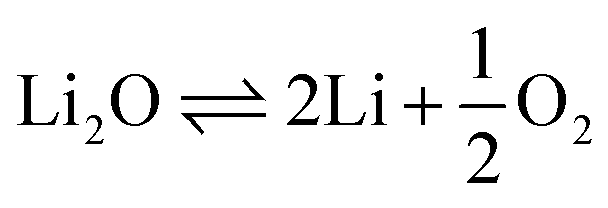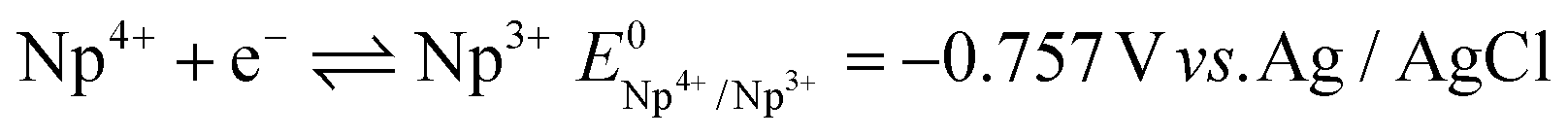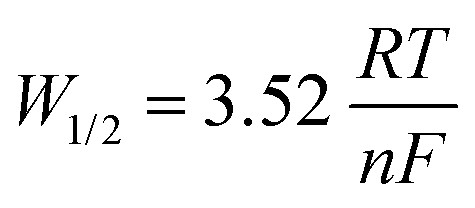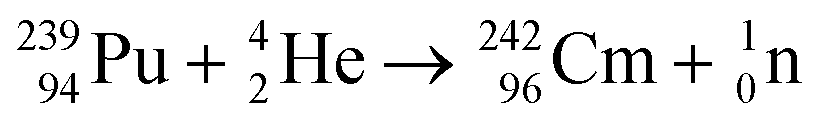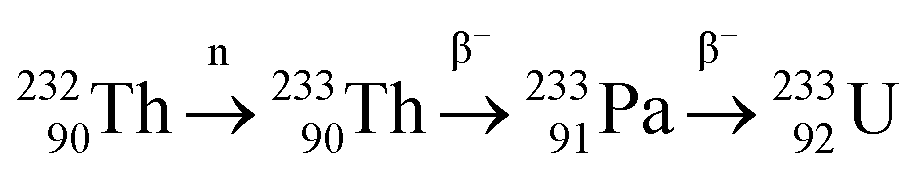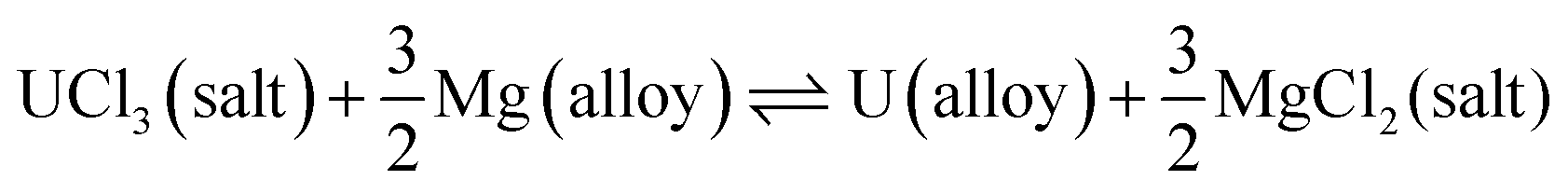DOI:
10.1039/D2EE02010F
(Review Article)
Energy Environ. Sci., 2023,
16, 952-982
Electrochemical processing in molten salts – a nuclear perspective
Received
24th June 2022
, Accepted 20th January 2023
First published on 17th February 2023
Abstract
Nuclear power provides a sustainable, long-term energy source that is an important technology in efforts to curb greenhouse gas emissions. However, issues related to construction, safety, risk of proliferation, poor public perception and the perpetual issue of end-of-life waste management remain of serious concern. A large body of work has employed molten salt-based electrochemical techniques as practical and viable methods to reprocess spent nuclear fuel and nuclear materials, in addition to producing refractory metals of broader technological relevance. This review summarises the essential aspects of the design of electrochemical reactors for electroreduction, -winning and -refining technologies. Specifically, a critique of materials choice in relation to temperature is presented alongside the effects of different metal oxide feed precursors and molten salt selection. A thorough discourse is provided on each of the actinides (Am, Cm, Np, Pu, Th and U) and lanthanides (Eu) with an evaluation of the performance of several precursor cell types and designs. The type of feed precursor, metal oxide-to-salt ratio and oxide ion concentration are factors that must be considered with regards to current efficiency and are critical to the scaling of current laboratory technologies to pilot-scale. However, a challenge remains in this transition due to potential irradiation hazards of materials under a neutron flux. There is still a need to understand the mechanism by which different radioisotopes are formed and their interaction with metallic compounds in molten salts, yet data in this area is scarce and further work is needed.
Broader context
From broad global consensus on the negative impact of CO2 emission, the world's industrial nations are moving to tackle climate change issues and transition to a low carbon energy era. As the global population continues to grow, so too does our need for more electricity and water, potentially leading to steep increments in anthropogenic greenhouse-gas emissions if alternative, low-carbon, sources of power are not adopted. Of all low carbon energy generation forms, nuclear power remains one of the few robust technologies that can be deployed on a large-scale to deliver ample ‘green’ electricity at the scale required. Arguably the greatest challenge for civilian nuclear power is the need to deal with spent fuel. This review examines the recycling strategies that have been employed from a civilian and defence perspective, with a focus on the use of molten salt technologies as a recycling process for the most significant lanthanides and actinides. Different models are explored to provide a comprehensive understanding of electrochemical reduction pathways and advanced experimental techniques to understand and optimise electrochemical processing in molten salts. The work shows that advances in molten salts research and development lie at the intersection between computation, electrochemistry, materials science and engineering and it is proposed that with a strong interdisciplinary approach we can continue to make headway in our commitment to achieving a net-zero economy.
|
1 Introduction
Ensuring the sustainable supply of future energy demands remains one of our greatest global challenges. With a growing world population aspiring to higher living standards and the expectation to achieve increasingly ambitious emissions targets, this places immense pressure on the transport, industry and power sectors. Nuclear energy is a well-established and reliable low-carbon energy source. The current 440 power reactors in operation generate 10% of the world's electricity supply.1 To increase this capacity, more must be done to maximise the operating lifetime and economics of nuclear power plants, as well as effectively and safely managing the spent fuel generated.2 However, there continues to be controversy surrounding the use of nuclear energy, namely due to operational safety, production and disposal procedures for radioactive waste, and public perception and acceptance. To mitigate some of these factors, there has been considerable work undertaken to understand how spent fuel can be reused using a combination of both chemical and electrochemical processes to reduce and/or refine the actinide metal oxides. The ability to reprocess spent fuel may lead to a much more sustainable and publicly-acceptable energy technology offering. But this relies on the careful selection of reactor systems and complementary fuel cycles which together can advance future nuclear fuel cycle strategies.
During the Cold War era, the US was regarded as the leader in spent fuel reprocessing. This included the extraction of weapons-grade plutonium from spent nuclear fuel; and took place via one of two processes: either precipitation or solvent extraction. Previously, during World War II, the bismuth phosphate (BiPO4) process was employed as part of the Manhattan Project as a method of coprecipitating pure plutonium with bismuth from irradiated nuclear fuel.3 Much of the large-scale work was carried out at the Hanford site near Richland, Washington.4 The principle behind this process lies in the dissimilar solubilities of uranium and plutonium. U6+ is far more soluble in sulfuric acid and will form sulfates, whereas plutonium (Pu3+ and Pu4+) is insoluble and co-precipitates with BiPO4 to form a compact cake.5 This product is then dissolved in nitric acid and oxidised by the addition of sodium or potassium dichromate to separate Pu6+ from BiPO4.6 Thereafter, several more purification steps are required to separate plutonium to a suitable degree of purity. Although a successful process it had the disadvantage of not allowing the recovery of uranium and generated a large amount of uranium containing radioactive waste.
In a later development, Herbert H. Anderson and Larned B. Asprey, who were participants of the Manhattan Project; an R&D programme comprising 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 workers to develop the first nuclear weapons during World War II,7,8 co-patented the plutonium–uranium reduction extraction (PUREX) process.9 The PUREX process is a mature liquid–liquid extraction technique that works by using an extractant, tri-n-butylphosphate (TBP) diluted in paraffin (kerosene) and a salting agent, nitric acid, from which separated uranium and plutonium can be recovered and purified.10 The desired actinide species are separated through a complexation process; in the aqueous phase (nitric acid) the preferential solubility of actinide complexes decreases with respect to the to the organic phase (TBP) as the molarity of the nitric acid increases.11 The facile recovery of nitric acid via distillation as well as the low volatility and high flash point of TBP offers two key advantages with respect to safety and waste requirements.10 The PUREX process has since been modified and adapted by the UK, France and Japan, who have implemented similar designs, resulting in the development of second-generation reprocessing plants.12 Third-generation civil processing plants began in the 1980s when the PUREX process went through another transformation resulting in the reduction of radioactive effluent waste.11 More recently both reprocessing facilities in Sellafield, UK, namely the Magnox and thermal oxide reprocessing plant (THORP) have now closed following the end of their design life.13Table 1 shows the current worldwide commercial spent nuclear fuel reprocessing capacity.
000 workers to develop the first nuclear weapons during World War II,7,8 co-patented the plutonium–uranium reduction extraction (PUREX) process.9 The PUREX process is a mature liquid–liquid extraction technique that works by using an extractant, tri-n-butylphosphate (TBP) diluted in paraffin (kerosene) and a salting agent, nitric acid, from which separated uranium and plutonium can be recovered and purified.10 The desired actinide species are separated through a complexation process; in the aqueous phase (nitric acid) the preferential solubility of actinide complexes decreases with respect to the to the organic phase (TBP) as the molarity of the nitric acid increases.11 The facile recovery of nitric acid via distillation as well as the low volatility and high flash point of TBP offers two key advantages with respect to safety and waste requirements.10 The PUREX process has since been modified and adapted by the UK, France and Japan, who have implemented similar designs, resulting in the development of second-generation reprocessing plants.12 Third-generation civil processing plants began in the 1980s when the PUREX process went through another transformation resulting in the reduction of radioactive effluent waste.11 More recently both reprocessing facilities in Sellafield, UK, namely the Magnox and thermal oxide reprocessing plant (THORP) have now closed following the end of their design life.13Table 1 shows the current worldwide commercial spent nuclear fuel reprocessing capacity.
Table 1 Worldwide commercial spent nuclear fuel reprocessing capacity14
| Type of reprocessing capacity |
Location (design) |
Reactor |
Capacity (t year−1) |
|
N.B. The reprocessing facilities based in Mayak, Russia are not strictly civilian as it was and still is a storage site for weapons-grade plutonium and highly-enriched uranium.15
Operation to start in 2024.
|
| Light-water reactor (LWR) fuel |
Russia, Ozersk (Mayak)a |
Thermal, oxide |
400 |
|
|
France, La Hague |
Thermal, oxide |
1700 |
|
|
Japan (Rokkasho) |
Thermal, oxide |
800b |
| Other nuclear fuels |
India (PHWR, 4 plants) |
Thermal, metal |
260 |
| Total civilian capacity in operation |
|
|
2360 |
2 Civilian nuclear power
2.1 Nuclear reactors and pyroprocessing
A nuclear reactor is a construction or device designed to sustain a long-term nuclear fission (chain reaction) process with a fissile material (235U and/or 239Pu) typically enriched to 3–5%. A set of conditions must be met for a required reaction to occur, these include: mass of fissile material (near to critical mass is optimal), geometry (reactor cores tend to approximate to spherical particles) and an appreciation of the neutron spectrum and the required kinetic energies for neutrons.16 Separate procedures are also in place to control the fission process according to the reactor design (for example: graphite moderators or chemical shim with boric acid) and the reactor is typically enclosed in a concrete edifice.17,18 The primary aim of a nuclear reactor is to therefore harness the heat energy generated from the fission process and convert it into electricity. The reactor types are designated according to the type of coolant, enrichment of the nuclear fuel, energy of the neutrons used in the fission reactions, and the purpose of the reactor. Three of the candidate reactors to be used in the next generation, Generation IV, of reactors use metal fuels, three use metal oxide fuels and one uses fluoride-based fuel. A thorough description of these designs is summarised in Table 2. The advantages of Generation IV reactors and the associated nuclear fuel reprocessing processes are that they are significantly more fuel-efficient compared with the previous generations and make the conversion of fissile material to weapons manufacture less attractive. This is because both uranium and plutonium are left mixed with several other elements rather than being purified as pure elements and while one might readily apply the PUREX process; this leads to the need for long-term storage, offsite transportation and the undesired proclivity of weapons manufacture.19
Table 2 A summary of generation IV reactor designs20
| Type of generation IV reactors |
Neutron spectrum |
Coolant |
Fuel |
Core outlet temperature (°C) |
Size (MWe) |
|
Fuel contains small quantities of 235U or 239Pu.
|
| High-temperature gas |
Thermal |
Helium |
UO2 |
900–1000 |
250–300 |
| Sodium-cooled fast |
Fast |
Sodium |
U-238 |
500–550 |
50–1500 |
| Supercritical water-cooled |
Thermal/fast |
Water |
UO2 |
510–625 |
300–1500 |
| Gas-cooled fast |
Fast |
Helium |
U-238a |
850 |
1200 |
| Lead-cooled fast |
Fast |
Lead or Pb/Si |
U-238a |
480–750 |
20–1200 |
| Molten salt fast |
Fast |
Fluoride salts |
UF4 |
700–800 |
1000 |
| Molten salt – advanced high temperature |
Thermal |
Fluoride salts |
UO2 |
750–1000 |
1000–1500 |
Of all the Generation IV designs (Table 2), molten salt reactors are arguably the most promising, with the ability to operate at low pressures and high temperatures. The carrier molten salts are well known for their high specific heat capacity per unit volume and relative radiation-resistant reprocessing media. Molten salt reactors have been designed such that they avoid the build-up of neutron poisoning gases, xenon and krypton, in the molten salt by employing gas extraction techniques.21 What is more, it is predicted that the civilian nuclear power sector will experience renewed interest in the thorium fuel cycle, spurred on by R&D progress that includes spent fuel reprocessing (reduction of metal oxides), and the process of electrochemical refining.22
With a wide range of reactor groups, it is essential that all, despite their technological advancements, size and capacities are developed to take into consideration the minimisation of radiotoxic elements produced as by-products of the nuclear fission process. After discharge from a reactor, the fuel rods contain a variety of natural and synthetic radiotoxic elements (Cm, Pu, U, Am, Np), which release between 50 (Magnox) to 5000 (Fast reactor) kW of heat per tonne in the first six months and contain radioactivity that lasts over many years;23 hence, a number of different strategies have been employed to ensure these elements are partitioned effectively both for storage and recycling through an appropriate reactor design. Overcoming these challenges remains difficult; and any of the aforementioned technologies which are implemented by different countries should aim to be environmentally friendly, resistant to nuclear weapons proliferation and economically viable.
Moreover, the spent metal fuels generated from nuclear reactors are incompatible with aqueous processing unless they are first oxidised followed by separation via the PUREX process, before ultimately being reduced. This, however, is needlessly circuitous and generates large volumes of waste. Pyroprocessing of spent nuclear fuel is the preferred option, incorporating a series of steps that utilises a high-temperature process and electrochemical reduction steps using molten salts, liquid media and/or gases to provide a safe and efficient route to metal production that is proliferation-resistant. It presents several advantages over conventional aqueous processes as it is able to handle a greater quantity of fissile materials. Unlike aqueous/organic reprocessing where the reagents can suffer from radiation damage there is no such problem with molten salts. The metal can be further purified by processes such as electrorefining.
Depending on the material to be treated, there are several different types of pyroprocessing techniques. Many of these processes try to avoid the troublesome use and handling of lithium metal, which is commonly found within metallothermic reduction (a displacement reaction of a reactive metal to form pure metals or alloys) using a molten lithium chloride pool.24 Additionally, the application of chloride and fluoride melts is far more beneficial than aqueous systems as it avoids the possibility of radiolysis-induced degradation and reduces the overall cooling time required before high-burnup spent nuclear fuel is processed when using aqueous solvents.25 Furthermore, the reduction process has drawbacks too; for example, the conventional ‘bomb reduction’ requires the use of hazardous fluorine, where there are high neutron doses associated with alpha-n reactions and inherently high temperatures and pressures.
Pyrochemical processing of spent nuclear fuel can be conducted such that pure plutonium metal or compounds are not isolated. The final steps in a typical pyro-process involve the recovery of uranium and/or plutonium, which are usually removed along with the other actinides (neptunium, americium and curium) as their reduction potentials lie close together. Plutonium, in this case, can form an intermetallic compound with its cathode crucible, e.g. when made from cadmium there is the formation of PuCd6 and the presence of lanthanide fission products may lead to contamination. The formation of PuCd6 occurred due to the reduction with lithium metal and an applied cathodic current density of 66 mA cm−2, leading to its deposition in the solid phase at the bottom of the crucible. The reason for the solid phase deposition may arise from a solubility limit or because of a vertical temperature gradient due to slower cooling above the molten salt.26 As a result, this intermetallic reaction process has two advantages; the first is that it prevents nuclear proliferation and the second is that the overall heat and radiation produced from the process is far less.25
3 Molten salts and their properties
Molten salts are inorganic compounds that are solid at standard room temperature and when heated to elevated temperatures, dissociate into cations and anions with high ionic conductivities and wide electroactivity ranges. They have attracted interest for a broad range of applications, and continue to find diverse new uses.27 For example, in the solar thermal industry, molten salts have been used as heat transfer media at high temperatures,28,29 as electrolytes for fuel cells and in the extraction of metals from spent lithium–ion batteries.30–32 Molten salts are used in metallurgy to reduce refractory metal oxides to metals where other more conventional methods of reducing oxides are impractical and difficult to commercialise.33–35 They have also been applied to the production of alloys with advanced properties.36,37 In most of these applications, molten salts are used as binary or ternary mixtures that may form a eutectic mixture with a lower combined melting point than each independent component.
With regards to the treatment of spent nuclear fuels, the field of pyroprocessing has changed dramatically. Since the 1980s and 1990s, countries across the world have employed different pyroprocessing (‘pyro’ or ‘high temperature’) approaches, with broad agreement that molten salt electrorefining is a promising approach for the processing and recovery of spent nuclear fuel and radioactive metals.38 In general, molten salts possess several key advantages in nuclear applications. As non-aqueous media, they are less likely to pose criticality hazards which are often found in aqueous solutions. Molten salts are able to operate at high temperatures without a direct fire risk. Furthermore, they have useful properties in processing high burn-up nuclear fuels with fewer risks post-irradiation, there is also reduced susceptibility to radiolysis compared to aqueous methods and they have the capability of dissolving large quantities of metal oxides compared with aqueous extraction methods.39 Despite these promising advantages, their inherently high operating temperature and corrosive nature (particularly fluorides) have acted as an impediment to technological progress and the subsequent advancement of industrial systems.
In Table 3, a variety of molten salt systems that have been used in the nuclear industry are considered. The reasons for the selection of one particular molten salt over another is often governed by temperature requirements, the solubility of oxide ions, issues of criticality limits and stability.38 For example, LiCl–KCl, NaCl–KCl and CaCl2 have vastly different working temperatures of 450 °C, 750 °C and 900 °C, respectively. LiCl has been used as a medium to extract metallic nuclear fuels, whereas NaCl–KCl (given its higher operating temperature) as well as LiCl–KCl eutectic, have been preferred for the electrowinning of spent oxide fuels. The method by which each of these molten salts has been used to reprocess the respective metal oxides/chlorides is described in this review. Moreover, molten salts are excellent heat transfer fluids and due to their advantageous characteristics, their application to industrial use, namely as a coolant and/or fuel in molten salt reactors and in pyroprocessing, appears promising. Given the wide array of molten salts on offer, research efforts must continue to explore their benefits and progress from the lab bench to industry.
Table 3 Molten salts and their properties40–42
| Properties |
LiCl–KCl |
NaCl–KCl |
CaCl2 |
LiF–CaF2 |
| Composition (A–B, mol% A) |
59 |
50 |
100 |
23 |
| Melting temperature (°C) |
352 |
658 |
772 |
769 |
| Molar mass (g mol−1) |
55.3 |
66.5 |
111 |
37.9 |
| Density at melting point (g cm−3) |
1.64 |
1.60 |
2.15 |
2.07 |
| Viscosity at melting point (g cm−1 s−1) |
2.80 |
2.13 |
|
|
| Electrical conductance (Ω−1 cm−1) |
0.637 |
2.20 |
2.48 |
6.25 |
4 Process chemistry
4.1 Electrochemical reduction reaction mechanisms
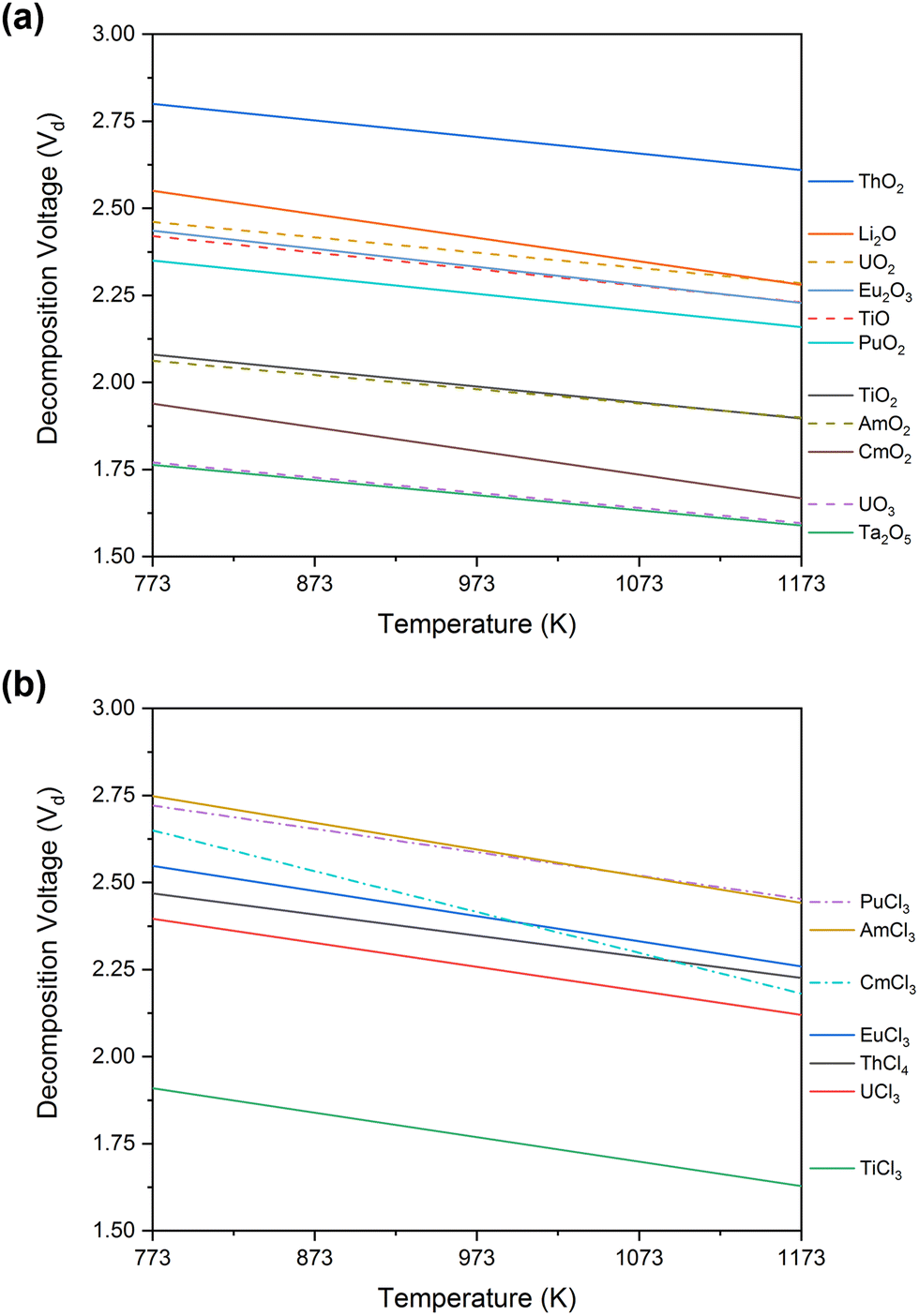
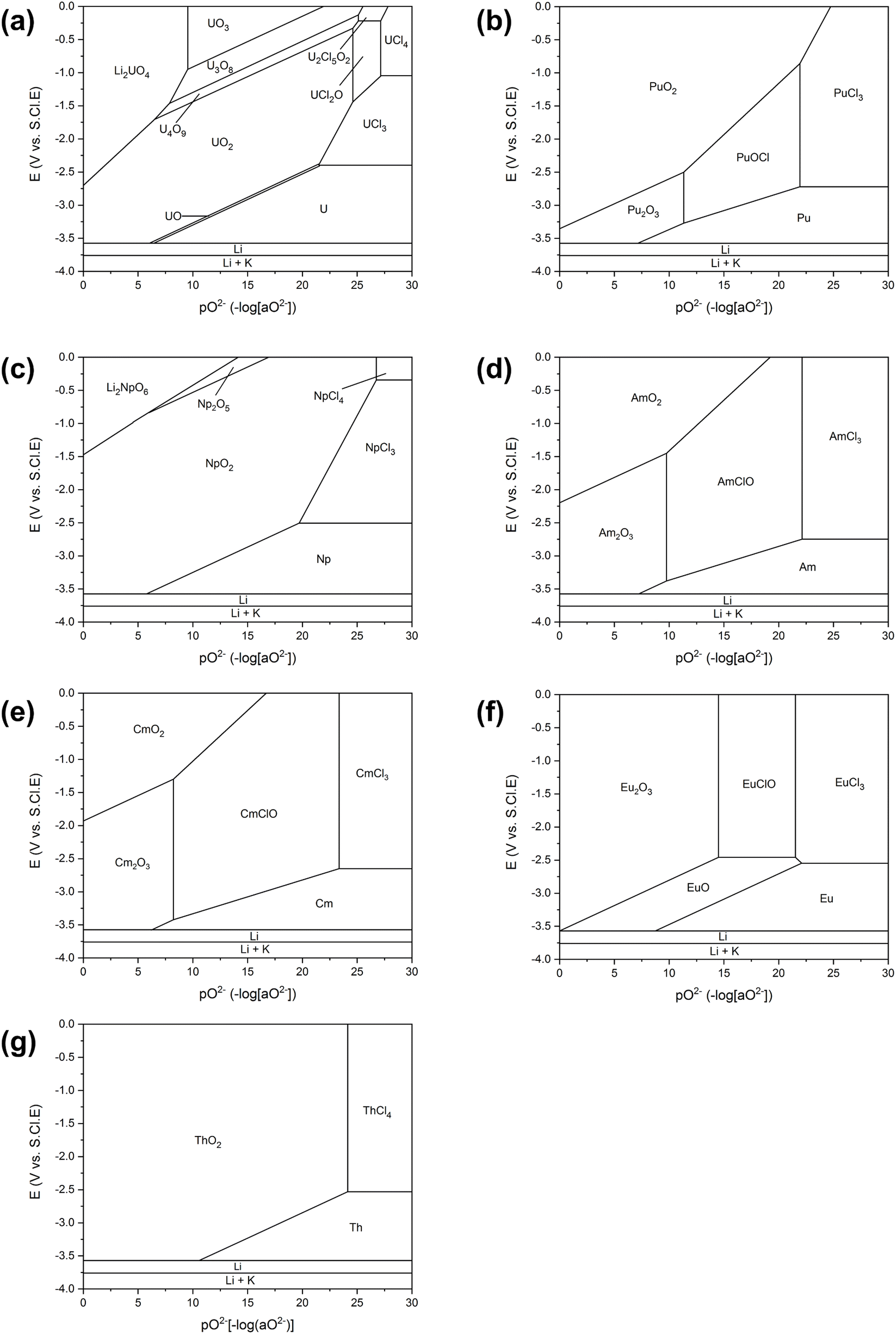
The FFC-Cambridge Process has demonstrated the process by which the direct electrochemical reduction of metal oxide to metal in a molten chloride system occurs.34 This process has been useful in enabling further understanding of the electrochemical reduction of spent oxide nuclear fuel to metal via an oxygen ionisation mechanism which is now one of the theories used to describe pyro-electrochemical processes. In this electro-deoxidation process, and upon the application of a suitable potential, electrochemical reduction of the metal oxide occurs at the cathode and the oxide ions generated migrate through the molten salt system to the anode. Here, the oxide ions react with (typically) a carbon anode to form CO or CO2 (eqn (1)). If a suitable overpotential is applied to the metal oxide at the cathode for a sufficient period, this leads to the metallisation of the surface of the cathode (eqn (2)).43 The exact process that occurs depends on the metal oxide, type of precursor, choice of molten salt and the activity of O2− and Cl− in the melt. Fig. 1 shows the decomposition voltages of M–O and M–Cl systems between a temperature range of 773–1173 K. The decomposition voltages of these systems must remain below that of molten salt systems to ensure a successful electrochemical reduction.| | | Anode (carbon): yO2− + C → COy + 2ye− | (1) |
| | | Cathode: MxOy + 2ye− → xM + yO2− | (2) |
Shortly after the invention of the FFC-Cambridge Process, the Ono-Suzuki process was presented.44 This study was originally demonstrated for TiO2 and can be applied to the study of nuclear materials reprocessing especially in terms of understanding the electrochemical reduction of UO2. Ti metal is recovered by a process called ‘calciothermic’ reduction, this is where, calcium metal is produced in situ from the reduction byproduct CaO. CaO is soluble in CaCl2 molten salt and leads to the formation of Ca+, liquid calcium (eqn (3) and (4)) and carbon dioxide (identical to the reaction described eqn (1) on the application of a suitable overpotential. Ti metal is formed by the thermochemical reaction between TiO2 and liquid Ca as shown in eqn (5). Further description of other electrolytic processes are described in Table 4 although they lie beyond the scope of this review article.| | | Cathode (1): Ca+ + e− → Ca | (3) |
| | | Cathode (2): Ca2+ + 2e− → Ca | (4) |
| | | Thermochemical reaction: TiO2 + 2Ca → Ti + 2CaO | (5) |
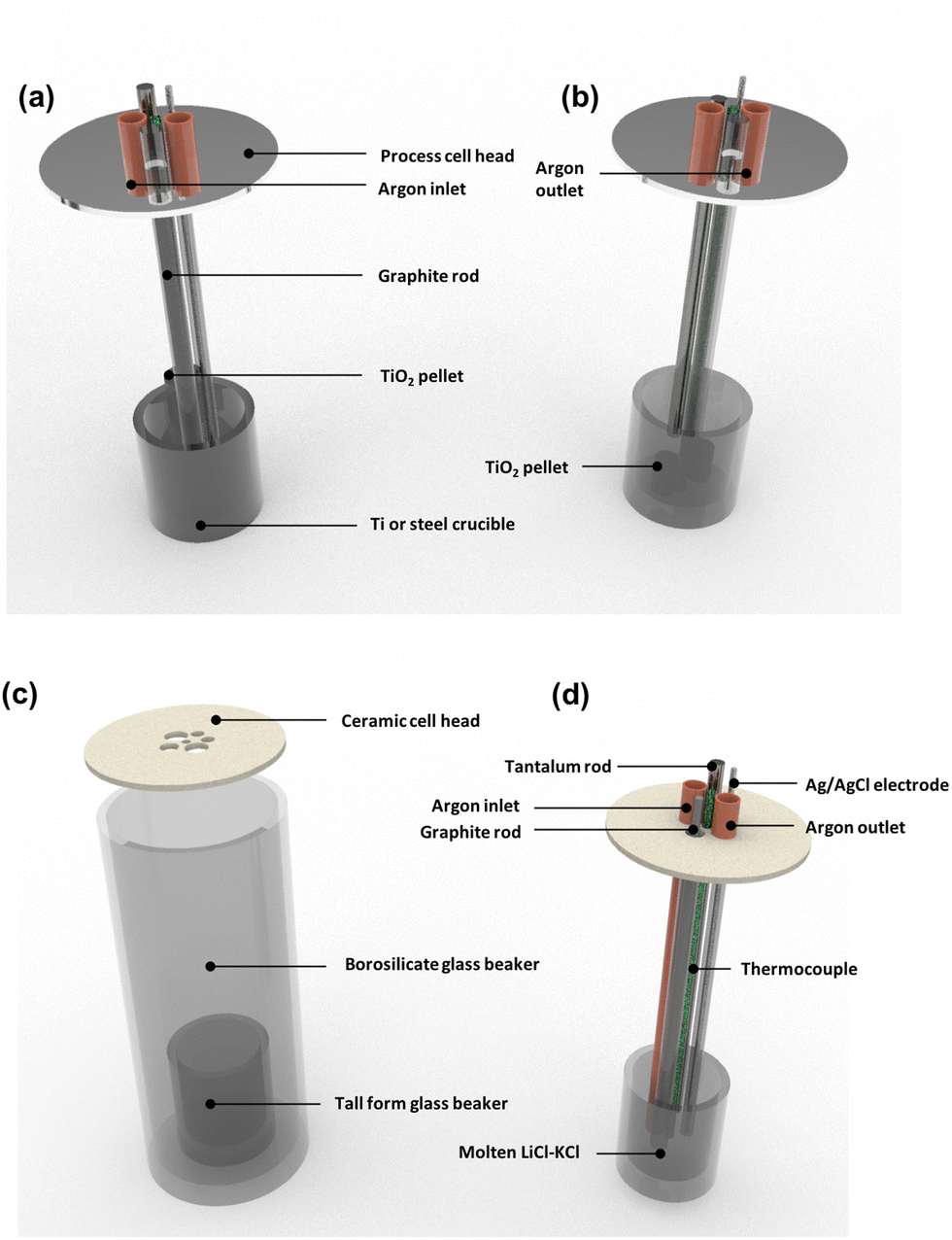
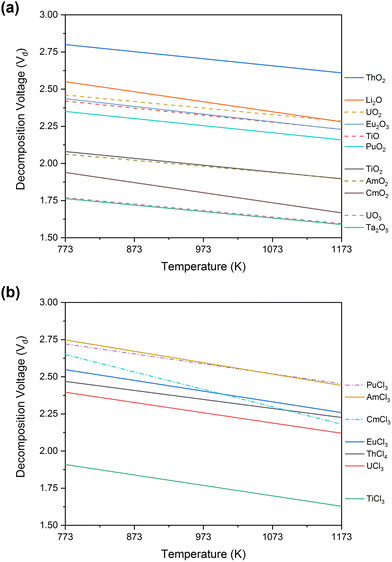 |
| | Fig. 1 Calculated values of decomposition voltage defined as the minimum voltage (V) vs. standard chlorine electrode needed for the reduction of the metal chloride or oxide systems stated above as determined from the Gibbs Energy of Formation derived between 773 and 1173 K for (a) metal–oxide and (b) metal–chloride systems; this can then be compared to the expected decomposition of inorganic salt systems to identify the feasibility of an electrochemical process. All data, relating to the Gibbs free energies, has been acquired from HSC Chemistry 6.12. | |
Table 4 A summary of the various electrochemical reaction schemes of molten salt processes. table reproduced from Xiao and Wang45 with permission from the Royal Society of Chemistry
| Reaction mechanism |
Cathodic products |
|
Direct electro-deoxidation of solid oxides
|
|
| One-step electro-deoxidation |
Metals |
| Continuous or stepwise electro-deoxidation |
Oxides or metals |
| Simultaneous electro-deoxidation and de-calcification |
Metals |
| Immobilisation of active liquid metal with formation of alloys |
Alloys |
| Cathodic passivation |
Oxides and metals |
| Direct electro-desulfidation of solid sulfides |
Metals or alloys |
|
Deposition of active metal together with electro-reduction of solid oxides
|
|
| Deposition of active metal followed by metallothermic reduction |
Metals |
| Electro-deoxidation under active metal deposition |
Metals |
| Under-potential deposition of active metals |
Alloys |
| Electro-inclusion of cations from molten salts into oxides |
Oxide composite |
|
In situ
dissolution–electrodeposition process
|
Nanostructured semiconductors |
4.2 Electrochemical reduction and refining of nuclear materials relevant to the pyrochemical process
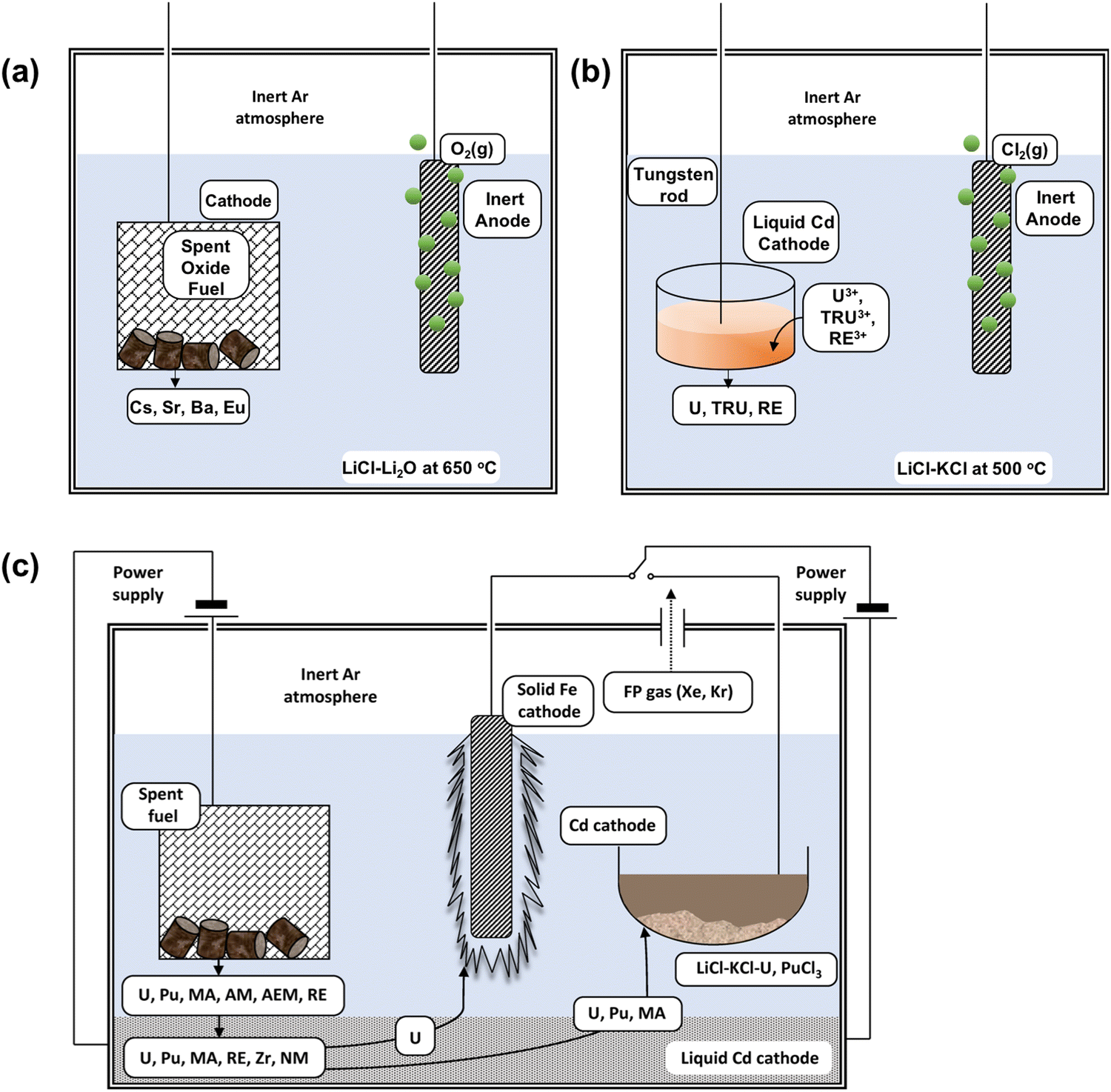
The electrochemical recovery processes mentioned before (e.g. FFC-Cambridge, Ono-Suzuki) are important processes that may be useful in closing the back end of the fuel cycle. Typically, this is where some or all of the spent nuclear fuel is reprocessed. It comprises the following steps: mining, milling, chemical conversion of uranium to an enriched version suitable for fuel rod fabrication and the subsequent disposal post-irradiation of the used fuel, including plutonium and fission products.16 There are several stages in the nuclear fuel cycle where electrochemical processes can feature (Fig. 2). Prior to electrochemical processing, the as-received fuel rods must be converted into a suitable form appropriate for use as a feed material which involves the disassembly and decladding of spent fuel assemblies into smaller segments and the subsequent voloxidation step where a high-temperature treatment (typically greater than 1000 °C) is applied to convert UO2 to U3O8. This latter process is imperative in improving the current efficiency for electrochemical reduction of the uranium and plutonium oxides generated and is an important fabrication step in controlling the stoichiometry and atomic arrangement of particles for subsequent steps. Heating the used fuel to high temperatures prior to addition to the molten salt removes the salt-soluble fission products such as caesium, iodine, rubidium and technetium, therefore preventing them from accumulating within the reduction process, although it does generate a separate waste stream containing fission products such as 137Cs and 99Tc. The electroreduction procedure involves the dissolution of the uranium and plutonium oxide granules or powder typically into a molten chloride system such as LiCl–Li2O at 650 °C (Fig. 3); the careful control of salt-soluble fission products is needed to prevent the slowing of the reduction process.46,47 It has been demonstrated that this initial step in the electrochemical reduction reduces the heat load (which is also described as the specific decay heat of fission products) and leads to a volumetric reduction by conversion of metal oxide to metal. For optimal results, this process must separate all fission products such as barium, caesium, and strontium from the fuel. The overall cathodic and anodic reduction is presented for the oxide fuel where M represents the actinide is described in eqn (1) and (2).
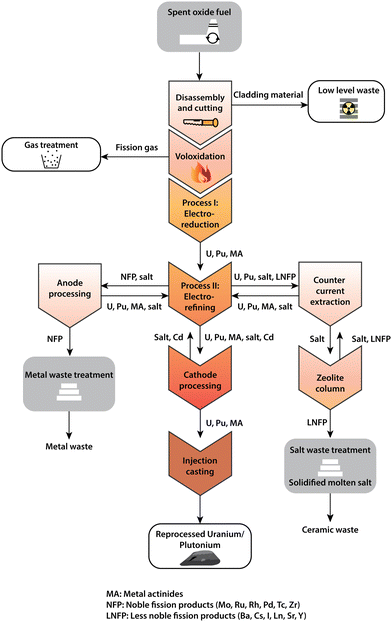 |
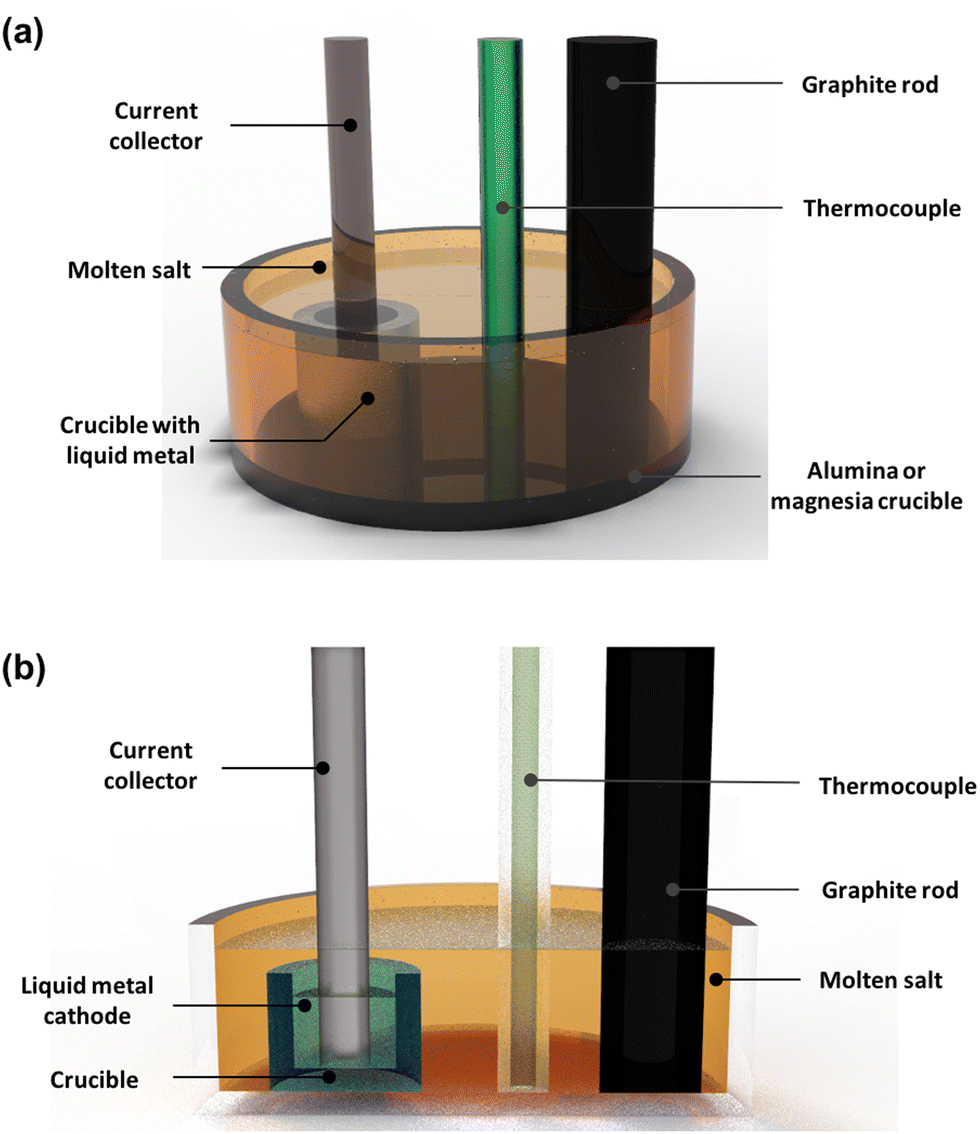
| | Fig. 2 Schematic recycle flow process showing the various steps involved in the reprocessing of spent nuclear (oxide) fuel. There are several different steps involved in the electrochemical processing of nuclear materials but the two most critical in the recovery of high purity uranium and plutonium are electrorefining and electroreduction. This schematic diagram has been reprinted from Koyama, with permission from Elsevier.47 | |
In general, the cathodic form of the process has been found to vary greatly and various studies have considered the performance when using rod-cut, powder, pellet form or crushed particles of actinide oxides which are loaded into a cathode basket.48–51 To improve the rate of reduction, an appropriate quantity of lithium oxide is added to prevent the anodic dissolution of the inert platinum anode enshrouded in magnesium oxide – a concentration which lies between 1–3 wt% is typically used to ensure there is enough present to accelerate the reduction without leading to any corrosion resistance or impediment to the extent of reduction.47 Uranium remains the most widely studied actinide in this field and comparisons have been made using differing oxidation states of feed materials (UO2 and U3O8); these studies have demonstrated that in an LiCl–KCl system the likelihood of other intermediates such as LiUO3, U4O9 and UO2 (if U3O8 is used as the feed material) forming in the initial stages is highly probable.52,53 The reduction of UO2 is therefore likely to proceed through two different routes, one of which involves the formation of a lithium-intercalated compounds and the other through direct cathodic reduction.
Electrorefining is the second process with potentially two feed streams. One of these streams comprises a spent metal feed and the other is a metal stream from the electrochemical reduction of the spent oxide fuel. For the former scenario, impure uranium metal is selectively separated from transuranium (TRU) and lanthanide elements at a solid cathode in various molten salt systems from the electrolytic reduction process. It is important to note that the steps described forthwith are in reference to the actinide separation process only and do not account for the initial decladding and final waste treatment processes which follow.
Electrorefining technology, since the early 1960s, has gone through several developmental phases and whilst most of the studies on UCl3 utilised BaCl2–KCl as the most appropriate molten salt, the realisation that LiCl–KCl offered a more practical and advantageous route, with a considerably lower working temperature, without compromising product or recovery, was found to be more suitable.54 There have been several reports on the electrorefining process for the LiCl–KCl–UCl3 eutectic solution. The uranium rod from the electroreduction step is subsequently reacted with cadmium chloride (in LiCl–KCl) to produce UCl3.55 The formation of UCl3 is advantageous because it stabilises the initial cell voltage according to eqn (8).56
| | | 2U + 3CdCl2(in LiCl–KCl) → 2UCl3(in LiCl–KCl) + 3Cd | (8) |
The electrorefining cell typically comprises a stainless-steel anode basket that contains the final reduction products, a solid iron cathode and a Ag/AgCl reference electrode. This setup is immersed in the aforementioned LiCl–KCl–UCl
3 system and since cadmium has a greater density this metal sinks to the bottom and captures any residual uranium deposit falling from the surface of the cathode. For the duration of the process, a current is supplied to avoid anodic dissolution of the stainless-steel, whilst both the cathode and anode assembly are simultaneously agitated between 10–20 rpm to enhance symmetrical uranium deposit growth at the cathode as well as improve the rate of anodic dissolution. The completion of the process occurs once the applied current decreases. Afterwards, the uranium metal in the anode basket is raised from the molten salt and left to cool. Then it is rinsed with distilled water to remove any adhering lithium chloride salt. This process, as explained in detail in
Fig. 2 and 3 (Mark-IV electrorefiner), results in the separation of pure uranium directly from the nuclear fuel onto the surface of the cathode and the resultant deposition of residual uranium, transuranic elements and lanthanides. The Mark-IV electrorefiner consists of four rectangular anode baskets and a stainless-steel cathodic rod. The reaction schemes for the electrorefining process are described as:
| | | Anode: U (in spent fuel) → U3+ + 3e− | (9) |
| | | Anode: Pu (in spent fuel) → Pu3+ + 3e− | (10) |
| | | Cathode (solid electrode): U3+ + 3e− → U | (11) |
| | | Cathode (liquid Cd): U3+ + 3e− → U (in Cd) | (12) |
| | | Cathode (liquid Cd): Pu3+ + 3e− + 6Cd → PuCd6 | (13) |
A large body of research on spent nuclear materials has been performed at the Argonne National Laboratory (ANL).
54,57,58 Their molten salt electrorefiner, illustrated in
Fig. 3, represents a striking innovation. Both cathodes are made from stainless-steel/iron and liquid cadmium. Initially, spent nuclear fuel is cut into metal ingots following a conversion and placed in a basket that forms the anode and undergoes electrochemical oxidation. The cadmium pool found at the bottom is where the reduction of actinides occurs. The uranium metal is transported to the stainless-steel cathode from the anode. All actinides migrate towards the cadmium electrode, where they are oxidised. The deposit of uranium in particular takes the form of a dendrite on the stainless-steel cathode which facilitates retrieval at the end of the process; however, when there is a mixture of fission products, especially with plutonium, the deposit does not adhere to the cathode surface and consequently use of a liquid cadmium cathode becomes far more fruitful than in uranium electrorefining cells.
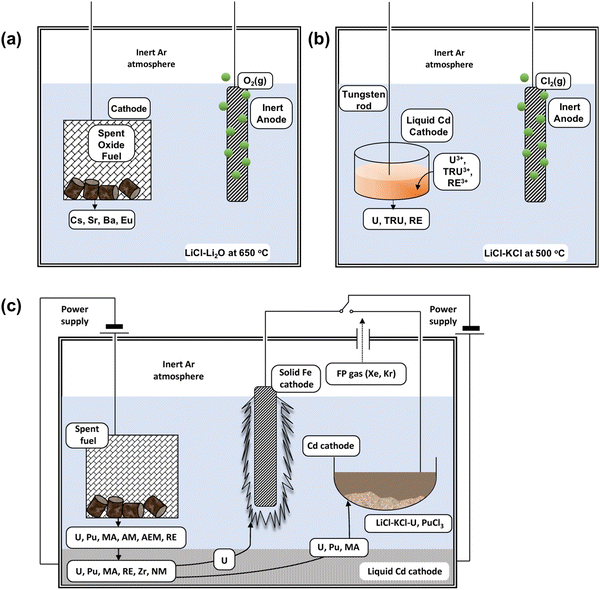 |
| | Fig. 3 Schematic of (a) electroreduction, (b) electrowinning and (c) electrorefining process. (a) Represents a typical schematic diagram for the electroreduction as a first step to reducing the feed material reprinted from Koyama with permission from Elsevier.47 (b) Represents a simplified drawing of the liquid cadmium cathode (LCC) electrowinning process. (c) Represents the Mark-IV electrorefiner developed by the Argonne National Laboratory operating at 500 °C, where AM is alkali metal, AEM is alkaline earth metal, MA is Np, Am and Cm, NM are the noble metals, RE are the rare earth elements and FP refers to the fission products. Reprinted by permission of the publisher, Taylor and Francis, on behalf of Atomic Energy Society of Japan from Koyama et al.59 | |
Koyama et al. extensively studied the application of an electrorefiner and his work led to the replacement of stainless-steel electrodes with an iron cathode (Fig. 3);59–61 Sakamura et al. and Roy et al. were able to determine the redox potentials which led to the reduction of actinides and cladding materials, such as those made from zirconium, from their chlorides after undergoing oxidation at the anode. Table 5 summarises these reported potentials.62,63 There is a discrepancy associated with each of the observed potentials (e.g. for the U3+/U0 couple, from Inman et al.,64 Kuznetsov et al.65 and Koyama et al.59). This occurs because of the dependence of a molten salt system on the concentration of O2− ion activities in molten salt systems.66–68
Table 5 Redox potentials of elements in LiCl–KCl eutectic salt (V vs. Ag/AgCl)59,62,69,70
| Elements |
Working temperature (°C) |
| 400 |
450 |
500 |
| Pu(III)/Pu(0) |
−1.591 |
−1.543 |
−1.497 |
| Np(III)/Np(0) |
−1.472 |
−1.434 |
−1.390 |
| Am(II)/Am(0) |
|
−1.592 |
|
| Zr(II)/Zr(0) |
|
−0.693 |
|
| U(III)/U(0) |
−1.274 |
−1.233 |
−1.190 |
After electrorefining, the obtained deposits can go through a subsequent purification step, cathode processing, where the deposit is heated to separate the actinide metal from the adhering cadmium and salt. As the vapour pressures between cadmium and the actinides differ quite significantly the separation between these species can proceed with ease. The final step is referred to as ‘injection casting,’ and is a method where the composition of the actinide metals is adjusted by melting U–Zr alloys at 1560 °C and Pu–U–MA–Zr alloy at 1480 °C under a vacuum.47 This is then injection-cast into a mould cavity which cools and hardens according to the shape of the mould (rod-shape). These rod-shaped moulds are then fabricated inside a steel cladding for use in a nuclear reactor.
Electrowinning is a separate procedure for the recovery of uranium and plutonium metal from TRU using a liquid cadmium cathode.71 This technology has gained traction through the Russian Institute of Atomic Reactors (RIAR) and the successful recovery of 7 tonnes of unirradiated fuel has been demonstrated.72 This system works by the electro-co-deposition of PuO2 and UO2 under the influence of a chlorine–oxygen sparge. In oxidising conditions, the conversion of PuO2 to PuO22+ becomes possible and by changing the redox potential of the system, all oxidation states of plutonium can form PuO2. Under the same oxidising conditions, uranium remains in the melt and plutonium is precipitated out. As the reduction of PuO22+ and UO22+ takes place at more positive potentials, purification takes place during electrodeposition. In other systems, the success of this technology lies in the distinct difference in reduction potential of the uranium and TRU, a diagram for which is shown in Fig. 3(c); it has been shown in studies that uranium metal has a preferentially close redox potential to solid working electrodes, some of which include tungsten and graphite, whereas the TRU have very close redox potentials to liquid metals such as cadmium and bismuth.59,73,74
5 The lanthanide and actinide materials
The exploratory work concerning the reduction of nuclear materials in molten salts began in the early 1960s. At this time nuclear materials had been used primarily for weapons production purposes and with this came a build-up of associated nuclear waste. For most of the elements detailed, consideration is given to the metal chloride systems which comprises a large part of the electrorefining process. A review of these elements is necessary because of the interesting outcomes observed in alkali halide eutectics and because they are commonly considered as part of the burn-up credit analysis conducted by the Nuclear Energy Agency with the exception of thorium metal.75 Additionally, progress is being made in the processing of metal oxide systems, although such systems are far less studied. Not all reduction processes described beforehand are mentioned in the following sub-sections. A general description of each chemical element, including its occurrence and earlier uses, is provided to help orientate readers.
5.1 Uranium
Uranium, atomic number 92, is one of the few naturally occurring actinides found within the earth's crust (<3 ppm) and is a constituent within many minerals such as: autunite, carnotite, monazite, samarskite and uraninite. Several extraction processes have been developed to industrially recover uranium from its ores. Uranium displays fluorescent properties, and was first used as a yellow-green colourant in glass from as early as the Roman period.76 This use remained unchanged for the next two millennia. In 1896, Henri Becquerel was the first to realise that uranium emitted penetrating rays;77 the important and ground-breaking discovery of nuclear fission was made in 1939 by Hahn and Strassman.78,79
Uranium dioxide, UO2, was first reported erroneously as an element in literature but this finding was corrected by Péligot who realised that it was in fact a pure oxide.80 For molten salt processes, UO2 is produced via the oxidation reaction of the higher oxides, U3O8, under the flow of hydrogen gas. This is commonly the feed material along with UCl3. To understand the electrorefining pathway, Inman et al. were the first to study this particular uranium species, UCl3, in LiCl–KCl eutectic.81 This study focused on uranium metal deposition on a uranium working electrode in accordance with the redox process described in eqn (14). The process was found to have a high faradaic efficiency (when current densities <100 mA cm−2) on a single electrode reaction.
In a separate experiment, the reduction potential was measured to be −1.398 V (
vs. Ag/AgCl). It was understood that uranium metal was formed by a chemical reduction with lithium present in the salt. Because of this, some of the final product appeared powdery whilst other product material formed at the working electrode (at lower working potentials) lead to dendritic growth. In comparison, Kuznetsov
et al. conducted their own study and determined that the reduction potential in a LiCl–KCl system was ∼−1.5 V
vs. Ag/AgCl.
82 Further details of redox potentials are described in
Table 6.
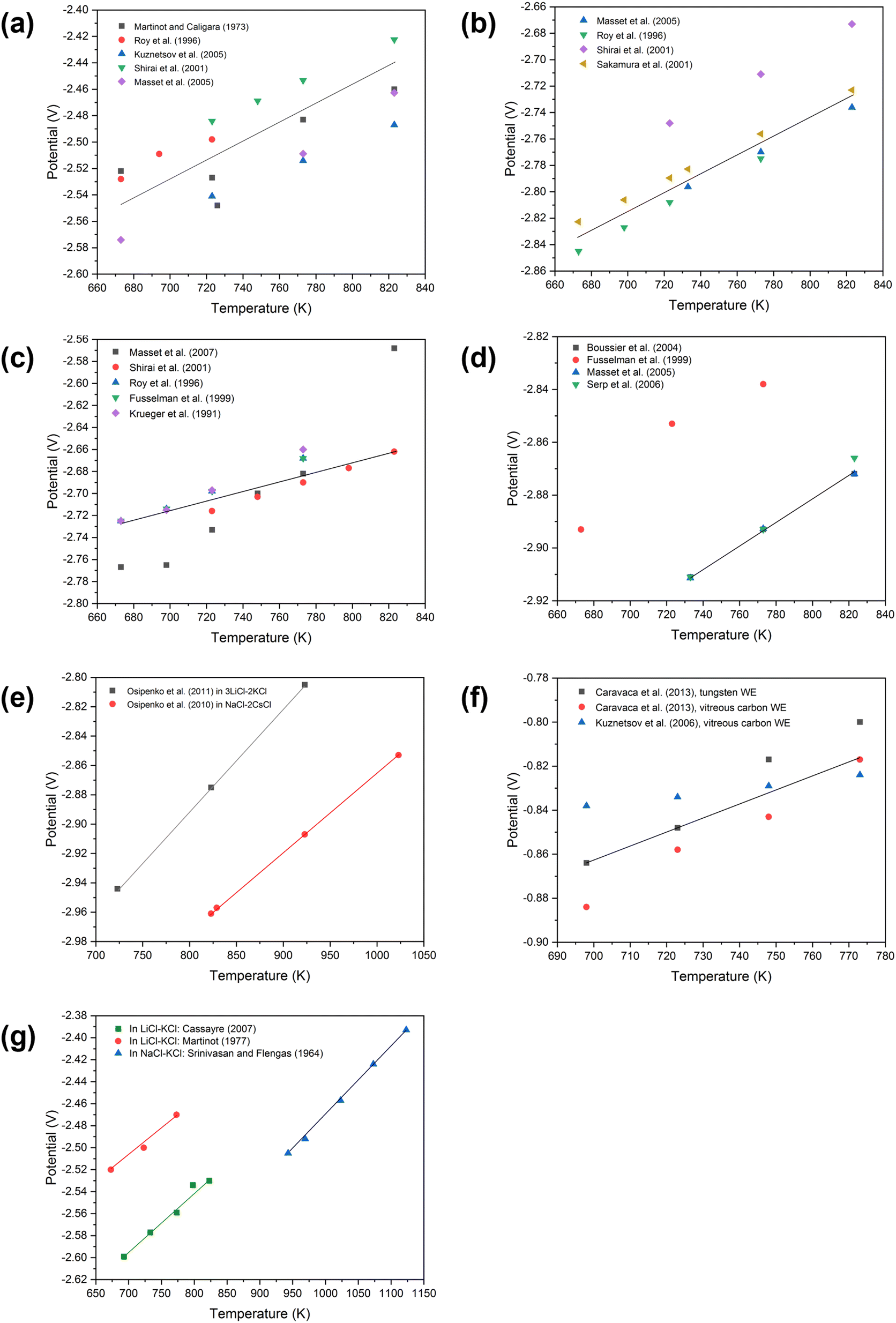
Table 6 Summary of reduction potentials of U3+/U0 in LiCl–KCl molten salt eutectic at various temperatures
|
T (K) |
Apparent experimental potential (V) of U3+/U0 |
| Martinot and Caligara83 |
Roy et al.69a |
Kuznetsov et al.84b |
Shirai et al.85b |
Masset et al.86b |
|
Reference electrode: Ag/AgCl.
Reference electrode: Cl2/2Cl−.
|
| 673 |
−2.522 |
−2.528 |
|
|
−2.5741 |
| −2.506 |
|
|
|
|
| −2.598 |
|
|
|
|
| 694 |
|
−2.509 |
|
|
|
| 723 |
−2.527 |
−2.498 |
−2.541 |
−2.4841 |
|
| −2.476 |
|
|
|
|
| −2.522 |
|
|
|
|
| −2.557 |
|
|
|
|
| 726 |
−2.548 |
|
|
|
|
| 748 |
|
|
|
−2.4689 |
|
| 773 |
−2.483 |
|
−2.514 |
−2.4533 |
−2.5088 |
| −2.453 |
|
|
|
|
| −2.491 |
|
|
|
|
| −2.527 |
|
|
|
|
| 823 |
−2.460 |
|
−2.487 |
−2.4224 |
−2.4627 |
There has also been a multitude of work conducted on spent fuel oxides in molten salts. Herrmann et al. placed crushed spent nuclear fuel into a LiCl-1 wt% Li2O at 650 °C.87 In this study, a platinum anode and a Ni/NiO reference electrode were used. A potential of −2.40 V vs. Ag/AgCl was calculated from the electrochemical reduction of uranium dioxide (eqn (15)) and the lithium deposition potential was −2.47 V vs. Ag/AgCl (eqn (16)). Given that the cathodic reduction potential of uranium dioxide and the decomposition of the salt lie very close together, this resulted in the in situ formation of lithium metal and presented an advantage in enabling the chemical reduction of uranium dioxide viaeqn (17). However, a disadvantage of this is that the formation of lithium resulted in the attack of the platinum anode and its dissolution. Similarly, Choi et al. studied uranium dioxide on a much larger scale reducing 17 kg of UO2 in LiCl–Li2O.88 Through their study they realised that to achieve higher current efficiencies a small pellet precursor should be suitably complemented with a larger anode surface area.
| | 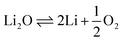 | (16) |
| | | 4Li + UO2 ⇌ U + 2Li2O | (17) |
There has also been work accomplished on the higher oxides of uranium such as U
3O
8. Seo
et al. studied the reduction mechanism of U
3O
8 within a porous magnesium oxide membrane and found the potential to be −2.27 V
vs. Pt.
89 Jeong
et al. studied this process on a larger scale reducing 20 kg of U
3O
8 and achieving a 99% conversion.
52 The reduction potentials of this process were found to vary from −2.47 to −3.46 V due to differences in O
2− ion concentration. These results were in agreement with Choi
et al. where they found the size of the pellet to be of hindrance. The larger the size of the pellet the more likely it was to inhibit the diffusion of the electrolyte leaving the pellet partially reduced.
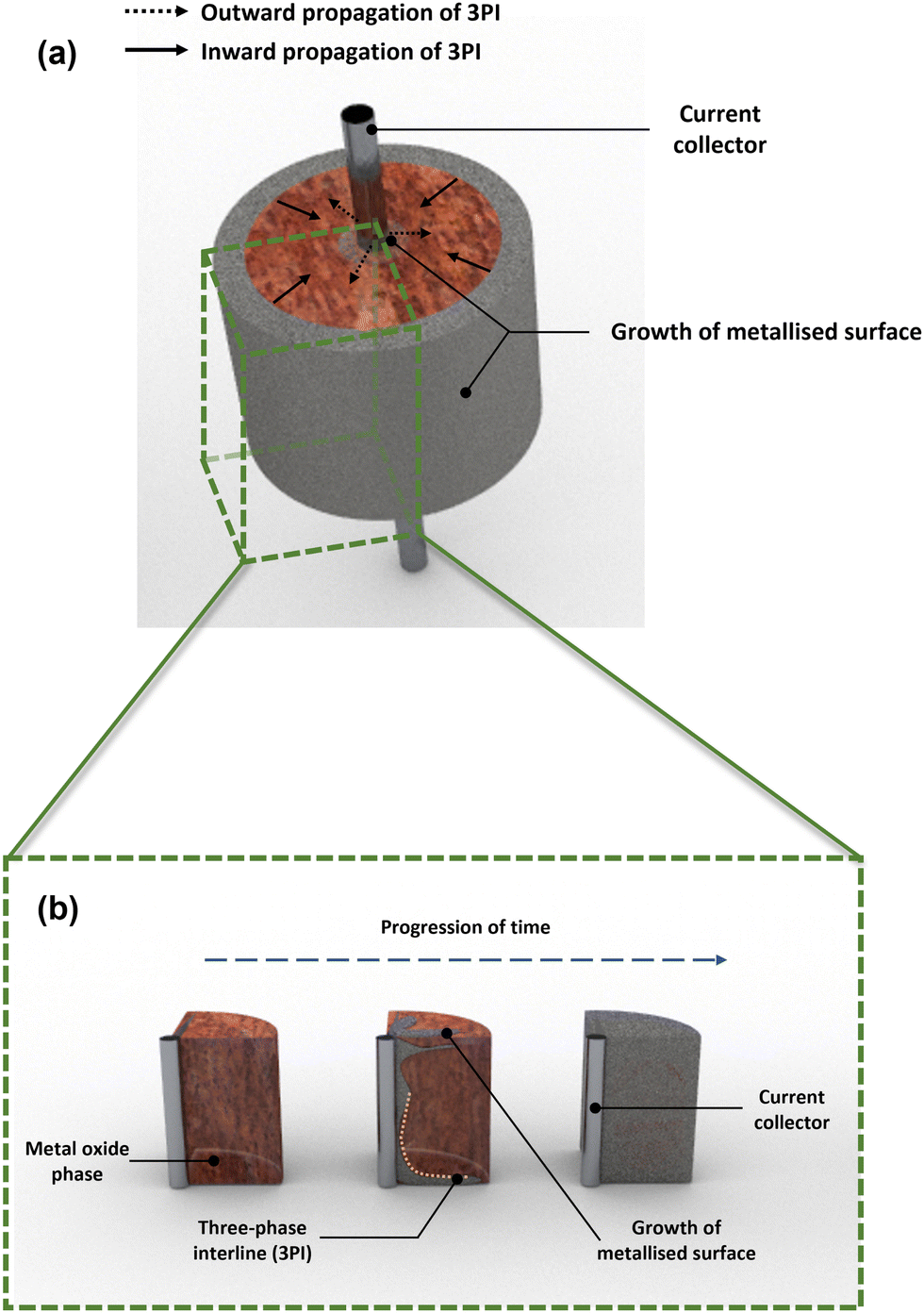
88 The three-phase interline (3PI) theory illustrated in
Fig. 4 can be used as an explanation of this occurrence. This model was initially conceived by Chen
et al. who were the first to propose a model for the 3PI before it being further developed by Deng
et al. to consider both static and dynamic 3PIs.
90,91 Kar and Evans then developed their own mathematical model accounting for metal oxide porosity, pellet radius and particle size distribution.
92 A major assumption made within this model is that microstructural changes during reduction are negligible. Furthermore, if we assume that there are two phases within a molten salt system then they can only be connected by a two-dimensional plane, however the majority of processes encompass three distinct phases where the point of intersection occurs at a single point (in one dimension), otherwise known as the 3PI. If we then take the example of a typical electrochemical system where we are reducing a metal oxide to a metal, the phases are as follows: metal phase (current passage path), metal oxide phase (reactant) and the molten salt phase (electrolyte). For pellet-based studies, the progress of forming a metal surface occurs inwardly from the ‘outer’ metal oxide surface and outwardly from the current collector. This relies on there being sufficient contact between the conductor and insulator for the transfer of electrons; transfer between oxide ions and the surrounding electrolyte.
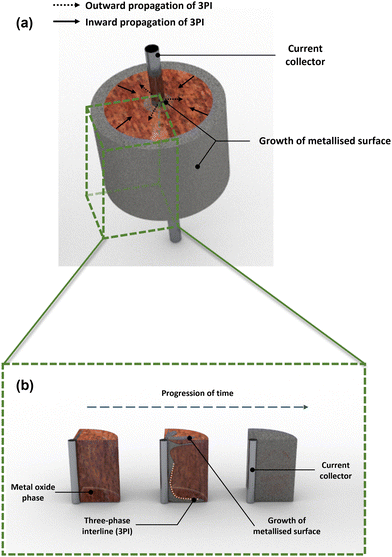 |
| | Fig. 4 3D rendering of the three-phase interline (3PI) or three-phase boundary (TPB) which highlights the metallization of the pellet surface showing inward and outward propagation of TPB in (a) and (b) where a cross-sectional area of the metal oxide phase changes with respect to time (blue dashed line). The 3PI (indicated by the orange dotted line) occurs between the current collector (central metal rod), metal oxide (red hue around the surface of a metal rod) and molten salt (surrounding region). | |
Sakamura et al. have also studied UO2 in both CaCl2 at 800 °C and LiCl at 650 °C. In the former system, the reduction occurred at <0.6 V vs. Ca/Ca2+.93 The reduction process however led to the formation of calcium metal on the metal oxide surface and prevented the formation of the TPB on the inside of the UO2 disk sample and impeding the reduction progress. In the latter system, the reduction potential was found to be <0.15 V vs. Li/Li+. This process yielded a much greater current efficiency as well as improved electrolyte diffusion in the precursor. Furthermore, Hur et al. then selected a system where a lower working temperature was needed as realised in LiCl–KCl–Li2O at 520 °C.51 The reduction potential of UO2 was determined to be −1.27 V vs. Li–Pd. The conversion of uranium dioxide to uranium metal was attributed directly to the chemical reduction facilitated by lithium as previously mentioned. The fluctuating activity of O2− ions in the molten salt is regarded as the reason behind the differences reported in reduction potentials and the same is found to be true here. The activity of O2− ions also has far-reaching effects when the reduction and decomposition potential are found to be in close proximity as this affects the kinetic pathways and rate-determining steps of a reaction. If the local O2− ion concentration increases, this impedes the withdrawal of the O2− ion from the electrolyte and thus leads to a lower reduction potential. However, the fluidised process provides a constant agitation of argon gas as well as sufficient electrical contact between the metal and metal oxide; it further enhances the mixing between these two phases and enables the process to take place at a more positive potential. Further discussion of the different electrochemical reactor types and the importance of the three-phase interline is presented in Section 7.45
Much of the headway made in improving the electrochemical reduction process of uranium chlorides is because the reaction proceeds far more easily at higher O2− ion concentrations. It is worth noting that uranium has three thermodynamically stable oxidation states, namely U3+, U4+ and U6+, and more often than not work on uranium oxides has attempted to determine the oxidation states of U4+ (UO2) and U6+ (UO3). The reduction potentials for the uranium chlorides has been summarised in Table 6 with small differences which have likely arisen due to differences in reference electrodes used and in uranium concentrations.
Progress on the electrochemical reduction of uranium has involved a range of molten chloride and fluoride systems. Nourry et al. considered the electrochemical behaviour of U3+ and U4+ in LiF–CaF2 to demonstrate a viable reduction pathway and confirmed that U3+ ions were reduced to uranium in a single step.94 Previous work has applied the same reduction to different molten salt systems such as LiF–NaF and LiF–BeF2–ZrF4.95,96 Using a nickel plate cathode, their group was able to separate U3+ from Gd3+ successfully, on the notion that the potential was carefully controlled between −0.16 and −0.38 V vs. Pt.
5.2 Plutonium
Plutonium-239, atomic number 94, is a synthetic isotope produced in nuclear reactors by neutron capture of 238U to 239U. This step is then followed by two successive beta emissions as a result of the natural instability of the radioisotopes. 239U has a half-life of approximately 20 minutes, decaying to 239Np with a half-life of approximately three days before decay to 239Pu. 239Pu has a relatively long half-life, 2.4 × 104 years and undergoes alpha decay to 235U. It exhibits unique chemical properties that have sparked interest particularly in light of pyroprocessing applications. Several studies have explored different methods of reduction that plutonium and its respective halides would follow given the rising concerns around plutonium metal production and its associated nuclear weapons proliferation risk.54 In a similar fashion to uranium studies, much of the earlier work was concerned with reducing PuCl3 as part of the Manhattan Project in the 1940s; this was done to re-use the large quantities of waste that had been generated from the production of the atomic bomb.
The correct determination of the melting point of plutonium metal was reported by comparison of different morphologies (droplets at higher temperature and powdery deposits at lower temperatures) before chemical determination at Los Alamos National Laboratory. Kolodney et al. were the first to successfully obtain plutonium metal from a melt containing PuCl3.97 A working temperature of 660 °C was applied to a Pyrex cell using a tantalum cathode and graphite anode. Once the process had been completed the final product was shown to contain minute quantities of various contaminants, including: beryllium, lithium, sodium and silicon, as a result of the interaction between the plutonium metal and Pyrex cell. Silicon has been shown to stabilise the lower density delta phase of plutonium. But no further tests were done to establish whether the addition of silicon led to an increase in plutonium stability. The Manhattan Project developed a process to extract plutonium from PuF4 using calcium metal, leaving CaF2 as a by-product, this technique being more commonly known as the ‘bomb reduction’ process.98
Subsequent work focused on obtaining higher purity depositions of plutonium without leading to the contamination that can occur from either an impure feed material or corrosion due to the electrochemical reactor design. In 1960, Blumenthal and Brodsky produced data suggesting that surface contamination was likely to occur with the molten plutonium and lead to hydrogen, carbon and uranium impurities.99 Their work focused on depositing plutonium as a solid. As a useful comparison, work on the reduction of U3O8 has found that the choice of electrochemical reactor material is important in avoiding corrosion which likely occurs in the cathode basket. In this study, an FeU6 peak was reported in an XRD pattern due to the interaction between iron present in the steel cathode basket and uranium.100 Therefore, the use of a Vycor cell (high-temperature silica resistant design) compared with Hastelloy (a superalloy of nickel, molybdenum and chromium) would prove to be far more appropriate in the design.54
Similarly, it was found that using tungsten rather than a tantalum cathode, leads to much less contamination in the final product despite earlier work indicating that a tungsten electrode was a far better choice.101 Work accomplished by Leary and Muslin found carburised tantalum cathodes to be far more promising because their heats of formation, 161 kJ mol−1, are much greater than using plutonium carbide (PuC), 7 kJ mol−1. This is indicative of stability as a cathode.102 These findings were directly useful to the Los Alamos molten plutonium electrorefining process (LAMEX) project which began in the late 1950s. Their preliminary work was channelled towards electrorefining a Pu–Fe alloy, otherwise known as ‘fissium,’103 to recover a high purity plutonium metal. In these experiments, impure plutonium was selected as the anode material and the high-purity metal formed was deposited at the cathode. Mullins, Leary and Bjorklund scaled up their process to operate with 100–300 g samples of Pu and Pu–Fe alloy before further development to a 3.5 kg scale.101 These experiments demonstrated a current efficiency of >70% and definitive reduction of impurities compared with previous reports. The final Fe concentration was found to be 0.02% compared with 2.54%. Their subsequent work involved selecting different crucible compositions such as alumina and magnesia (magnesium oxide) which were both able to tolerate high temperatures and were corrosion-resistant.
The choice of a suitable crucible material is vital to successfully processing and recovering plutonium metal. Most of these designs utilise alumina and magnesia due to their strong chemical and thermal resistance.104 Despite these two important qualities, they are only suitable for single use because their mechanical strength is far too low. Plutonium metal adherence to the surface of a ceramic is another significant issue; this may occur in the presence of other metals which changes the chemical characteristics of plutonium metal. Paget et al. reported this phenomenon in the presence of 2000 to 5000 ppm of titanium.105 It was later found that this could be overcome, on a smaller-scale, with the addition of CaCl2.106 A study has also been conducted on Si3N4 with results suggesting that this material has far superior corrosion and thermal resistance characteristics compared with magnesia, tantalum, tungsten, and other alloys.107
Considering the design of crucibles more closely, it was found that use of magnesia alone was far more stable and reduced hydrogen contamination. Magnesia is much more chemically inert when stabilised with yttria (Y2O3) and the same was also found to be true for fluoride molten salt systems. Yttria-stabilised magnesia was later found to have both mechanical and thermal shock-resistant properties which made it more suitable in electrolytic cell applications. The product was collected as an annular ring at the bottom of the crucible. Minor modifications to the crucible in the form of a magnesium titanate addition would lead to the formation of a flat disk of plutonium rather than a ring structure.108 Work conducted by Curtis considered different molten salt systems, where a mix of 30 wt% PuCl3, 28 wt% BaCl2 and 42 wt% KCl with the addition of PuCl3 as feed material.109 Their findings revealed several desirable properties pertaining to BaCl2; first, that it separated the oxides from the cathode ensuring the final product remained coalesced and second that it caused the oxides and less dense impurities to have a greater buoyancy induced by BaCl2.
Much of the later work focused on the electrochemical reduction of plutonium oxide with an overall aim of determining the feasibility of the process. In these experiments, the feed material, PuO2, was directly reduced in a molten fluoride salt system which was comprised of 50 wt% LiF, 20 wt% BaF2 and 30 wt% PuF3. The low current efficiency of 10–20% reported was most likely due to unwanted parasitic reactions taking place at the surface of the anode structure. These parasitic reactions could either have been the oxidation reaction of carbon with oxide anions or the reduction of Pu3+ and Pu4+ as described in eqn (18)–(20).
Anode reactions
It was found that a slow and constant addition of PuO
2 avoided the accumulation of the feed material at the bottom of the crucible. Addition of PuO
2 in bulk proved detrimental because it inhibited the coalescence of the final product and a substantial proportion of the oxide was not electrolysed. Brodsky and Carleson further investigated the potentially deleterious effects caused by a molten fluoride system.
110 Their work led to the understanding that the proximity of fluoride to plutonium would lead to a neutron flux in a NaCl–KCl system due to the
19F(α,n)
22Na reaction. In a LiCl–KCl system, such (α,n) reactions are less common. Alternatively, the use of PuCl
3 would also prove detrimental, given its highly hygroscopic nature resulting in the absorption of any moisture in the molten salt system. To avoid this, their molten salt system was chosen to be a dichloride Cs
2PuCl
6 salt which is non-hygroscopic, prevents any occurrence of neutron flux in the molten salt system and is readily produced.
104 They found that electrodeposition took place at the tantalum cathode at a temperature of 700 °C, albeit at a lower yield, but there was substantially less contamination by calcium, molybdenum and nickel in the final product. In the following years, there was much greater throughput of electrorefined plutonium alloy; overall, 653 batches, with the anode metal feed each consisting of approximately 3 kg of plutonium alloy, led to 1568 kg of plutonium metal recovery.
111 These batch weights were increased to 4 kg to increase product throughput and then finally to 6 kg after further anode feed optimisation in 1982.
112
Shortly after, Poa et al. conducted electrochemical studies into the deposition of UCl3 and PuCl3 within a melt consisting of BaCl2–CaCl2–LiCl–NaCl at 480 °C.113 This study used a three-electrode system where the working potential was compared to Ag/AgCl. Their work shed light on the effects of changing temperature and Pu3+ ion concentration on the reaction kinetics. It was reported that the reduction of Pu3+ took place in a single step, as indicated in eqn (21).
A similar study was conducted focusing on the reduction pathway adopted by Shirai
et al. using PuCl
3 in a LiCl–KCl eutectic using 0.544 wt% PuCl
3 at 450 °C.
114 A study where the temperature was varied revealed that there was a change in the magnitude of the cathodic current depending on the working electrode material and this was complemented by an increase in the diffusion coefficient with increasing temperature. Initially, Nissen reported a diffusion coefficient of 0.51 × 10
−5 cm
2 s
−1 at 400 °C, 1.11 × 10
−5 cm
2 s
−1 at 500 °C and 1.62 × 10
−5 cm
2 s
−1 at 600 °C.
115 Serp
et al. then determined that the diffusion coefficient of PuCl
3 at 460 °C fell between 1.3 × 10
−5 and 1.85 × 10
−5 cm
2 s
−1 agreeing with previous findings.
116 Due to difference in the working electrode surface area, diffusion coefficients are likely to vary. Although no observable difference was found by changing materials, the temperature had a direct effect on the potential of the cathode. The data indicated that the process occurred
via a reversible diffusion-controlled process; however, a chronopotentiometry study performed by Martinot and Duyckaerts indicated a retardation due to the formation of a coordination compound formed between Pu
3+ and the Cl
− ions in the electrolyte.
117Table 7 summarises the apparent standard potentials for the most stable oxidation state, Pu
3+, as reported in early studies.
63,118 The results are generally consistent, with the exception of the data obtained by Campbell and Leary and Martinot.
118,119
Table 7 Summary of reduction potentials of Pu3+/Pu0 at various temperatures in molten LiCl–KCl
|
T (K) |
Apparent experimental potential (V) of Pu3+/Pu0 |
| Campbell and Leary118a |
Martinot119a |
Masset120b |
Roy et al.63a |
Sakamura et al.121a |
Shirai et al.122b |
|
Reference electrode: Ag/AgCl.
Reference electrode: Cl2/2Cl−.
|
| 673 |
−2.1224 |
−2.0539 |
|
−2.845 |
−2.8227 |
|
| 698 |
−2.1199 |
−2.0273 |
|
−2.827 |
−2.8061 |
|
| 723 |
−2.1174 |
−2.0007 |
|
−2.808 |
−2.7895 |
−2.748 |
| 733 |
−2.1164 |
−1.9901 |
−2.7962 |
|
−2.7828 |
|
| 773 |
−2.1124 |
−1.9475 |
−2.7699 |
−2.775 |
−2.7562 |
−2.711 |
| 823 |
−2.1073 |
−1.8943 |
−2.7361 |
|
−2.7230 |
−2.673 |
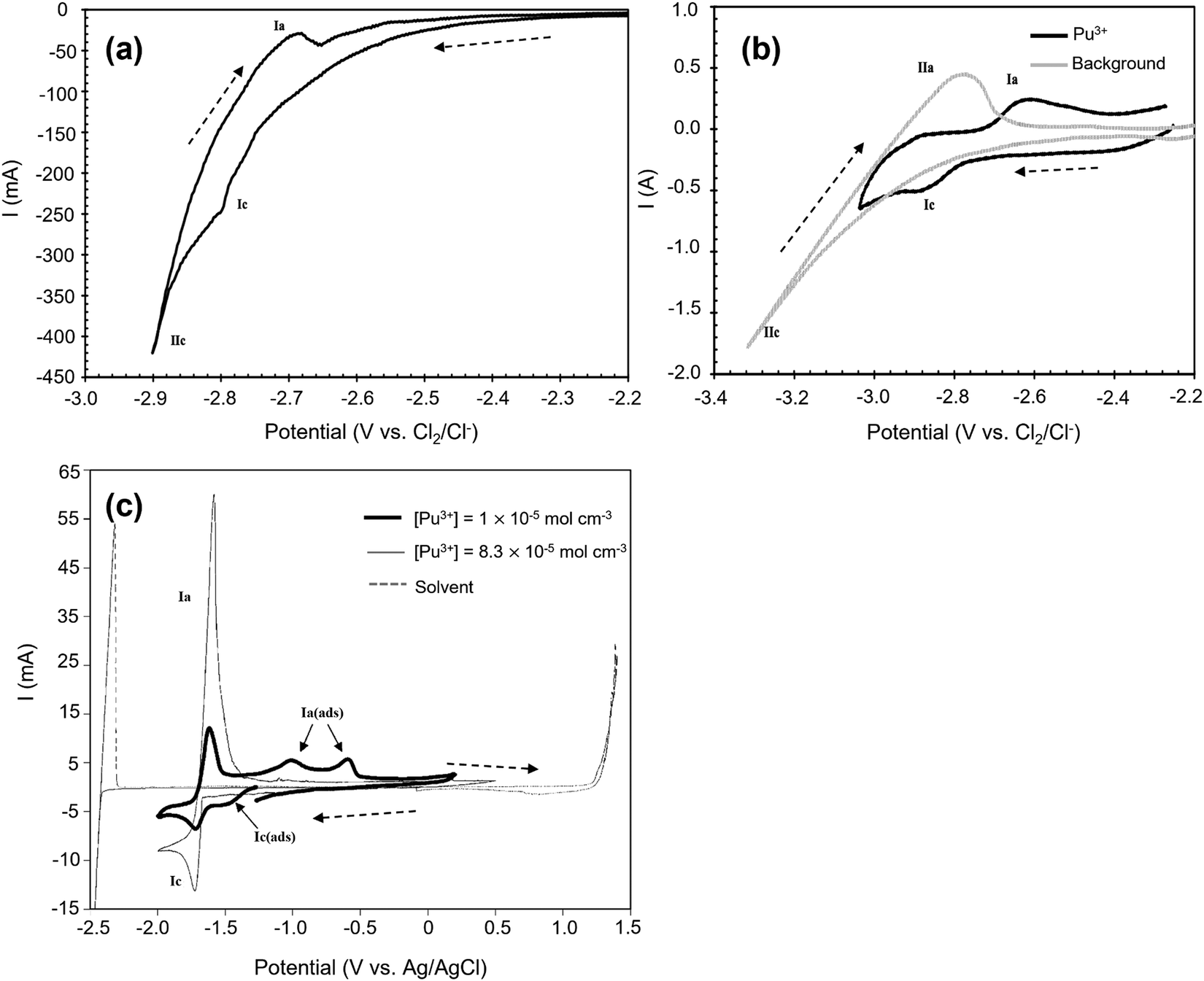
Fig. 5(a) and (b) are cyclic voltammograms of PuCl3 in equimolar NaCl–KCl and CaCl2 systems. In Fig. 5(a) there are two cathodic peaks, namely Ic and IIc. The first has been attributed to the direct reduction of Pu3+ ions to Pu and the second is ascribed to the electroplating of Na+ ions. However, the voltage separation between both processes is very close which makes them difficult to distinguish; whereas in the CaCl2, a difference of approximately 0.4 V makes the two processes easier to differentiate. McCurry et al. report that the addition of sodium chloride to a potassium chloride melt reduces the voltage separation difference making it difficult to separate the two processes without the possibility of electroplating sodium on the surface of the cathode.123Fig. 5(c) may be compared to the predominance diagram illustrated in Fig. 7(b) where the reduction potential is found to agree with theoretical prediction and occurs at −1.76 V. Predominance diagrams, as illustrated in Fig. 7, shed light on the electrochemical processes for various metal oxide, chloride and metal ion species (highlighting regions of ion stability) and are acknowledged as a preliminary step to understanding their reduction pathways in ideal conditions. Work by Brown et al. and Abdulaziz et al. has been undertaken on developing a comprehensive understanding of both uranium and plutonium oxide reduction steps and are described in Fig. 7(a) and (b).68,124 As before, this study confirms the findings in Fig. 5(a) and (b) where there is only one reduction step; it also proves that this process is feasible at a lower reduction potential which incurs a reduced energy penalty. However, at lower concentrations, there is an additional cathodic peak found at ∼−1.5 V which has been ascribed to the underpotential associated with the interaction of plutonium with the tungsten rod.116
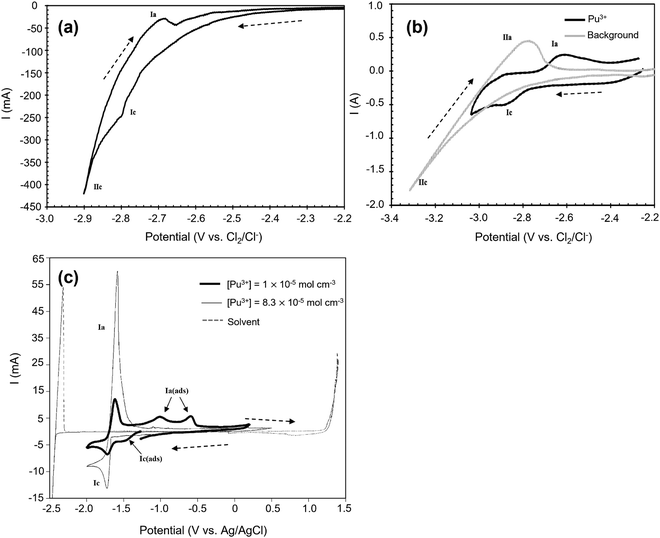 |
| | Fig. 5 Cyclic voltammetry of Pu3+ ion species in three different molten salt systems conducted on the tungsten working electrode. (a) Equimolar NaCl–KCl at 1073 K, scan rate 0.1 V s−1, surface area (SA) = 0.31 cm2. [PuCl3] = 1.1 × 10−3 mol. (b) CaCl2 at 1073 K, scan rate: 0.1 V s−1, SA = 0.31 cm2. [PuCl3] = 1.1 × 10−3 mol. (c) LiCl–KCl eutectic at 733 K, SA = 0.2 cm2. (a and b) Have been reprinted from Lambertin et al. © The Electrochemical Society. Reproduced by permission of IOP Publishing Ltd.125 (c) has been reprinted from Serp et al. with permission from Elsevier.116 | |
A small-scale study examined the electrochemical reduction of PuO2 in CaCl2–CaO by Jones et al. at 900 °C.126 Whereas previous studies had demonstrated that the reduction of MOX fuels (where a mixture of plutonium and uranium oxides are pressed into a nuclear fuel pellet) was viable, none had solely considered the use of PuO2 as a feed material. One of the reasons for not considering pure PuO2 for this was that the processing of feed material into nuclear fuel pellets could lead to increased radiation doses for operators as a result of ingrowth of the gamma emitting daughter radionuclide 241Am. These studies had considered different ratios of uranium and plutonium; Iizuka et al. showed that using LiCl-0.5 wt% Li2O ensured that the materials (47 wt% U, 35 wt% Pu, 4 wt% Np)O2 would eventually form an alloy.127 Kurata et al. demonstrated the reduction of (90.55 wt% U and 9.45 wt% Pu)O2 in LiCl-1.0 wt% Li2O.128 Both molten salts had a slightly lower working temperature of 650 °C. The findings revealed that the current efficiency of the process was poor, yielding 12.9% owing to charge losses in the reduction of calcium oxide and furthermore, the concentration of plutonium was found to be 3–5 wt%. Additionally, there were significant amounts of contamination, likely caused by the choice of basket material. The lack of cyclic voltammetry (CV) data also made it difficult to optimise the potential and overall charge that needed to be applied. From this, it was clear that there were no intermediate species and the formation of plutonium occurred in a one-step deoxidation from PuO2 with four electrons exchanged.
Recently, Rappleye et al. attempted the plutonium electrorefining process with a focus on reducing sub-kilogram quantities in CaCl2.129 Using NaCl–KCl eutectic can lead to pyrophoric sodium and potassium due to the co-reduction of these solvent ions, whereas the use of CaCl2 eliminates this. The separation potentials between Ca2+ and Pu3+ are greater than Na+/K+ and Pu3+ which avoids co-deposition with Pu3+. The reported efficiency and yield of this process was 71.4% and 78.2%, respectively, which is in line with previous studies by Mullins.112 Differences in results may arise because previous researchers used a lower concentration of MgCl2 which serves as an extractant (1.9 wt% compared with 3 wt%). Additionally, the use of a new tungsten cathode annulus led to a high concentration of tungsten in the final product (1200 ppm); this may have been caused by loosely adhering tungsten particles coalescing with the plutonium deposits or by the very nature of a small-scale process where there is a greater surface area of contact with the deposited plutonium.
5.3 Neptunium
Neptunium, atomic number 93, is a synthetic element that has been produced in significant quantities by the nuclear industry and may occur as a constituent in MOX fuel. It is commonly found as a product within radioactive waste streams and so understanding its interaction with other metallic species is critical. The development of compact and economic reprocessing plants is especially important and relies on an understanding of the actinide chemistry concerning neptunium species. The main production route for 237Np in a nuclear power plant is the capture of a neutron by 235U to form 236U. 236U subsequently captures a neutron to form 237U, which subsequently decays to 237Np with an approximately 7 day half-life. There is a minor production route via the n, 2n reaction with 238U to 237U, but this is more significant in a fast reactor than a thermal reactor. An n, 2n reaction is where 2 neutrons are emitted after the capture of one incoming neutron. The subsequent beta-decay leads to the formation of 237Np.130 Neptunium was the first of the actinide elements to be synthesised using a cyclotron in 1940 through the simple process of neutron bombardment of UO3. McMillan and Abelson,131 later questioned whether a decay product had formed from their new find and a year later Seaborg et al. published their work on the discovery of element 94 or plutonium.132 There are 22 known isotopes of neptunium that have been characterised; however, most radioanalytical chemists will be concerned with the three whose half-lives make them obtainable: 237Np (2.14 million years), 238Np (2.12 days) and 239Np (2.35 days).133 Tens of kilograms of neptunium, alongside associated waste, have been produced worldwide. Since the early 1980s, many of the electrochemical studies, in particular, voltammetric (where a potential is applied and a current is measured) and potentiometric (where a constant current is applied and the potential is measured) have been used to elucidate the governing kinetics and mass transfer processes and also to determine the formal working potential against a standard reference electrode. To complement this work, there has been a surge in effort towards reprocessing nuclear fuels, some of which have been reported in Table 8 and described in Fig. 7(c).
Table 8 Summary of reduction potentials of Np3+/Np0 at various temperatures
|
T (K) |
Apparent experimental potential (V) of Np3+/Np0 |
| Masset et al.134b |
Shirai et al.135a |
Martinot136a |
Roy et al.63a |
Krueger et al.137b |
Fusselman et al.138b |
|
Reference electrode: Ag/AgCl.
Reference electrode: Cl2/2Cl−.
|
| 673 |
−2.767 |
|
−2.39 |
−2.725 |
−2.725 |
−2.725 |
| 698 |
−2.765 |
|
|
−2.714 |
−2.715 |
−2.714 |
| 723 |
−2.733 |
−2.716 |
−2.35 |
−2.698 |
−2.697 |
−2.698 |
| 748 |
−2.700 |
−2.703 |
|
|
|
|
| 773 |
−2.682 |
−2.690 |
−2.31 |
−2.668 |
−2.660 |
−2.668 |
| 798 |
|
−2.677 |
|
|
|
|
| 823 |
−2.568 |
−2.662 |
−2.27 |
|
|
|
Neptunium has well-reported valence states in aqueous solution, which include: Np(H3O)63+ (III), Np(H2O)84+ (IV), NpO2(H2O)6+ (V), NpO2(H2O)62+ (VI) and NpO53− (VII). The first four oxidation states (III to VI inclusive) are stable in water and form hydrated ions. However, VII is only stable in dilute alkaline solution. From an electrochemical perspective, Np ions possess interesting redox properties where transfer of a single electron results in a fast equilibrium (eqn (22) and (23)) but a process where a Np–O bond is formed (eqn (24)) is much slower.
Standard potential (V)
| | | Np3+ ⇌ Np4+ + e− −0.152 | (22) |
| | | NpO2 ⇌ NpO2+ + e− 0.564 | (23) |
| | | Np4+ + 2H2O ⇌ NpO2+ + 4H+ + e− 0.749 | (24) |
Cassol
et al. were the first to study the reaction process in the presence of NaClO
4 at 298 K by potentiometric titrations.
139 In accordance with other literature, they found that the greater level of acidity was in the following order: PuO
22+ > NpO
22+ > UO
22+,
i.e. in reverse order of atomic number.
140,141 The unstable nature of NpO
22+ was confirmed by Martinot
et al. who reported a similar lack of stability in the presence of chloride ions in a LiCl–KCl eutectic at 450 °C.
136| | 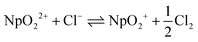 | (25) |
The first two reduction schemes from hexavalent NpO
22+ to pentavalent NpO
2+ in
eqn (25) and subsequently to tetravalent Np
4+ have been determined from
in situ absorption electron spectroscopy.
142,143 The reduction of Np
4+ to Np
3+ was reported by both CV and differential pulse polarography. It was found that Np
3+ and U
3+ share similar properties to Np
4+ and U
4+ in a LiCl–KCl molten salt system.
One of the first studies on neptunium, conducted in 1969, focused on the diffusion coefficients for the higher valence states.136 The starting material was made by the slow oxidation of Cs2NpCl6 to NpO2 (5+) before undergoing chronopotentiometry. The work revealed that the higher valent NpO2 (4+) had a lower diffusion coefficient of 2.0 × 10−6 cm2 s−1 compared with 4.8 × 10−6 cm2 s−1 for NpO2 (5+). This was because the ion in solution is more complexed when it comes into contact with chloride ions and thus it hinders the movement of the then formed coordination complex.136 Further work by Martinot revealed that the single-step reduction from NpCl3 and NpO2 was equally possible and highly reversible however the reduction of NpCl4 did not occur directly requiring reduction first to NpCl3.144 Use of higher valence states of neptunium was avoided because in the presence of chloride ions it would be reduced. The results proved positive with a powdery deposit of neptunium adhering to the surface of the tungsten cathode with some degree of impurity. These impurities mainly arose from the feed material itself with additional elements being drawn from the eutectic and silica crucible similar to the plutonium work.
Later, a more comprehensive study was conducted on the reduction of NpCl4 to Np in LiCl–KCl eutectic at 475 °C.134 It was concluded that the reduction took place in two separate steps with the single electron transfer of Np4+ to Np3+ taking place at a positive potential (eqn (22)). This experimental data is in good agreement with the predominance diagram for Np species in Fig. 7(c) where the electrochemical reduction of Np4+ to Np proceeds in two steps; the first is where one electron is exchanged and the second is where three electrons are required at a more negative cathodic potential (eqn (26) and (27)).
| | 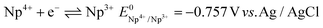 | (26) |
| | 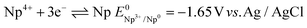 | (27) |
Square-wave voltammetry (SWV) was also used to determine the electron transfer and to compare the reduction potentials measured previous to the work undertaken by Masset.
134 The first peak indicated a single-electron transfer process at −0.757 V
vs. Ag/AgCl (surface area (SA) = 0.24 cm
2). However, the second cathodic peak was significantly sharper with a potential of −1.65 V
vs. Ag/AgCl associated with a three-electron transfer from Np
3+ to Np
0. The CV revealed that, at incremental scan rates, there was a noticeable negative shift in the cathodic peak (Np
3+ to Np
0). This finding is evidence that at higher scan rates the process is not reversible; a plot of current (
I) against scan rate potential (
ν1/2) further reveals that this is a diffusion-controlled process. No morphological or materials characterisation studies were performed in this paper. Other studies on NpCl
3 have considered its electrochemistry at the molybdenum and liquid metal electrodes with more positive potentials reported for the liquid metal electrodes.
73
5.4 Americium
Americium, atomic number 95, is a synthetic element, with two commonly occurring isotopes 241Am and 243Am. Applications of this radioisotope vary from those found in the nuclear industry to domestic applications e.g. smoke detectors. Americium was first discovered in 1944 by Seaborg et al. who realised that through the controlled bombardment of neutrons with 238U and 239Pu atoms, it was possible to produce other heavy elements.145 Americium was generated as a result of the irradiation of 238U with neutrons to form 241Pu. This subsequently led to the observation that 241Pu undergoes beta-decay according to the following process.| | | 238U → 239Pu (n,γ) 240Pu (n,γ) 241Pu → 241Am | (28) |
For domestic applications, americium, 241Am, is used in sub microgram quantities (approx. 0.3 μg) in domestic smoke alarm detectors. The 241Am radioisotope occurs as an oxide which is housed within a ventilated metal box and emits a small but constant stream of alpha particles which ionises the surrounding air molecules. A small charge is produced and is detected between two plates at different voltages in the metal box. In the presence of smoke, the ionised molecules will absorb the smoke particulates, impeding current flow and setting off the alarm.146 For industry applications, 241Am is mixed with beryllium and pressed into a pellet to provide a practical neutron source for purposes such as measuring the thickness of glass as it exits the production line. The neutrons are formed by (α,n) reactions with the beryllium. 241Am is also used in medical applications, such as radiography, and as a source for X-ray fluorescence spectroscopy.
Relatively little is known about the reduction pathway of AmClx (where x = 3 or 4) and published work on AmO2 is even more scarce. Fig. 6(d) describes the apparent standard potentials of Am2+ in LiCl–KCl eutectic, which is known to be stable along with Am3+. However, there is very little data published on the standard potential of Am3+, which undergoes a two-step reduction to Am0.120Fig. 6(d) indicates that there is good agreement in work conducted after the year 2000 and that the potential lies between −2.92 and −2.87 V in the range 723–823 K. There has been some headway made by Serp et al. who found, using CV, that the reduction pathway undergoes two separate steps.147Fig. 6(d) indicates that the reduction of Am3+ first involves a single electron exchange and the second involving the exchange of two electrons. Each of the two cathodic peaks were associated with two corresponding anodic peaks, the first cathodic peak being much flatter with a peak current of −6 mA as evidenced in the SWV. The reported peak current is associated with the exchange of a single electron during the reduction of Am3+ to Am2+ at the tungsten working electrode (SA = 0.2 cm2). The second cathodic peak with a current of −16 mA indicated a two-step electron transfer. The relationship between the number of exchanged electrons and the half-wave (Gaussian) peak can be determined by eqn (29).147
| | 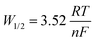 | (29) |
The transitions, which take place between the respective oxidation states of Am
3+ and Am
2+, have been confirmed in previous work.
148 Both of these species are the most stable in LiCl–KCl eutectic with the latter, Am
2+, being predominant. It must also be made clear, that the separation of americium from the lanthanides is the most difficult to achieve potentiometrically.
138 Additionally, using chronopotentiometry, the diffusion coefficients of species have been reported at two different temperatures (723 K and 743 K). Both studies report that Am
3+/Am
2+ remains stable at temperatures between 733–823 K in LiCl–KCl eutectic. With regard to understanding the reduction process of americium in LiCl–KCl eutectic the reduction proceeds in two separate steps on a tungsten working electrode. The first reduction of Am
3+ to Am
2+ occurs at −2.74 V
vs. Cl
2/Cl
− shortly followed by the reduction Am
2+ to Am at −2.82 V
vs. Cl
2/Cl
− at 504 °C.
149 Further details of the reduction potentials of Am
2+ to Am
0 may be found in
Table 9.
In addition to studies on an inert working electrode, there has been a focus on using liquid cadmium and results suggest that the reaction occurs in a single-step with the reduction of Am
3+ to Am; this occurred with an apparent potential of −2.56 V
vs. Cl
2/Cl
− at 504 °C (
eqn (32)).
149 Because of the small potential range (<100 mV), the complete separation of americium from lanthanum in this study would not yield sufficient recovery of americium. In general, greater current densities can be induced by installing a blade stirrer (impeller) to improve the mass transfer between the molten salt and metal chloride and ensuring that the evolution of chlorine gas does not contaminate the reduced product are steps that can be taken to increase the quantitative recovery of americium. Americium dispersion can be reduced by appropriate selection of cathode material.
The discoveries of the higher radiotoxic actinides (neptunium, plutonium and americium) have fuelled considerable work into partitioning and transmutation (P&T) studies, although the work has mostly focused on plutonium and uranium. As outlined in previous sections, the focus has been on the availability of thermochemical and electrochemical data in literature. Up till now, the rate of development and understanding of actinides such as americium and curium (as will be described in the following section) has been slow.
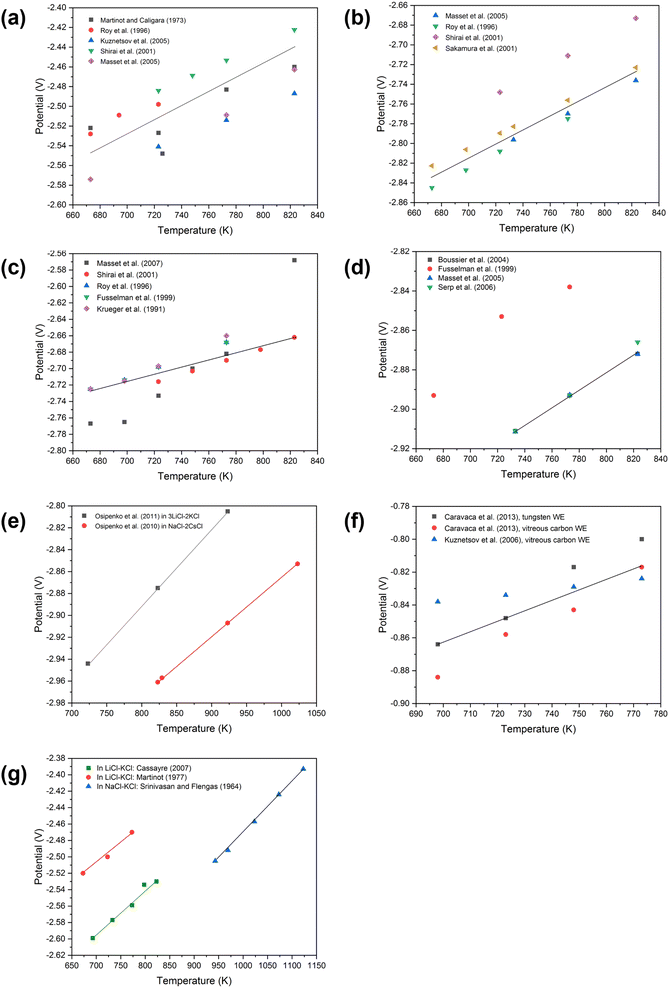 |
| | Fig. 6 Comparison of experimental data for certain actinide and lanthanide species. The following have been compared (the value in brackets refers to Tables 6–12 with accompanying references): (a) U (6), (b) Pu (7), (c) Np (8), (d) Am (9), (e) Cm (10), (f) Eu (11) and (g) Th (12). Reference electrodes are specified in the footnotes below Tables 6–12. | |
 |
| | Fig. 7 Predominance diagrams for the most relevant actinide elements; (a) U, (b) Pu, (c) Np, (d) Am, (e) Cm and (g) Th and lanthanide element; (f) Eu species in LiCl–KCl eutectic at 500 °C. All diagrams have been drawn using HSC Chemistry 6.12. | |
Table 9 Summary of reduction potentials of Am2+/Am0 at various temperatures in molten LiCl–KCl
|
T (K) |
Apparent experimental potential (V) of Am2+/Am0 |
| Boussier et al.150b |
Fusselman et al.138b |
Roy et al.69a |
Masset et al.120b |
Serp et al.147b |
Lambertin et al.148b |
|
Reference electrode: Ag/AgCl.
Reference electrode: Cl2/2Cl−.
|
| 673 |
|
−2.893 |
|
|
|
|
| 698 |
|
|
|
|
|
|
| 723 |
|
−2.853 |
−2.843 |
|
|
|
| 733 |
−2.911 |
|
|
−2.9114 |
−2.911 |
|
| 743 |
|
|
|
|
|
−2.945 |
| 773 |
−2.893 |
−2.838 |
|
−2.8927 |
−2.893 |
|
| 823 |
−2.872 |
|
|
|
−2.872 |
−2.866 |
5.5 Curium
Curium, atomic number 96, was first discovered in 1944 by Seaborg et al. who applied a similar method to that used for the discovery of americium and plutonium.151 In this case, rather than bombarding plutonium, 239Pu, with neutrons, alpha particles (helium cations) were used and the characterisation of curium was confirmed via the energy of the alpha particle emissions (eqn (33)). To achieve this, a 60-inch cyclotron at Berkeley was deployed to provide a controlled environment for such high-energy bombardment. Due to its size a much larger target was bombarded and a greater quantity of products formed. These products were then isolated and investigated to determine both chemical and physical properties. The isotopes of curium occupy a wide range in mass which extends from 238Cm to 251Cm. In general, the curium radioisotopes are difficult to work with, but two of its isotopes, 242Cm and 244Cm, have been used in power sources with amenable results.152 Moreover, 244Cm demonstrates remarkable incandescence, yielding 122 W g−1, on the other hand, 238Pu, which is more readily available, has been preferred in applications requiring this attribute.153 The third most commonly studied isotope, 248Cm, has also been used as an enriched target for the production of the transfermium nuclides (elements with atomic numbers greater than one hundred).154| | 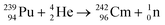 | (33) |
Curium has a much lower volatility than americium. 242Cm exhibits high levels of alpha, beta and gamma radiation rendering any laboratory-based work hazardous. As a result, extremely small quantities (on a milligram scale) of curium are used when conducting analytical investigations, with crystallographic examination being hampered because of the damage caused by the gamma rays to X-ray detectors.155 The high specific activity of curium isotopes make their partition from radioactive waste desirable. Isotopes of curium have large neutron-capture cross-sections which necessitate their partition from materials prior to processing to prevent contamination from capture products of curium in further experimental work.156 The high neutron capture cross sections of curium isotopes make them ideal for destruction by transmutation to radioisotopes with shorter half-lives.
In the aqueous phase, curium assumes a trivalent state as this is the most stable oxidation state. This stability is a result of the half-filled (5f7) configuration. In strictly anhydrous conditions, the 4+ oxidation state of curium occurs frequently as a compound, examples being CmO2 and CmF4. Much of the electrochemical work is focused on the 3+ oxidation state and there are very few studies that have been conducted in molten salt systems, with only a single report on the standard potential in Cm–Zn alloys.54,157,158 Two separate studies have been undertaken using different molten salt systems focusing on the reduction pathway adopted by Cm3+ in 3LiCl–2KCl and NaCl–2CsCl;156,159 the apparent standard potentials are shown in Table 10. Fig. 6(e) shows the difference between reduction potentials in two different molten salt systems. As the temperature increases, the reduction potential becomes more positive, suggesting the practical viability of this process at lower temperatures. In both, techniques such as CV and SWV, were used to elucidate the reduction pathway and determine the diffusion coefficient. It was found in both cases that the process remained reversible when the sweep rate was below 0.1 V s−1. Thus, the process was governed by mass transfer and was proven to be irreversible as shown by the observed cathodic shift (according to a higher sweep rate suggesting the process was charge transfer controlled).160
Table 10 Summary of reduction potentials of Cm3+/Cm0 at various temperatures
|
T (K) |
Apparent experimental potential (V) of Cm3+/Cm0 |
| Osipenko et al.156 in 3LiCl–2KCla |
Osipenko et al.159 in NaCl–2CsClb |
Osipenko et al.161 |
|
Reference electrode: Ag/AgCl.
Reference electrode: Cl2/2Cl−.
Value has been extrapolated between 829–1023 K using correlation: ECm3+/Cm0 = −3.285 + 5.48 × 10−4T.
|
| 723 |
−2.944 |
|
−2.924 (3LiCl–2KCl) |
| 823 |
−2.875 |
−2.961 |
|
| 829 |
|
−2.957 |
−2.996 (NaCl–2CsCl) |
| 923 |
−2.805 |
−2.907 |
|
| 1023 |
|
−2.853c |
−2.727 (NaCl–KCl) |
SWV of the NaCl–2CsCl system revealed a single independent peak indicating a single-step three electron transfer between −1.0 and −2.1 V vs. Ag/AgCl. According to eqn (29), the number of electrons exchanged is: 2.99 ± 0.15 electrons. Thus, eqn (34) describes the reduction found in this experiment.
The number of electrons exchanged was determined through SWV in both molten salt systems measuring the half-width of the peak recorded over a range of different frequencies: 12–30 Hz for the NaCl–2CsCl system and 6–80 Hz for the 3LiCl–2KCl system. There was good agreement between both systems; from the first, a total electron exchange of 2.99 ± 0.15 was reported compared with the second system where an exchange of 2.98 ± 0.06 electrons was calculated. Inspecting the cyclic voltammograms more closely reveals a difference in the cathodic potential between the two systems; in the 3LiCl–2KCl system, the single cathodic peak appears at ∼−1.95 V compared with ∼−2.05 V, at 723 and 823 K, respectively.
161 In both studies, the diffusion coefficient was found to increase with temperature according to Arrhenius-like behaviour. However, the associated activation energy was shown to be much lower in the 3LiCl–2KCl system (28.2 kJ mol
−1) compared with the NaCl–2CsCl system (44.5 kJ mol
−1); this is a result of the nature of the ionisation potential of the LiCl salt which is the smallest of the alkali metal cations.
162
5.6 Europium
Europium, atomic number 63, is a member of the lanthanide series and occurs naturally in minerals such as bastnaesite (La, Ce)CO3F and monazite (La, Ce)PO4. Typically, the concentration of europium is below 0.1%. This element is regarded as one of the most reactive metals of the lanthanide series being stored under an inert fluid to prevent oxidation. As a metal, it can fluctuate between a divalent and trivalent state with application of pressure, existing primarily in the 3+ oxidation state.163 Europium is not widely available and the method by which it is extracted from any mineral is dependent on its concentration in relation to other lanthanide like elements (cerium, yttrium and ytterbium) and the composition of the ore. This has led to the employment of different processing strategies. McCoy initially used zinc to extract europium from other lanthanide elements followed by the precipitation of europium (previously known as europous) sulfate with high recovery efficiency.164 However, europium is a commonly found byproduct of irradiated nuclear fuel and efforts have been made to focus on an extraction method in molten salt media focusing on the reduction of EuCl3 to EuCl2.165 Of all the lanthanide metals, there has been relatively little progress made in understanding the electrochemistry of the europium species, with most known about the trihalides.166,167
One of the first molten salt studies performed on europium was in a LiCl–KCl salt to determine the standard electrode potential in comparison with samarium and ytterbium; in this study europium was found to have the most positive potential.166,167 Other work has measured the redox potential for the Eu(II)/Eu(0) process, considering Eu–Pb alloys in NaCl–CsCl and the electrochemical behaviour of Eu(II) at low temperatures.168 Additional work has attempted to explain the behaviour of Eu3+/Eu2+ in LiF–NaF–KF molten salt; Kuznetsov et al. focused on the electrochemical behaviour of Eu3+ species in chloride melt, with studies in NaCl–KCl84,169 and CsCl at 973–1173 K.170 The apparent standard potentials measured are summarised in Table 11. A report of standard potential in NaCl–KCl was the first comprehensive study attempted by Kuznetsov et al. on Eu(III). It was found that the reduction to Eu(II) proceeded in a single step with the exchange of one electron.
Gibilaro
et al. studied the extraction of EuF
3 in LiF–CaF
2 at 840 °C.
171 They demonstrated that the reduction proceeds in two steps, the first is in accordance with
eqn (35); the second reduction occurs with the exchange of two electrons, as described in
eqn (36). However, this process had a significant issue; the Eu
2+ reduction potential was found to occur at a more negative potential than the deposition potential of lithium meaning that the final (direct) recovery of europium was difficult. To circumvent this issue, a reactive cathode substrate, copper, was employed leading to an intermetallic compound of the nature MN
y, where M is the metal substrate (copper) and N is the deposited metal species (europium). Compared with the inert metal electrode, the appearance of a large peak in the cyclic voltammogram is associated with the formation of an intermetallic compound shown in
eqn (37). The application of copper as a reactive cathode substrate was also reported, with success, in the extraction of neodymium and gadolinium.
172| | | Eu2+ + 2e− + yCu ⇌ EuCuy | (37) |
Linear sweep voltammetry and chronopotentiometry techniques were used to find the diffusion coefficient for both oxidation states and they showed good agreement. The diffusion coefficient was found to vary inversely with the oxidation state. Subsequent work considered the co-electrodeposition behaviour of both Eu
2+ and Al
3+ in a LiCl–KCl eutectic salt; it was found to be essential to maintain the concentration of Al
3+ below that of Eu
2+ to increase the size of the potential window and to prevent the dissolution of aluminium.
173 In the reaction process, however, the Al
3+ and Eu
2+ ions are reduced consecutively which leads to the formation of Al
xEu
y (
eqn (38)–(41)). On the counter electrode, aluminium metal is continuously oxidised; this then migrates to the working electrode to replenish depleting levels of Al
3+ on the working electrode. This subsequently reacts with Eu
2+ to form Al
xEu
y. Gibilaro
et al. also published results on the co-deposition process of europium and aluminium ions using an inert electrode.
171 The main principle behind forming an alloy is that there must be a distinct difference in the reactivities of both metals.
174,175 Using A to denote a reactive metal species and B to denote a less reactive metal; then the following can be stated:
| | | Reduction of reactive metal: γAx+ + xe− ⇌ γA | (38) |
| | | Reduction of less reactive metal: δBy+ + ye− ⇌ δB | (39) |
| | | Formation of alloy: γA + δB ⇌ AγBδ | (40) |
| | | Overall process: γAx+ + δBy++ 2xye− ⇌ AγBδ | (41) |
The work undertaken on the reduction of Eu
3+ to Eu
2+ in chloride melts revealed that the process occurs as a single electron transfer (
Fig. 6(f)) similar to the pathway outlined in the previous processes.
176 From this work, it was found that the extent of reduction was directly proportional to the square root of the potential scan rate and similarly to neptunium the potential was found to be dependent on sweep rate up to 0.1 V s
−1 only. Between 0.1–0.3 V s
−1, the process adopted a mixed diffusion and electron-controlled process and beyond this sweep rate the process observed was solely controlled by electron transfer kinetics.
177 However, the work by Schroll
et al. exhibited large discrepancies in data associated with differences in electrode surface area which induce different measured currents.
176 There are also differences associated with lower operating temperatures where the peak potential varies at sweep rates up to 0.6 V s
−1; up to this point, the reduction is limited by mass transfer, and beyond this point the process tends to be mostly electron-controlled.
178 In both of these experiments, the peak current was found to be dependent on the concentration of EuCl
3, the same results being found in fluoride melts.
179 In this process, at a temperature of 1073 K, the CV indicated that the reduction of Eu
3+ to Eu
2+ occurs at ∼0.18 V
vs. a Pt electrode.
Table 11 Summary of reduction potentials of Eu3+/Eu2+ at various temperatures
|
T (K) |
Apparent experimental potential (V) of Eu3+/Eu2+ |
| Schroll et al.176a |
Caravaca et al.180 (tungsten WE)b |
Caravaca et al.180 (vitreous carbon WE)b |
Kuznetsov et al.181b |
|
Reference electrode: Pt.
Reference electrode: Cl2/2Cl−.
|
| 698 |
|
−0.864 |
−0.884 |
−0.838 |
| 723 |
−0.355 |
−0.848 |
−0.858 |
−0.834 |
| 748 |
|
−0.817 |
−0.843 |
−0.829 |
| 773 |
−0.333 |
−0.800 |
−0.817 |
−0.824 |
| 823 |
−0.307 |
|
|
|
| 873 |
−0.282 |
|
|
|
| 923 |
−0.252 |
|
|
|
| 973 |
−0.228 |
|
|
|
| 1023 |
−0.203 |
|
|
|
5.7 Thorium
Until recently, advances in actinide chemistry were mainly concerned with plutonium and uranium species. More effort is now being given to understanding the reduction pathway adopted by the oxides and halides of thorium. Thorium, atomic number 90, has a high melting point (1755 °C) and good resistance to corrosion. Thorium has been widely used by industry compared to other actinides and lanthanides. The first most useful application, in the late nineteenth century, occurred as a result of its incandescent properties within a gas mantle.182 Thorium has been used in alloys such as Mg–Th and Mg–Th–Zr with markedly improved thermal resistance.183,184 More recently thorium has come to the fore as it exhibits several advantages as a nuclear fuel: (1) it can withstand greater burnup compared to uranium and plutonium fuel; (2) it is three to four times more abundant than uranium in the earth's crust, (3) it results in lower levels of waste compared to uranium and plutonium fuels, and (4) it promotes proliferation resistance due to the complexity in separating fissile isotopes. Thorium itself is not fissile in a thermal nuclear reactor but upon bombardment of thermal neutrons natural 232Th is converted to 233Th. 233Th is known to have a half-life of approximately 20 minutes and decays quickly to 233Pa. 233Pa has a half-life of approximately 30 days and decays to 233U (a fissionable radioisotope) according to eqn (42) where n refers to a neutron and β− refers to beta decay. For this reason, 232Th is described as ‘fertile’ as it does, upon neutron bombardment, form 233U, which is a fissile isotope.| |  | (42) |
232Th is the most abundant naturally occurring isotope of thorium, with trace quantities of other more radioactive isotopes. Therefore, it is a high value material and of significant interest as a nuclear fuel. There has been a wealth of work focused on understanding the reduction of thorium halides via both metallothermic and electrolytic processes using different feed materials. Some of the early work for thorium considered the reduction of ThCl4 in sodium with a focus to produce large quantities of high purity thorium metal; this method relied on a chlorination step prior to the formation of the feed material ThCl4.185
The production of ThCl4 incorporated either one of two reagents: sulfur monochloride or phosgene chloride. It was found after using sulfur monochloride that the product had large quantities of sulfur contaminant, in excess of 300 ppm, whereas phosgene chloride had much lower levels of impurity but needed nearly 60 h to produce the feed material, ThCl4. The reaction between hydrogen chloride gas with ThO2 has been reported to produce ThCl4 at 400 °C.186 In molten chloride systems, only Th4+ is stable.187 There is still limited literature on the stability of divalent, Th2+, and there has been no verification that divalent thorium occurs in molten chloride systems either.83,188 Furthermore, the reaction between Th4+ and hydrogen azide in an aqueous solution leading to the formation of Th3+ is still under question despite an experimental investigation by Klapötke and Schulz.189 Their means of demonstrating its existence was via UV-vis and electron paramagnetic resonance (ESR) spectra. Ionova et al. dispute the findings on grounds of the process being thermodynamically impossible and based upon a misinterpretation of the absorption spectra.190 The absorption band of the azide anion ligand lies between 320–680 nm and this complex would only exist with thorium in the 4+ oxidation state. Fig. 6(g) describes the range of potentials found experimentally for the reduction of Th4+ with a difference of 0.1 V between the temperatures for both LiCl–KCl systems. Table 12 summarises this data and there is some agreement in the potentials reported by Srinivasan and Flengas.191
Table 12 Summary of reduction potentials of Th4+/Th0 at various temperatures in molten LiCl–KCl and NaCl–KCl
|
T (K) |
Apparent experimental potential (V) of Th4+/Th0 |
T (K) |
Apparent experimental potential (V) of Th4+/Th0 |
| Cassayre et al.188b |
Martinot192b |
Srinivasan and Flengas191a |
|
Reference electrode: Ag/AgCl.
Reference electrode: Cl2/2Cl−.
|
| 673 |
−2.599 |
−2.52 |
943 |
−2.505 |
| 693 |
|
|
969 |
−2.492 |
| 723 |
|
−2.50 |
1023 |
−2.457 |
| 733 |
−2.577 |
|
1073 |
−2.424 |
| 773 |
−2.559 |
−2.47 |
1123 |
−2.393 |
| 798 |
−2.534 |
|
|
|
| 823 |
−2.53 |
|
|
|
| 798 |
|
|
|
|
| 823 |
|
|
|
|
Work had previously considered eliminating the halogenation step and instead focused on thorium oxide which would be easier to prepare. To the authors’ knowledge, Meyer's study is the first known example of the electrolytic reduction of thorium oxide in a fused metal chloride/fluoride system.193 Meyer's findings reveal that ThO2 responded far more amenably in a molten fluoride system, KF-ThF4, yielding a higher purity product and a large particle size <7–8 μm compared with a NaCl–KCl–ThF4 system, although this is one study alone.193 Even though the working temperature of the chloride melt was 200 °C less than the fluoride melt and promoted a greater solubility for the constituents in the leaching process, this led to the formation of an impure product with a smaller particle size. Clayton, Mamantov and Manning considered the reduction of both thorium and uranium in a mixed chloride/fluoride melt system at 500 °C.194 Their findings revealed a poorly defined peak at −2.03 V vs. a Ni(II)/Ni(0) reference electrode followed by a much larger and well-defined re-oxidation peak of the lithium metal at −1.83 V. This study implies that the reaction proceeds in a single step from Th(IV) to thorium metal with an exchange of four electrons, although there was no detailed CV/SWV work to support it. Comparison of different electrodes, in particular nickel, platinum and tungsten, revealed a discrepancy in the resolution of the peaks. The occurrence of this has been ascribed to the ability of platinum to form an alloy with lithium metal, thereby reducing the overall cathodic range.195
The electrochemical reduction of ThCl4 in LiCl–KCl,192 where a pure chloride electrolyte is used, indicates that the reduction process is electrochemically feasible at 500 °C (see Fig. 6(g)).196 This implies that it would be more sensible to work in a pure chloride electrolyte; furthermore, the reduction of ThF4 in NaCl–KCl at 800 °C yielded thorium metal with a reported purity of 96.6 to 97.8%. Later, Martinot studied the behaviour of ThCl4 in a pure chloride electrolyte finding that the process was near reversible.186 Compared with work accomplished on other actinides, the feed material varied in concentration between 5 and 20%; due to the much lower radiotoxicity of thorium, higher concentrations were safely handled, leading to an overall impurity of no more than 250 ppm.
6 Developing computational models for electrorefining
Computer-aided simulation can be used to model electrochemical processes taking place in a molten salt electrolytic cell. Experimental studies are time-consuming, difficult to monitor and control, especially with radioactive components and hence expensive to conduct and provide data. In comparison, computational models offer an accelerated/augmented means of understanding relevant processes. Models are also useful in steering the direction of future experimental work in more efficient directions. At present, various models exist in relation to molten salt work incorporating a variety of parameters and variables deemed suitable for each simulation. One of the very first models to be designed was that by Johnson et al. at the ANL; this thermodynamic model was developed based upon the distribution between uranium metal in a molten salt system and a binary alloy (eqn (43)).197| |  | (43) |
This theory can be applied to a uranium/plutonium system, where the transfer of uranium and plutonium occurs simultaneously in a single-phase liquid alloy, based upon the following equilibrium process (eqn (44)). It was realised that an alloy acceptor could be used in this process to result in two product streams, which are rich in uranium and plutonium, respectively.| | | UCl3 + Pu ⇌ U + PuCl3 | (44) |
Several other simulation codes were subsequently developed including the so-called TRAIL code. Kobayashi and Tokiwai incorporated data on the diffusion layer thickness which varied according to the uranium concentration in a LiCl–KCl eutectic.198 This study was designed to simulate the reduction process for a small-scale (10 g) electrorefiner (see Fig. 3). In a similar study, the same group then improved their model to consider how the various overvoltage components influence cell resistance and operating cell voltage.199 The model predicted that at a concentration of only 0.5 wt% for uranium the resistance should increase with cell voltage. Beyond 2 wt%, no difference in the resistance of a cell with change of voltage was anticipated. The DEVON model factored in the resistance of molten salt systems in two dimensions but failed to account for the activation overpotential.200
Fundamental studies considering current density distribution and the nature of metal ion movement were first reported by Lee et al.201 These studies used a primary current density model to investigate the nature of uranium deposition and molten salt flow through an anode basket. From this study, a cross-sectional area factor (CSAF) was implemented to predict the shape and size of each of the uranium particles where a value of unity corresponded to a perfectly spherical particle. In practice, estimating the CSAF value of uranium deposit was difficult due to the irregular dendritic geometry. This geometry was also found to change according to the speed of the stirrer which changes the volume fraction of the metal within the molten salt system. By increasing the degree of agitation (>100 rpm) and CSAF, which is proportional to the frictional force, the quantity of uranium deposited increased in the ‘stirrer’ domain. Here, the frictional force determines the motion of uranium dendrites once they spall from the surface of the current collector. Therefore, to recover high-purity uranium effectively an accurate value of the CSAF must be used together with knowledge of the friction force as this is presumed to affect the purity of the uranium product. Kim et al. developed a tertiary current density model to evaluate the separation behaviour and the nature of uranium deposition in a complex electrode system.202
Ghosh et al. modified the entire theory leading to the PRAGAMAN code which may be used to determine the results of changes in the solubility arising at the interface of the cathode and anode – leading to 16 different potential solubilities.203 These simulations were useful for making thermodynamic predictions of the outcome of a process but failed to consider the kinetic effects in relation to the reactant and product species that are found within an experimental process. Ghosh et al. then considered the nature of anodic dissolution for uranium and zirconium for a three-electrode system but failed to report the electrode surface area for the working electrodes meaning that the results are not comparable.204 Sequential work has focused on modelling the Mark-IV electrorefiner which works by simultaneously oxidising uranium at the anode and reducing uranium metal at the cathode (for further details see Section 4.2). Several components of the electrorefiner have been simulated such as the cathode, vessel wall and fuel basket using fundamental electrochemical theory.205 Prior to this, Ahluwalia et al. designed the General Purpose Electrochemical Simulator (GPEC) based upon the Butler–Volmer Equation and thermodynamically available data.206–208 This model was used to consider the behaviour of zirconium in the Experimental Breeder Reactor (EBR)-II. The REFIN model incorporates the diffusion layer model reported by Kobayashi and Tokiwai198 and simplifies the Butler–Volmer kinetics model.209 Moreover, in line with producing a multicomponent model which was able to simulate kinetic effects and fluid flow, Zhang's model captures the partial current of elements and their distribution within the cell.210 This model has several shortcomings; it does not consider nucleation during deposition or the process of diffusion at solid electrodes. It also does not enable the control of species at the interface between the molten salt and electrode and does not have the capability to model multiple oxidation states for different species.
From this, the Faradaic current for each element has been determined for a one-dimensional model where a solid cathode is used. Execution of this code is troublesome due to the long run times but a model developed by Cumberland and Yim, ERAD, was developed to extend the REFIN model and to focus specifically on simulating transient phenomena such as cyclic voltammetric peaks and rate-limiting steps in electrorefining (ER) processes.211 The same group used a least-squares fitting method to derive values for the apparent standard potential vs. Cl2/Cl−, diffusion coefficient and the exchange current density based on their ERAD model.212 This model does not account for kinetic factors, fluid flow characteristics, ohmic drop or metal deposition; it is purely one dimensional and is a useful method to fit/compare data and test assumptions without needing a prolonged period to converge.
7 Precursor types and electrochemical reactor design
7.1 Electroreduction techniques
Much of the success in molten salt technology lies in choosing appropriate operating parameters, electrode materials, electrochemical reactor designs and material selections. Choosing an appropriate molten salt, whether as a binary mixture or not, can also warrant a specific choice of electrochemical reactor to prevent vessel degradation, or electrode material to avoid corrosion. It is also well-known that each molten salt will have unique properties (summarised in Table 3) and thus govern the operation parameters and operational temperature. Moisture, for example, is an unavoidable contaminant within any cell design and different steps are taken to eschew this issue; for example, by vacuum drying salts for prolonged periods or conducting a pre-electrolysis against a sacrificial, inert electrode which might, in turn, lead to unwanted parasitic reactions.213 This section focuses specifically on precursor types of cell designs which lies within the experimentalist's control, offering a fresh outlook on previous work by exposing some of the advantages and disadvantages of each technique.
Several precursor types have been explored in molten salt processes including uniaxially pressed (sintered) pellets, metal cavity electrodes (MCEs), thin-film electrodes, the fluidised cathode and liquid metal cathodes. A pellet of metal oxide (MOx) is formed by combining the metal oxide powder with an appropriate lubricant or binder (often this is polymeric), such as bis-stearamide, before pressing the powder uniaxially at a pressure between 125 to 135 MPa. The addition of a lubricant is to improve the consistency and stability of the pellet during the reaction. The pressing step shortly precedes the sintering of the pellet which subsequently improves its mechanical strength for handling and integrity and removes any residual moisture. To date, there has been considerable work accomplished on TiO2,34,214–216 and Ta2O5,217,218 as part of pellet-based studies. In most studies, the authors have reported that the metal oxide pellet is left partially reduced; by this it is found that the outer surface of the pellet preferentially participates in the reaction and is reduced due to the O2− migration within the neighbouring electrolyte, leaving the inner-most part of the pellet unreduced or otherwise partially reduced. Studies have also explored the consequence of varying the porosity, density and pellet size, finding that a limit of 45% porosity normally yields the most beneficial results.218
To explore this outcome further, pellet reduction studies on TiO2 have been undertaken by Schwandt et al. who present a stepwise time-dependent analysis of the slow, progressive reduction of the TiO2 pellet.219 In the same study, due consideration is given to the volume of the pellet itself, with larger or denser pellets limiting the current efficiency and thus the degree of completion of the process. On the other hand, Fray, Farthing and Chen report that whilst the density of the pellet can impede the completion of the process, the use of a pellet can ensure oxidation inhibition after cooling.43 Because the pellet is withdrawn from the molten salt system, the residual salt that has penetrated within the pores will solidify, preventing any re-oxidation of the as-produced metal. Post-treatment processes are relatively simple, especially for CaCl2 molten salt systems where the reported solubility in water is 1.59 g ml−1 which results in a significantly more efficient process.43 Increasing the metal oxide-to-salt ratio results in a rise in measured current because there are more frequent interactions between the electrode and metal oxide particles. This may incur an iR drop, i.e., where voltage losses occur through the resistive elements, which manifests itself in cyclic voltametric and chronoamperometric studies where greater overvoltage potentials are usually reported.
Recent work has shed light on some of the electrochemical reduction processes of NiO/Cr2O3,220 Tb4O7,221 Ca2+/Ca,222 and MoS2.223 Studies on Nb2O5 in CaCl2 melt have found that although reduction of the oxide is able to take place, the subsequent formation of intermediate states as a result of intercalation between the salt and the metal oxide results in an expansion, due to formation of CaNb2O6 and Ca3Nb2O8, which impedes the subsequent reduction to the metal.224 In this study and other reports, MCEs are prepared by pressing a series of holes or cavities either by laser or drilling or through chemical etching within a molybdenum sheet before affixing to a working electrode such as tungsten using a conductive wire.220–223,225 This process alone can inhibit the number of pores available for the migration of O2− ions, and coupled with a potential that is too positive, explains the poor current efficiency and the long period required to reduce the metal oxide.
Moreover, experimental studies have confirmed that during the electrolysis it is quite likely that the metal oxide may fall from the cavity as a result of an associated volume change, thereby reducing the overall current efficiency.226 This can also cause the dissolution of the metal within the melt, depending on its solubility, which would affect the current efficiency. Furthermore, molybdenum metal cannot be re-used as frequently due to the high temperature and voltage in a harsh molten salt environment reducing its overall mechanical integrity and thereby increasing overall costs.
On a separate note, some drawbacks have been reported with the use of MCEs. The first concern relates to the control of the surface area in contact with the molten salt. Most experimental work will have a wire of low surface area in contact; however, for MCEs there is a much greater applied surface area which can often lead to smaller charging–discharging currents during double-layer charging.220 The second concern arises due to background current that may result through contact of the molten salt and substrate, hence the importance of selecting an appropriately inert electrode material. These currents may then mistakenly be reported as intrinsic electrode processes requiring that control experiments are undertaken to ensure robust results.227
‘Fluidisation’ or fluidised bed processes are where particulate solids come into close contact and agitation with fluids or gases results in a transformation where a pseudo-mixture is formed enhancing and improving the kinetics of a given process. Some of the earliest work in this field dates back to the 1960s and considers the use of fluidised bed electrodes for fuel cells.228 More recently, this technology has been established in several areas such as the synthesis of alkaline peroxide solutions,229,230 improving water purity by nitrate extraction and to facilitate copper deposition.231–234 It has had wide-reaching impact with applications to catalytic cracking and has been extended to consider the combustion phenomena of hydrocarbon fuels with inert particles within a fluidised bed system.235–238 Needless to say, this technology is versatile and offers many advantages: a large specific surface area, increased mass transfer, free-flowing character of the fluidised bed, constant temperature distribution (enhanced heat transfer) and uniformity of mixing. Progress has already been made in applying the theory of ‘fluidised bed electrodes’ to electrochemical reduction processes. Abdulaziz et al. first implemented this in the study of tungsten oxide in LiCl–KCl and in reducing UO2 (see Fig. 8 for details of the fluidised cathode process).239,240 In this context, argon gas is used to ‘fluidise’ the mixture through constant bubbling. In the reduction of tungsten oxide, several parameters were varied including metal oxide-to-salt ratio as well as the argon gas flowrate. It was found that increasing the metal oxide-to-salt ratio yielded larger currents because of an increase in particle collisions with the current collector; however, there was a limit to its efficiency as exceeding 100 g of tungsten oxide in 150 g LiCl–KCl proved difficult to sustain. This might have been due to the size of the system which prevented all the tungsten oxide particles from fluidising. A larger system with a greater working temperature may have yielded better results. More recently, this technique has been successful in the recovery of cobalt from both commercial and spent Li-ion battery materials with reported faradaic efficiencies of >70%.241
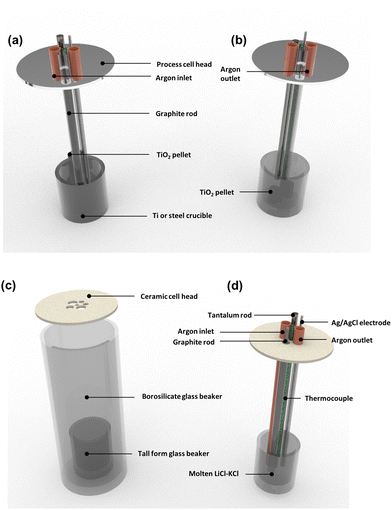 |
| | Fig. 8 3D rendering of the electrochemical cell for the FFC-Cambridge Process: in (a) the current collector is attached to the cathode and in (b) the pellets are electoreduced in situ at the bottom of the crucible. In (c) and (d) the fluidised cathode process is illustrated with the argon inlet sitting within the molten salt system which agitates the solution continuously throughout the process. | |
Fig. 9 illustrates the nature of particle collisions with a current collector for a fluidised cathode process. In this example, UO2 has been selected as the metal oxide particle. The success of this process, as demonstrated by the figure, lies in the initial contact between the uranium oxide and current collector surface; thereby facilitating the reduction process. The initial collision led to the formation of a deposit on the surface of the current collector (Fig. 9). The scanning electron micrograph in Fig. 9(b) indicates that after the process the particles agglomerate. Fig. 9(c) illustrates the initial increase in current versus time; this is in line with the reduction of uranium oxide where we expect the formation of extensive three-phase interlines (3PIs) enabling greater salt penetration and therefore an increased current density.
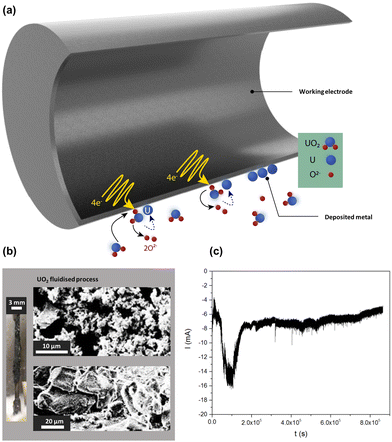 |
| | Fig. 9 A schematic diagram of the working electrode surface. (a) The reduction begins with the initial impingement of the metal oxide particles against the surface of the current collector. Thereafter, a direct transfer of electrons occurs resulting in the formation of uranium atoms and oxide ions which subsequently migrate towards the surface of the anode. (b) Scanning electron microscopy reveals the agglomeration of uranium particles following the fluidised cathode process. A photograph shows the deposited metal on the surface of the current collector. (c) Current vs. time curve of UO2 fluidised cathode in LiCl–KCl eutectic at 450 °C, argon flow rate: 600 cm3 min−1, voltage: −2.2 V vs. Ag/AgCl. (b) and (c) have been reprinted from Abdulaziz et al.239 | |
7.2 Applications of liquid metal cathodes
Liquid metal cathodes, shown in Fig. 10, are widely used in the field of molten salts, displaying excellent electrochemical properties: a constant surface area of contact with the active material, greater diffusion coefficients into the salt, typically a lower applied cell voltage and they enable the reduction of highly electronegative elements.59,242 The latter two properties are explained by the deposition potential, where the target metal ions behave differently depending on the state of the cathode (solid/liquid), thereby resulting in a more negative applied potential, which applies to the electrorefining process. This results in a lower cell voltage and reduces the overall energy consumption; several studies have reported these promising features for liquid metal cathodes.62,243,244 Advantageous electrochemical properties have also been realised for the single-step reduction of Ti3+ in NaCl–KCl melt on a liquid tin cathode where the multi-step reduction on tungsten electrodes is avoided, incurring a lower energy penalty.245 In the nuclear industry, liquid cathodes have been widely used in waste disposal and spent-fuel processing. They have shown merit in health and safety concerns because they allow (depending on solubility) the target metal ions to dissolve within the liquid metal cathode reducing the radiation dose to workers and reducing the overall quantity of radioactive waste.
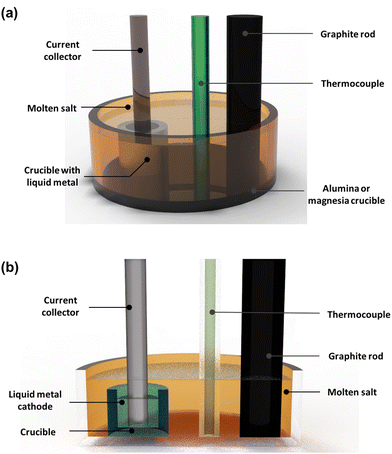 |
| | Fig. 10 A simplified arrangement of the liquid metal cathode electrochemical cell. (a) The current collector in this electrochemical cell sits in a liquid metal which is itself mixed with a molten salt electrolyte. This process works on the principle that the density of the liquid metal is greater than the surrounding electrolyte. (b) is a cross-sectional area representing these two different electrolytes (the molten salt medium and liquid metal cathode). | |
Gibilaro et al. investigated the use and application of liquid metal cathodes in molten fluoride systems.246 The work demonstrated successful studies on each of the following liquid metal cathodes: bismuth, gallium, lead, antimony and tin in two different melt systems LiF–NaF (750 °C) and LiF–CaF2 (850 °C). In this technique, the target metal ions are anodically dissolved within the melt before being selectively recovered on the surface of the metal cathode; any fission product isotopes are found within the molten salt after it solidifies. Using transient electrochemical techniques such as linear sweep voltammetry, a hierarchy of reactivities (gallium being the most reactive and bismuth the least) was found and subsequently, the rate of extraction was revealed from the data. Research programmes in Japan,59,60,243 US,58,63,247 and Korea,248 have made similar developments, exploring the electrochemical reduction of alkaline earths, lanthanides and some minor actinides in LiCl–KCl using a liquid Cd cathode. In 2007, the Idaho National Laboratory in the US reported engineering-scale (12–13 kg of depleted uranium processed) liquid Cd experiments in molten LiCl–KCl at 773 K.249
Notwithstanding the progress of liquid metal cathode work, appropriate selection and choice of design parameters must be made including: using a metal with low vapour pressure to avoid metal distillation, choosing a viable operating temperature range well below the boiling point of the metal, and ensuring that the density of the liquid metal is greater than the molten salt. Further work must also be undertaken on selecting an optimal material for crucible design. Vaden et al. chose a BeO crucible and upon product recovery found it to be damaged. Selecting a lower-cost material such as Al2O3 could work equally as well.249 On the other hand, many studies have used stainless steel baskets in the electrochemical reduction of uranium salts; XRD analysis in some cases has revealed iron impurities due to the homogeneous liquid alloy formation.100 For metals to remain good candidates in scaling-up processes they must be non-toxic and inexpensive.
8 Summary and future outlook
The energy supply industry is changing rapidly, not least accelerated by global net-zero targets. Nuclear energy is regarded as one of the few low carbon technologies that can meet future global energy demands, but concerns surrounding its safe use and spent fuel management remain a major challenge. Effective methods for spent fuel processing and metal recovery must be available so as to minimise nuclear waste. Molten salt technology has shown promising capabilities in the extractive metallurgy of uranium and plutonium from metal oxides, MOX fuels and in the production of high-grade alloys. This review has shown that singular, binary and ternary molten salts, in specified molar ratios, are amenable to the electro-deoxidation processes of metal oxide nuclear fuels, although most advances cover only the molten chloride and fluoride systems. This may relate to the efficacy of the final product morphology, ease of handling, optical transparency, solubility of oxide ions, stability at both elevated temperatures and pressure and ultimately the reduced hygroscopic nature. Despite these advantages, it is difficult to separate the metal deposit from the molten salt and in most, if not all processes, some residual salt remains, skewing the reported recovery efficiency. Whilst it might still be true that molten chlorides (LiCl–KCl and NaCl–KCl) and fluorides (LiF–CaF2) are optimal for electrochemical processes, there remains a void in experimental work on the lesser well-known molten salts which may also present favourable results depending on their interaction with transuranic and refractory elements and their physico-chemical properties.
The main body of this review comprises a thorough discourse of the most significant (defined in this context as the most abundant elements present in spent nuclear fuel) actinides and lanthanides (thorium, uranium, neptunium, plutonium, americium, curium and europium) concerning spent nuclear fuel reprocessing. Many experimental studies have been considered, some of which date back to the 1950s. Uranium and plutonium are two of the most widely studied elements and thus occupy a large section of this work. Both of these were studied by the US using precipitation, solvent extraction and electrorefining techniques for purposes of producing materials for nuclear weapons prior to civilian applications. Neptunium, americium and curium were present as by-products of nuclear decay or through neutron activation, and the extraction of these synthetic elements was necessary because they were impurities and were required to be removed from the uranium and plutonium either for further use in a nuclear reactor or for waste management purposes. Europium is the only lanthanide element studied in this work and the only viable means of extraction is via co-reduction with a reactive metal. Thorium is the last of the studied elements and perhaps the most promising for future applications; it is three to four times more abundant in the earth's crust than uranium and possesses a greater energy density compared to uranium as thorium reactor fuel theoretically can undergo a higher burn up. If the use of thorium in nuclear reactors, particularly molten salt reactors, is to come to fruition, investments will need to be directed towards understanding the operation and maintenance of such plant systems.
Much of the past work has sought to resolve the reduction pathways exhibited by the actinides and lanthanides and there has, to a large extent, been a wide range of success reported in a variety of operating conditions, cell designs and different molten salts. More studies have focused on chlorides in solution than on oxides. The selection of different electrode materials, a wide range of initial precursors (difference in oxidation states) and a variety of reference electrodes all contribute to many data inconsistencies in the literature. Whilst the proof-of-concept has demonstrated broad applicability, there needs to be a greater effort made towards reprocessing spent nuclear materials at the pilot-scale. Electroreduction, electrorefining and electrowinning are all processes that have yet to reach a mature stage. Improvements to their process operation and control are required. An understanding of criticality in the design process is also required. Some of the early indications as to whether the results associated with the transition from lab-scale to pilot-scale can be determined by use of computational fluid dynamic (CFD) models. Within these models, factors such as cell design, size, shape and arrangement of electrodes, operating current, liquid density and laminar viscosity all require consideration.
A variety of one, two and three-dimensional models for electrorefining have been reported in this review article and date back to the early 1990s. Each of these codes presents advantages and disadvantages in relation to how accurately a particular electrorefiner is modelled. Complete understanding of the operation of an electrorefiner is yet to be achieved; accurately predicting the fluid flow and appreciating the change of elemental oxidation states with time and current density distribution at the electrode/molten salt interface remain areas for future development.
A range of precursor types, cell designs and electrode configurations have been discussed. Initial electro-deoxidation studies of titanium dioxide pellets found that parameters such as porosity, pellet thickness and density are likely to affect the final product morphology and oxygen content. However, there are still lessons to be learnt from this process and others that focus on the production of refractory metals; in particular, whether the product morphology from different precursors is of relevance for industrial application. In light of this, more must be done to close the gap between the electrochemical and materials-based understanding of these processes. One of the key innovations for this technology remains in the use of the fluidised cathode process. It ensures both a constant and high surface area of contact and improves current efficiency. Continued study of its applicability to a broader range of materials and optimisation of process variables and conditions is required to assess the commercial promise for this process.
Molten salt electrochemistry provides a viable route to metal production, incurring a smaller energy penalty compared with harsh chemical reducing agents, and is anticipated to save on downstream costs in the production of powders and homogeneous alloys. The transition from laboratory to pilot-scale remains a challenge for this technology partly due to the chemical corrosion of electrochemical reactors at high temperatures, possible irradiation hazards with respect to materials under a neutron flux and parasitic reactions resulting in the formation of alloys. Consideration of different anode/cathode materials to improve selectivity, optimising the reduction potential and improving the current efficiency are some of the strategies that can be employed. For a completely successful scale-up, there must also be progress made in economic feasibility which, as of yet, remains understudied. Above all, the future growth of this field relies on sustaining strong partnerships between the nuclear industry and universities.
List of abbreviations
| 3PI | Three-phase interline |
| AGR | Advanced gas-cooled reactor |
| ANL | Argonne National Laboratory |
| BWR | Boiling water reactor |
| CFD | Computational fluid dynamics |
| CSAF | Cross-sectional area factor |
| CV | Cyclic voltammetry |
| EBR | Experimental breeder reactor |
| ER | Electrorefining |
| FFC | Fray–Farthing–Chen |
| FP | Fission products |
| HEP | High-enriched plutonium |
| HEU | High-enriched uranium |
| IAEA | International Atomic Energy Agency |
| ILW | Intermediate level waste |
| LAMEX | Los Alamos Molten Plutonium Electrorefining |
| LEU | Low-enriched uranium |
| LNFP | Less noble fission products |
| MA | Minor actinides |
| MCE | Metallic cavity electrode |
| MOX | Mixed oxide |
| NM | Noble metal |
| NFP | Noble fission products |
| NM | Noble metals |
| P&T | Partition and transmutation |
| PUREX | Plutonium uranium reduction extraction |
| SA | Surface area |
| S.Cl.E. | Standard chlorine electrode |
| SWV | Square wave voltammetry |
| TBP | Tri-n-butylphosphate |
| THORP | Thermal oxide reprocessing plant |
| TPB | Triple-phase boundary |
| TRU | Transuranium (elements) |
| RE | Rare earth (elements) |
| XRD | X-ray diffraction |
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors acknowledge the AWE for supporting the PhD and Research Fellowship of MM. PRS and DJLB acknowledge the Royal Academy of Engineering for supporting their respective Research Chairs [CiET1718/59 and RCSRF2021/13/53]. The EPSRC is acknowledged for funding pyroprocessing of nuclear materials research in the Electrochemical Innovation Lab [EP/T011386/1, EP/L018616/1, EP/R020973/1, EP/S018204/1]. The contribution from NNL was funded under the £46m Advanced Fuel Cycle Programme as part of the Department for Business, Energy and Industrial Strategy's (BEIS) £505m Energy Innovation Programme.
References
- World Nuclear Association, Nuclear Power in the World Today, https://world-nuclear.org/information-library/current-and-future-generation/nuclear-power-in-the-world-today.aspx, accessed 06/12/2022.
- D. J. Hill, Nat. Mater., 2008, 7, 680–682 CrossRef CAS PubMed
 .
.
-
S. G. Thompson and G. T. Seaborg, Modern Alchemy, World Scientific, 1994, vol. 2, pp. 363–371 Search PubMed .
- J. G. Reynolds, J. S. Page, G. A. Cooke and J. Pestovich, J. Radioanal. Nucl. Chem., 2015, 304, 1253–1259 CrossRef CAS .
-
D. L. Clark, S. S. Hecker, G. D. Jarvinen and M. P. Neu, The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Netherlands, Dordrecht, 2008, pp. 813–1264 Search PubMed .
-
S. Thompson and G. Seaborg, US Pat., US2785951A, 1957 Search PubMed .
-
B. C. Reed, The History and Science of the Manhattan Project, Springer, Berlin, 2014 Search PubMed .
-
B. C. Reed, The Physics of the Manhattan Project, Springer Nature, Switzerland, 4th edn, p. 2021 Search PubMed .
-
H. Anderson and B. Larned, US Pat., US2924506A, 1960 Search PubMed .
-
E. R. Irish and W. H. Reas, The Purex Process. A Solvent Extraction Reprocessing Method for Irradiated Uranium, HW-49483 A, Washington DC, 1957 Search PubMed .
-
R. S. Herbst, P. Baron and M. Nilsson, Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment, Woodhead Publishing, 2011, pp. 141–175 Search PubMed .
- P. Baron, S. M. Cornet, E. D. Collins, G. DeAngelis, G. Del Cul, Y. Fedorov, J. P. Glatz, V. Ignatiev, T. Inoue, A. Khaperskaya, I. T. Kim, M. Kormilitsyn, T. Koyama, J. D. Law, H. S. Lee, K. Minato, Y. Morita, J. Uhlíř, D. Warin and R. J. Taylor, Prog. Nucl. Energy, 2019, 117, 103091 CrossRef CAS .
- NDA, The Magnox Operating Programme (MOP9), available at: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/457808/The_Magnox_Operating_Programme__MOP9_.pdf, date accessed: 19/01/2022, Magnox Ltd, Sellafield Ltd, Dounreay Ltd, 2012.
- OECD, Nuclear Energy Data, available at: https://www.oecd-nea.org/jcms/pl_15082, date accessed: 19/01/2022, Boulogne-Billancourt, France, 2018.
-
P. Podvig and The International Panel on Fissile Materials, Consolidating Fissile Materials in Russia's Nuclear Complex, available at: https://fissilematerials.org/library/rr07.pdf, Princeton, 2009 Search PubMed.
-
P. D. Wilson, The nuclear fuel cycle from ore to wastes, Oxford University Press, Oxford, 1996 Search PubMed .
-
K. L. Murty and I. Charit, An introduction to nuclear materials: fundamentals and applications, John Wiley & Sons, Weinheim, 2013 Search PubMed .
- F. Kawakami, M. Tokiwai and Y. Fujii, Prog. Nucl. Energy, 2011, 53, 974–979 CrossRef CAS .
-
B. Zohuri, in Nuclear Reactor Technology Development and Utilization, ed. S. U.-D. Khan and A. Nakhabov, Woodhead Publishing, 2020, pp. 213–246 Search PubMed .
- OECD, Technology Roadmap Update for Generation IV Nuclear Energy Systems, 2014, available at: https://www.gen-4.org/gif/upload/docs/application/pdf/2014-03/gif-tru2014.pdf, date accessed: 19/01/2022 Search PubMed.
- J. Uhlíř, J. Nucl. Mater., 2007, 360, 6–11 CrossRef .
- C. Le Brun, J. Nucl. Mater., 2007, 360, 1–5 CrossRef CAS .
-
G. F. Hewitt and J. G. Collier, Introduction to nuclear power, CRC Press, 2nd edn, 2000 Search PubMed .
- E.-Y. Choi and S. M. Jeong, Prog. Nat. Sci.: Mater. Int., 2015, 25, 572–582 CrossRef CAS .
-
U. Basak, S. Bourg and A. Constantinet al., IAEA Tecdoc Series – Status and Trends in Pyroprocessing of Spent Nuclear Fuels, Vienna, 2015, available at: https://wwwpub.iaea.org/MTCD/Publications/PDF/TE-1967web.pdf, date accessed: 07/02/2023 Search PubMed .
- M. Iizuka, K. Uozumi, T. Inoue, T. Iwai, O. Shirai and Y. Arai, J. Nucl. Mater., 2001, 299, 32–42 CrossRef CAS .
- E. González-Roubaud, D. Pérez-Osorio and C. Prieto, Renewable Sustainable Energy Rev., 2017, 80, 133–148 CrossRef .
- R. W. Bradshaw and D. E. Meeker, Sol. Energy Mater., 1990, 21, 51–60 CrossRef CAS .
-
T. Bauer, N. Pfleger, D. Laing, W.-D. Steinmann, M. Eck and S. Kaesche, High-Temperature Molten Salts for Solar Power Application, in Molten Salts Chemistry: From Lab to Applications, Elsevier, 2013, pp. 415–438 Search PubMed .
- Q. Guo Zhao, S. Yu Miao, H. Guo and Y. Ting Wu, Appl. Therm. Eng., 2020, 171, 115084 CrossRef .
- B. Zhang, H. Xie, B. Lu, X. Chen, P. Xing, J. Qu, Q. Song and H. Yin, ACS Sustainable Chem. Eng., 2019, 7, 13391–13399 CrossRef CAS .
- A. M. Bonanos, M. C. Georgiou, K. G. Stokos and C. N. Papanicolas, Appl. Therm. Eng., 2019, 154, 294–301 CrossRef CAS .
- B. Mishra and D. L. Olson, J. Phys. Chem. Solids, 2005, 66, 396–401 CrossRef CAS .
- G. Chen, D. Fray and T. Farthing, Nature, 2000, 407, 361–364 CrossRef CAS PubMed .
- B. Claux, J. Serp and J. Fouletier, Electrochim. Acta, 2011, 56, 2771–2780 CrossRef CAS .
- D. Hu, A. Dolganov, M. Ma, B. Bhattacharya, M. T. Bishop and G. Z. Chen, JOM, 2018, 70, 129–137 CrossRef CAS PubMed .
- D. J. Fray, JOM, 2001, 53, 27–31 CrossRef .
-
A. V. Bychkov and O. V. Skiba, in Chemical Separation Technologies and Related Methods of Nuclear Waste Management, ed. G. R. Choppin and M. K. Khankhasayev, Springer, Netherlands, Dordrecht, 1999, pp. 71–98 Search PubMed .
-
S. Delpech, in Molten Salts Chemistry From Lab to Applications, ed. F. Lantelme and H. B. T. Groult, Elsevier, Oxford, 2013, pp. 497–520 Search PubMed .
- G. J. Janz, R. P. T. Tomkins, C. B. Allen, J. R. Downey Jr, G. L. Garner, U. Krebs and S. K. Singer, J. Phys. Chem. Ref. Data, 1975, 4, 871–1178 CrossRef CAS .
- E. A. Ukshe, Russ. Chem. Rev., 1965, 34, 141 CrossRef .
- G. J. Janz, G. L. Gardner, U. Krebs and R. P. T. Tomkins, J. Phys. Chem. Ref. Data, 1974, 3, 1–115 CrossRef CAS .
- D. J. Fray and G. Z. Chen, Mater. Sci. Technol., 2004, 20, 295–300 CrossRef CAS .
- K. Ono and R. O. Suzuki, JOM, 2002, 54, 59–61 CrossRef CAS .
- W. Xiao and D. Wang, Chem. Soc. Rev., 2014, 43, 3215–3228 RSC .
- Y. Sakamura, J. Nucl. Mater., 2011, 412, 177–183 CrossRef CAS .
-
T. Koyama, in Advanced Separation Techniques for Nuclear Fuel Reprocessing and Radioactive Waste Treatment, ed. K. L. Nash and G. J. Lumetta, Woodhead Publishing, 2011, pp. 269–310 Search PubMed .
- S. M. Jeong, H. S. Shin, S. H. Cho, J. M. Hur and H. S. Lee, Electrochim. Acta, 2009, 54, 6335–6340 CrossRef CAS .
- E.-Y. Choi, J. W. Lee, J. J. Park, J.-M. Hur, J.-K. Kim, K. Y. Jung and S. M. Jeong, Chem. Eng. J., 2012, 207, 514–520 CrossRef .
- E.-Y. Choi, J.-K. Kim, H.-S. Im, I.-K. Choi, S.-H. Na, J. W. Lee, S. M. Jeong and J.-M. Hur, J. Nucl. Mater., 2013, 437, 178–187 CrossRef CAS .
- J.-M. Hur, S.-S. Hong and H. Lee, J. Radioanal. Nucl. Chem., 2013, 295, 851–854 CrossRef CAS .
- S. M. Jeong, S.-B. Park, S.-S. Hong, C.-S. Seo and S.-W. Park, J. Radioanal. Nucl. Chem., 2006, 268, 349–356 CrossRef CAS .
- A. M. Abdelkader, K. T. Kilby, A. Cox and D. J. Fray, Chem. Rev., 2013, 113, 2863–2886 CrossRef CAS PubMed .
- J. L. Willit, W. E. Miller and J. E. Battles, J. Nucl. Mater., 1992, 195, 229–249 CrossRef CAS .
- Y. Sakamura and T. Omori, Nucl. Technol., 2010, 171, 266–275 CrossRef CAS .
- K.-C. Song, H.-S. Lee, J.-M. Hur, J.-G. Kim, D.-H. Ahn and Y.-Z. Cho, Nucl. Eng. Technol., 2010, 42, 131–144 CrossRef CAS .
- Z. Tomczuk, J. P. Ackerman, R. D. Wolson and W. E. Miller, J. Electrochem. Soc., 1992, 139, 3523 CrossRef CAS .
- J. J. Laidler, J. E. Battles, W. E. Miller, J. P. Ackerman and E. L. Carls, Prog. Nucl. Energy, 1997, 31, 131–140 CrossRef CAS .
- T. Koyama, M. Iizuka, Y. Shoji, R. Fujita, H. Tanaka, T. Kobayashi and M. Tokiwai, J. Nucl. Sci. Technol., 1997, 34, 384–393 CrossRef CAS .
- M. Iizuka, T. Koyama, N. Kondo, R. Fujita and H. Tanaka, J. Nucl. Mater., 1997, 247, 183–190 CrossRef CAS .
- T. Koyama, M. Iizuka, N. Kondo, R. Fujita and H. Tanaka, J. Nucl. Mater., 1997, 247, 227–231 CrossRef CAS .
- Y. Sakamura, T. Hijikata, K. Kinoshita, T. Inoue, T. S. Storvick, C. L. Krueger, L. F. Grantham, S. P. Fusselman, D. L. Grimmett and J. J. Roy, J. Nucl. Sci. Technol., 1998, 35, 49–59 CrossRef CAS .
- J. J. Roy, J. Electrochem. Soc., 1996, 143, 2487 CrossRef CAS .
- D. Inman, G. J. Hills, L. Young and J. Bockris, Trans. Faraday Soc., 1959, 55, 1904–1914 RSC .
- S. A. Kuznetsov, H. Hayashi, K. Minato and M. Gaune-Escard, J. Electrochem. Soc., 2005, 152, C203 CrossRef CAS .
- K. Dring, R. Dashwood and D. Inman, J. Electrochem. Soc., 2005, 152, D184 CrossRef CAS .
- R. Littlewood, J. Electrochem. Soc., 1962, 109, 525 CrossRef CAS .
- L. D. Brown, R. Abdulaziz, S. Simons, D. Inman, D. J. L. Brett and P. R. Shearing, J. Appl. Electrochem., 2013, 43, 1235–1241 CrossRef CAS .
- J. J. Roy, L. F. Grantham, D. L. Grimmett, S. P. Fusselman, C. L. Krueger, T. S. Storvick, T. Inoue, Y. Sakamura and N. Takahashi, J. Electrochem. Soc., 1996, 143, 2487 CrossRef CAS .
-
Encyclopedia of Electrochemistry of the Elements, Fused Salt Systems, ed. J. A. Plambeck and A. J. Bard, Marcel Dekker, Inc., 1976, vol. 10 Search PubMed .
- H. Lee, G.-I. Park, J.-W. Lee, K.-H. Kang, J.-M. Hur, J.-G. Kim, S. Paek, I.-T. Kim and I.-J. Cho, Sci. Technol. Nuclear Install., 2013, 2013, 1–11 CrossRef .
-
R. G. Lewin and M. T. Harrison, Woodhead Publishing Series in Energy, Woodhead Publishing, Oxford, 2015, pp. 373–413 Search PubMed .
- O. Shirai, H. Yamana and Y. Arai, J. Alloys Compd., 2006, 408, 1267–1273 CrossRef .
- Y. Sakamura, T. Hijikata, K. Kinoshita, T. Inoue, T. S. Storvick, C. L. Krueger, J. J. Roy, D. L. Grimmett, S. P. Fusselman and R. L. Gay, J. Alloys Compd., 1998, 271, 592–596 CrossRef .
- Nuclear Energy Agency, Spent Nuclear Fuel Assay Data for Isotopic Validation, State-of-the-art Report, 2011.
-
I. Grenthe, J. Drożdżynński, T. Fujino, E. C. Buck, T. E. Albrecht-Schmitt and S. F. Wolf, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Netherlands, Dordrecht, 2006, pp. 253–698 Search PubMed .
- M. Genet, Radiochim. Acta, 1995, 70–71, 3–12 CrossRef .
- O. Hahn and F. Strassmann, Naturwissenschaften, 1939, 27, 11–15 CrossRef CAS .
- H. L. Anderson, E. T. Booth, J. R. Dunning, E. Fermi, G. N. Glasoe and F. G. Slack, Phys. Rev., 1939, 55, 511–512 CrossRef CAS .
- E. Péligot, J. Prakt. Chem., 1841, 24, 442–451 CrossRef .
- D. Inman, G. J. Hills, L. Young and J. O. Bockris, Ann. N. Y. Acad. Sci., 1960, 79, 803–829 CrossRef CAS .
- S. A. Kuznetsov, H. Hayashi, K. Minato and M. Gaune-Escard, Electrochim. Acta, 2006, 51, 2463–2470 CrossRef CAS .
- L. Martinot and F. Caligara, At. Energy Rev., 1973, 11, 3–61 CAS .
- S. A. Kuznetsov, L. Rycerz and M. Gaune-Escard, J. Nucl. Mater., 2005, 344, 152–157 CrossRef CAS .
- O. Shirai, K. Uozumi, T. Iwai and Y. Arai, Anal. Sci., 2001, 17, i959–i962 CrossRef PubMed .
- P. Masset, D. Bottomley, R. Konings, R. Malmbeck, A. Rodrigues, J. Serp and J.-P. Glatz, J. Electrochem. Soc., 2005, 152, A1109 CrossRef CAS .
- S. D. Herrmann, S. X. Li, M. F. Simpson and S. Phongikaroon, Sep. Sci. Technol., 2006, 41, 1965–1983 CrossRef CAS .
- E.-Y. Choi, J.-M. Hur, I.-K. Choi, S. G. Kwon, D.-S. Kang, S. S. Hong, H.-S. Shin, M. A. Yoo and S. M. Jeong, J. Nucl. Mater., 2011, 418, 87–92 CrossRef CAS .
- C. S. Seo, S. Bin Park, B. H. Park, K. J. Jung, S. W. Park and S. H. Kim, J. Nucl. Sci. Technol., 2006, 43, 587–595 CrossRef CAS .
- G. Z. Chen, E. Gordo and D. J. Fray, Metall. Mater. Trans. B, 2004, 35, 223–233 CrossRef .
- Y. Deng, D. Wang, W. Xiao, X. Jin, X. Hu and G. Z. Chen, J. Phys. Chem. B, 2005, 109, 14043–14051 CrossRef CAS PubMed .
- P. Kar and J. W. Evans, Electrochim. Acta, 2008, 54, 835–843 CrossRef CAS .
- Y. Sakamura, M. Kurata and T. Inoue, J. Electrochem. Soc., 2006, 153, D31 CrossRef CAS .
- C. Nourry, P. Souček, L. Massot, R. Malmbeck, P. Chamelot and J.-P. Glatz, J. Nucl. Mater., 2012, 430, 58–63 CrossRef CAS .
- C. Hamel, P. Chamelot, A. Laplace, E. Walle, O. Dugne and P. Taxil, Electrochim. Acta, 2007, 52, 3995–4003 CrossRef CAS .
- G. Mamantov and D. L. Manning, Anal. Chem., 1966, 38, 1494–1498 CrossRef CAS .
-
M. Kolodney, Production of plutonium by electrolysis, Report LA-148, Los Alamos Scientific Laboratory, U.S. Atomic Energy Commission, 1944.
-
R. D. Baker and W. J. Maraman, in Extractive and Physical Metallurgy of Plutonium and Its Alloys, ed. W. D. Wilkinson, Interscience Publishers, New York, 1960, pp. 43–59 Search PubMed .
-
B. Blumenthal and M. B. Brodsky, in Plutonium, ed. E. Grison, W. B. H. Lord and R. D. Fowler, London, 1960 Search PubMed .
- S. M. Jeong, H.-S. Shin, S.-S. Hong, J.-M. Hur, J. B. Do and H. S. Lee, Electrochim. Acta, 2010, 55, 1749–1755 CrossRef CAS .
-
L. J. Mullins, J. A. Leary and C. W. Bjorklund, Large scale preparation of high purity plutonium metal by electrorefining. Preliminary Report, Los Alamos Scientific Lab., N. Mex., LAMS-2441, 1960.
-
J. A. Leary and L. J. Mullins, Preparation of Ultra-High Purity Plutonium, Los Alamos Scientific Lab., Univ. of California, N. Mex., 1965.
-
L. J. Mullins and A. N. Morgan, Large scale electrorefining of plutonium from plutonium-iron alloys, Los Alamos Scientific Laboratory of the University of California, New Mexico, 1964, vol. 3029.
-
T. Paget, J. Mcneese, K. Fife, M. Jackson and R. Watson, in Plutonium Handbook, ed. D. Clark, D. A. Geeson and R. J. Hanrahan Jr, La Grange Park, IL, 2nd edn, 2019, pp. 201–286 Search PubMed .
-
T. J. Paget, R. Watson and S. Slade, Plutonium Futures, Dijon, France, 2008 Search PubMed .
-
T. J. Paget, S. M. Slade and R. F. Watson, 42nd IUPAC Congress, Glasgow, Scotland, 2009 Search PubMed .
- D. F. Mclaughlin, C. E. Sessions and J. E. Marra, Nucl. Technol., 1992, 99, 242–251 CrossRef CAS .
-
T. S. Soine, Batch electrorefining of plutonium metal. Progress Report, General Electric Co., Richland, Wash. Hanford Atomic Products Operation, 1965.
-
M. H. Curtis, Continuous Electrowinning of Plutonium from Chloride Melts, General Electric Co. Hanford Atomic Products Operation, Richland, Wash., p. 1963 Search PubMed .
- M. B. Brodsky and B. G. F. Carleson, J. Inorg. Nucl. Chem., 1962, 24, 1675–1681 CrossRef CAS .
-
L. J. Mullins and A. N. Morgan, Review of operating experience at the Los Alamos Plutonium Electrorefining Facility, 1963–1977, Los Alamos National Lab., NM (USA), 1981.
-
L. J. Mullins, A. N. Morgan, S. A. Apgar III and D. C. Christensen, Six-kilogram-scale electrorefining of plutonium metal, Los Alamos National Lab., 1982.
- D. S. Poa, Z. Tomczuk and R. K. Steunenberg, J. Electrochem. Soc., 1988, 135, 1161 CrossRef CAS .
- O. Shirai, T. Iwai, Y. Suzuki, Y. Sakamura and H. Tanaka, J. Alloys Compd., 1998, 271–273, 685–688 CrossRef CAS .
- D. A. Nissen, J. Inorg. Nucl. Chem., 1966, 28, 1740–1743 CrossRef CAS .
- J. Serp, R. J. M. Konings, R. Malmbeck, J. Rebizant, C. Scheppler and J.-P. Glatz, J. Electroanal. Chem., 2004, 561, 143–148 CrossRef CAS .
- L. Martinot and G. Duyckaerts, Anal. Lett., 1971, 4, 1–11 CrossRef CAS .
-
G. M. Campbell and J. A. Leary, Thermodynamic Properties of Pu Compounds from EMF Measurements: I. Pu Vs. Ag in LiCl-KCl Eutectic, Technical Report LA-3399, United States Atomic Energy Commission, 1966.
- L. Martinot, J. Inorg. Nucl. Chem., 1975, 37, 2525–2528 CrossRef CAS .
- P. Masset, R. J. M. Konings, R. Malmbeck, J. Serp and J. P. Glatz, J. Nucl. Mater., 2005, 344, 173–179 CrossRef CAS .
- Y. Sakamura, O. Shirai, T. Iwai and Y. Suzuki, J. Alloys Compd., 2001, 321, 76–83 CrossRef CAS .
- O. Shirai, M. Iizuka, T. Iwai and Y. Arai, Anal. Sci., 2001, 17, 51–57 CrossRef CAS PubMed .
-
L. E. McCurry and G.-M. M. Moy, Trans Am. Nuc. Soc., Los Alamos National Lab., Los Angeles, CA, US, 1987, vol. 55, p. 248.
- R. Abdulaziz, L. D. Brown, D. Inman, S. Simons, P. R. Shearing and D. J. L. Brett, Int. J. Electrochem. Sci., 2016, 11, 10417–10435 CrossRef CAS .
- D. Lambertin, S. Ched homme, G. Bourges, L. Pescayre, S. Sanchez and G. Picard, ECS Proc. Vol., 2004, 24, 700 CrossRef .
- A. H. Jones, R. Watson, T. Paget, R. Campbell-Kelly, T. Caldwell and D. J. Fray, J. Nuclear Fuel Cycle Waste Technology, 2015, 13, 1–5 CrossRef .
- M. Iizuka, Y. Sakamura and T. Inoue, J. Nucl. Mater., 2006, 359, 102–113 CrossRef CAS .
- M. Kurata, T. Inoue, J. Serp, M. Ougier and J.-P. Glatz, J. Nucl. Mater., 2004, 328, 97–102 CrossRef CAS .
- D. Rappleye, J. McNeese, R. Torres, K. Holliday and J. R. Jeffries, J. Nucl. Mater., 2021, 552, 152968 CrossRef CAS .
-
J. G. Reavis, Experimental studies of actinides in molten salts, Los Alamos National Lab., LA-10340, New Mexico, 1985.
- E. McMillan and P. H. Abelson, Phys. Rev., 1940, 57, 1185–1186 CrossRef CAS .
- G. T. Seaborg, E. M. Mcmillan, J. W. Kennedy and A. C. Wahl, Phys. Rev., 1946, 69, 366–367 CrossRef CAS .
-
G. A. Burney and R. M. Harbour, Radiochemistry of Neptunium: Nuclear Science Series, NAS-NS-3060, Savannah River Laboratory, E. I. du Pont de Nemours & Co., South Carolina, 1974 Search PubMed .
- P. Masset, C. Apostolidis, R. J. M. Konings, R. Malmbeck, J. Rebizant, J. Serp and J.-P. Glatz, J. Electroanal. Chem., 2007, 603, 166–174 CrossRef CAS .
- O. Shirai, M. Iizuka, T. Iwai and Y. Arai, J. Appl. Electrochem., 2001, 31, 1055–1060 CrossRef CAS .
- L. Martinot and G. Duyckaerts, Inorg. Nuclear Chem. Lett., 1969, 5, 909–919 CrossRef CAS .
- C. L. Krueger, J. Electrochem. Soc., 1991, 138, 1186 CrossRef CAS .
- S. P. Fusselman, J. J. Roy, D. L. Grimmett, L. F. Grantham, C. L. Krueger, C. R. Nabelek, T. S. Storvick, T. Inoue, T. Hijikata, K. Kinoshita, Y. Sakamura, K. Uozumi, T. Kawai and N. Takahashi, J. Electrochem. Soc., 1999, 146, 2573–2580 CrossRef CAS .
- A. Cassol, L. Magon, G. Tomat and R. Portanova, Inorg. Chem., 1972, 11, 515–519 CrossRef CAS .
- R. M. Rush and J. S. Johnson, J. Phys. Chem., 1963, 67, 821–825 CrossRef CAS .
- A. Cassol, L. Magon, R. Portanova and E. Tondello, Radiochim. Acta, 1972, 17, 28–32 CrossRef CAS .
- R. Lysy and G. Duyckaerts, Anal. Chim. Acta, 1978, 96, 125–132 CrossRef CAS .
- I. B. Polovov, C. A. A. Sharrad, I. May, B. D. D. Vasin, V. A. Volkovich and T. R. R. Griffiths, ECS Trans., 2007, 3, 503–511 CrossRef CAS .
- L. Martinot, J. Less Common Metals, 1991, 170, 121–126 CrossRef CAS .
-
G. T. Seaborg, R. A. James and L. O. Morgan, Modern Alchemy, 1994, pp. 24–52 Search PubMed .
- B. Still, Nat. Chem., 2017, 9, 296 CrossRef CAS PubMed .
- J. Serp, P. Chamelot, S. Fourcaudot, R. J. M. Konings, R. Malmbeck, C. Pernel, J. C. Poignet, J. Rebizant and J. P. Glatz, Electrochim. Acta, 2006, 51, 4024–4032 CrossRef CAS .
- D. Lambertin, J. Lacquement, S. Sanchez and G. S. Picard, Plasmas Ions, 2000, 3, 65–72 CrossRef CAS .
-
A. Laplace, J. Lacquement, C. Maillard and L. Donnet, Proc. Atalante 2004: Advances for Future Nuclear Fuel Cycles, Nimes, France, 2004 Search PubMed .
-
H. Boussier, J. P. Glatz, R. J. M. Konings, R. Malmbeck, C. Pernel, J. Rebizant, C. Scheppler and J. Serp, PYRO Project Status, 2003, FIKW-CT-2000-00049.
-
G. T. Seaborg, R. A. James and A. Ghiorso, The New Element Curium (Atomic Number 96), AECD-2182, United States, 1948.
- H. J. Groh, R. T. Huntoon, C. S. Schlea, J. A. Smith and F. H. Springer, Nuclear Appl., 1965, 1, 327–336 CAS .
-
G. J. Lumetta, M. C. Thompson, R. A. Penneman and P. G. Eller, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Netherlands, Dordrecht, 2008, pp. 1397–1443 Search PubMed .
- Y. V. Lobanov, G. V. Buklanov, F. S. Abdullin, A. N. Polyakov, I. V. Shirokovsky, Y. S. Tsyganov and V. K. Utyonkov, Nucl. Instrum. Methods Phys. Res., Sect. A, 1997, 397, 26–29 CrossRef CAS .
-
R. A. Penneman and T. K. Keenan, The Radiochemistry of Americium and Curium, NAS-NS-3006, Washington DC, 1960 Search PubMed .
- A. Osipenko, A. Maershin, V. Smolenski, A. Novoselova, M. Kormilitsyn and A. Bychkov, J. Electroanal. Chem., 2011, 651, 67–71 CrossRef CAS .
- T. Koyama, T. R. Johnson and D. F. Fischer, J. Alloys Compd., 1992, 189, 37–44 CrossRef CAS .
- V. P. Kolesnikov, G. N. Kazantsev and O. V. Skiba, Radiokhimiya, 1977, 19, 545–548 CAS .
- A. Osipenko, A. Maershin, V. Smolenski, A. Novoselova, M. Kormilitsyn and A. Bychkov, J. Nucl. Mater., 2010, 396, 102–106 CrossRef CAS .
- A. J. Bard and L. R. Faulkner, Electrochem. Methods, 2001, 2, 580–632 Search PubMed .
-
A. Osipenko, A. Mayershin, V. Smolenski, A. Novoselova and M. Kormilitsyn, in Recent Trends in Electrochemical Science and Technology, ed. U. Kumar, Sur, IntechOpen, 2012 Search PubMed .
- T. T. Tsong, Prog. Surf. Sci., 2001, 67, 235–248 CrossRef CAS .
-
A. Ugale, T. N. Kalyani and S. J. Dhoble, in Lanthanide-Based Multifunctional Materials, ed. P. Martín-Ramos and M. B. T.-L.-B. M. M. Ramos Silva, Elsevier, 2018, pp. 59–97 Search PubMed .
- H. N. McCoy, J. Am. Chem. Soc., 1936, 58, 2279–2281 CrossRef CAS .
- T.-J. Kim, Y.-J. Jung, S.-H. Kim, S.-W. Paek and D.-H. Ahn, Bull. Korean Chem. Soc., 2011, 32, 863–866 CrossRef CAS .
- S. Bratsch and H. B. Silber, Polyhedron, 1982, 1, 219–223 CrossRef CAS .
- K. E. Johnson and J. R. Mackenzie, J. Electrochem. Soc., 1969, 116, 1697 CrossRef CAS .
- J. P. Schoebrechts, B. P. Gilbert and G. Duyckaerts, J. Electroanal. Chem. Interfacial Electrochem., 1983, 145, 139–146 CrossRef CAS .
- S. A. Kuznetsov and M. Gaune-Escard, Electrochim. Acta, 2001, 46, 1101–1111 CrossRef CAS .
- S. A. Kuznetsov and M. Gaune-Escard, J. Electroanal. Chem., 2006, 595, 11–22 CrossRef CAS .
- M. Gibilaro, L. Massot, P. Chamelot, L. Cassayre and P. Taxil, Electrochim. Acta, 2009, 55, 281–287 CrossRef CAS .
- C. Nourry, L. Massot, P. Chamelot and P. Taxil, J. Appl. Electrochem., 2009, 39, 927–933 CrossRef CAS .
- S. E. Bae, Y. J. Park, S. K. Min, Y. H. Cho and K. Song, Electrochim. Acta, 2010, 55, 3022–3025 CrossRef CAS .
- M. Gibilaro, L. Massot, P. Chamelot and P. Taxil, J. Nucl. Mater., 2008, 382, 39–45 CrossRef CAS .
- P. Taxil, P. Chamelot, L. Massot and C. Hamel, J. Min. Metall., Sect. B, 2003, 39, 177–200 CrossRef CAS .
- C. A. Schroll, S. Chatterjee, T. G. Levitskaia, W. R. Heineman and S. A. Bryan, Anal. Chem., 2013, 85, 9924–9931 CrossRef CAS PubMed .
- W. Huang, L. Tian, C. She, F. Jiang, H. Zheng, W. Li, G. Wu, D. Long and Q. Li, Electrochim. Acta, 2014, 147, 114–120 CrossRef CAS .
- M. R. Bermejo, F. de la Rosa, E. Barrado and Y. Castrillejo, J. Electroanal. Chem., 2007, 603, 81–95 CrossRef CAS .
- L. Massot, P. Chamelot, L. Cassayre and P. Taxil, Electrochim. Acta, 2009, 54, 6361–6366 CrossRef CAS .
-
C. Caravaca, G. Córdoba and M. J. Tomás, in 8th Information Exchange Meeting on P&T, Las Vegas, 2004.
- S. A. Kuznetsov, L. Rycerz and M. Gaune-Escard, J. New Mater. Electrochem. Syst., 2006, 9, 313 CAS .
-
M. S. Wickleder, B. Fourest and P. K. Dorhout, in The Chemistry of the Actinide and Transactinide Elements, ed. L. R. Morss, N. M. Edelstein and J. Fuger, Springer, Netherlands, Dordrecht, 2008, pp. 52–160 Search PubMed .
- I. Baghni, Y.-S. Wu, J.-Q. Li, C. Du and W. Zhang, Trans. Nonferrous Met. Soc. China, 2003, 13, 1253–1259 Search PubMed .
- T. E. Leontis, JOM, 1952, 4, 633–642 CrossRef CAS .
-
H. O. Poppleton and F. E. Block, A Process for the Preparation of Thorium by Sodium Reduction of Thorium Tetrachloride, US Department of the Interior, Bureau of Mines, 1964, vol. 6527 Search PubMed .
- L. Martinot, J. Less-Common Met., 1989, 147, 73–77 CrossRef CAS .
-
L. Martinot, Handbook on the Physics and Chemistry of the Actinides, Elsevier Science
Publishers, 1991 Search PubMed .
- L. Cassayre, J. Serp, P. Soucek, R. Malmbeck, J. Rebizant and J. P. Glatz, Electrochim. Acta, 2007, 52, 7432–7437 CrossRef CAS .
- T. M. Klapötke and A. Schulz, Polyhedron, 1997, 16, 989–991 CrossRef .
- G. Ionova, C. Madic and R. Guillaumont, Polyhedron, 1998, 17, 1991–1995 CrossRef CAS .
- R. Srinivasan and S. N. Flengas, Can. J. Chem., 1964, 42, 1315–1322 CrossRef CAS .
- L. Martinot, J. Bohet, G. Duyckaerts and W. Muller, Inorg. Nuclear Chem. Lett., 1977, 13, 315–319 CrossRef CAS .
- L. H. Meyer, J. Electrochem. Soc., 1960, 107, 43 CrossRef CAS .
- F. R. Clayton, G. Mamantov and D. L. Manning, J. Electrochem. Soc., 1974, 121, 86 CrossRef CAS .
-
R. P. Elliott, Constitution of binary alloys: first supplement, McGraw-Hill, New York, 1965 Search PubMed .
- L. Martinot and J. Fuger, J. Less-Common Met., 1986, 120, 255–266 CrossRef CAS .
- I. Johnson, J. Nucl. Mater., 1988, 154, 169–180 CrossRef CAS .
- T. Kobayashi and M. Tokiwai, J. Alloys Compd., 1993, 197, 7–16 CrossRef CAS .
- T. Kobayashi, R. Fujita, M. Fujie and T. Koyama, J. Nucl. Sci. Technol., 1995, 32, 653–663 CrossRef CAS .
- T. Kobayashi, R. Fujita, H. Nakamura and T. Koyama, J. Nucl. Sci. Technol., 1997, 34, 50–57 CrossRef CAS .
- J. H. Lee, Y. H. Kang, S. C. Hwang, H. S. Lee, E. H. Kim and S. W. Park, Nucl. Technol., 2008, 162, 107–116 CrossRef CAS .
- S. H. Kim, S. Bin Park, S. J. Lee, J. G. Kim, H. S. Lee and J. H. Lee, Nucl. Eng. Des., 2013, 257, 12–20 CrossRef CAS .
- S. Ghosh, B. Prabhakara Reddy, K. Nagarajan and P. R. Vasudeva Rao, Nucl. Technol., 2010, 170, 430–443 CrossRef CAS .
- S. Ghosh, S. Vandarkuzhali, N. Gogoi, P. Venkatesh, G. Seenivasan, B. P. Reddy and K. Nagarajan, Electrochim. Acta, 2011, 56, 8204–8218 CrossRef CAS .
- R. O. Hoover, S. Phongikaroon, M. F. Simpson, S. X. Li and T.-S. Yoo, Nucl. Technol., 2010, 171, 276–284 CrossRef CAS .
- R. K. Ahluwalia and T. Q. Hua, Nucl. Technol., 2002, 140, 41–50 CrossRef CAS .
- R. K. Ahluwalia, T. Q. Hua and H. K. Geyer, Nucl. Technol., 2001, 133, 103–118 CrossRef CAS .
- R. Ahluwalia, T. Q. Hua and H. K. Geyer, Nucl. Technol., 1999, 126, 289–302 CrossRef CAS .
-
B. G. Park and I. S. Hwang, Proc. of the Korean Nuclear Society autumn meeting, Seoul, Republic of Korea, 1999, p. 293 Search PubMed .
- J. Zhang, Prog. Nucl. Energy, 2014, 70, 279–286 CrossRef CAS .
- R. M. Cumberland and M.-S. Yim, Ann. Nucl. Energy, 2014, 71, 52–59 CrossRef CAS .
- R. M. Cumberland and M.-S. Yim, J. Electrochem. Soc., 2014, 161, D147 CrossRef CAS .
-
J. O. Bockris, B. E. Conway, E. Yeager and R. E. White, Comprehensive treatise of electrochemistry, Electrochemical Processing, Plenum Press, New York, 1st edn, 1981, vol. 2 Search PubMed .
- D. S. Maha Vishnu, N. Sanil, L. Shakila, R. Sudha, K. S. Mohandas and K. Nagarajan, Electrochim. Acta, 2015, 159, 124–130 CrossRef CAS .
- N. Xin-miao, D. Ling-yang, B. Chen-guang and C. Deng-fu, Trans. Nonferrous Met. Soc. China, 2006, 16, s723–s727 CrossRef .
- H. S. Shin, J. M. Hur, S. M. Jeong and K. Y. Jung, J. Ind. Eng. Chem., 2012, 18, 438–442 CrossRef CAS .
- R. Barnett, K. T. Kilby and D. J. Fray, Metall. Mater. Trans. B, 2009, 40, 150–157 CrossRef .
- T. Wu, X. Jin, W. Xiao, X. Hu, D. Wang and G. Z. Chen, Chem. Mater., 2007, 19, 153–160 CrossRef CAS .
- C. Schwandt and D. J. Fray, Electrochim. Acta, 2005, 51, 66–76 CrossRef CAS .
- G. Qiu, M. Ma, D. Wang, X. Jin, X. Hu and G. Z. Chen, J. Electrochem. Soc., 2005, 152, E328 CrossRef CAS .
- D. Wang, G. Qiu, X. Jin, X. Hu and G. Z. Chen, Angew. Chem., Int. Ed., 2006, 45, 2384–2388 CrossRef CAS PubMed .
- G. Qiu, K. Jiang, M. Ma, D. Wang, X. Jin and G. Z. Chen, Zeitschrift für Naturforschung A, 2007, 62, 292–302 CrossRef CAS .
- G. Li, D. Wang, X. Jin and G. Z. Chen, Electrochem. Commun., 2007, 9, 1951–1957 CrossRef CAS .
- G. Qiu, X. Feng, M. Liu, W. Tan and F. Liu, Electrochim. Acta, 2008, 53, 4074–4081 CrossRef CAS .
-
K. Rao, D. Brett, D. Inman and R. Dashwood, Molten Salt Discussion Group, London, 2008 Search PubMed .
-
L. D. Brown, PhD thesis, University College London, 2015 .
- J. Peng, Y. Deng, D. Wang, X. Jin and G. Z. Chen, J. Electroanal. Chem., 2009, 627, 28–40 CrossRef CAS .
- T. Berent, I. Fells and R. Mason, Nature, 1969, 223, 1054–1055 CrossRef CAS .
- C. Oloman and A. P. Watkinson, J. Appl. Electrochem., 1979, 9, 117–123 CrossRef CAS .
- Y. Xiong and H. T. Karlsson, Adv. Environ. Res., 2002, 7, 139–145 CrossRef CAS .
- S. Germain and F. Goodridge, Electrochim. Acta, 1976, 21, 545–550 CrossRef CAS .
- F. Goodridge, C. J. H. King and A. R. Wright, Electrochim. Acta, 1977, 22, 1087–1091 CrossRef CAS .
- M. Kalaruban, P. Loganathan, W. G. Shim, J. Kandasamy, G. Naidu, T. V. Nguyen and S. Vigneswaran, Sep. Purif. Technol., 2016, 158, 62–70 CrossRef CAS .
- M. J. Nelson, G. Nakhla and J. Zhu, Engineering, 2017, 3, 330–342 CrossRef .
- D. R. Chadeesingh and A. N. Hayhurst, Fuel, 2014, 127, 169–177 CrossRef CAS .
- A. N. Hayhurst, Combust. Flame, 1991, 85, 155–168 CrossRef CAS .
- D. Hu, X. Zeng, F. Wang, M. Haruna Adamu and G. Xu, Fuel, 2021, 290, 120038 CrossRef CAS .
- P. Chaiwang, H. Chitcharoenyoo, P. Piumsomboon and B. Chalermsinsuwan, Particuology, 2022, 67, 47–56 CrossRef CAS .
- R. Abdulaziz, L. Brown, D. Inman, C. A. Sharrad, A. Jones, P. R. Shearing and D. J. L. Brett, J. Electrochem. Soc., 2017, 164, H5280–H5285 CrossRef CAS .
- R. Abdulaziz, L. D. Brown, D. Inman, P. R. Shearing and D. J. L. Brett, Electrochim. Acta, 2017, 226, 18–28 CrossRef CAS .
- M. Mirza, R. Abdulaziz, W. C. Maskell, C. Tan, P. R. Shearing and D. J. L. Brett, Electrochim. Acta, 2021, 391, 138846 CrossRef CAS .
- S. Jiao, H. Jiao, W. Song, M. Wang and J. Tu, Int. J. Miner., Metall. Mater., 2020, 27, 1588–1598 CrossRef CAS .
- T. Kato, T. Inoue, T. Iwai and Y. Arai, J. Nucl. Mater., 2006, 357, 105–114 CrossRef CAS .
- K. Kinoshita, T. Inoue, S. P. Fusselman, D. L. Grimmett, J. J. Roy, R. L. Gay, C. L. Krueger, C. R. Nabelek and T. S. Storvick, J. Nucl. Sci. Technol., 1999, 36, 189–197 CrossRef CAS .
- H. Jiao, J. Wang, L. Zhang, K. Zhang and S. Jiao, RSC Adv., 2015, 5, 62235–62240 RSC .
- M. Gibilaro, S. Bolmont, L. Massot, L. Latapie and P. Chamelot, J. Electroanal. Chem., 2014, 726, 84–90 CrossRef CAS .
- J. P. Ackerman and J. L. Settle, J. Alloys Compd., 1991, 177, 129–141 CrossRef CAS .
- Y. S. Hwang, M. S. Jeong and S. W. Park, Prog. Nucl. Energy, 2007, 49, 463–472 CrossRef .
- D. Vaden, S. X. Li, B. R. Westphal, K. B. Davies, T. A. Johnson and D. M. Pace, Nucl. Technol., 2008, 162, 124–128 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a and
Dan J. L.
Brett
a and
Dan J. L.
Brett