
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
NacNac-zinc-pyridonate mediated ε-caprolactone ROP†
Jack W. J.
Hughes
,
Dawid J.
Babula
,
Findlay
Stowers-Veitch
,
Kang
Yuan
,
Marina
Uzelac
 ,
Gary S.
Nichol
,
Michael J.
Ingleson
* and
Jennifer A.
Garden
*
,
Gary S.
Nichol
,
Michael J.
Ingleson
* and
Jennifer A.
Garden
*
EaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: j.garden@ed.ac.uk
First published on 20th November 2023
Abstract
Herein we report the synthesis, isolation and polymerisation activity of two new zinc compounds based on a 2,6-diisopropylphenyl (Dipp) β-diiminate (NacNac) ligand framework with zinc also ligated by an amidate (2-pyridonate or 6-methyl-2-pyridonate) unit. The compounds crystallised as either monomeric (6-Me-2-pyridonate derivative) or dimeric (2-pyridonate) species, although both were found to be monomeric in solution via1H DOSY NMR spectroscopy, which was supported by DFT calculations. These observations suggest that both complexes initiate ring-opening polymerisation (ROP) through a single-site monometallic mechanism. High molecular weight poly ε-caprolactone (PCL) was achieved via exogenous initiator-free ROP conditions with both catalysts. An increase in the 2-pyridonate initiator steric bulk (6-Me- vs. 6-H-) resulted in an improved catalytic activity, facilitating complete monomer conversion within 1 h at 60 °C. Pyridonate end-groups were observed by MALDI-ToF mass spectrometry, contrasting with previous observations for DippNacNac-Zn acetate complexes (where no acetate end groups are observed), instead this more closely resembles the reactivity of DippNacNac-Zn alkoxide complexes in ROP (where RO end groups are observed). Additional major signals in the MALDI-ToF spectra were consistent with cyclic PCL species, which are attributed to back-biting ring-closing termination steps occuring in a process facilitated by the pyridonate unit being an effective leaving group. To the best of our knowledge, these complexes represent the first examples of pyridonate, and indeed amidate, initated ROP.
Introduction
Petrochemical-derived plastics are essential to everyday life but have led to the accumulation of vast amounts of poorly recycled, non-biodegradable materials, which have made their way into all eco-systems.1 As a result, there has been a push for bioderived and biodegradable alternatives to traditional polymers such as poly(ethylene) (PE) and poly(propylene) (PP).2,3 These have come in large part in the form of polyesters such as poly(lactic acid) (PLA) and poly(caprolactone) (PCL), prepared from lactide and ε-caprolactone (ε-CL) respectively via ring-opening polymerisation (ROP). A variety of enzymatic4 and organocatalytic5 routes have been reported for the ROP of cyclic esters to polyesters, however organometallic ROP catalysts are increasingly attractive due to their high activity, good molecular weight control and well-understood mechanisms.6,7 Toxicity concerns8 with the current industry standard, stannous octoate, have limited the implementation of materials such as PLA and PCL in tailored applications in biomedicine.9–11 This has furthered efforts into exploring ROP catalysis based on more bio-compatible, Earth abundant metals such as sodium, aluminium, iron and zinc.6 These catalysts have shown promising activity and selectivity, in some cases out-performing the industry standard.12–14Catalysts based on β-diiminate (NacNac) ligand frameworks have been studied extensively in polymerisation catalysis, including NacNac-Zn complexes.15 In the early examples by Coates and co-workers, a range of NacNac-Zn catalysts were prepared featuring monodentate alkoxide and bidentate carboxylate groups as co-ligands (Fig. 1).16–20 These studies included exploring the steric effect of the NacNac ligands upon polymerisation activity, highlighting the excellent efficiency of catalysts based on the bulky DippNacNac (CH{C(Me)N-Dipp}2, Dipp = 2,6-diisopropylphenyl) framework. The efficacy of various initiating groups also was explored, both for lactone ROP and for the ring-opening co-polymerisation (ROCOP) of epoxides with CO2. NacNac-Zn catalysts with monodentate alkoxide co-ligands (which act as initiating groups) can facilitate both lactone ROP and epoxide/CO2 ROCOP, affording polymers with low dispersity (Đ) and good control of both number average molecular weight (Mn) and chain-end fidelity.16,18,20–22 In contrast, NacNac-Zn catalysts with carboxylate (and structurally related sulfinate) co-ligands generally are only effective for ROCOP in which solution-state dimerisation effects are key to achieve good activity.19,24,25 Work by the groups of Coates, Schulz and Williams has shown acetate groups to be inefficient initiators for lactone ROP when combined with NacNac-Zn catalysts.16,26,27
 | ||
| Fig. 1 Select reported catalysts based on a DippNacNac-Zn framework.16,19 | ||
Indeed, the work by Coates and co-workers suggests that the Zn-carboxylate moieties require transformation by trace impurities to form species in low amounts which in turn initiate ROP.16 While alkoxide and carboxylate co-ligands are ubiquitous in this field, co-ligands that combine features of both these co-ligand classes are much less explored, despite the potential to access unique reactivity. 2-Pyridonates are bidentate co-ligands with a conjugate acid of intermediate Brønsted acidity (ca. 11.70–12.45)28 between that of the conjugate acid of an alkoxide (e.g. OiPr, pKa of HOiPr = 17.26, typically highly active ROP initiators) and a carboxylate (e.g. HOAc, pKa = 4.76, characteristically poor initiators in ROP) in H2O.29 Therefore we were interested in installing these as co-ligands onto NacNac-Zn and determining how they functioned in the ROP of a cyclic ester. Herein we describe the synthesis of two new DippNacNac-Zn complexes both featuring a bidentate pyridonate and show that this co-ligand is an effective initiating group towards ROP catalysis. While this positions 2-pyridonates closer to alkoxide co-ligands than carboxylate co-ligands in NacNac-Zn mediated ROP, the formation of a significant amount of cyclic polyesters confirm they represent a unique co-ligand class.
Results and discussion
Catalyst synthesis and characterisation
Complex 1 was synthesised via reaction of 2-pyridone with DippNacNac-ZnEt, with an isolated yield of 40% following fractional recrystallisation. The methyl-substituted analogue 2 also could be formed by this route, albeit in a crude mixture, and in this case 2 could not be isolated cleanly by fractional crystallisation. Compound 2 instead was prepared via a salt-metathesis route, employing DippNacNac-ZnI30 and the silver pyridonate 3. Insoluble AgI was formed as a by-product, which aided isolation of 2 and this approach afforded 2 in an 80% isolated yield (Scheme 1). Compound 3 is also novel and is presumably an oligo-/poly-meric material given its low solubility in common solvents – characterisation is given in the ESI.† Single crystals of 1 and 2 suitable for X-ray diffraction studies were grown from CH2Cl2. Complex 1 crystallised as a dimer; containing an 8-membered ring between two distorted tetrahedral zinc centres (Fig. 2). Complex 2, by comparison was monomeric, with a strained 4 membered metallacycle involving the distorted tetrahedral zinc centre. The crystallisation of 2 as a monomer was notable as no carboxylate NacNac-Zn(O2C-R) complexes have been reported as monomers with 4-membered rings; instead these dimerise to form an 8-membered ring (related to that seen for 1). This was the first indication of a significant disparity between carboxylates and these pyridonates in these NacNacZn systems. It should be noted that 2-pyridonates have been shown to form four membered metallacycles in other systems.31 The increased valency of nitrogen (present in the pyridonate) versus oxygen (in carboxylates) leads to the incorporation of more steric bulk proximal to the Zn–ligand bond, which will influence aggregation in both the solid and solution state. Indeed, work by Cundari, Holland and co-workers on NacNac-Fe complexes bearing N-R-amidinates (R = iPr) as co-ligands were observed to be solely monomeric, attributable to the N-steric bulk.32 It is likely that the greater steric crowding present using pyridonates (relative to carboxylates) helps disfavour dimerisation, in contrast to the analogous carboxylates where steric bulk can only be modified at the distal (relative to zinc) carbon position. An additional factor favouring dimerization with carboxylates will be the slightly larger O–C–O angle observed in [NacNac-Zn(O2C-R)] complexes (121–128°) relative to the N–C–O angles in 1 and 2 (vide infra).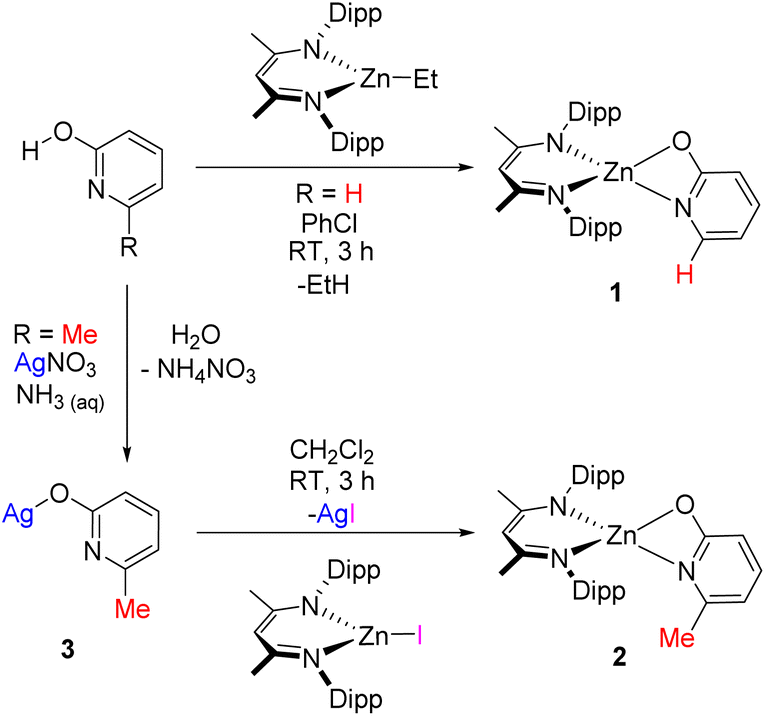 | ||
| Scheme 1 Synthetic routes to complexes 1 and 2 (the latter via3, which is presumably oligo-/poly-meric). | ||
 | ||
| Fig. 2 Molecular structures of complexes 1 (left) and 2 (right). H atoms and solvent omitted for clarity; ellipsoids are shown at the 30% probability level. | ||
The C–O bond lengths in both 1 and 2 (1.280(3) Å and 1.301(4) Å, respectively, Table 1) are intermediate between a single and a double C–O bond, and are greater than in the protonated ligands (1.250(0) Å and 1.262(5) Å respectively).33,34 Further, there was a reduction in C–N bond lengths for the pyridone unit (1.379(1) to 1.356(3) Å in 1, and 1.391(6) to 1.357(4) Å in 2), again relative to the protonated ligand, suggesting some multiple bond character. The lengthening of the CO bond and contraction of the CN bond in both instances indicates a significant degree of charge delocalisation in the amidate unit when bound to zinc, but with the oxypyridine form more dominate in 1 and 2 than in the protonated ligand (where the pyridone resonance form is more dominant). A narrow N1–Zn–O bond angle of 65.7(1)° was observed in 2, suggesting a significant degree of ring strain that is alleviated in 1 through dimerisation, which increases the N1–Zn–O bond angle to 113.3(3)°. In both complexes, a reduction in the O–C–N1 bond angle of the pyridone unit was observed relative to their protonated form, which was more pronounced in 2 (−5.5° cf. the protonated ligand). The reduction in the O–C–N bond angle upon metallation is in-line with other 4-membered pyridone metallacycles in the literature.31,35
| Bond | Bond angles/Å | |
|---|---|---|
| 1 | 2 | |
| Zn1–O1 | 1.896(2) | 2.038(2) |
| Zn1–N1 | 2.026(9) | 2.074(3) |
| Zn1–N2 | 2.0005(12) | 1.942(3) |
| Zn1–N3 | 1.9923(12) | 1.943(3) |
| O1–C1 | 1.280(3) | 1.301(4) |
| N1–C1 | 1.356(3) | 1.357(4) |
| N1–C2 | 1.355(4) | 1.356(4) |
| Bonds | Bond angles/° | |
|---|---|---|
| N1–Zn1–O1 | 113.3(3) | 65.7(1) |
| Zn1–O1–C1 | 153.3(4) | 91.6(2) |
| Zn1–N1–C1 | 122.0(5) | 88.5(2) |
| N2–Zn1–N3 | 96.52(5) | 99.41(12) |
| N1–C1–O1 | 118.5(4) | 114.2(3) |
The differing solid-state structures (monomeric vs. dimeric) meant that direct comparison of the effect of changing the co-ligand on the solid state structure of complexes 1 and 2 was not possible. Therefore the monomer and dimer structures were calculated in both cases at the B3PW91 level of theory using the LANL2DZ basis set for Zn and 6-311G(d,p) basis set for all other atoms in CH2Cl2 using a polarisation continuum model (PCM) at 298 K. A slight increase in the Zn–N bond length and decrease in Zn–O bond length was observed for the monomeric structure of 2 compared to 1 (Table S4†), however the majority of the bond lengths and angles were extremely similar. Comparison of the free energies of mono-/dimeric 1 and 2 suggest the dissolution of the dimer into two monomers is energetically favoured in both cases (by 24.2 and 36.9 kcal mol−1 respectively, Fig. S21†). The significantly energetically favoured conversion of the dimers into monomers was corroborated by 1H DOSY NMR spectroscopy. At the concentration of the polymerisation reactions (8.7 mM in catalyst, vide infra) in toluene-d8, 1 and 2 were observed to be monomeric. This was evidenced by a single diffusion coefficient, which was compared to a calibration plot in toluene-d8 (see Table S3 and Fig. S19 in ESI† for experimental set up and calibration), and gave estimated molecular weights of 572 and 581 g mol−1 for 1 and 2. These calculated values are within 2% error of the expected values for the monomeric complexes. Taken together, the DFT and DOSY NMR studies of 1 and 2 suggest that ROP takes place via a monometallic single-site mechanism. Analysis of the symmetry by 1H NMR spectroscopy (using the iPr methyl groups in DippNacNac) for both 1 and 2 revealed C2v solution symmetry as there were only two discrete environments observed (Fig. S1 and S6†). This suggests that the 4-membered metallacycles in 1 and 2 undergo ring-opening on the NMR time scale which is consistent with significant strain in the four membered metallacycle.
Polymerisation studies
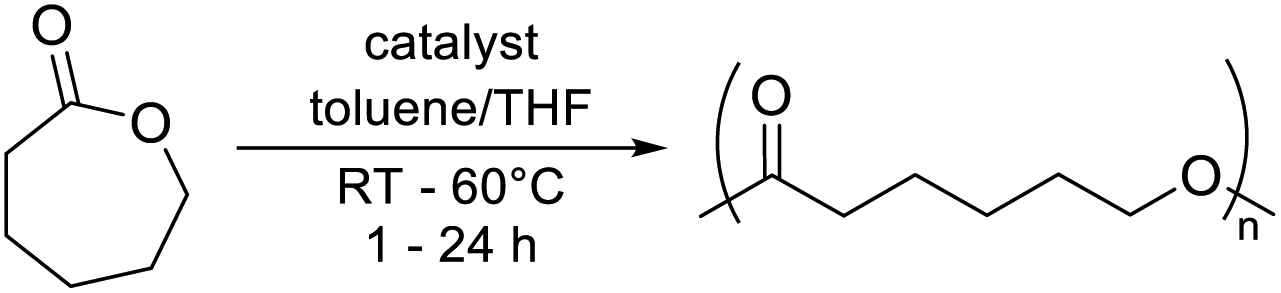
Complexes 1 and 2 were tested in the ROP of ε-caprolactone (ε-CL). Catalytic activity was investigated using a [catalyst]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[ε-CL] ratio of 1:100 between 20–60 °C in toluene; these are typical conditions for lactone ROP (Table 2).36 Significant polymerisation activity was observed for complex 2 at 60 °C, converting all ε-CL within 1 h (Table 2, entry 4). This was significantly faster than catalyst 1, which converted 64% of ε-CL under identical conditions and did not reach full conversion due to viscosity limitations (Table 2, entries 2 and 3). This trend was more pronounced at room temperature; complex 2 converted over seven times as much ε-CL in 4 h as 1 converted in 24 h (Table 2, entries 1 and 8). To confirm that ROP activity was due to complexes 1 and 2, zinc free control polymerisations with the two pyridone (2-pyridone for 1 and 6-methyl-2-pyridone for 2) ligands were conducted (Table 2, entries 4 and 9). These revealed 0% conversion after 4 h, suggesting that ROP activity was indeed due to zinc complexes 1 and 2 and not low quantities of free ligand.
:[ε-CL] ratio of 1:100 between 20–60 °C in toluene; these are typical conditions for lactone ROP (Table 2).36 Significant polymerisation activity was observed for complex 2 at 60 °C, converting all ε-CL within 1 h (Table 2, entry 4). This was significantly faster than catalyst 1, which converted 64% of ε-CL under identical conditions and did not reach full conversion due to viscosity limitations (Table 2, entries 2 and 3). This trend was more pronounced at room temperature; complex 2 converted over seven times as much ε-CL in 4 h as 1 converted in 24 h (Table 2, entries 1 and 8). To confirm that ROP activity was due to complexes 1 and 2, zinc free control polymerisations with the two pyridone (2-pyridone for 1 and 6-methyl-2-pyridone for 2) ligands were conducted (Table 2, entries 4 and 9). These revealed 0% conversion after 4 h, suggesting that ROP activity was indeed due to zinc complexes 1 and 2 and not low quantities of free ligand.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Temperature | Time (h) | Conversiona (%) | M n (kDa) | Đ | Initiation efficiencyd (%) | |
| Calculatedb | Observedc | |||||||
| 100:1 [ε-CL]:[cat], [ε-CL] = 0.87 M. ε-CL and the catalyst were both pre stirred in solvent for 5 minutes before mixing.a Conversion was calculated ex situ by 1H NMR spectroscopy.b Mn calc of polymers calculated from monomer conversion; Mn calc = ([ε-CL]0/[Cat]0) × (% conversion of ε-CL) × 114.14, assuming 1 polymer chain per catalyst centre.c Mn obs and Đ determined by size exclusion chromatography (SEC) using polystyrene standards in THF: values were corrected using a correction factor (0.56).37d Calculated according to eqn (S1)–(S3).†e Zinc free control reaction with the respective pyridone substrate.f Performed in THF rather than toluene. |
||||||||
| 1 | 1 | RT | 24 | 6 | — | — | — | — |
| 2 | 1 | 60 °C | 1 | 64 | 7.4 | 58.8 | 1.10 | 12.4 |
| 3 | 1 | 60 °C | 4 | 97 | 11.1 | 121.1 | 2.24 | 9.1 |
| 4e | Pyridone | 60 °C | 4 | 0 | — | — | — | — |
| 5 | 2 | 60 °C | 1 | >99 | 11.4 | 77.6 | 1.48 | 14.7 |
| 6 | 2 | RT | 1 | 2 | 0.2 | 9.5 | — | 2.5 |
| 7 | 2 | RT | 2 | 10 | 1.1 | 25.4 | 1.41 | 4.7 |
| 8 | 2 | RT | 4 | 43 | 4.9 | 45.8 | 1.47 | 11.2 |
| 9e | Me-pyridone | RT | 4 | 0 | — | — | — | — |
| 10f | 2 | RT | 1 | 0 | — | — | — | — |
| 11f | 2 | RT | 4 | 17 | 2.0 | 22.8 | 1.23 | 9.1 |
| 12f | 2 | RT | 23 | 81 | 9.3 | 51.0 | 1.32 | 19.1 |
Greater than calculated Mn values were observed using both complexes as catalyst, which was attributed to poor initiation resulting in kpropagation > kinitiation. The discrepancy between theoretical and experimental Mn was quantified by assigning an initiation efficiency value, which describes the theoretical % of active species which initiate ROP (eqn (S1)–(S3)†). While NacNac-Zn alkoxide complexes have been shown to exhibit high initiation efficiency, and therefore good Mn control, moving to less basic co-ligands (like carboxylate) results in a significant drop off in initiation efficiency, as evidenced in the work by Coates and co-workers (vide supra).16 The poor initiation efficiency of 1 resulted in very high MW PCL which formed a gel and hindered efficient mixing. This prevented full conversion from being achieved (Table 2, entry 3). An increase in Đ from 1.10 to 2.24 was noted (Table 2, entries 2 and 3), attributed to transesterification side reactions at high conversions.
Contrastingly, complete conversion was achieved with 2 accompanied by notably lower dispersity without mixing issues due to gelation (Table 2, entry 5). The slightly higher initiation efficiency for 2 is tentatively assigned to the greater steric encumbrance from the methyl group, favouring ring opening and forming a κ1-bound pyridonate. This is presumably required to enable the initiation step, which may proceed via monomer coordination. In any case, the initiation efficiency of both catalysts was noted to be poorer than the NacNac-Zn alkoxide complexes, which have demonstrated good control over Mn and Đ.16
Kinetic studies revealed that both complexes undergo an induction period, which was significantly reduced at elevated temperatures (Fig. 3). Induction periods have been observed for organometallic zinc-catalysed ROP and have been shown to vary with temperature, solvent and monomer.36,39–41 Post-induction, polymerisation proceeds via first order kinetics with respect to [ε-CL]. The near 4-fold reduction in kobs in THF cf. toluene is proposed to be due to coordinative competition between the THF solvent and the monomer, a phenomenon that has previously been observed in the literature.36,38,42,43 Note, with this bulky DippNacNac ligand the formation of 5-coordinate zinc complexes has extremely limited precedence therefore we disfavour their intermediacy in this process. Instead, we support the opening of the four membered strained metallacycle in monomeric 1 and 2 as a key step forming a three coordinate NacNac species that then binds an equivalent of monomer. We propose that this three coordinate zinc species is bonded through the oxygen of the pyridonate ligand, and that the increased sterics of 2 results in an increased propensity to ring-open the metallacycle due to steric repulsion, which could explain the increased rate relative to 1.
 | ||
| Fig. 3 1st order kinetic plot of ε-CL consumption by complexes 1 (60 °C, left, green) and 2 (RT, right, blue and purple). | ||
End-group elucidation
The high MW of the PCL produced by both catalysts made elucidation of end-groups by 1H NMR spectroscopy difficult, especially given the high MW at low polymer conversions (Table 2, entries 6 and 7). Therefore, matrix assisted laser desorption ionisation time-of-flight mass spectrometry (MALDI-ToF MS) analysis was used on both purified and crude PCL samples formed using catalysts 1 and 2. Major peaks corresponding to cyclic PCL were detected, with some minor peaks attributed to both pyridonate-capped chains and hydroxyl-capped chains (Fig. 4). This is strikingly different to that of NacNac-zinc acetate mediated lactone ROP reported in the literature, where polymer chains featuring acetate end groups have not been reported; this is presumably due to the greater nucleophilicity of the Zn-pyridonate unit enabling it to act as an initiator. | ||
| Fig. 4 MALDI-ToF MS of PCL made in the presence of complex 1 after 1 h at 60 °C (Table 2, entry 2), with a highlighted region shown to illustrate the polymeric species present. | ||
While the cyclic PCL signals may be overestimated when compared to their linear counterparts,44 their formation warrants discussion. This may occur through intramolecular back-biting/ring-closure termination reactions during polymerisation. It is possible that the leaving ability of the pyridonate group (consistent with the pKa of the pyridone units) facilitates this step, by enabling displacement of the relatively stable (compared to alkoxide) pyridonate anion and cyclisation of the PCL chain (refer to Fig. S14† for a proposed simplified schematic). Indeed, phenoxo-imine ligands complexed with group 1 metals have illustrated this ring-closing phenomenon, which also was attributed to the formation of more stable (than alkoxide) anionic leaving groups.45,46 While few studies have directly determined the pKa of the Schiff bases used in complexation, studies on structurally similar Schiff bases have revealed pKa values around 11, similar in magnitude to that of the pyridonates utilised herein.47 Cyclic polyesters have been shown to form during the ROP of cyclic esters with main group metal complexes of the TrenSal ligand, as reported previously by some of us.45 Comparison of the calculated pKa values for the TrenSal ligand and the pyridone ligands reported herein reveal they are between 9.11–11.91 in H2O, which tentatively supports a pKa based argument for cyclisation.48 NacNac-Zn alkoxide initiators, where the conjugate acid of the co-ligand typically has a much higher pKa, have not been reported to undergo analogous back-biting reactions to form cyclic polymers, and instead retain the alkoxide end-groups on linear polymer chains.23,49 In any case, the variety of PCL end-groups observed by MALDI-ToF is indicative of poor/stochastic initiation.
MALDI-ToF MS analysis of PCL samples prepared with 2, purified by quenching in wet CDCl3 and then precipitating in excess cold acidified MeOH revealed signals attributed to methoxy end groups (Fig. S13†) that were not observed in the crude samples. We attribute this to a combination of transesterification of the polymer end-groups and the ring-opening of macrocyclic caprolactone polymers. This phenomenon has been observed previously by Mehrkhodavandi and co-workers for PLA samples synthesised through zinc-catalysed ROP, that were quenched in wet chloroform and precipitated in cold MeOH,50 and by Sarkar, Chandrasekhar, Panda and co-workers, who used methanol to quench lactone polymerisations.51 In both instances, MeO end-groups were observed by either MALDI-ToF or 1H NMR spectroscopy, suggesting that this is not a feature of our catalytic system but rather an implication of the quenching and/or precipitation method.
Conclusions
Two new DippNacNac-Zn complexes with a pyridone-based co-ligand were synthesised and characterised in both the solid and solution-state. These complexes were active in the room temperature ROP of ε-CL to afford high MW PCL in the absence of exogenous co-initiators. Despite their different aggregation states in the solid state, both complexes are dominated by the monomeric form in the solution state under the polymerisation conditions. Therefore, these complexes are expected to act as single-site monometallic catalysts. MALDI-ToF MS analysis of the polymer products revealed a variety of end-groups with the formation of significant cyclic PCL species detected. This is distinct to DippNacNac-Zn-alkoxide derivatives and was attributed to a side-reaction involving back-biting of the growing polymer chains, facilitated by the relatively low (e.g. compared to iPrOH) pKa values of the pyridone groups enabling cleavage of the C(O)–Opyridone bond and formation of a pyridonate leaving group. Further, another series observed in the MALDI-ToF spectrum suggests that the pyridonate moiety can initiate ROP, furnishing pyridone end-capped PCL chains. These observations combined suggest that ROP proceeds by the ring-opening of the 4-membered Zn–N–C–O metallacycle to form a DippNacNac-Zn-aryloxide that is three coordinate at zinc and thus can bind a molecule of monomer to initiate polymerisation. This work shows that amidate groups can facilitate ROP of lactones under mild conditions, with distinct behaviour relative to acetate (which requires an initiator) and alkoxide (which forms no cyclic polymer), which suggests that amidate co-ligands warrant further exploration in other polymerisations.Experimental
General experimental details
All manipulations requiring inert conditions were performed under an argon atmosphere using standard Schlenk techniques or in a glove box. DippNacNac-ZnI30 and DippNacNac-ZnEt52 were synthesised using previously reported procedures. All reagents and solvents were obtained from Sigma-Aldrich, Fischer Scientific, Honeywell or Acros Organics and were used without further purification unless described otherwise. Dry THF, toluene and hexane were collected from a solvent purification system (Innovative Technologies), and stored over activated 4 Å molecular sieves under an argon atmosphere. Benzene-d6, chloroform-d, dimethylsulfoxide-d6 and toluene-d8 solvents were degassed by three freeze–pump–thaw cycles and stored over activated 4 Å molecular sieves under an argon atmosphere. ε-Caprolactone (ε-CL) was purified by vacuum distillation and was stored over activated 4 Å molecular sieves in the freezer. All manipulations involving silver compounds were kept in the dark with aluminium foil wrapping. 1H, 13C and 2D NMR (DOSY) spectra were recorded on Bruker AVA400, PRO500, AVA500 and AVA600 spectrometers at 298 K at 400 MHz, 500 MHz and 600 MHz and referenced to the residual solvent peaks (1H: δ 7.16 (C6D6), δ 7.26 (CDCl3), δ 2.50 ((CD3)2SO) and δ 2.08 (CD3C6D5)). The multiplicity of the signals are indicated as “s”, “d”, “t” “q” “pent”, “sept” or “m” for singlet, doublet, triplet, quartet, pentet, septet or multiplet, respectively. The reported DOSY masses (to the nearest whole number) and aggregation states were determined by comparison to a calibration plot made with a range of standards (adamantane, 2-phenylpyridine, pyrene, tri(o-tolyl)-phosphine, tetraphenylnaphthalene, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP)) with molecular weights varying from 136.2 to 622.7 g mol−1 in toluene-d8. More details are found in the ESI.† Elemental microanalyses of the silver salts for carbon, hydrogen, and nitrogen were performed by Elemental Microanalysis Ltd. SEC analyses of the filtered polymer samples were carried out in GPC grade THF at a flow rate of 1 mL min−1 at 35 °C on a 1260 Infinity II GPC/SEC single detection system with mixed bed C PLgel columns (300 × 7.5 mm). Suitable crystals of 1 or 2 were selected and mounted on a MITEGEN holder in oil on a Xcalibur, Eos diffractometer. The crystal was kept at 120.01(11) K during data collection. Using Olex2,53 the structure was solved with the SHELXT54 structure solution program using Intrinsic Phasing and refined with the SHELXL55 refinement package using Least Squares minimisation. Further experimental and refinement details are given in the CIF-files. CCDC 2285019–2285020 contains the supplementary crystallographic data for this paper.† MALDI-ToF MS analyses were performed using a Bruker Daltonics UltrafleXtreme™ MALDI-ToF/ToF MS instrument in either linear or reflectron mode. MALDI-ToF samples were made up either in a volume ratio of (a) 2:2:1 of polymer (10 mg mL−1), α-cyano-4-hydroxycinnamic acid (CHCA) (10 mg mL−1) and NaI (ionising agent, 10 mg mL−1) in THF or (b) 5:15:1 of polymer (10 mg mL−1), 2,5-dihydroxybenzoic acid (20 mg mL−1) and lithium iodide (10 mg mL−1) in THF. A droplet (2 μL) of the resultant mixture was spotted on to the sample plate and submitted for MALDI-ToF MS analysis.
Synthesis of 3
In a 100 mL round bottom flask, AgNO3 (6.6 mmol, 1.12 g) was added to 20 mL of distilled water with stirring, followed by 2-hydroxy-6-methylpyridine (6 mmol, 0.57 g). NH3 (aq) (11%, 6 mol L−1) in H2O was added dropwise with stirring until a neutral pH was achieved, affording an off-white suspension. The off-white suspension was filtered and washed, in order, with H2O (10 mL), EtOH (10 mL) and Et2O (10 mL). The resulting off-white solid was dried in vacuo (<0.28 mbar) at 50 °C for 5 h (1.22 g, 5.76 mmol, 48%).1H NMR (500 MHz, (CD3)2SO, 323 K): δ 7.23 (t, 1H, ArH), 6.19 (d, 1H, ArH), 6.16 (d, 1H, ArH), 2.37 (s, 3H, CH3).
13C{1H} NMR (126 MHz, (CD3)2SO, 323 K): δ 169.24, 153.76, 138.76, 112.39, 106.22, 25.96.
Analytical data (%), calculated: C, 33.37; H, 2.80; N, 6.49; found: C, 33.42; H, 2.79; N, 6.47.
Synthesis of 1
2-Pyridone (0.24 g, 2.5 mmol) was dissolved in dry PhCl (5 mL) and heated gently to fully dissolve the solid. In a separate flask DippNacNac-ZnEt (1.28 g, 2.5 mmol) was dissolved in dry PhCl (5 mL) and both solutions were stirred separately for 5 minutes. The DippNacNac-ZnEt solution was then added to the 2-pyridone solution dropwise. Upon addition the reaction mixture turned cloudy and began to effervesce. This solution was left to stir for 3 h at RT, after which the solution was concentrated under vacuum and placed in the freezer at −30 °C. After 16 h, a white powder formed which was isolated via filtration (0.57 g, 0.99 mmol, 40%).1H NMR (500 MHz, CD2Cl2, 298 K): δ 7.26 (m, 1H, ArH), 7.23 (m, 1H, ArH), 6.19 (m, 1H, ArH), 6.15 (m, 1H, ArH), 4.99 (s, 1H, NC(Me)CH), 3.20 (sept, 1H, CHMe2 (iPr)), 1.75 (s, 6 H, ArCH3), 1.19 (d, 12H, CHMe2), 1.08 (d, 12H, CHMe2).
13C{1H} NMR (126 MHz, CD2Cl2, 298 K): δ 172.98, 169.22, 144.27, 143.43, 142.39, 140.80, (134.12, 129.81, 128.54 and 126.56, residual PhCl solvent) 125.49, 123.49, 110.92, 109.67, 94.35, 27.95, 24.08, 23.75, 23.43.
APPI-MS: m/z [M]+: 575.2847 calculated [M]+: 575.2878.
Synthesis of 2
Ag-6-Methylpyridonate (3, 331 mg, 1.64 mmol) was added to a Schlenk flask followed by DippNacNac-ZnI (1.00 g, 1.64 mmol). Anhydrous CH2Cl2 (20 mL) was subsequently added to afford a cloudy yellow solution. The reaction was stirred in the dark at room temperature for 3 h, after which time stirring was stopped and the suspended solids were left to settle. The solution was filtered and the filtrate retained. CH2Cl2 was removed in vacuo from the filtrate to afford an off-white powder (0.766 g, 1.31 mmol, 80%). All crystals grown (from CH2Cl2 or hexane) were filtered and dried in vacuo at 50 °C for 6 h prior to NMR analysis and subsequent catalytic testing.1H NMR (500 MHz, CD3C6D5, 298 K): δ 6.73–6.70 (m, 1H, ArH), 5.88 (d, 1H ArH, J = 5 Hz), 5.78 (d, 1H ArH, J = 5 Hz), 4.87 (s, 1H, NC(Me)CH), 3.38 (sept, 1H, CHMe2 (iPr)), 2.02 (s, 3H, ArCH3), 1.68 (s, 6 H, NC(Me)CH), 1.21 (d, 12H, CHMe2), 1.17 (d, 12H, CHMe2).
13C{1H} NMR (126 MHz, CD3C6D5, 298 K): δ 174.29, 169.70, 153.02, 144.21, 142.93, 141.39, 126.56, 124.31, 109.33, 108.88, 95.39, 28.76, 25.14, 24.74, 24.09, 23.14.
APPI-MS: m/z [M]+: 590.31131. Calculated [M]+: 590.30833.
General polymerisation set up
In the glovebox, an air-tight 7 mL Supelco® glass vial was charged with 1 eq. of either 1 or 2 under argon and dissolved in dry solvent (toluene or THF, stored for >24 h over activated 4 Å molecular sieves). Another air-tight 7 mL Supelco® glass vial was charged with 100 eq. of ε-CL and dry solvent. Both vials were stirred at the target temperature for 5 minutes, after which time the monomer solution was transferred to the catalyst solution to start the polymerisation (the resultant polymerisation mixture has a monomer concentration of 0.87 M). The reaction was stirred at 300 rpm using a magnetic stirrer bar. After the desired amount of time, aliquots were taken from the reaction mixture and quenched in wet CDCl3 prior to 1H NMR analysis. Monomer conversion was calculated according to known monomer (4.15 ppm) and polymer (4.25 ppm) CH2 peaks in CDCl3. For PCL precipitated in MeOH, the reaction samples quenched in chloroform were dried using compressed air/nitrogen to remove all solvent. The crude PCL was then dissolved in a small amount (1–5 drops) of CHCl3 and added dropwise with stirring to an excess of cold (<0 °C) MeOH weakly acidified with HCl (approx. 2 drops of 12 M HCl per 50 mL of MeOH). The white precipitate was filtered and dried under air to give purified PCL.Author contributions
J. A. G. and M. J. I. supervised this project, and J. W. J. H. wrote the manuscript and performed the bulk of the reactions, with editing and revisions by both J. A. G. and M. J. I. Crystals were obtained by D. J. B. and solved by M. U. and G. S. N. 1 was tested by D. J. B. and F. S. and all computational calculations were performed by K. Y.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the UKRI Future Leaders Fellowship (J. A. G. MR\T042710\1), British Ramsay Memorial Trust (J. A. G.), British Royal Society (J. A. G. RSG\R1\180101) and the University of Edinburgh for funding. This project also has received funding from the European Union's Horizon 2020 Research and Innovation Programme (grant agreement No. 769599 to M. J. I.).References
- F. M. Windsor, I. Durance, A. A. Horton, R. C. Thompson, C. R. Tyler and S. J. Ormerod, Global Change Biol., 2019, 25, 1207–1221 CrossRef PubMed.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS PubMed.
- C. I. Gkountela and S. N. Vouyiouka, Macromolecules, 2022, 2, 30–57 CAS.
- N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- E. Fazekas, P. A. Lowy, M. A. Rahman, A. Lykkeberg, Y. Zhou, R. Chambenahalli and J. A. Garden, Chem. Soc. Rev., 2022, 51, 8793–8814 RSC.
- E. Fazekas, P. A. Lowy, M. A. Rahman, A. Lykkeberg, Y. Zhou, R. Chambenahalli and J. A. Garden, Chem. Soc. Rev., 2023, 52, 1157–1157 RSC.
- M. C. Tanzi, P. Verderio, M. G. Lampugnani, M. Resnati, E. Dejana and E. Sturani, J. Mater. Sci.: Mater. Med., 1994, 5, 393–396 CrossRef CAS.
- V. DeStefano, S. Khan and A. Tabada, Eng. Regen., 2020, 1, 76–87 Search PubMed.
- I. Manavitehrani, A. Fathi, H. Badr, S. Daly, A. Negahi Shirazi and F. Dehghani, Polymer, 2016, 8, 20 Search PubMed.
- A. Stjerndahl, A. Finne-Wistrand, A.-C. Albertsson, C. M. Bäckesjö and U. Lindgren, J. Biomed. Mater. Res., Part A, 2008, 87, 1086–1091 CrossRef CAS PubMed.
- M. Fuchs, P. M. Schäfer, W. Wagner, I. Krumm, M. Walbeck, R. Dietrich, A. Hoffmann and S. Herres-Pawlis, ChemSusChem, 2023, 16, e202300192 CrossRef CAS PubMed.
- R. D. Rittinghaus, P. M. Schäfer, P. Albrecht, C. Conrads, A. Hoffmann, A. N. Ksiazkiewicz, O. Bienemann, A. Pich and S. Herres-Pawlis, ChemSusChem, 2019, 12, 2161–2165 CrossRef CAS PubMed.
- P. V. S. Nylund, B. Monney, C. Weder and M. Albrecht, Catal. Sci. Technol., 2022, 12, 996–1004 RSC.
- R. L. Webster, Dalton Trans., 2017, 46, 4483–4498 RSC.
- B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed.
- M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1998, 120, 11018–11019 CrossRef CAS.
- L. R. Rieth, D. R. Moore, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 15239–15248 CrossRef CAS PubMed.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS PubMed.
- M. Cheng, A. B. Attygalle, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 1999, 121, 11583–11584 CrossRef CAS.
- F. Drouin, P. O. Oguadinma, T. J. J. Whitehorne, R. E. Prud'homme and F. Schaper, Organometallics, 2010, 29, 2139–2147 CrossRef CAS.
- M. Keram and H. Ma, Appl. Organomet. Chem., 2017, 31, e3893 CrossRef.
- H.-Z. Fan, X. Yang, J.-H. Chen, Y.-M. Tu, Z. Cai and J.-B. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202117639 CrossRef CAS PubMed.
- G. Trott, P. K. Saini and C. K. Williams, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef PubMed.
- R. Eberhardt, M. Allmendinger, G. A. Luinstra and B. Rieger, Organometallics, 2003, 22, 211–214 CrossRef CAS.
- C. Scheiper, C. Wölper, D. Bläser, J. Roll and S. Schulz, Z. Naturforsch., B: J. Chem. Sci., 2014, 69, 1365–1374 CrossRef CAS.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS PubMed.
- E. Spinner and J. C. B. White, J. Chem. Soc. B, 1966, 991–995 RSC.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, FL, 103rd edn, 2022 Search PubMed.
- M. P. Blake, N. Kaltsoyannis and P. Mountford, Chem. Commun., 2015, 51, 5743–5746 RSC.
- E. K. Brechin, L. M. Gilby, R. O. Gould, S. G. Harris, S. Parsons and R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 1998, 2657–2664 RSC.
- Y. Yu, A. R. Sadique, J. M. Smith, T. R. Dugan, R. E. Cowley, W. W. Brennessel, C. J. Flaschenriem, E. Bill, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2008, 130, 6624–6638 CrossRef CAS PubMed.
- G. S. Nichol and W. Clegg, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, o383–o385 CrossRef PubMed.
- H. W. Yang and B. M. Craven, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 912–920 CrossRef PubMed.
- B. Kozlevčar, M. Radišek, Z. Jagličić, F. Merzel, L. Glažar, A. Golobič and P. Šegedin, Polyhedron, 2007, 26, 5414–5419 CrossRef.
- W. Gruszka, H. Sha, A. Buchard and J. A. Garden, Catal. Sci. Technol., 2022, 12, 1070–1079 RSC.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114–2122 CrossRef CAS.
- W. Gruszka, L. C. Walker, M. P. Shaver and J. A. Garden, Macromolecules, 2020, 53, 4294–4302 CrossRef CAS.
- J. Chen, X. Wu, L. Zhang, Z. Duan and B. Liu, Polym. Chem., 2022, 13, 2971–2979 RSC.
- S. Lee, S. T. Baek, K. Anas, C.-S. Ha, D.-W. Park, J. W. Lee and I. Kim, Polymer, 2007, 48, 4361–4367 CrossRef CAS.
- J. A. Garden, A. J. P. White and C. K. Williams, Dalton Trans., 2017, 46, 2532–2541 RSC.
- H. Sheng, L. Zhou, Y. Zhang, Y. Yao and Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1210–1218 CrossRef CAS.
- W. Li, Z. Zhang, Y. Yao, Y. Zhang and Q. Shen, Organometallics, 2012, 31, 3499–3511 CrossRef CAS.
- S. M. Weidner and H. R. Kricheldorf, Rapid Commun. Mass Spectrom., 2020, 34, e8673 CrossRef CAS PubMed.
- Y. Zhou, G. S. Nichol and J. A. Garden, Eur. J. Org. Chem., 2021, 5557–5568 CrossRef CAS.
- F. M. García-Valle, R. Estivill, C. Gallegos, T. Cuenca, M. E. G. Mosquera, V. Tabernero and J. Cano, Organometallics, 2015, 34, 477–487 CrossRef.
- P. D. Bernardo, P. L. Zanonato, S. Tamburini, P. Tomasin and P. A. Vigato, Dalton Trans., 2006, 4711–4721 RSC.
- Q. Yang, Y. Li, J.-D. Yang, Y. Liu, L. Zhang, S. Luo and J.-P. Cheng, Angew. Chem., Int. Ed., 2020, 59, 19282–19291 CrossRef CAS PubMed.
- H.-Y. Chen, B.-H. Huang and C.-C. Lin, Macromolecules, 2005, 38, 5400–5405 CrossRef CAS.
- S. Soobrattee, X. Zhai, K. Nyamayaro, C. Diaz, P. Kelley, T. Ebrahimi and P. Mehrkhodavandi, Inorg. Chem., 2020, 59, 5546–5557 CrossRef CAS PubMed.
- S. Sagar, H. Karmakar, P. Nath, A. Sarkar, V. Chandrasekhar and T. K. Panda, Chem. Commun., 2023, 59, 8727–8730 RSC.
- M. Stender, R. J. Wright, B. E. Eichler, J. Prust, M. M. Olmstead, H. W. Roesky and P. P. Power, J. Chem. Soc., Dalton Trans., 2001, 3465–3469 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra and single crystal X-ray diffraction data. CCDC 2285019 and 2285020. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt03344a |
| This journal is © The Royal Society of Chemistry 2023 |