DOI:
10.1039/D3DT03177B
(Paper)
Dalton Trans., 2023,
52, 16492-16499
Mechanistic insights into nitric oxide oxygenation (NOO) reactions of {CrNO}5 and {CoNO}8†
Received
27th September 2023
, Accepted 13th October 2023
First published on 17th October 2023
Abstract
Here, we report the nitric oxide oxygenation (NOO) reactions of two distinct metal nitrosyls {Co–nitrosyl (S = 0) vs. Cr–nitrosyl (S = 1/2)}. In this regard, we synthesized and characterized [(BPMEN)Co(NO)]2+ ({CoNO}8, 1) to compare its NOO reaction with that of [(BPMEN)Cr(NO)(Cl−)]+ ({CrNO}5, 2), having a similar ligand framework. Kinetic measurements showed that {CrNO}5 is thermally more stable than {CoNO}8. Complexes 1 and 2, upon reaction with the superoxide anion (O2˙−), generate [(BPMEN)CoII(NO2−)2] (CoII–NO2−, 3) and [(BPMEN)CrIII(NO2−)Cl−]+ (CrIII–NO2−, 4), respectively, with O2 evolution. Furthermore, analysis of these NOO reactions and tracking of the N-atom using 15N-labeled NO (15NO) revealed that the N-atoms of 3 (CoII–15NO2−) and 4 (CrIII–15NO2−) derive from the nitrosyl (15NO) moieties of 1 and 2, respectively. This work represents a comparative study of oxidation reactions of {CoNO}8vs. {CrNO}5, showing different rates of the NOO reactions due to different thermal stability. To complete the NOM cycle, we reacted 3 and 4 with NO, and surprisingly, only 3 generated {CoNO}8 species, while 4 was unreactive towards NO. Furthermore, the phenol ring nitration test, performed using 2,4-di-tert-butylphenol (2,4-DTBP), suggested the presence of a proposed peroxynitrite (PN) intermediate in the NOO reactions of 1 and 2.
Introduction
Nitric oxide (NO) is a simple gas earlier thought to be an atmospheric pollutant and poison.1 In recent years, NO has been proven to be one of the essential signaling gases participating in a wide range of physiological processes, i.e., neurotransmission, vascular regulation, disaggregation of platelets, immune response towards multiple infections, etc.1,2 Inadequate NO generation causes biological dysfunctions (vide supra) and causes various diseases, such as diabetic hypertension,3 kidney disease,3 atherosclerosis,4 cognitive dysfunctions,5etc.6 Hence, to maintain biological homeostasis, two families of biological enzymes, i.e., nitric oxide synthases (NOSs)7,8 and nitrite reductases (NiRs),9 are involved in NO biosynthesis. When overproduced, NO leads to cytotoxicity by forming reactive nitrogen species (RNS), i.e., peroxynitrite (PN, OONO−)10 and nitrogen dioxide (˙NO2),11 upon reaction with dioxygen (O2),12 the superoxide anion (O2˙−),13 or hydrogen peroxide (H2O2).14 Thus, maintaining the optimal level of NO in the biosystem is necessary. Therefore, microbial/or mammalian systems oxidize excess NO to biologically benign nitrate (NO3−) using Fe-containing nitric oxide dioxygenase (NOD)15 enzymes via a plausible PN intermediate.16 In some bacteria and archaea, a unique di-iron protein carries out the process of NO detoxification by reducing it into N2O.17
Bio-mimetic modeling of NOD enzymes and their mechanistic investigation proposed the formation of a metal–dioxygen adduct upon reaction with O2, which then reacts with NO to generate NO3−via a proposed M–PN intermediate.15b,18 Several models of metal–dioxygen (M–O2) intermediates were developed to understand/establish the actual mechanism of the NOD reaction.19 In this regard, Kurtikyan et al. studied oxycoboglobin's NOD reaction that generates Co–NO3− at low temperatures.20 Also, CrIV–O22− and CoIII–O22− species produced CrIII–NO3− and CoII–NO3− species when reacted with NO, respectively.19c,21 In addition to NOD reaction products, NO-monooxygenation (NOM) products were also observed in several metal–dioxygen adduct reactions with NO. Karlin and coworkers observed CuII–NO2− in the reaction of CuII–O2˙− with NO via a PN intermediate.22 Nam and coworkers observed a NOM product formation (CrIII–NO2−) in the reaction of CrIII–O2˙− with NO via a CrIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species.19b Contrarily, the reaction of Fe–O22− with NO+ led to an FeIII–NO3− complex, a NOD reaction product.19a In another case, Fe–O2˙− and Mn–O22− bearing the TAML ligand led to the formation of FeIII–NO3− and MnIVO + NO2via a presumed PN intermediate, respectively.23 Thus, the metal center considerably controls the NO oxygenation reaction (NOD vs. NOM). In this regard, several comparative studies were performed to understand the role of the metal center. Recently, we have studied the comparative NO oxygenation reactivity of Co–O22− and Ni–O22− bearing a similar 12TMC ligand framework that generates CoII–NO2− and NiII–NO3− as the end products.24 Groves and coworkers reported the formation of FeIVO and NO2 in the reaction of metHb with the PN molecule.25 Considerable work is underway and has also already been performed to establish the presence of a PN intermediate in NO oxidation reactions, i.e., IR,19f,20,26 EPR,19fetc.; however, the same is under debate. In contrast, Pacheco and the group proposed that the NOD reaction with oxymyoglobin does not share the PN intermediate.27 Further supported by Moënne-Loccoz and coworkers’ work on the NOD reaction of oxymyoglobin, it is portentous that the millisecond intermediate is an FeIII–NO3− species and not a PN intermediate.28 Although the PN intermediate was not detected in the oxy-globin protein's NOD reaction, the experimental results proposed a short-lifetime intermediate before forming metal–NO3−.19e In biology, an additional pathway of the NOD reaction has also been presented, which suggests Fe–NO formation upon reaction of NO with the Fe-center of Hb, and then it reacts with O2, resulting in NO3− formation.29 However, a reverse pathway was investigated in detail by Stuehr and coworkers by taking a series of different Fe–NO species.30
O species.19b Contrarily, the reaction of Fe–O22− with NO+ led to an FeIII–NO3− complex, a NOD reaction product.19a In another case, Fe–O2˙− and Mn–O22− bearing the TAML ligand led to the formation of FeIII–NO3− and MnIVO + NO2via a presumed PN intermediate, respectively.23 Thus, the metal center considerably controls the NO oxygenation reaction (NOD vs. NOM). In this regard, several comparative studies were performed to understand the role of the metal center. Recently, we have studied the comparative NO oxygenation reactivity of Co–O22− and Ni–O22− bearing a similar 12TMC ligand framework that generates CoII–NO2− and NiII–NO3− as the end products.24 Groves and coworkers reported the formation of FeIVO and NO2 in the reaction of metHb with the PN molecule.25 Considerable work is underway and has also already been performed to establish the presence of a PN intermediate in NO oxidation reactions, i.e., IR,19f,20,26 EPR,19fetc.; however, the same is under debate. In contrast, Pacheco and the group proposed that the NOD reaction with oxymyoglobin does not share the PN intermediate.27 Further supported by Moënne-Loccoz and coworkers’ work on the NOD reaction of oxymyoglobin, it is portentous that the millisecond intermediate is an FeIII–NO3− species and not a PN intermediate.28 Although the PN intermediate was not detected in the oxy-globin protein's NOD reaction, the experimental results proposed a short-lifetime intermediate before forming metal–NO3−.19e In biology, an additional pathway of the NOD reaction has also been presented, which suggests Fe–NO formation upon reaction of NO with the Fe-center of Hb, and then it reacts with O2, resulting in NO3− formation.29 However, a reverse pathway was investigated in detail by Stuehr and coworkers by taking a series of different Fe–NO species.30
Hence, the NO activation using metal ions has been an active field of research for chemists and biochemists for many years to understand its coordination chemistry and reactivity.31 In various biological enzymatic reactions, i.e., nitrogen fixation,32 NiR reaction,24,33 NOD reactions,15a,34etc., M–NOs are the key intermediates. In recent years, very few M–NOs have been prepared and explored for their various reactions to understand and mimic the biological M–NOs’ reactivity.15c,31d,35 Among them, only a few M–NOs were examined for NO-oxidation reactions, i.e., reactions with dioxygen,12 superoxides,13 base,36 and H2O.37 Oxidation of M–NOs generates NO mono- or di-oxygenated products, usually depending on the type of M–NO and stability of the intermediate involved. {CoNO}8 produced NO2− when reacted with O2;38 in another example, {FeNO}7 formed NO2− from NO oxidation.39 Recently, Nam and coworkers showed the oxidation of {CoNO}8 to CoII–NO3− and CoII–NO2− + O2 upon reaction with O2 and O2˙−, respectively.35b Also, Mondal and coworkers reported the NO3− generation from {CoNO}8 and {CuNO}10 species upon reaction with hydrogen peroxide (H2O2) via a proposed PN intermediate.40 In contrast to {CoNO}8 reactivity towards O2˙−, {MnNO}6 upon reaction with O2˙− generated MnIII–NO3−via a presumed PN intermediate.41 As the oxidized products of M–NOs depend upon the choice of the metal center, their oxidation state, and the intermediate involved in the reaction, a deep study is required to establish the actual mechanism of the M–NO oxidation reactions.
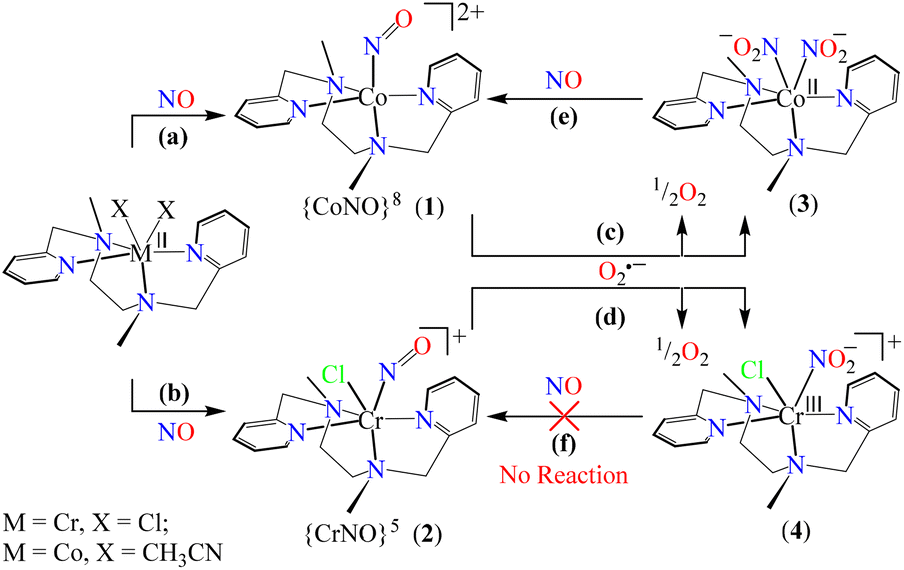
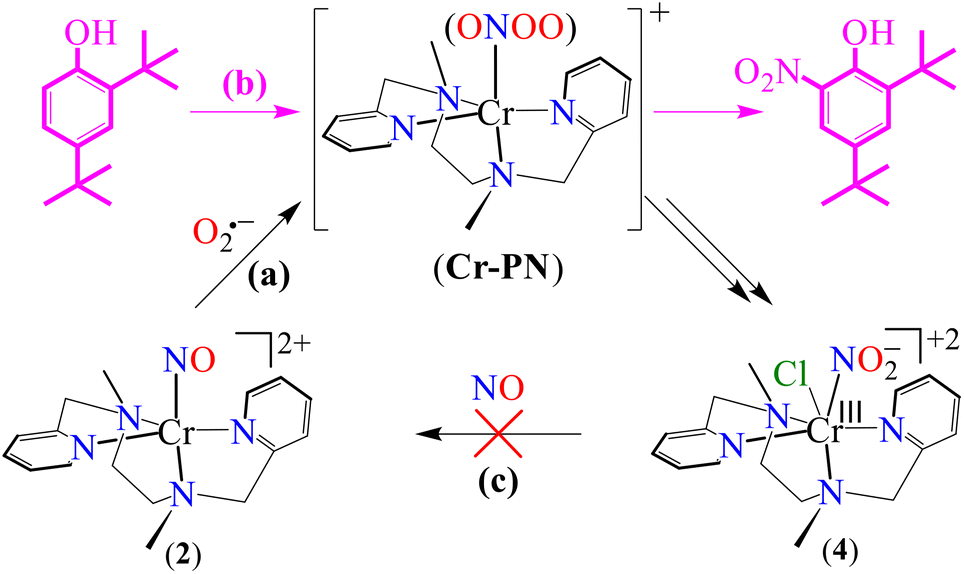
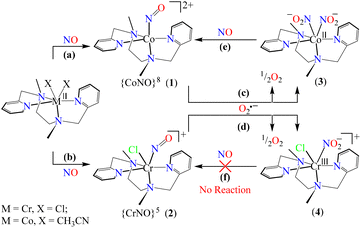
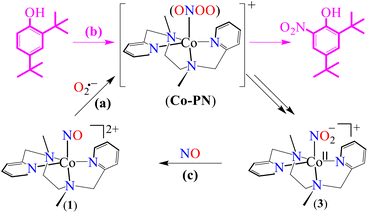
Among various metal–nitrosyl complexes, {CrNO}5 and {CoNO}8 species are widely explored and known to be reasonably stable with linear and bent metal–NO coordination, respectively.24,33e,35b,42 Thus, to understand the NO oxidation reactions of metal–nitrosyl complexes, we prepared {CrNO}5 (S = 1/2) and {CoNO}8 (S = 0), having different spin states/magnetic properties with similar ligand frameworks. Therefore, [(BPMEN)Co(NO)]2+ ({CoNO}8, 1) and [(BPMEN)Cr(NO)(Cl−)]+ ({CrNO}5, 2) complexes43 (BPMEN = N,N′-bis(2-pyridylmethyl)-1,2-diaminoethane) were explored for their reactivity towards O2˙− (KO2/18-crown-6), to understand the effect of the metal center and the spin state/magnetic properties (Scheme 1). Following our previous reports,43,44 we synthesized new {CoNO}8 and {CrNO}5 complexes and calculated various physical parameters for 1 and 2 to determine their thermal stability, the NO oxidation reactions and the intermediates involved (Scheme 1, reactions a and b). Complex 1 generates a CoII–nitrito complex [(BPMEN)CoII(NO2−)2] (CoII–NO2−, 3) + O2 in the presence of O2˙−via a proposed thermally unstable [Co–PN]+ species (Scheme 1, reaction c). However, 2 generates an oxidized CrIII–nitrito complex [(BPMEN)CrIII(NO2−)Cl−]+ (CrIII–NO2−, 4) + O2 upon reaction with O2˙−via a proposed [Cr–PN]+ intermediate species (Scheme 1, reaction d). The phenol ring nitration test performed using 2,4-DTBP suggested the presence of the proposed PN intermediate in the NOM reactions of 1 and 2. Mechanistic studies using 15N-labeled nitric oxide (15NO) revealed that the N-atoms of 3 (CoII–15NO2−) and 4 (CrIII–15NO2−) were derived from the 15NO moieties of 1 and 2, respectively. In addition, to complete the NOM cycle, we reacted 3 and 4 with NO, which showed the formation of {CoNO}8 from 3, while 4 was unreactive to NO (Scheme 1, reactions e and f). The equilibrium constant (Keq) of the formation of 2 is ∼25 times that of 1, suggesting that 2 is more stable than 1; hence, it also explains why the reaction of 2 with O2˙− is slower than that of 1. In both reactions, we observed NO2− (NOM) formation; however, only complex 3 could generate the initial M–NO (1) while 4 was unreactive towards NO.
 |
| | Scheme 1 NO activation at Cr and Co centers and NOO reactions of 1 and 2. | |
Results and discussion
Synthesis of Co–nitrosyl, [(BPMEN)Co(NO)]2+ ({CoNO}8, 1)
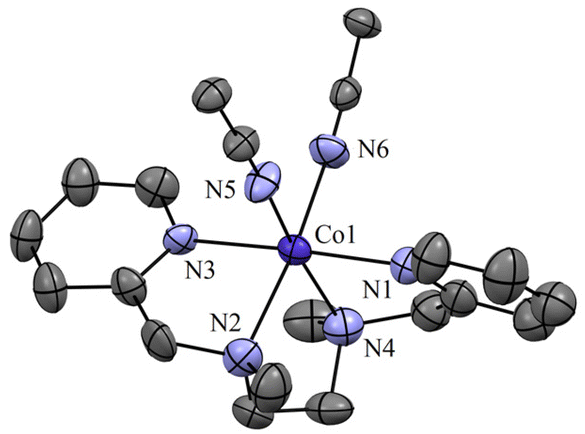
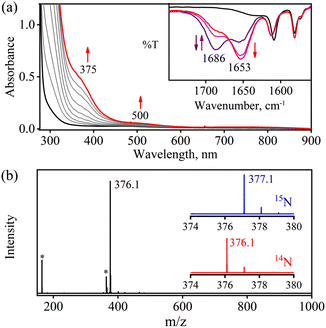
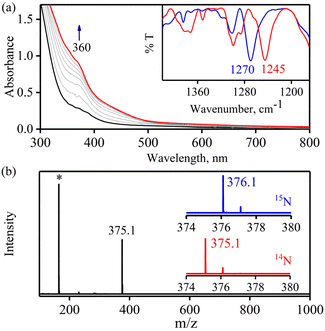
The initial CoII-complex [(BPMEN)CoII(CH3CN)2]2+ (Co-1) was synthesized by adding the BPMEN ligand to a stirring solution of [CoII(H2O)6](BF4)2 and characterized with various spectroscopic measurements (Fig. 2, see the ESI and Experimental section (ES), Fig. S1†). The addition of excess NO to the CH3CN solution of Co-1 at 233 K under an Ar atmosphere resulted in the generation of [(BPMEN)Co(NO)]2+ ({CoNO}8, 1) (λmax = 375, ε = 956 M−1 cm−1, red line) within one hour (Fig. 1a and ESI Fig. S2a†) (Scheme 1, reaction a). The FT-IR spectrum also reveals that the NO moiety is bound to a Co-center, suggesting a bent NO with a typical Co–NO stretching at 1653 cm−1 (inset: Fig. 1a; ESI, Fig. S2b†).33e,35a,b Electrospray ionization mass spectrometry (ESI-MS) of 1 showed a prominent ion peak at m/z 376.1, whose mass and isotope distribution patterns correspond to [(BPMEN)Co(NO)(OH−)]+ (calcd m/z 376.1) (ESI,†Fig. 1b). Upon substitution of the NO moiety with 15N-labeled 15NO in 1,24,45 the mass peak corresponding to [Co(BPMEN)(15NO)(OH−)]+ appears at m/z 377.1 (calcd m/z 377.1) (inset: Fig. 1b; ESI, Fig. S2c†), suggesting that the NO moiety is bound to the Co-center. The Evans’ method established a high-spin CoII-center (S = 3/2) in Co-1 (ESI, Fig. S1e†);46 hence, its 1H NMR does not show any signal for aromatic/aliphatic protons in the normal range (ESI, Fig. S3a†). However, we observed these signals in complex 1, confirming a diamagnetic Co-center (ESI, Fig. S3b†). The redox potential of 1 was determined using a cyclic voltammogram (ESI, Fig. S3c†). To perform NOO experiments, we prepared/isolated 1 by purging Co-1 with excess NO gas in CH3CN at 233 K under Ar (ESI, ES;† yield: 76%).
 |
| | Fig. 1 (a) UV-vis spectral change of Co-1 (0.5 mM, black line) upon addition of NO(g) in CH3CN under Ar at 233 K. Inset: Solution IR spectra of the formation of 1 (red line). (b) ESI-MS spectra of 1. The peak at 376.1 is assigned to [(BPMEN)CoII(NO)(OH−)]+ (calcd m/z 376.1). Inset: Isotopic distribution patterns of 1-14NO (red line) and 1-15NO (blue line). The peaks at m/z 364.1 and 164.5 marked with asterisks are assigned to [(BPMEN)Co(Cl−)]+ and [(BPMEN)Co]2+, respectively. | |
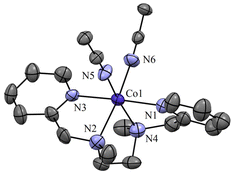 |
| | Fig. 2 Displacement ellipsoid plot (30% probability) of Co-1 at 298 K. Anions and H-atoms have been removed for clarity. | |
NO oxygenation reaction of the {CoNO}8 complex (1)
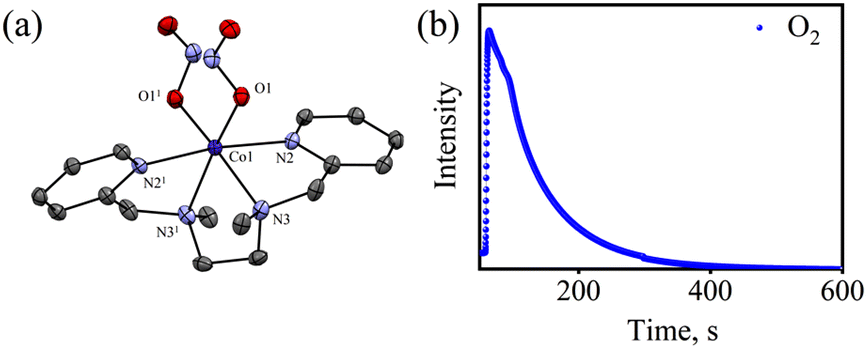
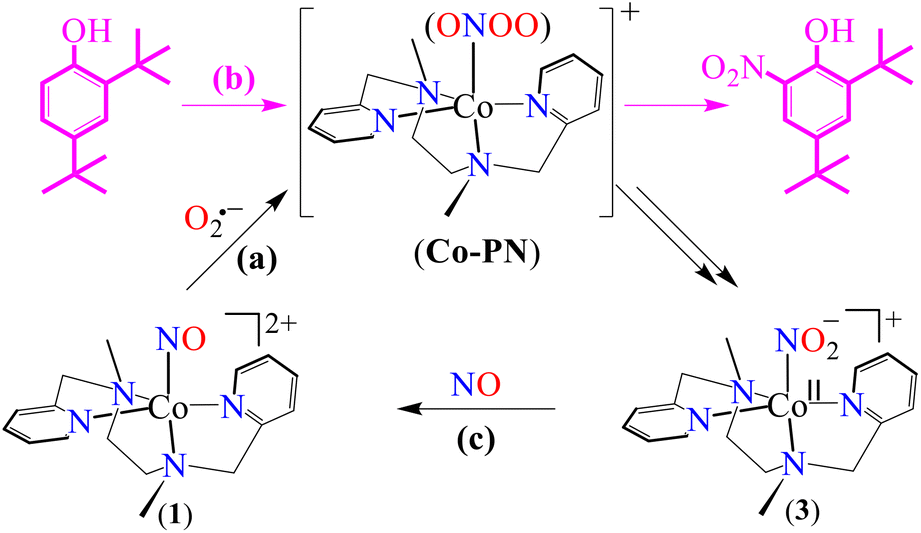
To explore the NO oxygenation (NOO) reaction of {CoNO}8, we reacted the new Co–NO complex [(BPMEN)Co(NO)]2+ ({CoNO}8, 1; S = 0) with O2˙−. The addition of O2˙− to a solution of 1 resulted in the generation of [(BPMEN)CoII(NO2−)2] (CoII–NO2−, 3). The characteristic UV-vis absorption bands of 1 (λmax = 375 nm, ε = 956 M−1 cm−1) changed to a new band (λmax = 360 nm, ε = 4040 M−1 cm−1), which corresponds to 3, within 5 minutes in CH3CN at 298 K under Ar (Fig. 3a; ESI, Fig. S5†). However, 1 does not show any spectral changes in the absence of O2˙−, ruling out the natural decomposition of 1 generating free NO followed by the reaction with O2˙− (ESI, Fig. S5b†). Complex 3 was determined to be [(BPMEN)CoII(NO2−)2] based on various spectroscopic and single-crystal X-ray structural analyses (vide infra). The FT-IR spectrum of 3 showed a characteristic peak for CoII-bound NO2− stretching at 1270 cm−1, which shifted to 1245 cm−1 (15NO2−) when 3 was generated in the reaction of 15N-labeled NO {Co15NO}8 and O2˙− (inset: Fig. 3a; ESI, Fig. S6a and b†), suggesting that the N-atom in the 15NO2− moiety is derived from {Co15NO}8. The ESI-MS spectrum of 3 showed a prominent peak at m/z 375.1, [(BPMEN)Co(14NO2−)]+ (calcd m/z 375.1), which shifted to 376.1, [(BPMEN)Co(15NO2−)]+ (calcd m/z 376.1), when the reaction was performed using {Co15NO}8 (Fig. 3b; ESI, Fig. S7†), indicating that the NO2− derived from the NO moiety of 1. We did not observe the characteristic signal of aliphatic protons of the BPMEN ligand for 3 in the 1H-NMR spectrum, signifying a bivalent cobalt center.35a,45c In addition, Evans’ method confirmed a low-spin CoII-center (S = 1/2), as the magnetic moment of 3 was found to be 1.77 BM (ESI; ES, Fig. S8†).46 Electrochemical measurement of 3 showed a non-reversible cyclic voltammogram (ESI, Fig. S9†). The exact conformation of 3 was determined by single-crystal X-ray crystallographic structural analysis (Fig. 4a, ESI, ES, and Tables T1 and T2†). The two NO2− ligands are coordinated to a Co-center in an end-on fashion with a distorted octahedral geometry. The Co–O–N and O–N–O bond angles were 118.91 and 116.12, respectively. The Griess reagent test confirmed the amount of NO2− generated in the above reaction and was determined to be 93(±5)% (ESI; SI, Fig. S10†).35a,47 Various spectral and structural analyses of 3 undoubtedly showed that the reaction of 1 with O2˙− generated CoII–NO2− (3) as the NOM product (Scheme 1c). The side product of the NOM reaction of 1 was determined to be O2, which is believed to be formed via a proposed transient [Co–PN]+ intermediate (Scheme 2). The PN intermediate is known to be a source of the reactive oxygen atom, which can produce O2;35a also, in an aqueous medium, PN was found to generate the NO2− anion with O2.48 Recently, one of our reports on the CoIII–peroxo reaction with NO showed the generation of NO2− + O2via a [Co–PN]+ intermediate,24 and similarly Nam and coworkers demonstrated the formation of CoII–NO2− and O2via a [Co–PN]+ intermediate.35a Likewise, the generation of CuII–NO2− with the evolution of O2 was observed from a [Cu–PN]+ intermediate.22,49 Hence, in the above reaction, the generation of CoII–NO2− with O2 evolution supports our assumption of the proposed [Co–PN]+ intermediate, as described in the previous reports on aqueous PN chemistry,48 non-aqueous CoII–PN,35a and CuII–PN22,49 chemistry. We followed and trapped the evolved O2 by following its generation from the reaction solution to support our assumption as proposed in [Co–PN]+ chemistry19f,20,26 and aqueous PN chemistry.48 To confirm the formation of O2, we carried out the reaction of 1 with O2˙− and followed the generation of gases by reaction flask headspace analysis using a gas-mass analyzer and observed the formation of O2 (Fig. 4b). In addition, we attempted to characterize the proposed [Co–PN]+ intermediate to elucidate the mechanism of its conversion to 3 in the above NOM reaction (Scheme 2). However, our efforts to characterize the [Co–PN]+ were futile due to its unstable nature. However, indirectly, the PN intermediate was detected by a DTBP ring-nitration test, as reported in the previous literature19c,f,41,42c and as explained in the chemistry we have described earlier (vide supra).12 The generation of NO2-2,4-DTBP (∼52%) and 2,4-DTBP-D (∼12%) (ESI, Fig. S11†) actively supports the proposed reaction mechanism in the above NOM reaction and, therefore, the formation of a [Co–PN]+ intermediate in the reactions of 1 with O2˙− (Scheme 2, reactions a and b). The formation of a [Co(BPMEN)(NO)(O2˙−)]+ species before PN formation, as reported earlier, can't be ruled out,42c,50 but such an intermediate further undergoes rearrangement to generate the PN intermediate ultimately. We did not observe the formation of such species spectroscopically; however, indirect proof from the phenol ring nitration test confirms the PN formation.
 |
| | Fig. 3 (a) UV-vis spectral change of 1 (0.25 mM, black line) upon addition of KO2/18-crown-6 in CH3CN under Ar at 298 K. Inset: IR spectra of 3-14NO2− (blue line) and 3-15NO2− (red line) in KBr. (b) ESI-MS spectra of 3. The peak at 375.1 is assigned to [(BPMENCo(NO2−)]+ (calcd m/z 375.1). Inset: Isotopic distribution patterns of 3-14NO2− (red line) and 3-15NO2− (blue line). | |
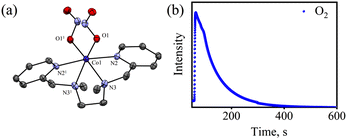 |
| | Fig. 4 (a) Displacement ellipsoid plot (30% probability) of 3 at 100 K. H-atoms have been removed for clarity. (b) Mass spectra of the formation of O2 in the reaction of 1 (20.0 mM) with O2˙−. | |
 |
| | Scheme 2 Phenol ring nitration to trap the [Co–PN]+ intermediate. | |
NO oxygenation reaction of the {CrNO}5 complex (2)
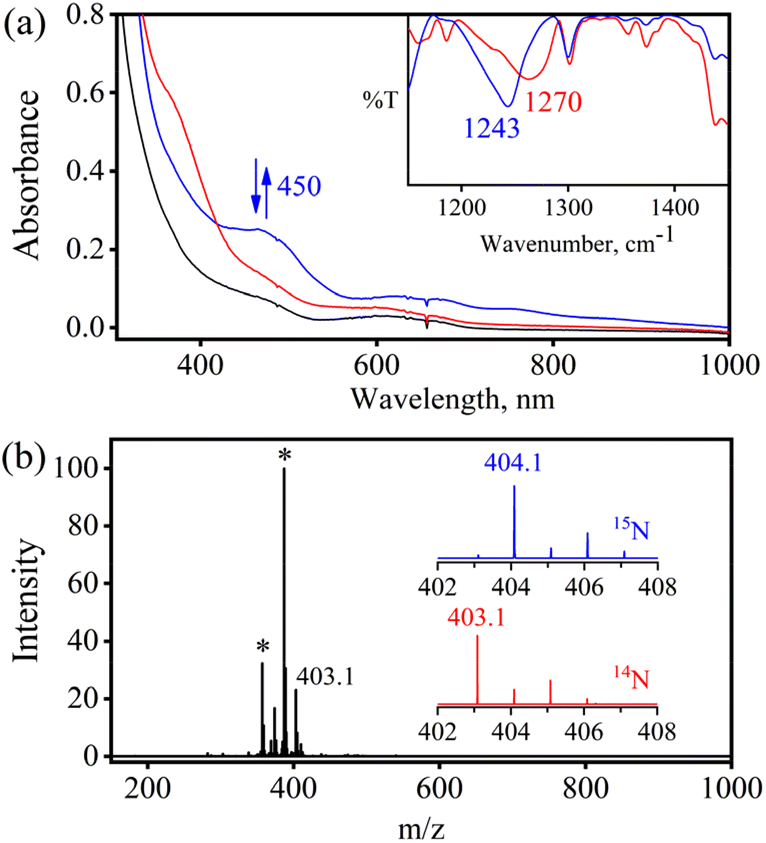
In order to find the influence of the metal center on oxidation of metal-bound nitrosyls, we explored the reaction of [(BPMEN)Cr(NO)(Cl−)]+ ({CrNO}5, 2) with O2˙− (yield: 70%). Complex 2 was synthesized and isolated by following our previous report.43 The addition of one equivalent of O2˙− (KO2/18-crown-6) to a CH3CN solution of 2 under Ar showed a color change from light green to dark red at 298 K. Upon reaction of 2 with O2˙−, the characteristic UV-vis absorption bands of 2 (black line, λmax = 600 nm) changed to a new band (blue line, λmax = 450 nm) within ∼one minute (Fig. 5a), which gradually changed to the red line (NO-oxidized product, 4) in ∼five minutes (Fig. 5a, ESI, Fig. S12a†) at 298 K under an Ar atmosphere. It is worth noting that 2 does not show any spectral changes in the absence of O2˙− under similar reaction conditions, suggesting that 2 is thermally stable; therefore, we can rule out the natural decomposition of 2 (ESI, Fig. S12b†).51 In addition to the UV-vis spectral analysis, we investigated the NOO reaction of 2 with different spectral measurements and tried to follow or characterize the proposed [Cr–PN]+ intermediate. However, being a thermally unstable species, our efforts to spectroscopically characterize the proposed [Cr–PN]+ failed.
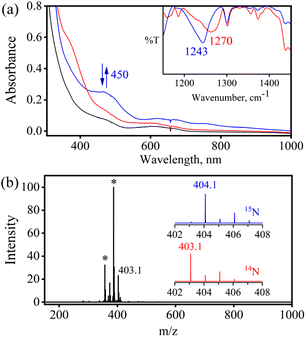 |
| | Fig. 5 (a) UV-vis spectral change of 2 (1 mM, black line) upon addition of KO2/18-crown-6 showing the formation of the blue line, and it further decomposes to form 4 (red line) in CH3CN under Ar at 298 K. Inset: IR spectra of 4-14NO2− (red line) and 4-15NO2− (blue line) in KBr. (b) ESI-MS of 4 formed in the reaction of 2 with KO2/18-crown-6 recorded in CH3CN. The peak at m/z 403.1 is assigned to [(BPMEN)Cr(14NO2−)(Cl−)]+ (calcd: m/z 403.1). The peaks at 387.1, 357.1, and 374.1 marked with asterisks are assigned to [(BPMEN)Cr(NO)(Cl−)]+ (calcd: m/z 387.1), [(BPMEN)Cr(Cl−)]+ (calcd: m/z 357.1) and [(BPMEN)Cr(OH−)(Cl−)]+ (calcd: m/z 374.1), respectively. Inset: Isotopic distribution patterns of 4-14NO2− (red line) and 4-15NO2− (blue line). | |
Further, to understand the NOO product of 2, we characterized the reaction products with different spectral measurements. The FT-IR spectrum of the isolated product (4) from the reaction of 2 with O2˙− showed a new peak at 1270 cm−1, characteristic of NO2− stretching frequency (inset in Fig. 5a; ESI, Fig. S13a†).19b The NO2− stretching frequency shifted to 1243 cm−1 (15N16O2−) when reacting 15NO-labeled 2 (i.e., [(BPMEN)Cr(15NO)(Cl−)]+) with O2˙− (inset in Fig. 5b; ESI, Fig. S13b†). The negative shifting of NO2− stretching frequency (Δ = 27 cm−1) denoted that the N atom in the NO2− anion came from the NO moiety of 2. The ESI-MS spectrum of 4 exhibited a prominent ion peak at m/z 403.1, [(BPMEN)CrIII(NO2−)(Cl−)]+ (calcd. m/z 403.1), which shifted to m/z 404.1, [(BPMEN)CrIII(15NO2−)(Cl−)]+ (calcd. m/z 404.1), when the reaction was performed with 15N-labeled 2 ({Cr15NO}5) (Fig. 5b; ESI, Fig. S14†), indicating clearly that the NO2− in 4 is derived from the NO moiety. Also, we compared the UV-vis spectrum of 4 with that of independently prepared CrIII–NO2− (ESI, Fig. S15†), which further confirmed the formation of the CrIII–NO2− complex in the NOO reaction of 2. In addition, we calculated the magnetic moment of 4 by Evans’ method and found it to be 3.54 BM (theoretical μs = 3.87 BM), confirming a CrIII center (d3) (ESI, Fig. S16†). Additionally, formation of 4 from the NOM reaction of 2 was also confirmed with EPR spectra as the spectrum of 4 showed a peak at g = 3.5 and 4.90 (ESI, Fig. S17†).52 Cyclic voltammetric measurements of 4 showed a quasi-reversible cyclic voltammogram, clearly different from that of 2, suggesting a completely new species (ESI, Fig. S16b and c†). For the exact validation of the NOO product of 2, different spectroscopic data of an authentic sample of 4 were compared with those of the product obtained in the reaction of 2 with O2˙−. This comparison confirmed that the oxidized product of 2 is a CrIII–metal complex bound to the NO2− anion.19b Complex 4 was found to be thermally stable, showing no natural decay in the UV-Vis measurements (ESI, Fig. S18a†). Finally, we determined the amount of NO2− ions by a Griess reagent assay34a,47 in the reaction of 2 with NO and found it to be 87(±5)% (ESI, Fig. S10†). Various spectroscopic characterization studies of 4 showed that the reaction of {CrNO}5 with O2˙− yielded CrIII–NO2− (4) as the NOM product (Scheme 3). Using a gas-mass analyzer we observed the formation of O2 (ESI, Fig. S19b†).
 |
| | Scheme 3 Phenol ring nitration to trap the [Cr–PN]+ intermediate. | |
Isomerization of the PN moiety is usually possible via O–O bond homolysis to form NO3−,19c,20,41 or NO2− + O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 48a,b,53 upon rearrangement. It is known that M–PN intermediates are highly unstable, and there are only a few reports on the spectral characterization of metal-bound PN intermediates.19f,20,26b,54 However, alternatively, PN can also be confirmed using its phenol ring nitration chemistry when reacted with 2,4-DTBP vide supra.19a–c,f,26b,40b,50a,54a,55 We observed the formation of NO2-2,4-DTBP (yield: 65%) when 2 was reacted with O2˙− in the presence of 2,4-DTBP (ESI, Fig. S20†). This phenol ring nitration test using 2,4-DTBP supports that the reaction of 1 with O2˙− is going through a proposed [Cr–PN]+ intermediate and generates CrIII–NO2− + O2 (Scheme 3).
48a,b,53 upon rearrangement. It is known that M–PN intermediates are highly unstable, and there are only a few reports on the spectral characterization of metal-bound PN intermediates.19f,20,26b,54 However, alternatively, PN can also be confirmed using its phenol ring nitration chemistry when reacted with 2,4-DTBP vide supra.19a–c,f,26b,40b,50a,54a,55 We observed the formation of NO2-2,4-DTBP (yield: 65%) when 2 was reacted with O2˙− in the presence of 2,4-DTBP (ESI, Fig. S20†). This phenol ring nitration test using 2,4-DTBP supports that the reaction of 1 with O2˙− is going through a proposed [Cr–PN]+ intermediate and generates CrIII–NO2− + O2 (Scheme 3).
In addition, to determine the driving forces for the NOO reaction of 1 and 2, the equilibrium constants (Keq) for the formation of {CoNO}8 and {CrNO}5 were determined and found to be 88 M−1 and 2270 M−1 (ESI, Fig. S21a and b†), respectively, which suggest that the Keq for 2 is ∼25 times larger than that of 1. This comparison of Keq values undoubtedly suggests that {CrNO}5 is more stable than {CoNO}8. Hence, the NOO reaction of {CoNO}8 was found to be faster than that of {CrNO}5 in the presence of O2˙−.
NO activation of NOM products (3 and 4)
To further explore the chemistry of CoII–NO2− (3) and CrIII–NO2− (4) complexes, we investigated their reactions with excess NO. In this regard, we reacted isolated 3 (obtained in the reaction of 1 with one equivalent of O2˙−) with excess NO to explore its NO activation chemistry. In the reaction, we observed the decomposition of the band 355 nm and the formation of a new absorption band (λmax = 375 nm) in ∼2 hours in CH3CN under an Ar atmosphere at 233 K, suggesting the generation of a new species, believed to be {CoNO}8 (Fig. 6) (Scheme 2, step c). This indicates that complex 1 first reacts with one equivalent of O2˙− to generate the corresponding CoII–NO2−, which reacts further with NO to produce {CoNO}8. In contrast to the NO activation of 3, the reaction of 4 with excess NO did not yield Cr–NO, [(BPMEN)Cr(NO)(Cl−)]+ (2), under similar reaction conditions (Scheme 3, step d and ESI, Fig. S22†) which is so obvious as the end product of the NOO reaction is the CrIII system. The exploration of the NO activation chemistry of CoII (d7, S = 3/2 or d7, S = 1/2), CrII (d4, S = 2), and CrIII (d3, S = 3/2) complexes with similar ligand frameworks suggests that M–nitrosyl formation depends on the oxidation states and physical parameters of the metal center.
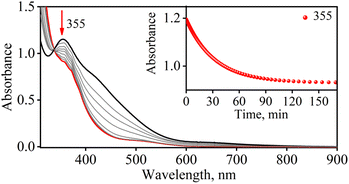 |
| | Fig. 6 UV-Vis spectral changes observed in the reaction of 3 (0.5 mM) with NO in CH3CN under Ar at 233 K. Inset: Time course of decomposition of 3 monitored at 355 nm (red circles). | |
Conclusion
In this report, we have demonstrated that the nitric oxide oxygenation (NOO) reactions of Co–nitrosyl, [(BPMEN)Co(NO)]2+ ({CoNO}8, 1), and Cr–nitrosyl, [(BPMEN)Cr(NO)(Cl−)]+ ({CrNO}5, 2), complexes, bearing a common BPMEN ligand, are regulated by the stability of metal–nitrosyls and the intermediate species involved in the reactions. Here, we observed that the reaction of 1 with O2˙− generates a CoII–nitrite complex in the same oxidation state, [(BPMEN)CoII(NO2−)2] (3), with O2 evolution via a proposed [Co–PN]+ intermediate, as observed in other examples of NOM chemistry.22,24 In contrast, when 2 reacted with O2˙−, it generated an oxidized CrIII–nitrite complex, [(BPMEN)CrIII(NO2−)Cl−]+ (4), and O2via a putative [Cr–PN]+ intermediate, similar to the chemistry of CoII–PN and CuII–PN intermediates.22,35a,49a The proposed PN intermediates in the NOM reaction of 1 and 2 were supported by the phenol ring nitration test. Studies using 15N-labeled 15NO revealed that the N-atoms of CoII–NO2− and CrIII–NO2− derived from the NO moieties of 1 and 2, respectively. Both the complexes, 1 and 2, generate the NOM products (3 and 4) when reacted with O2˙−; however, we were only able to regenerate {CoNO}8 from 3, in contrast, {CrNO}5 from 4. In conclusion, the rate of NOM depends on the thermal stability of M–NOs. In contrast, the regeneration of initial M–NOs from NOM products depends on the oxidation states of the metal center of final products.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Grants-in-Aid (Grant No. CRG/2021/003371 & EEQ/2021/000109) from SERB-DST. AK thanks the SERB-DST for her fellowship (Grant No. CRG/2021/003371). SD, K, and PB thank IISER, Tirupati, for their fellowships.
References
-
(a) G. B. Richter-Addo, P. Legzdins and J. Burstyn, Chem. Rev., 2002, 102, 857–860 CrossRef CAS PubMed;
(b) C. Bogdan, Nat. Immunol., 2001, 2, 907–916 CrossRef CAS PubMed;
(c) L. Jia, C. Bonaventura, J. Bonaventura and J. S. Stamler, Nature, 1996, 380, 221–226 CrossRef CAS PubMed.
-
(a) R. F. Furchgott, Angew. Chem., Int. Ed., 1999, 38, 1870–1880 CrossRef CAS;
(b) L. J. Ignarro, Biosci. Rep., 1999, 19, 51–71 CrossRef CAS PubMed;
(c)
L. J. Ignarro, Nitric Oxide: Biology and Pathobiology, Academic press, 2000 Search PubMed;
(d) N. Lehnert, E. Kim, H. T. Dong, J. B. Harland, A. P. Hunt, E. C. Manickas, K. M. Oakley, J. Pham, G. C. Reed and V. S. Alfaro, Chem. Rev., 2021, 121(24), 14682–14905 CrossRef CAS PubMed.
- C. S. Wilcox, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 2005, 289, R913–R935 CrossRef CAS PubMed.
- H. Li and U. Förstermann, Curr. Opin. Pharmacol., 2013, 13, 161–167 CrossRef CAS PubMed.
- C. Hölscher, L. McGlinchey, R. Anwyl and M. J. Rowan, Learn. Mem., 1996, 2, 267–278 CrossRef PubMed.
-
(a) F. Vargas, J. M. Moreno, R. Wangensteen, I. Rodriguez-Gomez and J. Garcia-Estan, Eur. J. Endocrinol., 2007, 156, 1–12 CAS;
(b) S. Cook, O. Hugli, M. Egli, P. Vollenweider, R. Burcelin, P. Nicod, B. Thorens and U. Scherrer, Swiss Med. Wkly., 2003, 133, 360–363 Search PubMed.
-
(a) L. Castillo, T. C. deRojas, T. E. Chapman, J. Vogt, J. F. Burke, S. R. Tannenbaum and V. R. Young, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 193–197 CrossRef CAS PubMed;
(b) R. M. Palmer, D. S. Ashton and S. Moncada, Nature, 1988, 333, 664–666 CrossRef CAS PubMed.
-
(a) Q. Liu and S. S. Gross, Methods Enzymol., 1996, 268, 311–324 CAS;
(b) C. Nathan and Q. W. Xie, J. Biol. Chem., 1994, 269, 13725–13728 CrossRef CAS PubMed.
-
(a) B. A. Averill, Chem. Rev., 1996, 96, 2951–2964 CrossRef CAS PubMed;
(b) J. O. Lundberg, E. Weitzberg and M. T. Gladwin, Nat. Rev. Drug Discovery, 2008, 7, 156–167 CrossRef CAS PubMed.
- R. Radi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4003–4008 CrossRef CAS PubMed.
- C. H. Lim, P. C. Dedon and W. M. Deen, Chem. Res. Toxicol., 2008, 21, 2134–2147 Search PubMed.
- R. S. Lewis and W. M. Deen, Chem. Res. Toxicol., 1994, 7, 568–574 Search PubMed.
-
(a) S. Goldstein, J. Lind and G. Merenyi, Chem. Rev., 2005, 105, 2457–2470 CrossRef CAS PubMed;
(b) P. C. Dedon and S. R. Tannenbaum, Arch. Biochem. Biophys., 2004, 423, 12–22 CrossRef CAS PubMed;
(c) P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315–424 CrossRef CAS PubMed.
- B. Kalyanaraman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11527–11528 CrossRef CAS PubMed.
-
(a) P. R. Gardner, A. M. Gardner, L. A. Martin and A. L. Salzman, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10378–10383 CrossRef CAS PubMed;
(b) M. P. Doyle and J. W. Hoekstra, J. Inorg. Biochem., 1981, 14, 351–358 CrossRef CAS PubMed;
(c) P. C. Ford and I. M. Lorkovic, Chem. Rev., 2002, 102, 993–1018 CrossRef CAS PubMed.
- D. A. Nnate and N. K. Achi, J. Sci. Res. Rep., 2016, 1, 1–19 Search PubMed.
-
(a) N. Pal, M. Jana and A. Majumdar, Chem. Commun., 2021, 57, 8682–8698 RSC;
(b) P. C. Mills, G. Rowley, S. Spiro, J. C. D. Hinton and D. J. Richardson, Microbiology-SGM, 2008, 154, 1218–1228 CrossRef CAS PubMed;
(c) A. M. Gardner, R. A. Helmick and P. R. Gardner, J. Biol. Chem., 2002, 277, 8172–8177 CrossRef CAS PubMed.
-
(a) R. F. Eich, T. Li, D. D. Lemon, D. H. Doherty, S. R. Curry, J. F. Aitken, A. J. Mathews, K. A. Johnson, R. D. Smith, G. N. Phillips and J. S. Olson, Biochemistry, 1996, 35, 6976–6983 CrossRef CAS PubMed;
(b) R. E. Huie and S. Padmaja, Free Radical Res. Commun., 1993, 18, 195–199 CrossRef CAS PubMed;
(c) S. Herold, FEBS Lett., 1999, 443, 81–84 CrossRef CAS PubMed.
-
(a) A. Yokoyama, J. E. Han, K. D. Karlin and W. Nam, Chem. Commun., 2014, 50, 1742–1744 RSC;
(b) A. Yokoyama, K. B. Cho, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2013, 135, 14900–14903 CrossRef CAS PubMed;
(c) A. Yokoyama, J. E. Han, J. Cho, M. Kubo, T. Ogura, M. A. Siegler, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2012, 134, 15269–15272 CrossRef CAS PubMed;
(d) T. S. Kurtikyan, S. R. Eksuzyan, J. A. Goodwin and G. S. Hovhannisyan, Inorg. Chem., 2013, 52, 12046–12056 CrossRef CAS PubMed;
(e) T. S. Kurtikyan and P. C. Ford, Chem. Commun., 2010, 46, 8570–8572 RSC;
(f) S. K. Sharma, A. W. Schaefer, H. Lim, H. Matsumura, P. Moënne-Loccoz, B. Hedman, K. O. Hodgson, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2017, 139, 17421–17430 CrossRef CAS PubMed;
(g) S. K. Sharma, P. J. Rogler and K. D. Karlin, J. Porphyrins Phthalocyanines, 2015, 19, 352–360 CrossRef CAS PubMed.
- T. S. Kurtikyan, S. R. Eksuzyan, V. A. Hayrapetyan, G. G. Martirosyan, G. S. Hovhannisyan and J. A. Goodwin, J. Am. Chem. Soc., 2012, 134, 13861–13870 CrossRef CAS PubMed.
- S. Saha, S. Ghosh, K. Gogoi, H. Deka, B. Mondal and B. Mondal, Inorg. Chem., 2017, 56, 10932–10938 CrossRef CAS PubMed.
- D. Maiti, D. H. Lee, A. A. Narducci Sarjeant, M. Y. Pau, E. I. Solomon, K. Gaoutchenova, J. Sundermeyer and K. D. Karlin, J. Am. Chem. Soc., 2008, 130, 6700–6701 CrossRef CAS PubMed.
- S. Hong, P. Kumar, K. B. Cho, Y. M. Lee, K. D. Karlin and W. Nam, Angew. Chem., Int. Ed., 2016, 55, 12403–12407 CrossRef CAS PubMed.
- M. Yenuganti, S. Das, Kulbir, S. Ghosh, P. Bhardwaj, S. S. Pawar, S. C. Sahoo and P. Kumar, Inorg. Chem. Front., 2020, 7, 4872–4882 RSC.
-
(a) J. Lee, J. A. Hunt and J. T. Groves, J. Am. Chem. Soc., 1998, 120, 7493–7501 CrossRef CAS;
(b) J. Su and J. T. Groves, J. Am. Chem. Soc., 2009, 131, 12979–12988 CrossRef CAS PubMed.
-
(a) J. J. Liu, M. A. Siegler, K. D. Karlin and P. Moenne-Loccoz, Angew. Chem., Int. Ed., 2019, 58, 10936–10940 CrossRef CAS PubMed;
(b) N. G. Tran, H. Kalyvas, K. M. Skodje, T. Hayashi, P. Moënne-Loccoz, P. E. Callan, J. Shearer, L. J. Kirschenbaum and E. Kim, J. Am. Chem. Soc., 2011, 133, 1184–1187 CrossRef CAS PubMed.
- K. J. Koebke, D. J. Pauly, L. Lerner, X. Liu and A. A. Pacheco, Inorg. Chem., 2013, 52, 7623–7632 CrossRef CAS PubMed.
- E. T. Yukl, S. de Vries and P. Moënne-Loccoz, J. Am. Chem. Soc., 2009, 131, 7234–7235 CrossRef CAS PubMed.
-
(a) M. T. Forrester and M. W. Foster, Free Radicals Biol. Med., 2012, 52, 1620–1633 CrossRef CAS PubMed;
(b) P. Ascenzi and M. Brunori, J. Porphyrins Phthalocyanines, 2016, 20, 134–149 CrossRef CAS.
- J. Tejero, A. P. Hunt, J. Santolini, N. Lehnert and D. J. Stuehr, J. Biol. Chem., 2019, 294, 7904–7916 CrossRef CAS PubMed.
-
(a)
G. B. Richter-Addo and P. Legzdins, Metal nitrosyls, Oxford University Press, 1992 Search PubMed;
(b) S. Hematian, I. Garcia-Bosch and K. D. Karlin, Acc. Chem. Res., 2015, 48, 2462–2474 CrossRef CAS PubMed;
(c) J. Fitzpatrick and E. Kim, Acc. Chem. Res., 2015, 48, 2453–2461 CrossRef CAS PubMed;
(d) A. P. Hunt and N. Lehnert, Acc. Chem. Res., 2015, 48, 2117–2125 CrossRef CAS PubMed;
(e) M.-L. Tsai, C.-C. Tsou and W.-F. Liaw, Acc. Chem. Res., 2015, 48, 1184–1193 CrossRef CAS PubMed;
(f) T. C. Berto, A. L. Speelman, S. Zheng and N. Lehnert, Coord. Chem. Rev., 2013, 257, 244–259 CrossRef CAS;
(g) M. Sarma, A. Kalita, P. Kumar, A. Singh and B. Mondal, J. Am. Chem. Soc., 2010, 132, 7846–7847 CrossRef CAS PubMed;
(h) P. Kumar, A. Kalita and B. Mondal, Dalton Trans., 2011, 40, 8656–8663 RSC;
(i) A. Kalita, P. Kumar, R. C. Deka and B. Mondal, Inorg. Chem., 2011, 50, 11868–11876 CrossRef CAS PubMed;
(j) B. Mondal, P. Kumar, P. Ghosh and A. Kalita, Chem. Commun., 2011, 47, 2964–2966 RSC.
- A. Boscari, E. Meilhoc, C. Castella, C. Bruand, A. Puppo and R. Brouquisse, Front. Plant Sci., 2013, 4, 384 Search PubMed.
-
(a) C. E. Sparacino-Watkins, J. Tejero, B. Sun, M. C. Gauthier, J. Thomas, V. Ragireddy, B. A. Merchant, J. Wang, I. Azarov, P. Basu and M. T. Gladwin, J. Biol. Chem., 2014, 289, 10345–10358 CrossRef CAS PubMed;
(b) C. Gherasim, P. K. Yadav, O. Kabil, W. N. Niu and R. Banerjee, PLoS One, 2014, 9, e85544 CrossRef PubMed;
(c) J. L. Zweier, P. Wang, A. Samouilov and P. Kuppusamy, Nat. Med., 1995, 1, 804–809 CrossRef CAS PubMed;
(d) Kulbir, S. Das, T. Devi, S. Ghosh, S. Chandra Sahoo and P. Kumar, Chem. Sci., 2023, 14, 2935–2942 RSC;
(e) Kulbir, S. Das, T. Devi, M. Goswami, M. Yenuganti, P. Bhardwaj, S. Ghosh, S. C. Sahoo and P. Kumar, Chem. Sci., 2021, 12, 10605–10612 RSC.
-
(a) H. Subedi and N. E. Brasch, Inorg. Chem., 2013, 52, 11608–11617 CrossRef CAS PubMed;
(b) C. M. Frech, O. Blacque, H. W. Schmalle and H. Berke, Dalton Trans., 2006, 4590–4598, 10.1039/B604858G.
-
(a) P. Kumar, Y. M. Lee, Y. J. Park, M. A. Siegler, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2015, 137, 4284–4287 CrossRef CAS PubMed;
(b) P. Kumar, Y. M. Lee, L. Hu, J. Chen, Y. J. Park, J. Yao, H. Chen, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2016, 138, 7753–7762 CrossRef CAS PubMed;
(c) M. A. Puthiyaveetil Yoosaf, S. Ghosh, Y. Narayan, M. Yadav, S. C. Sahoo and P. Kumar, Dalton Trans., 2019, 48, 13916–13920 RSC;
(d) S. Das, S. A. C. Kulbir, S. Singh, S. Roy, R. Singh, S. Ghosh and P. Kumar, Dalton Trans., 2023, 52, 5095 RSC;
(e) A. Kalita, P. Kumar, R. C. Deka and B. Mondal, Chem. Commun., 2012, 48, 1251–1253 RSC;
(f) A. Kalita, P. Kumar and B. Mondal, Chem. Commun., 2012, 48, 4636–4638 RSC;
(g) P. Kumar, A. Kalita and B. Mondal, Inorg. Chim. Acta, 2013, 404, 88–96 CrossRef CAS;
(h) P. Kumar, A. Kalita and B. Mondal, Dalton Trans., 2012, 41, 10543–10548 RSC;
(i) P. Kumar, A. Kalita and B. Mondal, Dalton Trans., 2011, 40, 8656–8663 RSC.
-
(a) S. Das, Kulbir, S. Ghosh, S. Chandra Sahoo and P. Kumar, Chem. Sci., 2020, 11, 5037–5042 RSC;
(b) F. Roncaroli, J. A. Olabe and R. van Eldik, Inorg. Chem., 2002, 41, 5417–5425 CrossRef CAS PubMed.
- A. Kalita, P. Kumar, R. C. Deka and B. Mondal, Chem. Commun., 2012, 48, 1251–1253 RSC.
-
(a) S. G. Clarkson and F. Basolo, J. Chem. Soc., Chem. Commun., 1972, 670–671, 10.1039/c39720000670;
(b) S. G. Clarkson and F. Basolo, Inorg. Chem., 1973, 12, 1528–1534 CrossRef CAS.
- L. Cheng, D. R. Powell, M. A. Khan and G. B. Richter-Addo, Chem. Commun., 2000, 2301–2302, 10.1039/b006775j.
-
(a) B. Mondal, S. Saha, D. Borah, R. Mazumdar and B. Mondal, Inorg. Chem., 2019, 58, 1234–1240 CrossRef CAS PubMed;
(b) A. Kalita, P. Kumar and B. Mondal, Chem. Commun., 2012, 48, 4636–4638 RSC.
- B. Mondal, D. Borah, R. Mazumdar and B. Mondal, Inorg. Chem., 2019, 58, 14701–14707 CrossRef CAS PubMed.
-
(a) C. P. Brock, J. P. Collman, G. Dolcetti, P. H. Farnham, J. A. Ibers, J. E. Lester and C. A. Reed, Inorg. Chem., 2002, 12, 1304–1313 CrossRef;
(b) C. H. Chuang, W. F. Liaw and C. H. Hung, Angew. Chem., Int. Ed., 2016, 55, 5190–5194 CrossRef CAS PubMed;
(c) K. Gogoi, S. Saha, B. Mondal, H. Deka, S. Ghosh and B. Mondal, Inorg. Chem., 2017, 56, 14438–14445 CrossRef CAS PubMed;
(d) Y. Guo, J. R. Stroka, B. Kandemir, C. E. Dickerson and K. L. Bren, J. Am. Chem. Soc., 2018, 140, 16888–16892 CrossRef CAS PubMed;
(e) H. Kruszyna, J. S. Magyar, L. G. Rochelle, M. A. Russell, R. P. Smith and D. E. Wilcox, J. Pharmacol. Exp. Ther., 1998, 285, 665–671 CAS;
(f) A. Keller, Jeż and B. Owska-Trzebiatowska, Inorg. Chim. Acta, 1981, 51, 123–130 CrossRef CAS;
(g) A. Døssing, Rev. Inorg. Chem., 2013, 33, 129–137 Search PubMed.
- S. Das, Kulbir, S. Ray, T. Devi, S. Ghosh, S. S. Harmalkar, S. N. Dhuri, P. Mondal and P. Kumar, Chem. Sci., 2022, 13, 1706–1714 RSC.
- Kulbir, C. S. A. Keerthi, S. Beegam, S. Das, P. Bhardwaj, M. Ansari, K. Singh and P. Kumar, Inorg. Chem., 2023, 62, 7385–7392 CrossRef CAS PubMed.
-
(a) J. Cho, H. Y. Kang, L. V. Liu, R. Sarangi, E. I. Solomon and W. Nam, Chem. Sci., 2013, 4, 1502–1508 RSC;
(b) J. Du, D. Xu, C. Zhang, C. Xia, Y. Wang and W. Sun, Dalton Trans., 2016, 45, 10131–10135 RSC;
(c) Y. Jo, J. Annaraj, M. S. Seo, Y.-M. Lee, S. Y. Kim, J. Cho and W. Nam, J. Inorg. Biochem., 2008, 102, 2155–2159 CrossRef CAS PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005, 10.1039/jr9590002003.
- E. Tatsch, G. V. Bochi, R. D. S. Pereira, H. Kober, V. A. Agertt, M. M. Anraku de Campos, P. Gomes, M. M. M. F. Duarte and R. N. Moresco, Clin. Biochem., 2011, 44, 348–350 CrossRef CAS PubMed.
-
(a) J. W. Coddington, J. K. Hurst and S. V. Lymar, J. Am. Chem. Soc., 1999, 121, 2438–2443 CrossRef CAS;
(b) S. Pfeiffer, A. C. Gorren, K. Schmidt, E. R. Werner, B. Hansert, D. S. Bohle and B. Mayer, J. Biol. Chem., 1997, 272, 3465–3470 CrossRef CAS PubMed.
-
(a) G. Y. Park, S. Deepalatha, S. C. Puiu, D. H. Lee, B. Mondal, A. A. Narducci Sarjeant, D. del Rio, M. Y. Pau, E. I. Solomon and K. D. Karlin, J. Biol. Inorg. Chem., 2009, 14, 1301–1311 CrossRef CAS PubMed;
(b) O. A. Babich and E. S. Gould, Res. Chem. Intermed., 2002, 28, 575–583 CrossRef CAS.
-
(a) R. Cao, L. T. Elrod, R. L. Lehane, E. Kim and K. D. Karlin, J. Am. Chem. Soc., 2016, 138, 16148–16158 CrossRef CAS PubMed;
(b) R. Mazumdar, B. Mondal, S. Saha, B. Samanta and B. Mondal, J. Inorg. Biochem., 2022, 228, 111698 CrossRef CAS PubMed.
- J. Cho, R. Sarangi, J. Annaraj, S. Y. Kim, M. Kubo, T. Ogura, E. I. Solomon and W. Nam, Nat. Chem., 2009, 1, 568–572 CrossRef CAS PubMed.
- S. Das, Kulbir, S. Ray, T. Devi, S. Ghosh, S. S. Harmalkar, S. N. Dhuri, P. Mondal and P. Kumar, Chem. Sci., 2022, 13, 1706–1714 RSC.
- W. H. Koppenol, P. L. Bounds, T. Nauser, R. Kissner and H. Ruegger, Dalton Trans., 2012, 41, 13779–13787 RSC.
-
(a) M. P. Schopfer, B. Mondal, D. H. Lee, A. A. Sarjeant and K. D. Karlin, J. Am. Chem. Soc., 2009, 131, 11304–11305 CrossRef CAS PubMed;
(b) J. J. Liu, M. A. Siegler, K. D. Karlin and P. Moenne-Loccoz, Angew. Chem., Int. Ed., 2019, 58, 10936–10940 CrossRef CAS PubMed.
-
(a) M. S. Ramezanian, S. Padmaja and W. H. Koppenol, Chem. Res. Toxicol., 1996, 9, 232–240 Search PubMed;
(b) A. Vandervliet, J. P. Eiserich, C. A. Oneill, B. Halliwell and C. E. Cross, Arch. Biochem. Biophys., 1995, 319, 341–349 CrossRef CAS PubMed;
(c) A. van der Vliet, C. A. O'Neill, B. Halliwell, C. E. Cross and H. Kaur, FEBS Lett., 1994, 339, 89–92 CrossRef CAS PubMed;
(d) A. Kalita, R. C. Deka and B. Mondal, Inorg. Chem., 2013, 52, 10897–10903 CrossRef CAS PubMed;
(e) S. Kim, M. A. Siegler and K. D. Karlin, Chem. Commun., 2014, 50, 2844–2846 RSC;
(f) L. Qiao, Y. Lu, B. Liu and H. H. Girault, J. Am. Chem. Soc., 2011, 133, 19823–19831 CrossRef CAS PubMed;
(g) K. M. Skodje, P. G. Williard and E. Kim, Dalton Trans., 2012, 41, 7849–7851 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Kulbir
a,
Kulbir