
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of solvent on the electronic structure and the photochemistry of nitrophenols†
Avery B.
Dalton
 a,
Scott M.
Le
a,
Natalia V.
Karimova
a,
R. Benny
Gerber
ab and
Sergey A.
Nizkorodov
*a
a,
Scott M.
Le
a,
Natalia V.
Karimova
a,
R. Benny
Gerber
ab and
Sergey A.
Nizkorodov
*a
aDepartment of Chemistry, University of California, Irvine, Irvine, CA 92697, USA. E-mail: nizkorod@uci.edu
bInstitute of Chemistry, Fritz Haber Research Center, Hebrew University of Jerusalem, Jerusalem 91904, Israel
First published on 4th January 2023
Abstract
Previous studies have suggested that the photochemistry of nitroaromatics in organic solvents can vary significantly from the photochemistry in aqueous solutions. This work compares the photodegradation of 2-nitrophenol (2NP), 4-nitrophenol (4NP), 2,4-dinitrophenol (24DNP), and 2,4,6-trinitrophenol (246TNP) in 2-propanol and water to better understand the photochemical loss of nitrophenols in atmospheric organic particles and aqueous droplets. Polychromatic quantum yields were determined by monitoring the loss of absorbance of each nitrophenol with UV/vis spectroscopy in the presence of an acid (undissociated nitrophenol) or base (nitrophenolate). There was no orderly variation between loss rates in the organic and aqueous phases: 2NP and 4NP had similar yields in the two solvents. 246TNP was an outlier in these results as it dissociated in both acidified 2-propanol and water due to its exceptionally strong acidity. A notable result is that only for 24DNP was a dramatically increased reactivity found in 2-propanol compared to that in water. Time-dependent density functional theory calculations were carried out to characterize the excited state energies and absorption spectra with a conductor-like polarizable continuum model or explicit solvation by a few solvent molecules. Explicit solvent calculations suggest the enhanced reactivity of 24DNP in 2-propanol is due to the strong interaction between a 2-propanol molecule and an –NO2 group in the excited state. For the other nitrophenols, the solvent effects on electronic structure were minimal. Overall, the observations in this work suggest that solvent effects on the electronic structure and condensed-phase photochemistry of nitrophenols are minimal, with the exception of 24DNP.
Environmental significanceNitrophenols are produced during fossil fuel combustion and biomass burning, and they are widespread in the atmospheric environment. They affect plants because of their phytotoxicity, and they affect climate because they are moderately strong absorbers of solar radiation. Previous research largely focused on photochemical reactions of nitrophenols in the gaseous phase and in the aqueous phase, representing fog and cloud water. This study examines the photochemistry of four different nitrophenols in the presence of an organic solvent – an environment that more closely mimics the conditions in atmospheric aerosol particles. One of the nitrophenols examined in this work, 2,4-dinitrophenol, exhibited dramatically increased reactivity in isopropanol compared to that in water, suggesting that it should have short photodegradation lifetimes in atmospheric organic aerosol particles. |
1 Introduction
Nitrophenols first garnered scientific interest upon the discovery that these molecules exhibit phytotoxic characteristics and could potentially lead to forest decline.1,2 This prompted questions about the fate of these molecules in the atmosphere, and the overall lifetimes and chemical mechanisms of their atmospheric degradation.3–12 Atmospheric sources of nitrophenols include both primary sources, such as automobile engines and biomass burning, and secondary sources, through reactions between phenols and NO2 or NO3 radicals.3,13–18 The type of atmospheric loss that the molecule undergoes (i.e., reaction with an atmospheric oxidant or a photochemical reaction), as well as the phase in which the molecule is encapsulated, has a strong effect on both the lifetime and the degradation products. While some processes change nitrophenols into phenols and catechols, the oxidation of these products in the presence of NOx leads back to the formation of nitrophenols.6,16,17,19Much of the previous study of nitrophenol photochemistry has revolved around the production of HONO, which is itself a contributor to the oxidative potential of the atmosphere.7,11,20 The chemistry of nitrophenols in the gas phase has been studied extensively – but considering the relatively low volatility of these species, evolution of these molecules in the condensed phases that are representative of atmospheric aerosol particles requires further investigation. While the release of HONO could have significant atmospheric implications, the importance of other photochemical pathways should not be overlooked. For instance, dimerization and functionalization have been shown to happen with these nitrophenol compounds.9,21,22 The products of these reactions are less volatile and would be more likely to partition into, or remain trapped in, atmospheric particles.22,23
The importance of the chemical environment in the fate of nitrophenols and other nitroaromatics has been exhibited through experiments in both aqueous and organic phases.9,19,20,24–26 Gas phase studies have indicated that the photochemistry of nitrophenols is comparable to the aging driven by hydroxyl or nitrate radicals, which is reported to have a lifetime of around 330 h for 2-nitrophenol (2NP).7,16 This resistance to gas-phase loss, in combination with the low volatility and high Henry constants of nitrophenols, grants more importance to condensed-phase processes. In the presence of organic compounds, for example, in organic aerosol particles, nitrophenols are thought to react through excited triplet states, abstracting a hydrogen atom from a neighboring solvent molecule.27–29 The product of the H-atom abstraction will then go on to react further with nearby molecules. The influence of the chemical complexity of atmospheric organic aerosols creates potential for numerous reactive partners and products which could have a wide variety of environmental impacts.
In the condensed phase, photochemistry will also be impacted by the ionization of the molecule through acid–base processes, which causes changes in molecular extinction.20 Barsotti et al. (2017) found that the anionic forms of a series of nitrophenols exhibited a greater yield toward photochemical loss of NO2, in the form of NO2− or HONO, in the aqueous phase, accounting for 10–30% of the total photochemical yield of these molecules.20 Importantly, this result also supports other photochemical studies of 4-nitrophenol (4NP) and 2,4-dinitrophenol (24DNP), in which multiple photodegradation pathways were observed.6,8
In laboratory experiments, photochemical degradation via hydrogen abstraction is generally slower in water than in an organic matrix, prompting the need for a better understanding of photoreactivity in the organic phase.9 In contrast, in the aqueous phase photochemistry of nitrophenols, the excited-state nitrophenol is expected to undergo a hydrolysis reaction with a neighboring water molecule, which is a major step in the production of HONO. These differing reactive pathways make predicting the reactivity of a nitrophenol in real atmospheric particles difficult.26 Further, nitrophenols have relatively poor solubility in water, potentially amplifying the effects of photochemical loss in the organic phase of atmospheric particles. This work aims to investigate the photochemistry of a series of nitrophenols in an organic solvent under simulated sunlight conditions, combining experimental results with ab initio calculations to provide insights into the relative reactivities of each nitrophenol.
2 Systems and methods
A. Experimental methods
The photochemistry experiments with 2-nitrophenol (2NP, TCI, 98%), 4-nitrophenol (4NP, Chem-Impex, 99.6%), 2,4-dinitrophenol (24DNP, Aldrich, 98%) and 2,4,6-trinitrophenol (246TNP, commonly referred to as picric acid, Sigma-Aldrich, ≥98%) were performed without further purification of the stock chemicals. The solvents used in these experiments included 2-propanol (or isopropanol, Fisher, HPLC grade) and water (Milli-Q Ultrapure). Although 2-propanol is a small molecule with only a single hydroxyl group, previous studies have shown that nitroaromatics have comparable rates of photodegradation in simple alcohols and in more complex secondary organic aerosol material.19 While the rate of photodegradation is suppressed by the high viscosity of the secondary organic aerosol matrix, this suppression is counteracted by the higher diversity of reactive functional groups in secondary organic aerosol compounds.To better understand the difference in photochemistry between undissociated nitrophenols and nitrophenolates, the experiments were carried out under acidic and basic conditions. To change the pH of samples, small amounts of 1 N hydrochloric acid (Fisher) and 1 N potassium hydroxide (Fisher) were used. In the case of 246TNP, which has a pKa of 0.4, an additional solution was made with 37% hydrochloric acid (Fisher) to reach [HCl] ≈ 5 M to record the absorption spectrum of the undissociated form.
Photochemistry experiments were carried out using radiation from a xenon arc lamp, which was filtered and then directed onto the sample via a liquid light guide. The optical filters included a 280–400 nm dichroic mirror, followed by Schott BG1 (UV bandpass) and WG295 (295 nm longpass) filters. The overall power of the radiation reaching the sample ranged from 28 to 30 mW. With this setup, the near-UV spectrum of the radiation reaching the sample extended to lower wavelength (down to 280 nm) than that of the ambient solar radiation (the lamp's spectral flux density is compared to that of solar radiation reaching the Earth's surface in Fig. S1†). Liquid phase photochemistry experiments were conducted directly inside a Shimadzu UV-vis spectrophotometer, irradiating the sample vertically through the square opening in the top of a standard 10 mm UV/vis cuvette (Starna Cells, 21-Q-10-MS). The cuvettes were filled to ∼80% of their volume, and the samples were not sealed but exposed to air permitting oxygen to dissolve in the solution. The oxygen concentration was not explicitly measured. The light guide was removed during each UV-vis scan to eliminate the interference scattering by the solution in the cuvette. In the case of 2NP, the sample was irradiated without the light guide since the degradation was much slower, subjecting the sample to ∼140 mW from the side of the cuvette.
Rate constants, k, for total photochemical degradation were obtained from fitting the loss in normalized absorbance to the fit shown in eqn (1).21
 | (1) |
This equation accounts for the formation a single absorbing product, though it is possible that multiple absorbing products could be formed. The absorption properties of the product are accounted for in the value C, which is calculated as the extinction by the product divided by the extinction by the starting compound, εNP/εproduct.
B. Models & computational methods
All quantum chemical calculations were carried out using the Q-Chem quantum chemistry package with the goal of giving context to the absorptivity and reactivity of the different nitrophenols.30 All geometry optimizations were performed at the B3LYP/6-31+G* level of theory. Time-dependent density functional theory (TDDFT)31 calculations were done to generate simulated excitation spectra using combinations of the B3LYP32 or PBE0 (ref. 33) hybrid-exchange functionals, which have both been shown to work well in other nitroaromatic systems.34,35 With the variation of chemical features within this series of nitrophenols, each of the 6-311+G* and 6-311++G**, aug-cc-pVDZ and aug-cc-pVTZ, and def2-TZVPD and def2-TZVPPD basis sets was evaluated. The Tamm–Dancoff approximation (TDA),36 which has been shown to produce good results in similar aromatic systems,37 was employed for all results reported in this work. Excitation energies were calculated in the gas-phase, as well as in water and 2-propanol using the conductor-like polarizable continuum model (C-PCM),38,39 employing the dielectric constants for each solvent at 25 °C. Simulated excitation spectra were produced by a convolution of Lorentzian functions calculated from the oscillator strengths at each excitation wavelength. A half-width at half-maximum of 20 nm was used in all the presented data. Natural transition orbitals (NTOs)40,41 were calculated for the triplet states in 24DNP to evaluate the differences in electronic character between 2-propanol and water.A combination of C-PCM and an explicit solvation model was required to obtain more accurate spectra for 24DNP in 2-propanol. Explicit solvation by one and three 2-propanol molecules was tested. The initial geometries were set up with the solvent molecules relatively far away to try to mimic the liquid-like solution, however geometry optimization inevitably brought these solvent molecules closer. The 2-propanol molecules were placed next to the functional groups to allow for hydrogen bonding to occur. In the case of solvation by one molecule, two scenarios were tested: one with the 2-propanol acting as a hydrogen-bond acceptor for the alcohol group and one with the 2-propanol acting as a hydrogen-bond donor to the para –NO2. No change in the excitation spectrum was observed in the latter case, and those results are not reported. For solvation by three 2-propanol molecules, the previous two models were combined, and the third 2-propanol molecule was oriented with the alcoholic hydrogen near the ortho –NO2 group. This same scheme was employed with water as the solvent.
3 Results and discussion
A. Experimental absorption spectra
The shape of the absorption spectra changed drastically for each of the nitrophenols in 2-propanol solutions with added KOH (Fig. 1). The absorption spectra of nitrophenols are known to depend on a variety of experimental conditions,42,43 especially on changes in pH.20 The pKa values of 7.23 (2NP), 7.14 (4NP), 4.09 (24DNP), and 0.42 (246TNP) make it possible for undissociated nitrophenols and nitrophenolates to co-exist in atmospheric particles. The nitrophenolates examined in this work all exhibit a significant redshift in absorption upon the addition of a base, as anticipated based on other studies.20,44–46 Even though the autoprotolysis constant for 2-propanol is several orders of magnitude lower than that for water,47 all of the nitrophenols studied in this work ionized significantly in 2-propanol with the added base. As shown in Fig. 1(d), it was difficult to prevent 246TNP from ionizing due to its strong acidity. The predominant species of 246TNP in 2-propanol solutions was the anion, even after acidifying with HCl at 10−3 M and 10−1 M concentrations. However, in a sample with HCl at a concentration of 5 M, the spectrum did shift significantly toward the UV region. | ||
| Fig. 1 Spectrally resolved molar absorption coefficients of (a) 2NP, (b) 4NP, (c) 24DNP, and (d) 246TNP in a solution of 2-propanol with either co-dissolved KOH or co-dissolved HCl present, as indicated using color. | ||
These spectral differences between nitrophenol and nitrophenolates could be an important factor towards photodegradation since there is more actinic flux at these longer wavelengths, potentially amplifying the importance of the degradation of these species relative to the neutral counterparts. The solar flux between 400 and 500 nm is nearly two times stronger than the flux in the near-UV region (300–400 nm), making nitrophenolates potentially more photolyzable. However, their photochemical quantum yields in the aqueous phase are lower than for the undissociated nitrophenols.20 While most aerosol particles in the atmosphere are acidic, they do show a wide range of acidities,48 and given the relatively high acidities of these molecules, it is reasonable to expect the nitrophenolate form to be present under some environmental conditions. Moreover, these nitrophenolates can potentially undergo a different photochemical pathway.43
Each of the undissociated nitrophenols exhibited absorption in the 200–400 nm range and showed no remarkable differences between the absorption spectra in acidified water and acidified 2-propanol, except for 24DNP. The absorption spectra of 24DNP in these two solvents are shown in Fig. 2, with the most notable difference being that the 260 nm peak in water is less intense and blue-shifted to 250 nm in 2-propanol. This difference can be explained by solute–solvent interactions, as discussed in more detail in Section 3C on calculated absorption spectra.
 | ||
| Fig. 2 Molar absorption coefficients of 24DNP in acidified water (blue trace) and acidified 2-propanol (red trace) solutions. | ||
B. Photochemical quantum yields and lifetimes
The reactivity of these nitrophenols in acidified 2-propanol was typically greater than under basic conditions. Quantum yields and estimated atmospheric lifetimes for all of the nitrophenols are presented in Table 1. Under acidic conditions, 24DNP was by far the most reactive, with a polychromatic quantum yield greater than 10−3, comparable to previous reports in octanol. The absorption spectra collected over 3 h of irradiation, along with the normalized absorbance and its decay, are shown in Fig. 3. Similar plots for each of the other nitrophenols and nitrophenolates are presented in the ESI, Fig. S2–S8.† In the discussion below, references are made to various values for aqueous photochemical yields from the literature which have been aggregated in Table S1.†| Average quantum yield,c [HCl] = 10−3 M | Atmospheric lifetimea/h | Average quantum yield,c [KOH] = 10−4 M | Atmospheric lifetimea/h | |
|---|---|---|---|---|
| a Scaled to the 24 h averaged Los Angeles solar flux. b 246TNP has a very low pKa and was in its anionic form under both reported conditions. c A description of how photochemical quantum yields were calculated is available in the ESI. | ||||
| 2NP | (4.7 ± 0.2) × 10−6 | 1000 | (1.6 ± 0.8) × 10−6 | 3000 |
| 4NP | (1.04 ± 0.02) × 10−4 | 37 | (1.2 ± 0.1) × 10−4 | 4 |
| 24DNP | (2.50 ± 0.06) × 10−3 | 3.8 | (2.65 ± 0.05) × 10−5 | 19 |
| 246TNPb | (8.5 ± 0.6) × 10−5 | 3 | (2.5 ± 0.4) × 10−5 | 3 |
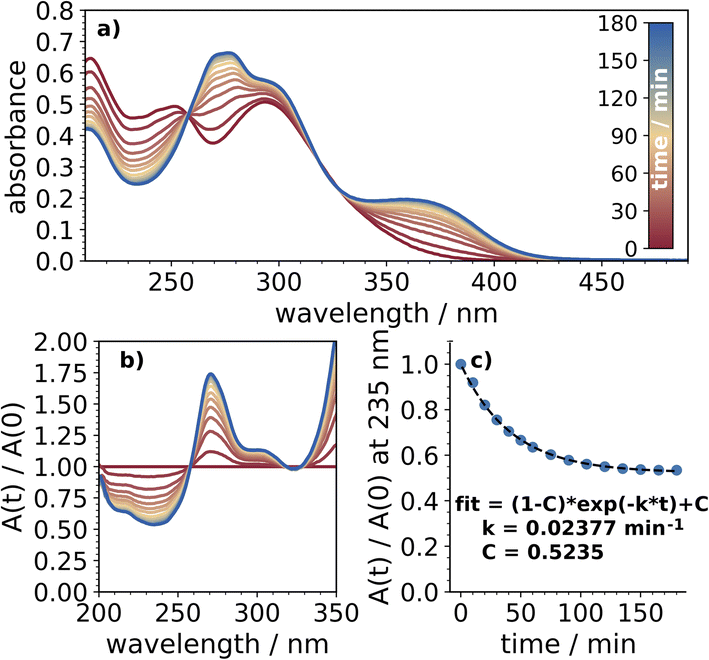 | ||
| Fig. 3 (a) The absorption of 24DNP collected over three hours of UV exposure, (b) the absorption of 24DNP normalized to the absorption spectrum obtained before photolysis began, and (c) the decrease in absorption at 235 nm, indicating loss of 24DNP, fit to eqn (1). | ||
In the condensed phase, both aqueous and organic, 2NP appears to be the least reactive nitrophenol. Our result of very low quantum yield in acidified 2-propanol greatly differs from a reported aqueous quantum yield of 1 × 10−4 in acidified solution with a very similar radiation source.20 We conducted a similar aqueous experiment with [HCl] = 10−3 M and found the averaged quantum yield to be (6.8 ± 0.3) × 10−6. These results are in closer agreement with those reported by Alif et al. (1991), who reported a monochromatic quantum yield of 4.7 × 10−6 at 365 nm.49
The quantum yield for 4NP in 2-propanol is in better agreement with the range of values previously reported under aqueous conditions. Again, the reactivity in the organic matrix is not much different than in aqueous solution. In an aqueous experiment we obtained a quantum yield of (9.9 ± 0.1) × 10−5, which is within a factor of ∼5–8 from values reported in other works.20,50–52 To put into perspective the results in the organic phase, the lifetime of 4NP in an aqueous solution containing co-dissolved α-pinene ozonolysis SOA compounds was ∼11 h.50 Our result of ∼37 h in pure 2-propanol is reasonable given that 2-propanol is a less reactive partner compared to the molecules found in SOA.
No trends were observed for the anions of each NP. In the experiment for the 2NP anion there was a rapid shift in the spectrum during the first 45 min that appears to be caused by some re-formation of neutral 2NP (see Fig. S5d†). To account for this, the fit was applied only to the data after the first 45 min. Similar types of double fitting have been employed for other systems when it is apparent that there is a fast process and some other slower process.26 This resulted in a more reasonable quantum yield of (1.6 ± 0.8) × 10−6, which should be regarded as a lower limit due to the difficulty in decoupling photodegradation from acid/base equilibrium. For 2NP and 24DNP dissolved in 2-propanol, the yields were lower for the anions, which agrees with their photochemistry in aqueous solutions.20,53 4NP reacted more under basic conditions but the difference was negligible. Since 246TNP remained in the ionized form both in the presence of acid and base, no comparison can be made regarding its reactivity relative to that of the undissociated molecule. The reactivity of the anion of 246TNP did change depending on the acidity of the solution. The effective quantum yield for 246TNP loss under acidic conditions is ∼3 times larger than that under basic conditions, however, the estimated ambient lifetime is about the same, largely due to the impact that the presence of HCl had on decreasing the amount of absorption at wavelengths greater than 300 nm.
It should be recognized that if multiple absorbing products are formed then eqn (1) will likely underestimate the loss rate constant, so the quantum yields listed in Table 1 likely represent lower limits. The presence of isosbestic points in these normalized spectra indicates minimal secondary photochemical processes occurred throughout the timescales of the experiments. Normally this would also indicate that only one product is formed, however, previous studies have observed multiple products from the photolysis of 24DNP and a similar molecule, 4-nitrocatechol, even with distinct isosbestic points in the absorption spectra.9,21 Product analysis was outside the scope of this work and the quantum yields reported here represent total photochemical quantum yields of NP + hv → products.
The relatively low reaction rates indicate that the photochemistry has not gone to completion (except for 24DNP) within the short time scales of these experiments. This was anticipated, as experiments with 4NP in the aqueous phase reported by Braman et al. (2020) did not reach completion until around 10 h of irradiation.50 The exception to this was 24DNP, which is the only NP where the loss appeared to have plateaued within the timescale of the experiment. 24DNP also provided the most notable difference between organic and aqueous phase photochemistry. Under acidic conditions, reported values for aqueous polychromatic quantum yields range from 10−6 to 10−4.9,20,52,53 The value of (2.50 ± 0.06) × 10−3 reported in this work strengthens the position that organic-phase photochemistry is significantly faster for 24DNP. This yield is slightly higher than the previously reported value (1.7 × 10−3) in an octanol film, potentially due to the lower viscosity of 2-propanol.9
C. Simulated absorption spectra
Each of the nitrophenols exhibited absorption bands in the 200–400 nm range (Fig. 1). In the following discussion, the experimentally observed absorption bands will be referred to as ‘A’, ‘B’, ‘C’, etc. in the order of increasing excitation energy. Table 2 shows the calculated excitation energies for each nitrophenol, which bands they fall into, and their respective oscillator strengths. All results shown in Table 2 account for solvent effects by using the C-PCM for 2-propanol. Simulated spectra for aqueous solutions are shown in Fig. S9.† Generally, the simulated spectra in 2-propanol are in good agreement with the experimental data. The effect of the chosen basis set on the accuracy of each excitation spectrum was minimal in comparison to the effect of the exchange functional used. For the sake of consistency, all results reported in this work come from 6-311++G**, and data from the other basis sets are presented in the ESI.† The shapes of the respective spectra are similar between B3LYP and PBE0, but in every case, B3LYP produced lower excitation energies than PBE0. Previous studies with similar objectives of predicting absorption spectra have benchmarked these TDDFT methods against higher-level methods such as the Algebraic Diagrammatic Construction (ADC(n)) with good success.54–57 The use of TDDFT/TDA worked relatively well for all of the systems in this work, so due to computational expense higher-level methods were not applied for these systems.| Method | Band (λexp/nm) | λ sim/nm | Oscillator strength (f) | Δλ/nm |
|---|---|---|---|---|
| a Explicit solvation with three 2-propanol solvent molecules. | ||||
| 2-Nitrophenol (2NP) | ||||
| PBE0/6-311++G** | A (346) | 352 | 0.099 | +6 |
| B (272) | 271 | 0.388 | −1 | |
| C (210) | 215 | 0.016 | +5 | |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| 4-Nitrophenol (4NP) | ||||
| B3LYP/6-311++G** | A (312) | 311 | 0.502 | −1 |
| B (225) | 227 | 0.067 | +2 | |
|
||||
| 2,4-Dinitrophenol (24DNP) | ||||
| PBE0/6-311++G** without explicit solvation | A (350) | 329 | 0.097 | −21 |
| B (293) | 287 | 0.332 | −6 | |
| C (250) | 261 | 0.376 | +11 | |
| 254 | 0.172 | +4 | ||
| D (212) | 204 | 0.010 | −8 | |
| 190 | 0.160 | −22 | ||
| PBE0/6-311++G** with explicit solvationa | A (350) | 335 | 0.086 | −15 |
| 333 | 0.027 | −17 | ||
| B (293) | 296 | 0.206 | +3 | |
| 294 | 0.192 | +1 | ||
| C (250) | 264 | 0.263 | +14 | |
| 256 | 0.304 | +6 | ||
| D (212) | 207 | 0.011 | −5 | |
| 204 | 0.034 | −8 | ||
|
||||
| 2,4,6-Trinitrophenol (246TNP) | ||||
| PBE0/6-311++G** | A (342) | 325 | 0.130 | −17 |
| 312 | 0.025 | −30 | ||
| B (285) | 280 | 0.124 | −5 | |
| 278 | 0.055 | −7 | ||
| 273 | 0.025 | −12 | ||
| 269 | 0.120 | −16 | ||
| C (245) | 258 | 0.248 | +13 | |
| 249 | 0.113 | +4 | ||
| 236 | 0.067 | −9 | ||
| 232 | 0.077 | −13 | ||
| 228 | 0.071 | −17 | ||
The excitation spectrum of 4NP was reproduced well with B3LYP, however, the excitation energies of 2NP and 24DNP were better served using PBE0. It is noteworthy that 2NP and 24DNP both have an intramolecular hydrogen bond while 4NP does not. The discrepancy between the methods may be due to the ability of PBE0 to yield more accurate energy values for hydrogen bonded systems.58 This, and the fact that 4NP also exhibits significantly different properties (such as with respect to solubility and vapor pressure) from the others are the reasons for treating 2NP and 24DNP differently from 4NP in this work.
With 246TNP being an anion under all experimental conditions, the results for simulated spectra of the undissociated acid and the anion are presented only for the sake of completeness. We do not report calculations for the other NP anions as it would require a different computational approach. Some theoretical calculations have been done with the nitrophenolates of 2NP, 3-nitrophenol, and 4NP with the CC2 coupled clustered model, providing varying levels of accuracy between each isomer.43,59 It would be important, especially for the easily ionizable 246TNP, to expand on this work in follow-up studies. In the following sections the specific computation results for each molecule are discussed individually.
 | ||
| Fig. 4 Simulated (blue) and experimental (red) absorption spectra of 2-nitrophenol (a) and 4-nitrophenol (b). The insets show the second derivative of the spectra to help identify the energy and breadth of each band. All spectra are normalized to the most intense band. | ||
Ultimately, the spectral differences between 24DNP in aqueous and alcohol solutions could not be completely recreated by the theory used in this work. While the errors in relative intensities could be due to the use of the Tamm–Dancoff approximation,61 the values for the excitation energies themselves should be more accurate. It would be of interest to try more accurate methods to try to characterize the major blue-shift in the C band that is observed when 24DNP is in an alcohol matrix. With regard to the atmospheric chemistry applications, where band A is the most critical transition to describe correctly, the results from these methods are sufficiently accurate. This excitation is poorly resolved in experimental spectra and has been roughly assigned to 350 nm in this work. Explicit solvation slightly red-shifted this excitation to give better agreement with experimental spectra.
D. Simulated triplet states
Ultimately, theoretical calculations were done to better understand the role of excited triplet states in the photochemistry of these nitrophenols in organic matrices. It has generally been assumed that the photochemistry of nitrophenols goes through a triplet state, given that relaxation processes from excited singlet states are very fast in the condensed phase, as previously shown for nitrobenzene.27,28 Triplet state lifetimes of nitrophenols are short and difficult to measure experimentally.62 Previous studies found that 2NP is likely to undergo intersystem crossing via S1 → T2 followed by rapid (less than 0.1 ps) internal conversion to T1.63 Takezaki et al. found that the T1 lifetimes (in benzene solvent) of 2NP and 4NP were 900 and 500 ps, respectively.62 These ultrashort excited-state lifetimes are a common characteristic of these nitroaromatic type molecules, and it is surprising that they have enough time to react before returning to the ground state via a nonreactive and nonradiative relaxation pathway.10,29,64–67 While there is no information about the photophysics of 24DNP, it is reasonable to expect the excited state lifetimes would be comparable to those of 2NP and 4NP. Given that 24DNP has the most efficient photochemistry, it would be of interest to directly study the photophysics of 24DNP with time resolved laser spectroscopy.The calculated vertical excitation triplet state energies, ET1, for 2NP, 4NP, and 24DNP are presented in Table 3. The energies for 2NP and 4NP agree with those reported from calculations with the CAS-SCF method.62 Though it is likely for these NPs to each enter the triplet manifold via an S1 → Tn>1 ISC process, the T1 state should still be the longest lived and most likely to react photochemically.63,68 The role of the triplet state in hydrogen abstraction by nitrophenols is not clear, though it is noteworthy that the energies of T1 and the average quantum yields both increase in the order of 2NP < 4NP < 24DNP. The values of these energies are comparable to those found from chromophore dissolved organic matter, where the “high-energy” triplets were shown to be 2.6–3.1 eV.69 The differences in energy, ΔEST, between T1 and S1 are also shown, to aid in providing insight into the stability of the T1 states.
| Molecule | E T1 (eV) | ΔEST (T1–S1, eV) | ||
|---|---|---|---|---|
| C-PCM | 2-Propanol | Water | 2-Propanol | Water |
| a Calculations done with PBE0a or B3LYPb and the 6-311++G** basis set. Explicit solvation with three solvent molecules used for 24DNPc. | ||||
| 2NPa | 2.721 | 2.708 | −0.804 | −0.808 |
| 4NPb | 2.839 | 2.812 | −1.141 | −1.163 |
| 24DNPa | 3.077 | 3.071 | −0.689 | −0.690 |
| 24DNPac | 3.035 | 3.018 | −0.669 | −0.692 |
| 246TNPa | 3.194 | 3.191 | −0.622 | −0.624 |
The reactivity of the triplet state in nitroaromatics was reported early on for nitrobenzene, and the reactive part of the molecule was proposed to be the –NO2 group.27,28 Theoretical studies of nitrobenzene and nitrophenols have observed similar importance of the –NO2 group toward photochemistry.10,68,70 Though it has been difficult experimentally to trap the reaction in this state, studies have found photoreduction products after irradiation.9,71,72Fig. 5 displays the highest-strength natural transition orbital (NTO) pairs for the T1 state of 24DNP. In these π → π* transitions, most of the electron density gets shifted to the –NO2 groups upon excitation, particularly in the orbitals for pair #2, supporting the suggestion that this is where the H-atom abstraction likely occurs. The same transition in water (as compared to 2-propanol) results in less electron density being shifted into the –NO2 group, with the hydroxyl group retaining some electron density. In addition to the fact that 2-propanol has less strongly bound hydrogen atoms than water, this shift in electronic structure of the T1 state of 24DNP could be playing a role in its enhanced reactivity towards alcohols.
 | ||
| Fig. 5 Natural transition orbitals for the first excited triplet state of 2,4-dinitrophenol calculated at the PBE0/6-311++G** level of theory and depicted with an isovalue of 0.05. A combination of explicit solvation (shown) and C-PCM was used to simulate a 2-propanol (red box) or water (blue box) solution. | ||
4 Conclusions
This work has compared the photochemistry of nitrophenols in 2-propanol to that in water to better understand their photochemical transformations in atmospheric organic particles. Experimental measurements of the quantum yields for polychromatic photodegradation showed nitrophenols do not follow a well-defined trend with the number of nitro groups in the molecule. Of the molecules studied, 24DNP with two nitro groups turned out to be far more photolabile than nitrophenols with one or three nitro groups. Theoretical calculations were performed to characterize the excited states of these nitrophenols, and how they vary between aqueous and organic solutions. Theoretical absorption spectra were in good agreement with experimental spectra, with the predicted excitation energies typically falling within 5 nm from the experimentally observed bands. The relative energies of each nitrophenol's T1 state did not show a strong dependence on solvent.While 2NP and 4NP showed little difference in reactivity in water versus 2-propanol, 24DNP showed a significant (factor of 100 to 1000) increase in photochemical quantum yield in 2-propanol, in agreement with previous studies of 24DNP photochemistry. Therefore, 24DNP should be the most reactive nitrophenol among the primary and secondary atmospheric organic aerosol particles.
The undissociated nitrophenols generally had higher quantum yields than their deprotonated counterparts (as measured by comparing photochemistry in the presence of acid and base), with the exception of 4NP which had effectively the same quantum yield in acidic and basic solutions. In light of this, it is fair to say that the condensed-phase photochemical loss of 2NP, 4NP, and 246TNP is not highly dependent on the chemical environment: 2NP degrades minimally in either case, 4NP shows minimal change between water and 2-propanol, and 246TNP is always in the anion form in solution. The most drastic solvation difference is observed in 24DNP, resulting in a significant change to its absorption spectrum and reducing its ambient lifetime to just a few hours.
In summary, organic-phase photochemistry deviates the most from that in the aqueous phase for 24DNP, which appears to be a special case in the nitrophenol family. Analysis of the natural transition orbitals suggests that, at least for the lowest triplet state of 24DNP, the presence of alcohol solvent molecules results in more electron density on the –NO2 groups, potentially accounting for its enhanced reactivity. It would be of interest to investigate these solvent effects further, perhaps with more viscous solvent systems where the –NO2 groups have less of an ability to reorganize after the excitation.
Author contributions
The experiments and data analysis were conceived by ABD and SAN, and carried out by SL and ABD. Computational work was carried out by ABD with guidance from NK and RBG. The manuscript was written by ABD and edited by NK, RBG, and SAN. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This experimental work was supported by the US National Science Foundation grant AGS-1853639. The authors would also like to acknowledge US National Science Foundation grant CHE-0840513 for funding the cluster used for theoretical calculations. RBG acknowledges partial support of this research by grant 593/20 from the Israel Science Foundation.References
- G. Rippen, E. Zietz, R. Frank, T. Knacker and W. Klöpffer, Do airborne nitrophenols contribute to forest decline?, Environ. Technol. Lett., 1987, 8, 475–482 CrossRef CAS.
- M. Natangelo, S. Mangiapan, R. Bagnati, E. Benfenati and R. Fanelli, Increased concentrations of nitrophenols in leaves from a damaged forestal site, Chemosphere, 1999, 38, 1495–1503 CrossRef CAS.
- J. Tremp, P. Mattrel, S. Fingler and W. Giger, Phenols and nitrophenols as tropospheric pollutants: emissions from automobile exhausts and phase transfer in the atmosphere, Water, Air, Soil Pollut., 1993, 68, 113–123 CrossRef CAS.
- R. Belloli, B. Barletta, E. Bolzacchini, S. Meinardi, M. Orlandi and B. Rindone, Determination of toxic nitrophenols in the atmosphere by high-performance liquid chromatography, J. Chromatogr. A, 1999, 846, 277–281 CrossRef CAS.
- Y. Dubowski and M. R. Hoffmann, Photochemical transformations in ice: implications for the fate of chemical species, Geophys. Res. Lett., 2000, 27, 3321–3324 CrossRef CAS.
- D. Vione, V. Maurino, C. Minero and E. Pelizzetti, Aqueous Atmospheric Chemistry: Formation of 2,4-Dinitrophenol upon Nitration of 2-Nitrophenol and 4-Nitrophenol in Solution, Environ. Sci. Technol., 2005, 39, 7921–7931 CrossRef CAS PubMed.
- I. Bejan, Y. A. E. Aal, I. Barnes, T. Benter, B. Bohn, P. Wiesen and J. Kleffmann, The photolysis of ortho-nitrophenols: a new gas phase source of HONO, Phys. Chem. Chem. Phys., 2006, 8, 2028–2035 RSC.
- D. Vione, V. Maurino, C. Minero, M. Duncianu, R.-I. Olariu, C. Arsene, M. Sarakha and G. Mailhot, Assessing the transformation kinetics of 2- and 4-nitrophenol in the atmospheric aqueous phase. Implications for the distribution of both nitroisomers in the atmosphere, Atmos. Environ., 2009, 43, 2321–2327 CrossRef CAS.
- H. Lignell, M. L. Hinks and S. A. Nizkorodov, Exploring matrix effects on photochemistry of organic aerosols, PNAS, 2014, 111, 13780–13785 CrossRef CAS PubMed.
- H. A. Ernst, T. J. A. Wolf, O. Schalk, N. González-García, A. E. Boguslavskiy, A. Stolow, M. Olzmann and A.-N. Unterreiner, Ultrafast Dynamics of o-Nitrophenol: An Experimental and Theoretical Study, J. Phys. Chem. A, 2015, 119, 9225–9235 CrossRef CAS PubMed.
- L. Vereecken, H. K. Chakravarty, B. Bohn and J. Lelieveld, Theoretical Study on the Formation of H- and O-Atoms, HONO, OH, NO, and NO2 from the Lowest Lying Singlet and Triplet States in Ortho-Nitrophenol Photolysis, Int. J. Chem. Kinet., 2016, 48, 785–795 CrossRef CAS.
- R. F. Hems and J. P. D. Abbatt, Aqueous Phase Photo-oxidation of Brown Carbon Nitrophenols: Reaction Kinetics, Mechanism, and Evolution of Light Absorption, ACS Earth Space Chem., 2018, 2, 225–234 CrossRef CAS.
- Y. Iinuma, E. Brüggemann, T. Gnauk, K. Müller, M. O. Andreae, G. Helas, R. Parmar and H. Herrmann, Source characterization of biomass burning particles: the combustion of selected European conifers, African hardwood, savanna grass, and German and Indonesian peat, J. Geophys. Res.: Atmos., 2007, 112, D08209 CrossRef.
- Y. Iinuma, M. Keywood and H. Herrmann, Characterization of primary and secondary organic aerosols in Melbourne airshed: the influence of biogenic emissions, wood smoke and bushfires, Atmos. Environ., 2016, 130, 54–63 CrossRef CAS.
- P. Lin, N. Bluvshtein, Y. Rudich, S. A. Nizkorodov, J. Laskin and A. Laskin, Molecular Chemistry of Atmospheric Brown Carbon Inferred from a Nationwide Biomass Burning Event, Environ. Sci. Technol., 2017, 51, 11561–11570 CrossRef CAS PubMed.
- R. Atkinson, S. M. Aschmann and J. Arey, Reactions of hydroxyl and nitrogen trioxide radicals with phenol, cresols, and 2-nitrophenol at 296 +- 2 K, Environ. Sci. Technol., 1992, 26, 1397–1403 CrossRef CAS.
- E. Bolzacchini, M. Bruschi, J. Hjorth, S. Meinardi, M. Orlandi, B. Rindone and E. Rosenbohm, Gas-Phase Reaction of Phenol with NO3, Environ. Sci. Technol., 2001, 35, 1791–1797 CrossRef CAS PubMed.
- D. Vione, V. Maurino, C. Minero and E. Pelizzetti, Phenol photonitration upon UV irradiation of nitrite in aqueous solution I: effects of oxygen and 2-propanol, Chemosphere, 2001, 45, 893–902 CrossRef CAS PubMed.
- M. L. Hinks, M. V. Brady, H. Lignell, M. Song, J. W. Grayson, A. K. Bertram, P. Lin, A. Laskin, J. Laskin and S. A. Nizkorodov, Effect of viscosity on photodegradation rates in complex secondary organic aerosol materials, Phys. Chem. Chem. Phys., 2016, 18, 8785–8793 RSC.
- F. Barsotti, T. Bartels-Rausch, E. De Laurentiis, M. Ammann, M. Brigante, G. Mailhot, V. Maurino, C. Minero and D. Vione, Photochemical Formation of Nitrite and Nitrous Acid (HONO) upon Irradiation of Nitrophenols in Aqueous Solution and in Viscous Secondary Organic Aerosol Proxy, Environ. Sci. Technol., 2017, 51, 7486–7495 CrossRef CAS PubMed.
- A. B. Dalton and S. A. Nizkorodov, Photochemical Degradation of 4-Nitrocatechol and 2,4-Dinitrophenol in a Sugar-Glass Secondary Organic Aerosol Surrogate, Environ. Sci. Technol., 2021, 55, 14586–14594 CrossRef CAS PubMed.
- R. J. Mayorga, Z. Zhao and H. Zhang, Formation of secondary organic aerosol from nitrate radical oxidation of phenolic VOCs: implications for nitration mechanisms and brown carbon formation, Atmos. Environ., 2021, 244, 117910 CrossRef CAS.
- J. H. Kroll and J. H. Seinfeld, Chemistry of secondary organic aerosol: formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 2008, 42, 3593–3624 CrossRef CAS.
- R. Zhao, A. K. Y. Lee, L. Huang, X. Li, F. Yang and J. P. D. Abbatt, Photochemical processing of aqueous atmospheric brown carbon, Atmos. Chem. Phys., 2015, 15, 6087–6100 CrossRef CAS.
- K. Grygoryeva, J. Kubečka, A. Pysanenko, J. Lengyel, P. Slavíček and M. Fárník, Photochemistry of Nitrophenol Molecules and Clusters: Intra- vs. Intermolecular Hydrogen Bond Dynamics, J. Phys. Chem. A, 2016, 120, 4139–4146 CrossRef CAS PubMed.
- A. L. Klodt, M. Adamek, M. Dibley, S. A. Nizkorodov and R. E. O'Brien, Effects of the sample matrix on the photobleaching and photodegradation of toluene-derived secondary organic aerosol compounds, Atmos. Chem. Phys., 2022, 22, 10155–10171 CrossRef CAS.
- R. Hurley and A. C. Testa, Photochemical n → π* Excitation of Nitrobenzene, J. Am. Chem. Soc., 1966, 88, 4330–4332 CrossRef CAS.
- R. Hurley and A. C. Testa, Nitrobenzene photochemistry. II. Protonation in the excited state, J. Am. Chem. Soc., 1967, 89, 6917–6919 CrossRef CAS.
- D. Döpp, in Triplet States II, ed. U. P. Wild, D. Döpp and H. Dürr, Springer, Berlin, Heidelberg, 1975, pp. 49–85 Search PubMed.
- Software for the frontiers of quantum chemistry: an overview of developments in the Q-Chem 5 package, J. Chem. Phys., 2021, 155, 084801 Search PubMed.
- D. P. Chong, Recent Advances in Density Functional Methods, Part I, World Scientific, 1995 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: the PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- J.-P. Cornard, Rasmiwetti and J.-C. Merlin, Molecular structure and spectroscopic properties of 4-nitrocatechol at different pH: UV-visible, Raman, DFT and TD-DFT calculations, Chem. Phys., 2005, 309, 239–249 CrossRef CAS.
- C. Reichardt, R. A. Vogt and C. E. Crespo-Hernández, On the origin of ultrafast nonradiative transitions in nitro-polycyclic aromatic hydrocarbons: excited-state dynamics in 1-nitronaphthalene, J. Chem. Phys., 2009, 131, 224518 CrossRef PubMed.
- S. Hirata and M. Head-Gordon, Time-dependent density functional theory within the Tamm–Dancoff approximation, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- M. J. G. Peach, M. J. Williamson and D. J. Tozer, Influence of Triplet Instabilities in TDDFT, J. Chem. Theory Comput., 2011, 7, 3578–3585 CrossRef CAS PubMed.
- V. Barone, M. Cossi and J. Tomasi, A new definition of cavities for the computation of solvation free energies by the polarizable continuum model, J. Chem. Phys., 1997, 107, 3210–3221 CrossRef CAS.
- T. N. Truong and E. V. Stefanovich, A new method for incorporating solvent effect into the classical, ab initio molecular orbital and density functional theory frameworks for arbitrary shape cavity, Chem. Phys. Lett., 1995, 240, 253–260 CrossRef CAS.
- R. L. Martin, Natural transition orbitals, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- E. D. Glendening, C. R. Landis and F. Weinhold, NBO 6.0: Natural bond orbital analysis program, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- R. Z. Hinrichs, P. Buczek and J. J. Trivedi, Solar Absorption by Aerosol-Bound Nitrophenols Compared to Aqueous and Gaseous Nitrophenols, Environ. Sci. Technol., 2016, 50, 5661–5667 CrossRef CAS PubMed.
- M. Wanko, J. Houmøller, K. Støchkel, M.-B. S. Kirketerp, M. Å. Petersen, M. B. Nielsen, S. B. Nielsen and A. Rubio, Substitution effects on the absorption spectra of nitrophenolate isomers, Phys. Chem. Chem. Phys., 2012, 14, 12905–12911 RSC.
- J. Houmøller, M. Wanko, A. Rubio and S. B. Nielsen, Effect of a Single Water Molecule on the Electronic Absorption by o- and p-Nitrophenolate: A Shift to the Red or to the Blue?, J. Phys. Chem. A, 2015, 119, 11498–11503 CrossRef PubMed.
- N. C. Michenfelder, H. A. Ernst, C. Schweigert, M. Olzmann and A.-N. Unterreiner, Ultrafast stimulated emission of nitrophenolates in organic and aqueous solutions, Phys. Chem. Chem. Phys., 2018, 20, 10713–10720 RSC.
- J. Leier, N. C. Michenfelder, A.-N. Unterreiner and M. Olzmann, Indications for an intermolecular photo-induced excited-state proton transfer of p-nitrophenol in water, Mol. Phys., 2021, 119, e1975051 CrossRef.
- S. Rondinini, P. Longhi, P. R. Mussini and T. Mussini, Autoprotolysis constants in nonaqueous solvents and aqueous organic solvent mixtures, Pure Appl. Chem., 1987, 59, 1693–1702 CrossRef CAS.
- A. P. Ault, Aerosol Acidity: Novel Measurements and Implications for Atmospheric Chemistry, Acc. Chem. Res., 2020, 53, 1703–1714 CrossRef CAS PubMed.
- A. Alif, J.-F. Pilichowski and P. Boule, Photochemistry and environment XIII: phototransformation of 2-nitrophenol in aqueous solution, J. Photochem. Photobiol., A, 1991, 59, 209–219 CrossRef CAS.
- T. Braman, L. Dolvin, C. Thrasher, H. Yu, E. Q. Walhout and R. E. O'Brien, Fresh Versus Photo-recalcitrant Secondary Organic Aerosol: Effects of Organic Mixtures on Aqueous Photodegradation of 4-Nitrophenol, Environ. Sci. Technol. Lett., 2020, 7, 248–253 CrossRef CAS.
- J. Lemaire, J. A. Guth, O. Klais, J. Leahey, W. Merz, J. Philp, R. Wilmes and C. J. M. Wolff, Ring test of a method for assessing the phototransformation of chemicals in water, Chemosphere, 1985, 14, 53–77 CrossRef CAS.
- F. S. G. Einschlag, L. Carlos, A. L. Capparelli, A. M. Braun and E. Oliveros, Degradation of nitroaromatic compounds by the UV–H2O2 process using polychromatic radiation sources, Photochem. Photobiol. Sci., 2002, 1, 520–525 CrossRef PubMed.
- A. Albinet, C. Minero and D. Vione, Phototransformation processes of 2,4-dinitrophenol, relevant to atmospheric water droplets, Chemosphere, 2010, 80, 753–758 CrossRef CAS PubMed.
- S. A. Epstein, D. Shemesh, V. T. Tran, S. A. Nizkorodov and R. B. Gerber, Absorption Spectra and Photolysis of Methyl Peroxide in Liquid and Frozen Water, J. Phys. Chem. A, 2012, 116, 6068–6077 CrossRef CAS PubMed.
- P. H. P. Harbach, M. Wormit and A. Dreuw, The third-order algebraic diagrammatic construction method (ADC(3)) for the polarization propagator for closed-shell molecules: efficient implementation and benchmarking, J. Chem. Phys., 2014, 141, 064113 CrossRef PubMed.
- N. V. Karimova, M. Luo, V. H. Grassian and R. Benny Gerber, Absorption spectra of benzoic acid in water at different pH and in the presence of salts: insights from the integration of experimental data and theoretical cluster models, Phys. Chem. Chem. Phys., 2020, 22, 5046–5056 RSC.
- N. V. Karimova, M. Luo, I. Sit, V. H. Grassian and R. B. Gerber, Absorption Spectra and the Electronic Structure of Gallic Acid in Water at Different pH: Experimental Data and Theoretical Cluster Models, J. Phys. Chem. A, 2022, 126, 190–197 CrossRef CAS PubMed.
- L. Rao, H. Ke, G. Fu, X. Xu and Y. Yan, Performance of Several Density Functional Theory Methods on Describing Hydrogen-Bond Interactions, J. Chem. Theory Comput., 2009, 5, 86–96 CrossRef CAS PubMed.
- M.-B. Suhr Kirketerp, M. Åxman Petersen, M. Wanko, L. Andres Espinosa Leal, H. Zettergren, F. M. Raymo, A. Rubio, M. Brøndsted Nielsen and S. Brøndsted Nielsen, Absorption Spectra of 4-Nitrophenolate Ions Measured In Vacuo and in Solution, ChemPhysChem, 2009, 10, 1207–1209 CrossRef CAS PubMed.
- E. Sim, S. Song and K. Burke, Quantifying Density Errors in DFT, J. Phys. Chem. Lett., 2018, 9, 6385–6392 CrossRef CAS PubMed.
- A. Chantzis, A. D. Laurent, C. Adamo and D. Jacquemin, Is the Tamm-Dancoff Approximation Reliable for the Calculation of Absorption and Fluorescence Band Shapes?, J. Chem. Theory Comput., 2013, 9, 4517–4525 CrossRef CAS PubMed.
- M. Takezaki, N. Hirota and M. Terazima, Nonradiative Relaxation Processes and Electronically Excited States of Nitrobenzene Studied by Picosecond Time-Resolved Transient Grating Method, J. Phys. Chem. A, 1997, 101, 3443–3448 CrossRef CAS.
- C. Xu, L. Yu, C. Zhu, J. Yu and Z. Cao, Intersystem crossing-branched excited-state intramolecular proton transfer for o-nitrophenol: an ab initio on-the-fly nonadiabatic molecular dynamic simulation, Sci. Rep., 2016, 6, 26768 CrossRef CAS PubMed.
- R. Hurley and A. C. Testa, Triplet-state yield of aromatic nitro compounds, J. Am. Chem. Soc., 1968, 90, 1949–1952 CrossRef CAS.
- R. A. Vogt, C. Reichardt and C. E. Crespo-Hernández, Excited-State Dynamics in Nitro-Naphthalene Derivatives: Intersystem Crossing to the Triplet Manifold in Hundreds of Femtoseconds, J. Phys. Chem. A, 2013, 117, 6580–6588 CrossRef CAS PubMed.
- C. E. Crespo-Hernández, G. Burdzinski and R. Arce, Environmental Photochemistry of Nitro-PAHs: Direct Observation of Ultrafast Intersystem Crossing in 1-Nitropyrene, J. Phys. Chem. A, 2008, 112, 6313–6319 CrossRef PubMed.
- J. S. Zugazagoitia, E. Collado-Fregoso, E. F. Plaza-Medina and J. Peon, Relaxation in the Triplet Manifold of 1-Nitronaphthalene Observed by Transient Absorption Spectroscopy, J. Phys. Chem. A, 2009, 113, 805–810 CrossRef CAS PubMed.
- M. Takezaki, N. Hirota, M. Terazima, H. Sato, T. Nakajima and S. Kato, Geometries and Energies of Nitrobenzene Studied by CAS-SCF Calculations, J. Phys. Chem. A, 1997, 101, 5190–5195 CrossRef CAS.
- K. McNeill and S. Canonica, Triplet state dissolved organic matter in aquatic photochemistry: reaction mechanisms, substrate scope, and photophysical properties, Environ. Sci.: Processes Impacts, 2016, 18, 1381–1399 RSC.
- K. J. Blackshaw, B. I. Ortega, N.-K. Quartey, W. E. Fritzeen, R. T. Korb, A. K. Ajmani, L. Montgomery, M. Marracci, G. G. Vanegas, J. Galvan, Z. Sarvas, A. S. Petit and N. M. Kidwell, Nonstatistical Dissociation Dynamics of Nitroaromatic Chromophores, J. Phys. Chem. A, 2019, 123, 4262–4273 CrossRef CAS PubMed.
- S. Hashimoto, J. Sunamoto, H. Fujii and K. Kano, Photochemical Reduction of Nitrobenzene and Its Reduction Intermediates. III. The Photochemical Reduction of Nitrobenzene, Bull. Chem. Soc. Jpn., 1968, 41, 1249–1251 CrossRef CAS.
- S. Hashimoto and K. Kano, The Photochemical Reduction of Nitrobenzene and Its Reduction Intermediates. X. The Photochemical Reduction of the Monosubstituted Nitrobenzenes in 2-Propanol, Bull. Chem. Soc. Jpn., 1972, 45, 549–553 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ea00144f |
| This journal is © The Royal Society of Chemistry 2023 |