Liu, Nano Energy, 2022, 94, 106918 CrossRef CAS.
- X. Peng, T. Wang, B. Liu, Y. Li and T. Zhao, Energy Environ. Sci., 2022, 15, 5350–5361 RSC.
- H. Liang, Z. Gu, X. Zhao, J. Guo, J. Yang, W. Li, B. Li, Z. Liu, W. Li and X. Wu, Angew. Chem., Int. Ed., 2021, 60, 26837–26846 CrossRef CAS PubMed.
- X. Ren, L. Zou, S. Jiao, D. Mei, M. H. Engelhard, Q. Li, H. Lee, C. Niu, B. D. Adams, C. Wang, J. Liu, J.-G. Zhang and W. Xu, ACS Energy Lett., 2019, 4, 896–902 CrossRef CAS.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed.
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. Niu, B. E. Matthews, Z. Zhu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J.-G. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef CAS.
- J. Zhang, H. Zhang, L. Deng, Y. Yang, L. Tan, X. Niu, Y. Chen, L. Zeng, X. Fan and Y. Zhu, Energy Storage Mater., 2023, 54, 450–460 CrossRef.
- X. Liu, Y. Yang, Y. Li, L. Wu, H. Yu, J. Zhang, Y. Liu and Q. Zhao, Energy Adv., 2022, 1, 872–876 RSC.
- M. M. Ottakam Thotiyl, S. A. Freunberger, Z. Peng and P. G. Bruce, J. Am. Chem. Soc., 2013, 135, 494–500 CrossRef CAS PubMed.
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563–566 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee02606f |
| ‡ These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2023 |