
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn unexpected feasible route for the formation of organosulfates by the gas phase reaction of sulfuric acid with acetaldehyde catalyzed by dimethylamine in the atmosphere†
Ju-Rui
Yang
a,
Ai
Liu
b and
Bo
Long
 *ab
*ab
aDepartment of Physics, Guizhou University, Guiyang 550025, China. E-mail: wwwltcommon@sina.com
bSchool of Materials Science and Engineering, Guizhou Minzu University, Guiyang, 550025, China
First published on 3rd February 2023
Abstract
An understanding of the formation of organosulfates is required for elucidating the formation of secondary organic aerosols in atmosphere. Herein, we report a new feasible reaction pathway for the formation of organosulfates in the reaction of sulfuric acid (H2SO4) with acetaldehyde (CH3CHO) catalyzed by dimethylamine ((CH3)2NH) using quantum chemical methods and reaction rate theory. We found that dimethylamine has a strong catalytic effect in the CH3CHO + H2SO4 + (CH3)2NH reaction because the energy barriers of the CH3CHO + H2SO4 + (CH3)2NH reaction are reduced by 18.28–23.08 kcal mol−1 as compared to the CH3CHO + H2SO4 reaction. The calculated results show that the CH3CHO + H2SO4 + (CH3)2NH reaction can compete well with the traditional sink for CH3CHO by hydroxyl radicals (OH), when the OH, H2SO4, and (CH3)2NH concentrations are 104 molecules per cm3 during the night and 1.0 × 106 and 3.2 × 109 molecules per cm3 at below 240 K, respectively. The calculated results also show that the CH3CHO + H2SO4 + (CH3)2NH reaction makes a limited contribution to the sink for sulfuric acid in the atmosphere. The present findings provide a new insight into the generation of organosulfates, which could be extended to other aldehydes with sulfuric acid catalyzed by amines in the atmosphere.
Environmental significanceOrganosulfates are key components in secondary organic aerosols. This study presents a new feasible route for the formation of organosulfates via the gas phase reactions of acetaldehyde with sulfuric acid catalyzed by dimethylamine. This mechanistic route could be extended to the reactions of other aldehydes with sulfuric acid catalyzed by amines in the atmosphere. However, organosulfates are often obtained by heterogeneous processes. Our findings also show that the CH3CHO + H2SO4 + (CH3)2NH reaction can make an important contribution to the sink for acetaldehyde during the night at low temperatures. This study is of great significance for the accurate evaluation of the atmospheric environmental effects of CH3CHO, particularly, air quality and atmospheric modeling and policies. |
1. Introduction
Secondary organic aerosols (SOAs) are formed by the atmospheric oxidation of volatile organic compounds (VOCs),1–8 which have important effects on the climate and environment, human health, and cloud condensation nuclei (CCN).2,9–17 Although there are extensive studies on secondary organic aerosols, the formation process of secondary organic aerosols is still not completely clear because their formation is significantly influenced by many factors such as the complex atmospheric oxidation processes of VOCs and different sources of atmospheric particles.2,9,18,19The formation of secondary organic aerosols occurs by atmospheric nucleation,20 which is determined by the formation and decomposition of molecular clusters of different sizes and chemical compositions.21 Previous studies have shown that sulfuric acid is the most common nucleating precursor and dimethylamine is an important component of atmospheric aerosol precursors in acid-base cluster reactions.22–25 The strong interactions between dimethylamine and sulfuric acid can effectively form clusters,26–29 which further contribute to the formation of secondary organic aerosols.27,28,30
Acetaldehyde (CH3CHO) is an important aldehyde with atmospheric concentrations of about 15.9 ppb and 45.60 ppb in major urban areas of China and Brazil, respectively.31,32 CH3CHO has serious impacts on the air environment,33–36 and is emitted from anthropogenic and natural sources.32,35,37–41 Additionally, the photochemical degradation of VOCs has been considered to be the main source of acetaldehyde in the atmosphere.31,37,42 Previous studies have shown that acetaldehyde makes an important contribution to the formation of secondary organic aerosols.36,41 Organosulfates are formed via the heterogeneous reaction process of aldehydes as reported in the literature.43,44 However, the formation of organosulfates by acetaldehyde has not been reported in the gas phase of the atmosphere.
| CH3CHO + H2SO4 + (CH3)2NH → CH3CHO + H2SO4⋯(CH3)2NH → CH3HC(OH)OSO3H + (CH3)2NH | (R1) |
Herein, we have investigated the reaction of sulfuric acid with acetaldehyde catalyzed by dimethylamine in reaction (R1) using theoretical methods. We obtained the energy barriers and reaction rates of reaction (R1) to show the catalytic ability of dimethylamine. We propose a new mechanistic pathway for the formation of organosulfates via the homogeneous reaction process, whereas previous investigations considered the formation of organosulfates by heterogeneous reaction processes.45 The present findings of this work not only provide a new source of organosulfates but are also likely to be applied in other aldehyde reactions with sulfuric acid catalyzed by dimethylamine.
2. Computational methods
In this article, all geometries and the corresponding frequencies of reactants, transition states, products, and complexes were optimized using the M06-2X46 functional with the MG3S47 basis set in the CH3CHO + H2SO4 + (CH3)2NH reaction. The reliability of the M06-2X functional and its wide application have been demonstrated in previous studies on the atmospheric reactions of sulfuric acid and sulfuric acid molecular clusters.48–53 The calculated results of the corresponding frequencies of reactants, complexes, transition states, and products using M06-2X/MG3S suggest that all stable structures have positive frequencies, while the transition state has only one imaginary frequency. The stability of the density functional theory method has been tested by using the keyword (stable = opt).54,55 Intrinsic reaction coordinate (IRC)56,57 calculations were utilized to determine whether the transition state corresponds to the reactants and products.To obtain the relative energies more reliably, we did single-point energy calculations for these stationary points in the present investigation. Based on previous studies, the accuracy of CCSD(T)-F12a/jun-cc-pV(T+d)Z calculation results can be approximated to CCSD(T)/CBS.58,59 Therefore, we have used the CCSD(T)-F12a60,61 theoretical method and the jun-cc-pV(T+d)Z62 basis set to calculate the single-point energies to obtain reliable relative energies based on the optimization geometric structure of M06-2X/MG3S. The reliability of CCSD(T) has been confirmed by the T1 diagnostic values of all species with the upper limit of 0.02 in the closed-shell systems.63 The T1 diagnostic values of all species in this article are below the upper limit of 0.02 (see Table S1†).
We calculated rate constants by using conventional transition state theory64–66 with Eckart tunneling67 for each reaction studied in this article. All electronic structure calculations were done using the Gaussian 16 (ref. 68) and Molpro 2019 (ref. 69) codes, while the rate constants were calculated using The Rate70 code.
3. Results and discussion
The optimized geometries and calculated relative energies with zero-point vibrational correction for the reaction (R1) are shown in Fig. 1. Concerning the complexes and transition states reported here, we used the binding energies and energy barriers, which are equal to the relative energies obtained by CCSD(T)-F12a/jun-cc-pV(T+d)Z//M06-2X/MG3S, plus the relative zero-point vibrational correction by M06-2X/MG3S, respectively. The rate constants and equilibrium constants relative to the reaction (R1) are listed in Table 1. The relative Gibbs free energy for the reaction of acetaldehyde with sulfuric acid catalyzed by dimethylamine at 298 K is shown in Fig. S1.† We discuss the relative energies with zero-point vibrational correction based on CCSD(T)-F12a/jun-cc-pV(T+d)Z//M06-2X/MG3S calculated results unless otherwise stated.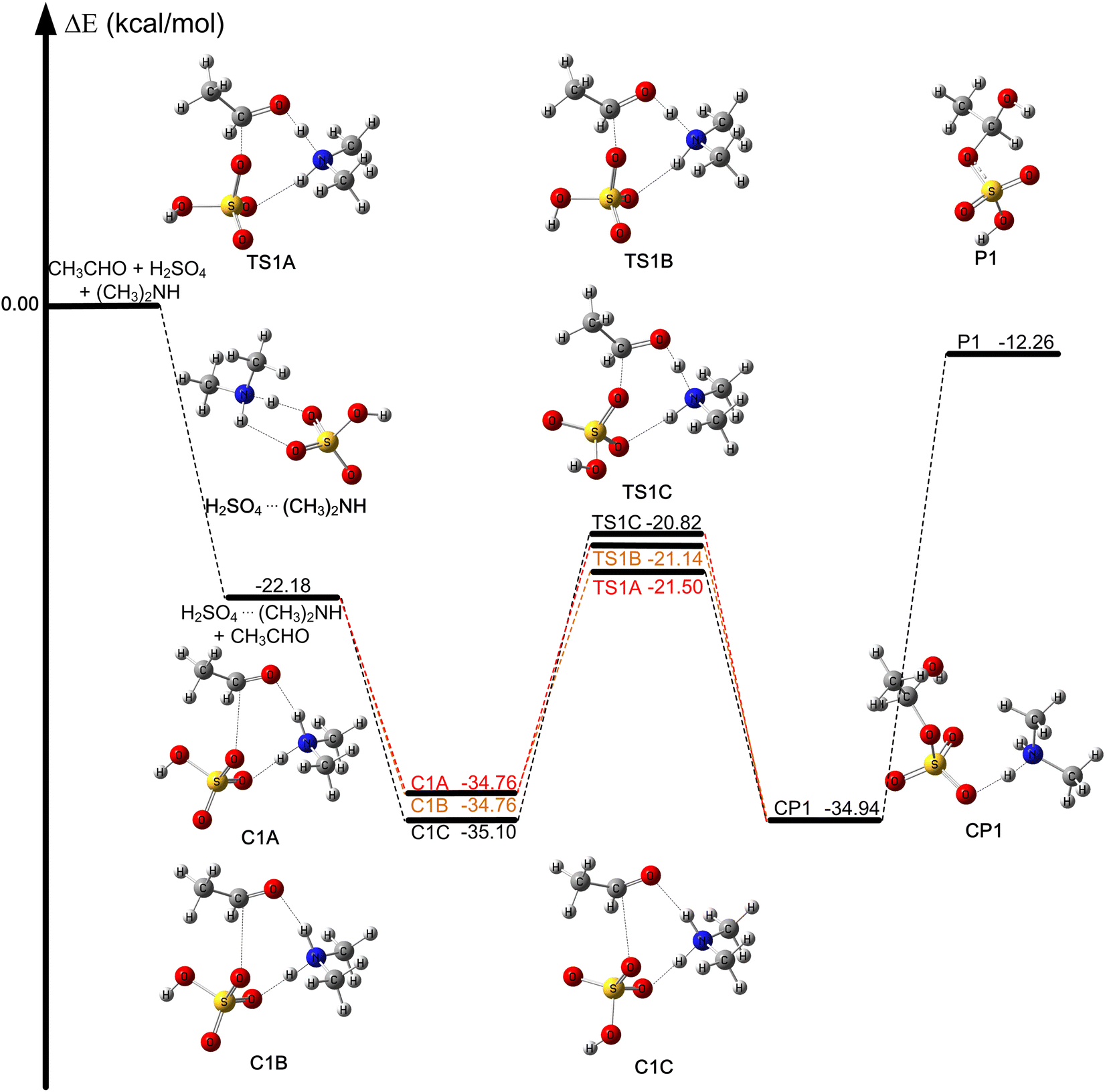 | ||
| Fig. 1 Calculated relative energies with zero-point vibrational correction for the reaction CH3CHO + H2SO4 + (CH3)2NH → CH3HC(OH)OSO3H + (CH3)2NH at the CCSD(T)-F12a/jun-cc-pV(T+d)Z//M06-2X/MG3S level (in kcal mol−1). | ||
| 200 K | 220 K | 240 K | 260 K | 280 K | 298 K | |
|---|---|---|---|---|---|---|
| a The equilibrium constants of the H2SO4⋯(CH3)2NH complex with respect to H2SO4 and (CH3)2NH. b The bimolecular rate constants of the H2SO4⋯(CH3)2NH + CH3CHO reaction. | ||||||
| K eq1 | 2.33 × 10−3 | 1.56 × 10−5 | 2.43 × 10−7 | 7.27 × 10−9 | 3.63 × 10−10 | 3.47 × 10−11 |
| k 1 | 1.55 × 10−16 | 1.70 × 10−16 | 1.90 × 10−16 | 2.13 × 10−16 | 2.40 × 10−16 | 2.68 × 10−16 |
3.1. The CH3CHO + H2SO4 reaction catalyzed by (CH3)2NH
The reaction of acetaldehyde with sulfuric acid catalyzed by dimethylamine is shown in Fig. 1, where there are three molecular reaction systems. In trimolecular reaction systems, there are three different entrance channels, and the probability of the three molecules colliding simultaneously in the gas phase of the atmosphere is very low, which has been reported in the literature.36,71–75 Therefore, when the three molecules CH3CHO, H2SO4, and (CH3)2NH first collide, two molecules of the three form a dimer, and then the dimer collides with the third molecule to form the ternary complex: H2SO4⋯(CH3)2NH + CH3CHO, CH3CHO⋯H2SO4 + (CH3)2NH, and CH3CHO⋯(CH3)2NH + H2SO4.We compared the calculated binding energies of the dimer complexes with the values from previous investigations in the literature. The computed binding energy of H2SO4⋯(CH3)2NH (M1A) is −22.18 kcal mol−1 (see Table S2†), which is consistent with the value of −21.99 kcal mol−1 at the CCSD(T)/aug-cc-pVTZ//ωB97XD/6-311++(2d,2p) level.76 The binding energy of CH3CHO⋯H2SO4 was computed to be −11.91 kcal mol−1, which is slightly different from the value of CH3CHO⋯H2SO4 (−12.59 kcal mol−1) at the CCSD(T)-F12a/cc-pVTZ-F12//M06-2X/MG3S level (see Table S2 and Fig. S2†). The calculated binding energy of CH3CHO⋯(CH3)2NH (−3.43 kcal mol−1) is consistent with the value of −3.42 kcal mol−1 calculated by the CCSD(T)-F12a/VTZ-F12//M06-2X/6-311++G(d,p).36 Because the binding energies of the binary complexes in the entrance channel were obtained using different theoretical methods, they do not exactly match the values in the literature.77 However, the differences are still small (around 0.5 kcal mol−1).
The binding energy of H2SO4⋯(CH3)2NH (−22.18 kcal mol−1) is much lower than those of CH3CHO⋯H2SO4 (−11.91 kcal mol−1) and CH3CHO⋯(CH3)2NH (−3.43 kcal mol−1); this resulted in the equilibrium constant of H2SO4⋯(CH3)2NH being much larger than those of CH3CHO⋯H2SO4 and CH3CHO⋯(CH3)2NH in Table S3.† For example, the equilibrium constant of H2SO4⋯(CH3)2NH is at least 106 and 1012 larger than those of CH3CHO⋯H2SO4 and CH3CHO⋯(CH3)2NH at 298 K, respectively (see Table S3†). A larger equilibrium constant of the complex leads to a higher concentration in the atmosphere. For example, when the concentrations of CH3CHO,32,34 H2SO4,78 and (CH3)2NH79 are 1.12 × 1012, 4.0 × 108, and 3.2 × 109 molecules per cm3, respectively, the calculated the concentrations of H2SO4⋯(CH3)2NH, CH3CHO⋯H2SO4, and CH3CHO⋯(CH3)2NH are 2.98 × 1015 to 4.97 × 105, 7.34 × 106 to 5.14 × 101 and 2.09 × 10−1 to 1.47 × 10−2 molecules per cm3, respectively at 200–340 K (see Table S3†). In particular, the concentrations of H2SO4 and (CH3)2NH were 4.0 × 108 and 3.2 × 109 molecules per cm3, respectively, while the corresponding concentration of the H2SO4⋯(CH3)2NH dimer was calculated to be 2.98 × 1015 to 4.64 × 108 molecule per cm3 over the 200–280 K. The larger concentration of the H2SO4⋯(CH3)2NH dimer was caused by the larger equilibrium constant at low temperatures in Table S3.† Moreover, the concentration of the H2SO4⋯(CH3)2NH complex was much higher than those of CH3CHO···H2SO4 and CH3CHO⋯(CH3)2NH. In addition, the peak concentration of sulfuric acid in the atmospheric boundary layer during the daytime is about 106–3 × 107 molecules per cm3 and the addition of dimethylamine can stabilize clusters and reduce evaporation.25,80 Therefore, we only considered the entrance channel CH3CHO + H2SO4⋯(CH3)2NH in the present investigation.
The CH3CHO + H2SO4⋯(CH3)2NH reaction occurs in one elementary step as depicted in Fig. 1, which is similar to the hydrolysis of acetaldehyde catalyzed by sulfuric acid, as well as the reaction of formaldehyde with sulfuric acid catalyzed by formic acid and other catalysts.34,36,50,51,53,81–83 It can be seen that when the H2SO4⋯(CH3)2NH complex and CH3CHO act as reactants, the reaction occurs via the formation of pre-reaction complexes and then undergoes a unimolecular isomerization through these corresponding transition states responsible for the formation of the post-reaction complex. In the CH3CHO + H2SO4⋯(CH3)2NH reaction, there is a concerted reaction mechanism where the hydrogen atom of the OH group in sulfuric acid is transferred to the oxygen atom of the carbonyl group in CH3CHO by the amino group in dimethylamine, and the oxygen atom of the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in sulfuric acid is simultaneously added to the carbon atom in CH3CHO, where dimethylamine acts as a catalyst; this leads to the formation of the most stable post-reaction complex (CP1), which is a nucleation precursor of secondary organic aerosols.31 We also note that the formation of P1 is endothermic with respect to the H2SO4⋯(CH3)2NH and CH3CHO reactants and, therefore, the reverse reaction is possible.
O group in sulfuric acid is simultaneously added to the carbon atom in CH3CHO, where dimethylamine acts as a catalyst; this leads to the formation of the most stable post-reaction complex (CP1), which is a nucleation precursor of secondary organic aerosols.31 We also note that the formation of P1 is endothermic with respect to the H2SO4⋯(CH3)2NH and CH3CHO reactants and, therefore, the reverse reaction is possible.
The pre-reaction complexes (CH3CHO⋯H2SO4⋯(CH3)2NH) have an eight-membered ring structure with two hydrogen-bonded interactions and a van der Waals interactions. As can be seen from Fig. S4,† the bond lengths of the C1–O16 bond, the N5–H3 bond and the O15–H17 bond in C1A are the same as those in C1B. From C1A and C1B to TS1A and TS1B, the C1–O16 bond length decreased from 2.731 Å to 1.846 Å and 1.848 Å, the N5–H3 bond distance increased from 1.034 Å to 1.259 Å and 1.282 Å, and the O15–H17 bond was lengthened to 1.989 Å and 1.979 Å from 1.551 Å respectively. The O15–H18 bond in sulfuric acid was lengthened to 1.967 Å from 1.565 Å, and the N5–H3 bond increased to 1.283 Å from 1.033 Å and the bond distance between the C1 atom of the aldehyde group in CH3CHO and the O17 atom of H2SO4 was shortened to 1.837 Å from 2.793 Å from C1C to TS1C. From a geometrical point of view, dimethylamine plays a crucial bridging role in the (CH3)2NH-catalyzed reaction of sulfuric acid with acetaldehyde.
The binding energies of the pre-reaction complexes (C1A, C1B, and C1C) were computed to be −34.76, −34.76 and −35.10 kcal mol−1 with respect to the separate reactants. Additionally, the binding energies of the CH3CHO⋯H2SO4⋯(CH3)2NH complexes are also 11.60–13.76 kcal mol−1 lower than those of the HCHO⋯H2SO4⋯NH3 complexes at the M08-SO/maug-cc-pVTZ level.81 This shows that dimethylamine has a stronger ability to form molecular clusters, compared with ammonia.
In the CH3CHO + H2SO4 + (CH3)2NH reaction, we found six different transition states (TS1A, TS1B, TS1C, TS1D, TS1E, and TS1F) as shown in Fig. S3.† The differences among them are produced by the relative orientation of the dangling OH group in sulfuric acid and the attack of CH3CHO on the H2SO4⋯(CH3)2NH complex from different directions. The six transition states are three isomers, where each isomer has two conformations (TS1A and TS1D, TS1B and TS1E, TS1C and TS1F). Additionally, the energy barriers for TS1A, TS1B, TS1C, TS1D, TS1E and TS1F were computed to be −21.50, −21.14, −20.82, −18.78, −18.32 and −17.87 kcal mol−1, respectively, shown in Fig. S3.† The energy barriers of TS1D, TS1E, and TS1F are higher than those of TS1A, TS1B, and TS1C by about 2.04–3.63 kcal mol−1, therefore, we did not consider TS1D, TS1E, and TS1F for kinetics calculations.
From Fig. S2,† there are three different reaction pathways and the energy barriers of the CH3CHO + H2SO4 reaction were computed to be 0.41, 0.43 and 1.58 kcal mol−1 for TS2A, TS2B and TS2C in the CH3CHO + H2SO4 reaction. Therefore, the energy barriers of the CH3CHO + H2SO4 + (CH3)2NH reaction decreased by about 21.23–23.08 kcal mol−1, as compared with the CH3CHO + H2SO4 reaction. These results show that dimethylamine plays a remarkable catalytic role in the CH3CHO + H2SO4 + (CH3)2NH reaction. Furthermore, some similar reactions have been studied previously. For example, the energy barrier of the HCHO + HNO3 reaction was reduced by 21.97 kcal mol−1 with the assistance of (CH3)2NH.84 The energy barrier of the reaction between H2SO4 and HCHO decreased by 16–21 kcal mol−1 when NH3 was added to the catalyst.81 The energy barrier of the HCHO + H2SO4 reaction catalyzed by HCOOH is about 14.81 kcal mol−1 lower than that of the HCHO + H2SO4 reaction.53 Compared with these similar reactions, it can be seen that (CH3)2NH has an excellent catalytic effect. Thus, the above conclusions also suggest that dimethylamine plays a crucial catalytic role in the CH3CHO + H2SO4 + (CH3)2NH reaction, and the reaction of acetaldehyde with sulfuric acid catalyzed by (CH3)2NH accelerates the formation of nucleation precursors in the atmosphere.
3.2. Kinetics
Because the temperature of the earth's atmosphere varies with the different regions, we considered the rate constants at different temperatures. Herein, we have calculated the rate constants for the three pathways of the trimolecular reaction using conventional transition state theory with Eckart tunneling in the temperature range between 200 and 298 K.With regard to the CH3CHO + H2SO4 + (CH3)2NH reaction, we only considered H2SO4…(CH3)2NH and CH3CHO as reactants, and the reaction mechanism is written as follows in (R2) and (R3):
| (CH3)2NH + H2SO4 ↔ H2SO4⋯(CH3)2NH | (R2) |
| H2SO4⋯(CH3)2NH + CH3CHO → CH3HC(OH)OSO3H + (CH3)2NH | (R3) |
Thus, the total reaction rate of (R1) is computed as shown in the following eqn (1):
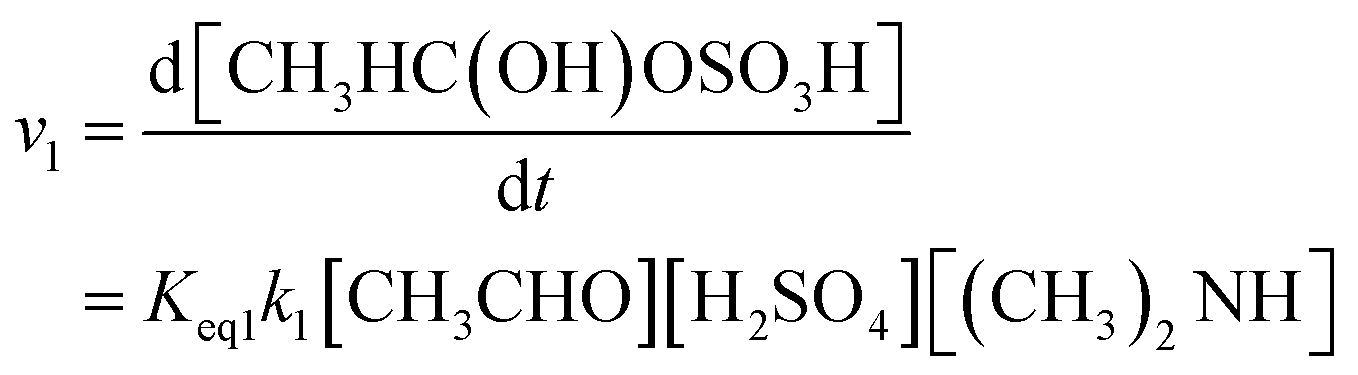 | (1) |
| CH3CHO + H2SO4 → CH3HC(OH)OSO3H | (R4) |
The reaction rate of acetaldehyde with sulfuric acid is expressed as follows:
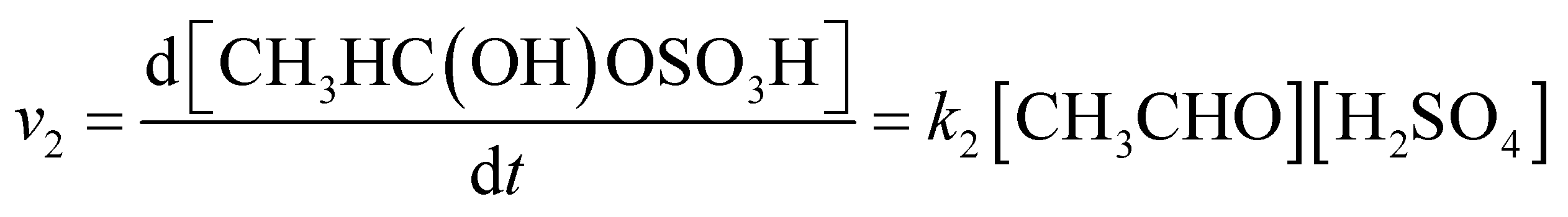 | (2) |
The rate constant k2 of the reaction was computed to be 3.88 × 10−16 to 5.77 × 10−16 cm3 per molecule per s at 200–298 K, as shown in Table S4;† this is still a slightly positive temperature dependence.
3.3 Atmospheric implications
To estimate the catalytic ability of dimethylamine in the H2SO4 + (CH3)2NH + CH3CHO reaction (R1), we considered the rate ratio of v1/v2, as written in eqn (3): | (3) |
The rate ratio v1/v2 depends on both the concentration of dimethylamine and temperature. When the concentration of dimethylamine is 3.2 × 109 molecules per cm3, measured in a contaminated environment,79 the values of v1/v2 are in the range 2.98 × 106 to 9.95 × 100 at 200–260 K (see Table S5†). These results show that dimethylamine makes a significant contribution in the reaction of acetaldehyde with sulfuric acid at low temperatures. The results also indicate the strong catalytic ability of dimethylamine in the H2SO4 + (CH3)2NH + CH3CHO reaction.
The main sink pathway of acetaldehyde is its reaction with OH with an atmospheric lifetime of about one day.37,85 To further investigate the important atmospheric contribution of the H2SO4 + (CH3)2NH + CH3CHO reaction, we compared the H2SO4 + (CH3)2NH + CH3CHO reaction with CH3CHO + OH. The rate ratio is expressed in eqn (4):
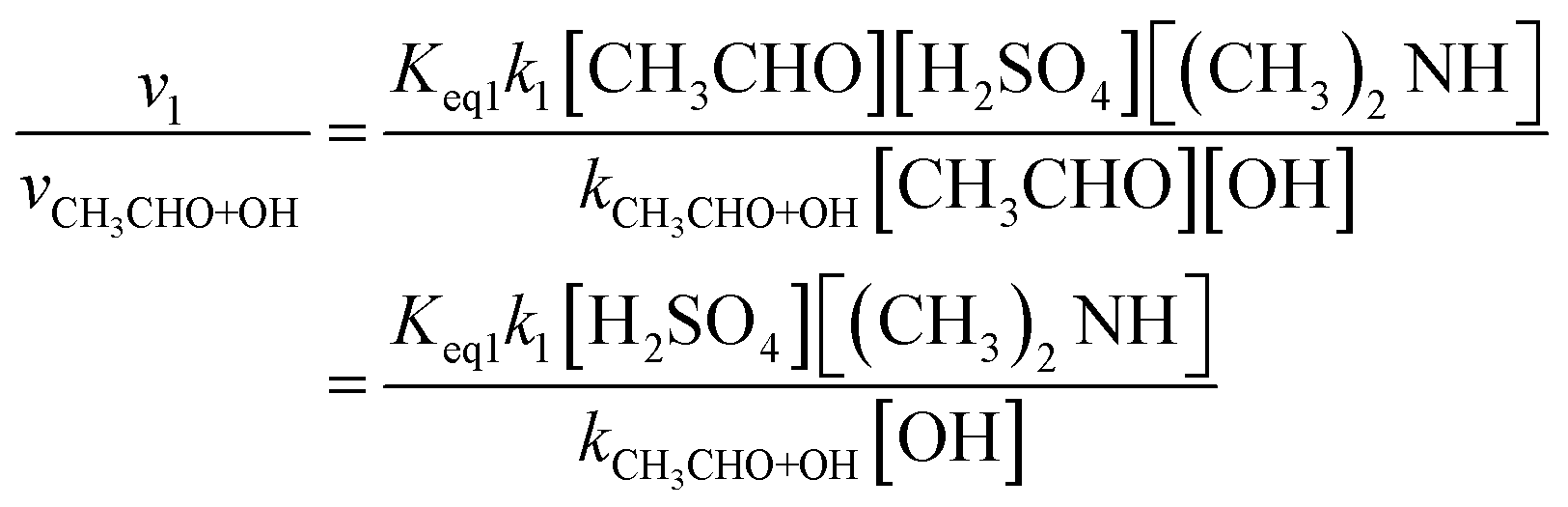 | (4) |
From eqn (4), it can be seen that v1/vCH3CHO+OH is related to the concentrations of sulfuric acid, dimethylamine, and OH. Atmospheric sulfuric acid concentrations are in the range of 104 to 4.0 × 108 molecules per cm3 in different areas.87–89 The concentration of (CH3)2NH is often 3.2 × 109 molecules per cm3.79 However, the concentration of OH is remarkably varied during the daytime and nighttime, with values in the range of 104–106 molecules per cm3.90,91 When the concentration of (CH3)2NH is 3.2 × 109 molecules per cm3 and the concentration of sulfuric acid is 106, 107, 4.0 × 108 and 5.1 × 109 molecules per cm3,92 the values of v1/vCH3CHO+OH at the different concentrations of OH are as listed in Tables S6–S9† at 200–298 K. When [(CH3)2NH] and [H2SO4] are 3.2 × 109, and 1.0 × 106 molecules per cm3, respectively, the rate ratio v1/vCH3CHO+OH as the function of OH concentration is shown in Fig. 2 at 200–298 K. When [OH], [CH3CHO],32,34 [H2SO4], and [(CH3)2NH] are 1.0 × 106, 1.12 × 1012, 1.0 × 106, and 3.2 × 109 molecules per cm3, respectively, the rate ratio of v1/vCH3CHO+OH is 3.67 at 220 K (listed in Table S6†); this shows that even if the OH concentration is very high during the daytime, the H2SO4 + (CH3)2NH + CH3CHO reaction can make some contribution to the sink for CH3CHO. Furthermore, when the OH concentration is further decreased to 1.0 × 104 molecules per cm3 during the night, the three-molecule reaction of acetaldehyde with sulfuric acid catalyzed by dimethylamine can compete well with the CH3CHO + OH reaction with the concentrations of H2SO4 (1.0 × 106 molecules per cm3) and (CH3)2NH (3.2 × 109 molecules per cm3) below 240 K in Table S6;† this shows that the H2SO4 + (CH3)2NH + CH3CHO reaction contributes significantly to the formation of organosulfates and the sink for acetaldehyde. In particular, previous studies have shown that carbonyl compounds can promote the formation of secondary organic aerosols through oxidation, hydration, and other reactions.39,93,94 Therefore, the H2SO4 + (CH3)2NH + CH3CHO reaction may be expected to be like the other reactions of other aldehydes with sulfuric acid catalyzed by dimethylamine, responsible for the formation of organosulfates in the gas phase reaction of the atmosphere.
 | ||
| Fig. 2 Rate ratio v1/vCH3CHO+OH at [H2SO4] = 1.0 × 106 molecules per cm3, and [(CH3)2NH] = 3.2 × 109 molecules per cm3 at different temperatures and different OH concentrations. | ||
We also compared the H2SO4 + (CH3)2NH + CH3CHO reaction with the H2SO4 + OH reaction. The rate ratio is given in eqn (5):
 | (5) |
The atmospheric lifetimes of sulfuric acid determined by heterogeneous processes are estimated to be approximately 102 s in polluted air and 104 s in clean air.96 In the H2SO4 + (CH3)2NH + CH3CHO reaction, the corresponding atmospheric lifetime of sulfuric acid was estimated mainly based on the concentrations of acetaldehyde and dimethylamine and the corresponding reaction rates as listed in Table S11.† In previous studies, acetaldehyde concentrations of 0.10 ppb–45.60 ppb were measured in some rural areas and in ambient air.97–99 With [(CH3)2NH] = 3.2 × 109 molecules per cm3 and [CH3CHO] = 2.46 × 109 and 1.12 × 1012 molecules per cm3, respectively, the atmospheric lifetimes of H2SO4 were calculated to be about 3.52 × 10−1 to 1.37 × 107 and 7.74 × 10−4 to 3.01 × 104 s at 200–298 K (see Tables S11 and S12†). The atmospheric lifetime of sulfuric acid was 3.52 × 10−1 and 7.74 × 10−4 s at 200 K, when the concentration of dimethylamine was 3.2 × 109 molecules per cm3 and the concentration of acetaldehyde was 2.46 × 109 and 1.12 × 1012 molecules per cm3, respectively. Therefore, the H2SO4 + (CH3)2NH + CH3CHO reaction only makes a limited contribution to the sulfuric acid sinks in the atmosphere.
4. Conclusions
In this article, the reaction mechanisms and kinetics of the trimolecular reaction of sulfuric acid with acetaldehyde catalyzed by dimethylamine have been investigated using high-level quantum chemical methods and conventional transition state theory with Eckart tunneling. The calculated results show that dimethylamine has a strong catalytic effect in the H2SO4 + (CH3)2NH + CH3CHO reaction responsible for the formation of organosulfates in the gas phase of the atmosphere, which was previously considered via heterogeneous processes. The calculated kinetic results reveal that the H2SO4 + (CH3)2NH + CH3CHO reaction can be an important sink for acetaldehyde in the atmospheric gas-phase reactions below 240 K when the OH concentration is about 104 molecules cm−3 and the H2SO4 concentration is about 1.0 × 106 molecules per cm3. Moreover, the H2SO4 + (CH3)2NH + CH3CHO reaction makes only a limited contribution to the sink for sulfuric acid. The present findings show that organosulfates can be formed via the reaction of CH3CHO with sulfuric acid catalyzed by dimethylamine, which provides a new insight into the initial nucleation process for sulfuric acid, amines, and aldehydes.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported in part by the National Natural Science Foundation of China and (4211001056, 41775125 and 91961123), by Guizhou Provincial Science and Technology Projects, China (CXTD [2022]001), and by the Science and Technology Foundation of Guizhou Provincial Department of Education, China (No. KY [2021]014 and KY[2021]107).References
- J. H. Kroll and J. H. Seinfeld, Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS
.
- P. J. Ziemann and R. Atkinson, Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 2012, 41, 6582–6605 RSC
- J. Wang, J. Ye, Q. Zhang, J. Zhao, Y. Wu, J. Li, D. Liu, W. Li, Y. Zhang, C. Wu, C. Xie, Y. Qin, Y. Lei, X. Huang, J. Guo, P. Liu, P. Fu, Y. Li, H. C. Lee, H. Choi, J. Zhang, H. Liao, M. Chen, Y. Sun, X. Ge, S. T. Martin and D. J. Jacob, Aqueous production of secondary organic aerosol from fossil-fuel emissions in winter Beijing haze, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2022179118 CrossRef CAS PubMed
- S. M. Charan, Y. Huang and J. H. Seinfeld, Computational Simulation of Secondary Organic Aerosol Formation in Laboratory Chambers, Chem. Rev., 2019, 119, 11912–11944 CrossRef CAS PubMed
- A. Akherati, Y. He, M. M. Coggon, A. R. Koss, A. L. Hodshire, K. Sekimoto, C. Warneke, J. de Gouw, L. Yee, J. H. Seinfeld, T. B. Onasch, S. C. Herndon, W. B. Knighton, C. D. Cappa, M. J. Kleeman, C. Y. Lim, J. H. Kroll, J. R. Pierce and S. H. Jathar, Oxygenated Aromatic Compounds are Important Precursors of Secondary Organic Aerosol in Biomass-Burning Emissions, Environ. Sci. Technol., 2020, 54, 8568–8579 CrossRef CAS PubMed
- Y. Zhang, Y. Chen, Z. Lei, N. E. Olson, M. Riva, A. R. Koss, Z. Zhang, A. Gold, J. T. Jayne, D. R. Worsnop, T. B. Onasch, J. H. Kroll, B. J. Turpin, A. P. Ault and J. D. Surratt, Joint Impacts of Acidity and Viscosity on the Formation of Secondary Organic Aerosol from Isoprene Epoxydiols (IEPOX) in Phase Separated Particles, ACS Earth Space Chem., 2019, 3, 2646–2658 CrossRef CAS
- P. H. Chowdhury, Q. He, R. Carmieli, C. Li, Y. Rudich and M. Pardo, Connecting the Oxidative Potential of Secondary Organic Aerosols with Reactive Oxygen Species in Exposed Lung Cells, Environ. Sci. Technol., 2019, 53, 13949–13958 CrossRef CAS PubMed
- I. M. Al-Naiema, J. H. Offenberg, C. J. Madler, M. Lewandowski, J. Kettler, T. Fang and E. A. Stone, Secondary organic aerosols from aromatic hydrocarbons and their contribution to fine particulate matter in Atlanta, Georgia, Atmos. Environ., 2020, 223, 117227 CrossRef CAS PubMed
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prévôt, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS
- M. Mahilang, M. K. Deb and S. Pervez, Biogenic secondary organic aerosols: A review on formation mechanism, analytical challenges and environmental impacts, Chemosphere, 2021, 262, 127771 CrossRef CAS PubMed
- G. McFiggans, T. F. Mentel, J. Wildt, I. Pullinen, S. Kang, E. Kleist, S. Schmitt, M. Springer, R. Tillmann, C. Wu, D. Zhao, M. Hallquist, C. Faxon, M. Le Breton, Å. M. Hallquist, D. Simpson, R. Bergström, M. E. Jenkin, M. Ehn, J. A. Thornton, M. R. Alfarra, T. J. Bannan, C. J. Percival, M. Priestley, D. Topping and A. Kiendler-Scharr, Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 2019, 565, 587–593 CrossRef CAS PubMed
- J. Wei, T. Fang, C. Wong, P. S. J. Lakey, S. A. Nizkorodov and M. Shiraiwa, Superoxide Formation from Aqueous Reactions of Biogenic Secondary Organic Aerosols, Environ. Sci. Technol., 2021, 55, 260–270 CrossRef CAS PubMed
- H. Tong, P. S. J. Lakey, A. M. Arangio, J. Socorro, F. Shen, K. Lucas, W. H. Brune, U. Pöschl and M. Shiraiwa, Reactive Oxygen Species Formed by Secondary Organic Aerosols in Water and Surrogate Lung Fluid, Environ. Sci. Technol., 2018, 52, 11642–11651 CrossRef CAS PubMed
- J. H. Slade, A. P. Ault, A. T. Bui, J. C. Ditto, Z. Lei, A. L. Bondy, N. E. Olson, R. D. Cook, S. J. Desrochers, R. M. Harvey, M. H. Erickson, H. W. Wallace, S. L. Alvarez, J. H. Flynn, B. E. Boor, G. A. Petrucci, D. R. Gentner, R. J. Griffin and P. B. Shepson, Bouncier Particles at Night: Biogenic Secondary Organic Aerosol Chemistry and Sulfate Drive Diel Variations in the Aerosol Phase in a Mixed Forest, Environ. Sci. Technol., 2019, 53, 4977–4987 CrossRef CAS PubMed
- Y. Li and M. Shiraiwa, Timescales of secondary organic aerosols to reach equilibrium at various temperatures and relative humidities, Atmos. Chem. Phys., 2019, 19, 5959–5971 CrossRef CAS
- X. Qi, S. Zhu, C. Zhu, J. Hu, S. Lou, L. Xu, J. Dong and P. Cheng, Smog chamber study of the effects of NOx and NH3 on the formation of secondary organic aerosols and optical properties from photo-oxidation of toluene, Sci. Total Environ., 2020, 727, 138632 CrossRef CAS PubMed
- S.-H. Lee, H. Gordon, H. Yu, K. Lehtipalo, R. Haley, Y. Li and R. Zhang, New Particle Formation in the Atmosphere: From Molecular Clusters to Global Climate, J. Geophys. Res.: Atmos., 2019, 124, 7098–7146 CrossRef CAS
- M. Shrivastava, C. D. Cappa, J. Fan, A. H. Goldstein, A. B. Guenther, J. L. Jimenez, C. Kuang, A. Laskin, S. T. Martin, N. L. Ng, T. Petaja, J. R. Pierce, P. J. Rasch, P. Roldin, J. H. Seinfeld, J. Shilling, J. N. Smith, J. A. Thornton, R. Volkamer, J. Wang, D. R. Worsnop, R. A. Zaveri, A. Zelenyuk and Q. Zhang, Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 2017, 55, 509–559 CrossRef
- J. Li, Z. Liu, W. Gao, G. Tang, B. Hu, Z. Ma and Y. Wang, Insight into the formation and evolution of secondary organic aerosol in the megacity of Beijing, China, Atmos. Environ., 2020, 220, 117070 CrossRef CAS
- M. Kulmala, J. Kontkanen, H. Junninen, K. Lehtipalo, H. E. Manninen, T. Nieminen, T. Petäjä, M. Sipilä, S. Schobesberger, P. Rantala, A. Franchin, T. Jokinen, E. Järvinen, M. Äijälä, J. Kangasluoma, J. Hakala, P. P. Aalto, P. Paasonen, J. Mikkilä, J. Vanhanen, J. Aalto, H. Hakola, U. Makkonen, T. Ruuskanen, R. L. Mauldin, J. Duplissy, H. Vehkamäki, J. Bäck, A. Kortelainen, I. Riipinen, T. Kurtén, M. V. Johnston, J. N. Smith, M. Ehn, T. F. Mentel, K. E. J. Lehtinen, A. Laaksonen, V.-M. Kerminen and D. R. Worsnop, Direct Observations of Atmospheric Aerosol Nucleation, Science, 2013, 339, 943–946 CrossRef CAS PubMed
- M. Kulmala, I. Riipinen, M. Sipilä, H. E. Manninen, T. Petäjä, H. Junninen, M. D. Maso, G. Mordas, A. Mirme, M. Vana, A. Hirsikko, L. Laakso, R. M. Harrison, I. Hanson, C. Leung, K. E. J. Lehtinen and V.-M. Kerminen, Toward Direct Measurement of Atmospheric Nucleation, Science, 2007, 318, 89–92 CrossRef CAS PubMed
- N. Myllys, J. Elm, R. Halonen, T. Kurtén and H. Vehkamäki, Coupled Cluster Evaluation of the Stability of Atmospheric Acid–Base Clusters with up to 10 Molecules, J. Phys. Chem. A, 2016, 120, 621–630 CrossRef CAS PubMed
- F. Bianchi, A. P. Praplan, N. Sarnela, J. Dommen, A. Kürten, I. K. Ortega, S. Schobesberger, H. Junninen, M. Simon, J. Tröstl, T. Jokinen, M. Sipilä, A. Adamov, A. Amorim, J. Almeida, M. Breitenlechner, J. Duplissy, S. Ehrhart, R. C. Flagan, A. Franchin, J. Hakala, A. Hansel, M. Heinritzi, J. Kangasluoma, H. Keskinen, J. Kim, J. Kirkby, A. Laaksonen, M. J. Lawler, K. Lehtipalo, M. Leiminger, V. Makhmutov, S. Mathot, A. Onnela, T. Petäjä, F. Riccobono, M. P. Rissanen, L. Rondo, A. Tomé, A. Virtanen, Y. Viisanen, C. Williamson, D. Wimmer, P. M. Winkler, P. Ye, J. Curtius, M. Kulmala, D. R. Worsnop, N. M. Donahue and U. Baltensperger, Insight into Acid–Base Nucleation Experiments by Comparison of the Chemical Composition of Positive, Negative, and Neutral Clusters, Environ. Sci. Technol., 2014, 48, 13675–13684 CrossRef CAS PubMed
- L. Yao, O. Garmash, F. Bianchi, J. Zheng, C. Yan, J. Kontkanen, H. Junninen, S. B. Mazon, M. Ehn, P. Paasonen, M. Sipilä, M. Wang, X. Wang, S. Xiao, H. Chen, Y. Lu, B. Zhang, D. Wang, Q. Fu, F. Geng, L. Li, H. Wang, L. Qiao, X. Yang, J. Chen, V.-M. Kerminen, T. Petäjä, D. R. Worsnop, M. Kulmala and L. Wang, Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity, Science, 2018, 361, 278–281 CrossRef CAS PubMed
- J. Almeida, S. Schobesberger, A. Kürten, I. K. Ortega, O. Kupiainen-Määttä, A. P. Praplan, A. Adamov, A. Amorim, F. Bianchi, M. Breitenlechner, A. David, J. Dommen, N. M. Donahue, A. Downard, E. Dunne, J. Duplissy, S. Ehrhart, R. C. Flagan, A. Franchin, R. Guida, J. Hakala, A. Hansel, M. Heinritzi, H. Henschel, T. Jokinen, H. Junninen, M. Kajos, J. Kangasluoma, H. Keskinen, A. Kupc, T. Kurtén, A. N. Kvashin, A. Laaksonen, K. Lehtipalo, M. Leiminger, J. Leppä, V. Loukonen, V. Makhmutov, S. Mathot, M. J. McGrath, T. Nieminen, T. Olenius, A. Onnela, T. Petäjä, F. Riccobono, I. Riipinen, M. Rissanen, L. Rondo, T. Ruuskanen, F. D. Santos, N. Sarnela, S. Schallhart, R. Schnitzhofer, J. H. Seinfeld, M. Simon, M. Sipilä, Y. Stozhkov, F. Stratmann, A. Tomé, J. Tröstl, G. Tsagkogeorgas, P. Vaattovaara, Y. Viisanen, A. Virtanen, A. Vrtala, P. E. Wagner, E. Weingartner, H. Wex, C. Williamson, D. Wimmer, P. Ye, T. Yli-Juuti, K. S. Carslaw, M. Kulmala, J. Curtius, U. Baltensperger, D. R. Worsnop, H. Vehkamäki and J. Kirkby, Molecular understanding of sulphuric acid–amine particle nucleation in the atmosphere, Nature, 2013, 502, 359–363 CrossRef CAS PubMed
- J. Elm, Elucidating the Limiting Steps in Sulfuric Acid–Base New Particle Formation, J. Phys. Chem. A, 2017, 121, 8288–8295 CrossRef CAS PubMed
- G. Duporté, M. Riva, J. Parshintsev, E. Heikkinen, L. M. F. Barreira, N. Myllys, L. Heikkinen, K. Hartonen, M. Kulmala, M. Ehn and M.-L. Riekkola, Chemical Characterization of Gas- and Particle-Phase Products from the Ozonolysis of α-Pinene in the Presence of Dimethylamine, Environ. Sci. Technol., 2017, 51, 5602–5610 CrossRef PubMed
- A. Kürten, T. Jokinen, M. Simon, M. Sipilä, N. Sarnela, H. Junninen, A. Adamov, J. Almeida, A. Amorim, F. Bianchi, M. Breitenlechner, J. Dommen, N. M. Donahue, J. Duplissy, S. Ehrhart, R. C. Flagan, A. Franchin, J. Hakala, A. Hansel, M. Heinritzi, M. Hutterli, J. Kangasluoma, J. Kirkby, A. Laaksonen, K. Lehtipalo, M. Leiminger, V. Makhmutov, S. Mathot, A. Onnela, T. Petäjä, A. P. Praplan, F. Riccobono, M. P. Rissanen, L. Rondo, S. Schobesberger, J. H. Seinfeld, G. Steiner, A. Tomé, J. Tröstl, P. M. Winkler, C. Williamson, D. Wimmer, P. Ye, U. Baltensperger, K. S. Carslaw, M. Kulmala, D. R. Worsnop and J. Curtius, Neutral molecular cluster formation of sulfuric acid–dimethylamine observed in real time under atmospheric conditions, Proc. Natl. Acad. Sci., 2014, 111, 15019–15024 CrossRef PubMed
- J. Elm, P. Norman and K. V. Mikkelsen, Rayleigh light scattering properties of atmospheric molecular clusters consisting of sulfuric acid and bases, Phys. Chem. Chem. Phys., 2015, 17, 15701–15709 RSC
- M. Jang and R. M. Kamens, Atmospheric Secondary Aerosol Formation by Heterogeneous Reactions of Aldehydes in the Presence of a Sulfuric Acid Aerosol Catalyst, Environ. Sci. Technol., 2001, 35, 4758–4766 CrossRef CAS PubMed
- R. Zhang, A. Khalizov, L. Wang, M. Hu and W. Xu, Nucleation and Growth of Nanoparticles in the Atmosphere, Chem. Rev., 2012, 112, 1957–2011 CrossRef CAS PubMed
- S. M. Corrêa, E. M. Martins and G. Arbilla, Formaldehyde and acetaldehyde in a high traffic street of Rio de Janeiro, Brazil, Atmos. Environ., 2003, 37, 23–29 CrossRef
- J. F. Doussin, B. Picquet-Varrault, R. Durand-Jolibois, H. Loirat and P. Carlier, A visible and FTIR spectrometric study of the nighttime chemistry of acetaldehyde and PAN under simulated atmospheric conditions, J. Photochem. Photobiol., A, 2003, 157, 283–293 CrossRef CAS
- X.-F. Tan, B. Long, D.-S. Ren, W.-J. Zhang, Z.-W. Long and E. Mitchell, Atmospheric chemistry of CH3CHO: the hydrolysis of CH3CHO catalyzed by H2SO4, Phys. Chem. Chem. Phys., 2018, 20, 7701–7709 RSC
- Y. Cheng, S. C. Lee, Y. Huang, K. F. Ho, S. S. H. Ho, P. S. Yau, P. K. K. Louie and R. J. Zhang, Diurnal and seasonal trends of carbonyl compounds in roadside, urban, and suburban environment of Hong Kong, Atmos. Environ., 2014, 89, 43–51 CrossRef CAS
- Z.-G. Dong, F. Xu and B. Long, The energetics and kinetics of the CH3CHO + (CH3)2NH/CH3NH2 reactions catalyzed by a single water molecule in the atmosphere, Comput. Theor. Chem., 2018, 1140, 7–13 CrossRef CAS
- D. B. Millet, A. Guenther, D. A. Siegel, N. B. Nelson, H. B. Singh, J. A. de Gouw, C. Warneke, J. Williams, G. Eerdekens, V. Sinha, T. Karl, F. Flocke, E. Apel, D. D. Riemer, P. I. Palmer and M. Barkley, Global atmospheric budget of acetaldehyde: 3-D model analysis and constraints from in-situ and satellite observations, Atmos. Chem. Phys., 2010, 10, 3405–3425 CrossRef CAS
- K. Yamada, R. Hattori, Y. Ito, H. Shibata and N. Yoshida, Carbon isotopic signatures of methanol and acetaldehyde emitted from biomass burning source, Geophys. Res. Lett., 2009, 36, L18807 CrossRef
- Z. Li, A. N. Schwier, N. Sareen and V. F. McNeill, Reactive processing of formaldehyde and acetaldehyde in aqueous aerosol mimics: surface tension depression and secondary organic products, Atmos. Chem. Phys., 2011, 11, 11617–11629 CrossRef CAS
- H. A. Rypkema, A. Sinha and J. S. Francisco, Carboxylic Acid Catalyzed Hydration of Acetaldehyde, J. Phys. Chem. A, 2015, 119, 4581–4588 CrossRef CAS PubMed
- S. Sarkar, B. K. Oram and B. Bandyopadhyay, Ammonolysis as an important loss process of acetaldehyde in the troposphere: energetics and kinetics of water and formic acid catalyzed reactions, Phys. Chem. Chem. Phys., 2019, 21, 16170–16179 RSC
- L. Benning and A. Wahner, Measurements of Atmospheric Formaldehyde (HCHO) and Acetaldehyde (CH3CHO) during POPCORN 1994 Using 2.4-DNPH Coated Silica Cartridges, J. Atmos. Chem., 1998, 31, 105–117 CrossRef CAS
- J. D. Surratt, J. H. Kroll, T. E. Kleindienst, E. O. Edney, M. Claeys, A. Sorooshian, N. L. Ng, J. H. Offenberg, M. Lewandowski, M. Jaoui, R. C. Flagan and J. H. Seinfeld, Evidence for Organosulfates in Secondary Organic Aerosol, Environ. Sci. Technol., 2007, 41, 517–527 CrossRef CAS PubMed
- J. Liggio and S.-M. Li, Organosulfate formation during the uptake of pinonaldehyde on acidic sulfate aerosols, Geophys. Res. Lett., 2006, 33, L13808 CrossRef
- M. Brüggemann, R. Xu, A. Tilgner, K. C. Kwong, A. Mutzel, H. Y. Poon, T. Otto, T. Schaefer, L. Poulain, M. N. Chan and H. Herrmann, Organosulfates in Ambient Aerosol: State of Knowledge and Future Research Directions on Formation, Abundance, Fate, and Importance, Environ. Sci. Technol., 2020, 54, 3767–3782 CrossRef PubMed
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed
- B. J. Lynch, Y. Zhao and D. G. Truhlar, Effectiveness of Diffuse Basis Functions for Calculating Relative Energies by Density Functional Theory, J. Phys. Chem. A, 2003, 107, 1384–1388 CrossRef CAS
- J. Elm, M. Bilde and K. V. Mikkelsen, Assessment of Density Functional Theory in Predicting Structures and Free Energies of Reaction of Atmospheric Prenucleation Clusters, J. Chem. Theory Comput., 2012, 8, 2071–2077 CrossRef CAS PubMed
- B. Long, X.-F. Tan, C.-R. Chang, W.-X. Zhao, Z.-W. Long, D.-S. Ren and W.-J. Zhang, Theoretical Studies on Gas-Phase Reactions of Sulfuric Acid Catalyzed Hydrolysis of Formaldehyde and Formaldehyde with Sulfuric Acid and H2SO4···H2O Complex, J. Phys. Chem. A, 2013, 117, 5106–5116 CrossRef CAS PubMed
- F.-Y. Liu, X.-F. Tan, Z.-W. Long, B. Long and W.-J. Zhang, New insights in atmospheric acid-catalyzed gas phase hydrolysis of formaldehyde: a theoretical study, RSC Adv., 2015, 5, 32941–32949 RSC
- L. Zhang and B. Long, Hydrolysis of Formyl Fluoride Catalyzed by Sulfuric Acid and Formic Acid in the Atmosphere, ACS Omega, 2019, 4, 18996–19004 CrossRef CAS PubMed
- Z.-G. Dong, F. Xu, E. Mitchell and B. Long, Trifluoroacetaldehyde aminolysis catalyzed by a single water molecule: An important sink pathway for trifluoroacetaldehyde and a potential pathway for secondary organic aerosol growth, Atmos. Environ., 2021, 249, 118242 CrossRef CAS
- J.-Y. Liu, Z.-W. Long, E. Mitchell and B. Long, New Mechanistic Pathways for the Reactions of Formaldehyde with Formic Acid Catalyzed by Sulfuric Acid and Formaldehyde with Sulfuric Acid Catalyzed by Formic Acid: Formation of Potential Secondary Organic Aerosol Precursors, ACS Earth Space Chem., 2021, 5, 1363–1372 CrossRef CAS
- R. Seeger and J. A. Pople, Self-consistent molecular orbital methods. XVIII. Constraints and stability in Hartree–Fock theory, J. Chem. Phys., 1977, 66, 3045–3050 CrossRef CAS
- R. Bauernschmitt and R. Ahlrichs, Stability analysis for solutions of the closed shell Kohn–Sham equation, J. Chem. Phys., 1996, 104, 9047–9052 CrossRef CAS
- K. Fukui, The path of chemical reactions - the IRC approach, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS
- C. Gonzalez and H. B. Schlegel, Reaction path following in mass-weighted internal coordinates, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS
- B. Long, J. L. Bao and D. G. Truhlar, Kinetics of the Strongly Correlated CH3O + O2 Reaction: The Importance of Quadruple Excitations in Atmospheric and Combustion Chemistry, J. Am. Chem. Soc., 2019, 141, 611–617 CrossRef CAS PubMed
- Y. Xia, B. Long, S. Lin, C. Teng, J. L. Bao and D. G. Truhlar, Large Pressure Effects Caused by Internal Rotation in the s-cis-syn-Acrolein Stabilized Criegee Intermediate at Tropospheric Temperature and Pressure, J. Am. Chem. Soc., 2022, 144, 4828–4838 CrossRef CAS PubMed
- J. M. L. Martin and M. K. Kesharwani, Assessment of CCSD(T)-F12 Approximations and Basis Sets for Harmonic Vibrational Frequencies, J. Chem. Theory Comput., 2014, 10, 2085–2090 CrossRef CAS PubMed
- G. Knizia, T. B. Adler and H.-J. Werner, Simplified CCSD(T)-F12 methods: Theory and benchmarks, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed
- E. Papajak and D. G. Truhlar, Convergent Partially Augmented Basis Sets for Post-Hartree-Fock Calculations of Molecular Properties and Reaction Barrier Heights, J. Chem. Theory Comput., 2011, 7, 10–18 CrossRef CAS PubMed
- T. J. Lee and P. R. Taylor, A diagnostic for determining the quality of single-reference electron correlation methods, Int. J. Quantum Chem., 1989, 36, 199–207 CrossRef
- H. Eyring, The Activated Complex in Chemical Reactions, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS
- M. G. Evans and M. Polanyi, Some applications of the transition state method to the calculation of reaction velocities, especially in solution, Trans. Faraday Soc., 1935, 31, 875–894 RSC
- D. G. Truhlar, B. C. Garrett and S. J. Klippenstein, Current Status of Transition-State Theory, J. Phys. Chem., 1996, 100, 12771–12800 CrossRef CAS
- C. Eckart, The Penetration of a Potential Barrier by Electrons, Phys. Rev., 1930, 35, 1303–1309 CrossRef CAS
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
-
H. J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, S. J. Bennie, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, S. J. R. Lee, Y. Liu, A. W. Lloyd, Q. Ma, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, T. F. Miller III, M. E. Mura, A. Nicklaß, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and M. Welborn, MOLPRO 2019.2, a Package of Ab Initio Programs; Cardiff, U.K. 2018 Search PubMed
- W. Duncan, R. Bell and T. Truong, TheRate: Program for ab initio direct dynamics calculations of thermal and vibrational-state-selected rate constants, J. Comput. Chem., 1998, 19, 1039–1052 CrossRef CAS
- F. T. Smith, Three-body collision rates in atomic recombination reactions, Discuss. Faraday Soc., 1962, 33, 183–188 RSC
- R. T Pack, R. B. Walker and B. K. Kendrick, Three-body collision contributions to recombination and collision-induced dissociation. I. Cross sections, J. Chem. Phys., 1998, 109, 6701–6713 CrossRef
- P. A. Whitlock, J. T. Muckerman and R. E. Roberts, Classical mechanics of recombination via the resonance complex mechanism: H + H + M → H2 + M for M = H, H2, He, and Ar, J. Chem. Phys., 1974, 60, 3658–3673 CrossRef CAS
- J. Pérez-Ríos, S. Ragole, J. Wang and C. H. Greene, Comparison of classical and quantal calculations of helium three-body recombination, J. Chem. Phys., 2014, 140, 044307 CrossRef PubMed
- J. Elm, M. Bilde and K. V. Mikkelsen, Influence of Nucleation Precursors on the Reaction Kinetics of Methanol with the OH Radical, J. Phys. Chem. A, 2013, 117, 6695–6701 CrossRef CAS PubMed
- P. Sebastianelli, P. M. Cometto and R. G. Pereyra, Systematic Characterization of Gas Phase Binary Pre-Nucleation Complexes Containing H2SO4 + X, [ X = NH3, (CH3)NH2, (CH3)2NH, (CH3)3N, H2O, (CH3)OH, (CH3)2O, HF, CH3F, PH3, (CH3)PH2, (CH3)2PH, (CH3)3P, H2S, (CH3)SH, (CH3)2S, HCl, (CH3)Cl)]. A Computational Study, J. Phys. Chem. A, 2018, 122, 2116–2128 CrossRef CAS PubMed
- H. R. Leverentz, J. I. Siepmann, D. G. Truhlar, V. Loukonen and H. Vehkamäki, Energetics of Atmospherically Implicated Clusters Made of Sulfuric Acid, Ammonia and Dimethyl Amine, J. Phys. Chem. A, 2013, 117, 3819–3825 CrossRef CAS PubMed
- Z. B. Wang, M. Hu, D. Mogensen, D. L. Yue, J. Zheng, R. Y. Zhang, Y. Liu, B. Yuan, X. Li, M. Shao, L. Zhou, Z. J. Wu, A. Wiedensohler and M. Boy, The simulations of sulfuric acid concentration and new particle formation in an urban atmosphere in China, Atmos. Chem. Phys., 2013, 13, 11157–11167 CrossRef CAS
- X. Ge, A. Wexler and S. Clegg, Atmospheric amines–Part I. A review, Atmos. Environ., 2011, 45, 524–546 CrossRef CAS
- J. Kirkby, J. Curtius, J. Almeida, E. Dunne, J. Duplissy, S. Ehrhart, A. Franchin, S. Gagné, L. Ickes, A. Kürten, A. Kupc, A. Metzger, F. Riccobono, L. Rondo, S. Schobesberger, G. Tsagkogeorgas, D. Wimmer, A. Amorim, F. Bianchi, M. Breitenlechner, A. David, J. Dommen, A. Downard, M. Ehn, R. C. Flagan, S. Haider, A. Hansel, D. Hauser, W. Jud, H. Junninen, F. Kreissl, A. Kvashin, A. Laaksonen, K. Lehtipalo, J. Lima, E. R. Lovejoy, V. Makhmutov, S. Mathot, J. Mikkilä, P. Minginette, S. Mogo, T. Nieminen, A. Onnela, P. Pereira, T. Petäjä, R. Schnitzhofer, J. H. Seinfeld, M. Sipilä, Y. Stozhkov, F. Stratmann, A. Tomé, J. Vanhanen, Y. Viisanen, A. Vrtala, P. E. Wagner, H. Walther, E. Weingartner, H. Wex, P. M. Winkler, K. S. Carslaw, D. R. Worsnop, U. Baltensperger and M. Kulmala, Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation, Nature, 2011, 476, 429–433 CrossRef CAS PubMed
- X.-F. Tan, L. Zhang and B. Long, New mechanistic pathways for the formation of organosulfates catalyzed by ammonia and carbinolamine formation catalyzed by sulfuric acid in the atmosphere, Phys. Chem. Chem. Phys., 2020, 22, 8800–8807 RSC
- F. Xu, X.-F. Tan, Z.-G. Dong, D.-S. Ren and B. Long, Hydrolysis of ketene catalysed by nitric acid and water in the atmosphere, Environ. Chem., 2020, 17, 457–467 CrossRef CAS
- N. Lin, Z.-W. Long and B. Long, Important Routes for Methanediol Formation by Formaldehyde Hydrolysis Catalyzed by Iodic Acid and for the Contribution to an Iodic Acid Sink by the Reaction of Formaldehyde with Iodic Acid Catalyzed by Atmospheric Water, ACS Earth Space Chem., 2022, 6, 1890–1898 CrossRef CAS
- G.-B. Li, S.-H. Cai and B. Long, New reactions for formation of organic nitrate in the atmosphere, ACS Omega, 2022, 7, 39671–39679 CrossRef CAS PubMed
- B. Long, Y. Xia and D. G. Truhlar, Quantitative Kinetics of HO2 Reactions with Aldehydes in the Atmosphere: High-Order Dynamic Correlation, Anharmonicity, and Falloff Effects Are All Important, J. Am. Chem. Soc., 2022, 144, 19910–19920 CrossRef CAS PubMed
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi, J. Troe and I. Subcommittee, Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 2006, 6, 3625–4055 CrossRef CAS
- F. L. Eisele and D. J. Tanner, Measurement of the gas phase concentration of H2SO4 and methane sulfonic acid and estimates of H2SO4 production and loss in the atmosphere, J. Geophys. Res.: Atmos., 1993, 98, 9001–9010 CrossRef CAS
- T. Petäjä, I. R. L. Mauldin, E. Kosciuch, J. McGrath, T. Nieminen, P. Paasonen, M. Boy, A. Adamov, T. Kotiaho and M. Kulmala, Sulfuric acid and OH concentrations in a boreal forest site, Atmos. Chem. Phys., 2009, 9, 7435–7448 CrossRef
- S. Mikkonen, S. Romakkaniemi, J. N. Smith, H. Korhonen, T. Petäjä, C. Plass-Duelmer, M. Boy, P. H. McMurry, K. E. J. Lehtinen, J. Joutsensaari, A. Hamed, R. L. Mauldin Iii, W. Birmili, G. Spindler, F. Arnold, M. Kulmala and A. Laaksonen, A statistical proxy for sulphuric acid concentration, Atmos. Chem. Phys., 2011, 11, 11319–11334 CrossRef CAS
-
G. P. Brasseur and S. Solomon, Aeronomy of the Middle Atmosphere: Chemistry and Physics of the Stratosphere and Mesosphere, Springer Netherlands, 2005 Search PubMed
- M. A. H. Khan, M. J. Ashfold, G. Nickless, D. Martin, L. A. Watson, P. D. Hamer, R. P. Wayne, C. E. Canosa-Mas and D. E. Shallcross, Night-time NO3 and OH radical concentrations in the United Kingdom inferred from hydrocarbon measurements, Atmos. Sci. Lett., 2008, 9, 140–146 CrossRef
- M. Torrent-Sucarrat, J. S. Francisco and J. M. Anglada, Sulfuric Acid as Autocatalyst in the Formation of Sulfuric Acid, J. Am. Chem. Soc., 2012, 134, 20632–20644 CrossRef CAS PubMed
- M. M. Galloway, M. H. Powelson, N. Sedehi, S. E. Wood, K. D. Millage, J. A. Kononenko, A. D. Rynaski and D. O. De Haan, Secondary Organic Aerosol Formation during Evaporation of Droplets Containing Atmospheric Aldehydes, Amines, and Ammonium Sulfate, Environ. Sci. Technol., 2014, 48, 14417–14425 CrossRef CAS PubMed
- A. A. Rodriguez, A. de Loera, M. H. Powelson, M. M. Galloway and D. O. De Haan, Formaldehyde and Acetaldehyde Increase Aqueous-Phase Production of Imidazoles in Methylglyoxal/Amine Mixtures: Quantifying a Secondary Organic Aerosol Formation Mechanism, Environ. Sci. Technol. Lett., 2017, 4, 234–239 CrossRef CAS
- J. M. Anglada, S. Olivella and A. Solé, Hydrogen Transfer between Sulfuric Acid and Hydroxyl Radical in the Gas Phase: Competition among Hydrogen Atom Transfer, Proton-Coupled Electron-Transfer, and Double Proton Transfer, J. Phys. Chem. A, 2006, 110, 1982–1990 CrossRef CAS PubMed
- M. Dal Maso, M. Kulmala, K. E. J. Lehtinen, J. M. Mäkelä, P. Aalto and C. D. O'Dowd, Condensation and coagulation sinks and formation of nucleation mode particles in coastal and boreal forest boundary layers, J. Geophys. Res., 2002, 107, 8097–8107 CrossRef
- J. Slemr, W. Junkermann and A. Volz-Thomas, Temporal variations in formaldehyde, acetaldehyde and acetone and budget of formaldehyde at a rural site in Southern Germany, Atmos. Environ., 1996, 30, 3667–3676 CrossRef CAS
- Y.-N. Lee, X. Zhou and K. Hallock, Atmospheric carbonyl compounds at a rural southeastern United States site, J. Geophys. Res.: Atmos., 1995, 100, 25933–25944 CrossRef
- S. M. Corrêa, E. M. Martins and G. Arbilla, Formaldehyde and acetaldehyde in a high traffic street of Rio de Janeiro, Brazil, Atmos. Environ., 2003, 37, 23–29 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: The T1 diagnostic values for all the species as listed in Table S1; the calculated relative energies with zero-point vibrational correction of the dimer complexes as listed in Table S2; the calculated equilibrium constants and the concentration of the dimer complexes when the [(CH3)2NH] = 3.2 × 109, [H2SO4] = 4 × 108, [CH3CHO] = 1.12 × 1012 molecule per cm3 for the H2SO4 + (CH3)2NH + CH3CHO in the temperature range of 200–298 K as listed in Table S3; the rate constants of the CH3CHO + H2SO4 reaction as listed in Table S4; the rate ratio of each reaction as listed in Table S5; the rate ratio of the v1/vCH3CHO+OH at different OH concentrations in the temperature range of 200–298 K ([(CH3)2NH] = 3.2 × 109, [H2SO4] = 106, 107, 4.0 × 108 and 5.1 × 109 molecule per cm3 respectively) as listed in Tables S6–S9; the rate ratio of the v1/vH2SO4+OH at different OH concentrations in the temperature range of 200–298 K([(CH3)2NH] = 3.2 × 109, [CH3CHO] = 1.12 × 1012 molecule per cm3 respectively) as listed in Table S10; calculated atmospheric lifetime of H2SO4 ([(CH3)2NH] = 3.2 × 109, [CH3CHO] = 2.46 × 109 and 1.12 × 1012 molecule per cm3 respectively) as listed in Tables S11 and S12; molecular coordinates and molecular frequencies as listed in Table S13; Gibbs free energy reaction profile of CH3CHO + H2SO4 + (CH3)2NH reaction at 298 K as listed in Fig. S1; relative energies with zero point correction for the CH3CHO + H2SO4 reaction at 0 K as listed in Fig. S2; relative energies with zero point correction for CH3CHO + H2SO4 + (CH3)2NH reaction as listed in Fig. S3; selected geometrical parameters as shown in Fig. S4; intrinsic reaction coordinate results of TS1A, TS1B, TS1C, TS1D, TS1E and TS1F as listed in Fig. S5–S10. See DOI: https://doi.org/10.1039/d2ea00159d |
| This journal is © The Royal Society of Chemistry 2023 |