
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColor-controlled nonstoichiometric spinel-type cobalt gallate nanopigments prepared by supercritical hydrothermal synthesis†
Bo
Xie
 a,
Chiya
Numako
b,
Takashi
Naka
c and
Seiichi
Takami
*a
a,
Chiya
Numako
b,
Takashi
Naka
c and
Seiichi
Takami
*a
aDepartment of Materials Process Engineering, Graduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan. E-mail: xie.bo.d7@s.mail.nagoya-u.ac.jp; takami.seiichi@material.nagoya-u.ac.jp
bDepartment of Chemistry, Graduate School of Science, Chiba University, Chiba 263-8522, Japan
cResearch Center for Materials Nanoarchitectonics, National Institute for Materials Science (NIMS), Tsukuba, Ibaraki 305-0047, Japan
First published on 15th October 2023
Abstract
Spinel-type inorganic pigments with intensive color and chemical/thermal stability are showing extensive applications that could be further broadened by color manipulation and improvement of the material properties through nanosizing. In this study, we report the supercritical hydrothermal synthesis of nonstoichiometric spinel-type cobalt gallate nanoparticles (Co–Ga NPs) with controlled color. Without the conventional calcination procedure, NPs with greenish-blue, blue, and yellowish-green colors were synthesized from precursor solutions at pH 7, 9, and 11, respectively, with a low Co/Ga molar ratio of 0.25. X-ray diffraction, scanning/transmission electron microscopy, and inductively coupled plasma-atomic emission spectroscopy methods suggest that the products were spinel-type cobalt gallate NPs with high crystallinity and a nonstoichiometric composition. Based on an X-ray absorption fine structure investigation, the prepared nonstoichiometric Co–Ga NPs were found to have different cationic configurations from stoichiometric CoGa2O4 produced by a solid-state reaction during calcination. Meanwhile, the degrees of distortions at tetrahedral and octahedral sites in the NPs were evaluated by Raman spectroscopy. In particular, nonstoichiometric Co–Ga NPs with a blue color were prepared without calcination for the first time and were found to have lower tetrahedral cobalt occupancy but comparable octahedral cobalt occupancy and larger polyhedral distortions at tetrahedral sites when compared to calcined CoGa2O4. We also discuss strategies that could realize Co–Ga NPs with a more brilliant blue color using the present technique based on an investigation of the growth process.
1. Introduction
Inorganic pigments with a spinel-type crystal structure show great utilization value owing to their high thermal/chemical stability, high mechanical resistance,1 good compatibility with organic modifiers and optical absorption performance.2,3 Spinel-type pigments exhibit a wide range of intrinsic colors with a high intensity by accommodating various types of transition metal cations, including Fe2+, Co2+, Cu2+, Fe3+, and Cr3+.4 Therefore, color manipulation can be realized by doping with transition or rare-earth metal ions.5–7 Other methods for color manipulation include the use of capping agents,8 use of different polymorphs of the starting material,9 and control of the calcination temperature.10 A change in the calcination temperature was thought to realize different cationic configurations at tetrahedral and octahedral sites in the spinel structure, which could control the color of the products.11–13Compared to the normal spinel (IV(A)VI[B2]O4) and inverse spinel (IV(B)VI[AB]O4) structures, CoGa2O4 has a partially inverse structure expressed as IV(Co1−xGax)VI[CoxGa2−x]O4 (x: inversion parameter), as shown in Fig. 1. The inversion parameter of intensive blue CoGa2O4 calcined at 1300 °C for 24 h was estimated to be 0.664.15 When using different calcination temperatures, Mathur et al. successfully produced CoGa2O4 with dark green, grass green, and blue colors by calcining intermediate species at 400–500 °C, 600–800 °C, and 1000–1200 °C, respectively.16 The emergence of the blue color at 1000–1200 °C was considered the result of an increased number of tetrahedrally coordinated Co2+ ions (Co2+–O4) and deoxidation of Co3+ to Co2+ during the temperature increase, similar to CoAl2O4.17 However, blue CoGa2O4 nanoparticles have not been synthesized without calcination.
 | ||
| Fig. 1 Crystal structure of a partially inverse CoGa2O4 (IV(Co1−xGax)VI[CoxGa2−x]O4) created by VESTA.14 | ||
Apart from color manipulation, inorganic pigments with improved material properties could be realized by producing their nanosized counterparts. In the field of painting, nanoscale ceramic pigments could show improved brilliance with the help of a higher surface coverage and a higher number of reflection points, as well as enhanced mechanical strength owing to better dispersibility with binders.18 Meanwhile, nanoscale pigments also show negligible visible light scattering, and therefore, applications related to high transparency are expected.19,20 Furthermore, nanopigments could also exhibit enhanced dispersibility in aqueous/organic solutions after encapsulation and surface modification.21–23 In regard to the traditional preparation of CoGa2O4, calcination at 600–1200 °C favors the production of CoGa2O4 with high crystallinity, and color manipulation from green to blue is easily achievable. However, the high synthesis temperature and long calcination time could result in not only unexpected agglomeration that makes nanosizing harder but also large energy consumption. To our knowledge, there are still no studies that have reported the synthesis of spinel-type metal oxides with different colors at a fixed low temperature of approximately 300–500 °C without calcination, which could favor the production of metal oxides with a small crystallite size, suppress agglomeration and serve the purpose of energy savings.
In this work, we studied the supercritical hydrothermal synthesis of spinel-type cobalt gallate nanoparticles (Co–Ga NPs) with controlled color. Supercritical hydrothermal synthesis was conducted at 400 °C without additional calcination in air. As seen in CoAl2O4,24,25 octahedrally coordinated Co3+ ions (Co3+–O6) could be generated from the oxidization process of Co2+ to Co3+ at 400–600 °C, and they could hamper color manipulation due to the exhibition of an intense green color.17,26,27 Therefore, we conducted supercritical hydrothermal synthesis at 400 °C for 10 min, and we consider that the rapid synthesis time of 10 min could alleviate the previous oxidization process. Moreover, we also used a precursor solution with a low Co/Ga molar ratio (0.25) to further reduce the generation of Co3+ during the synthesis. To achieve color manipulation, we changed the pH of the precursor solution to realize different cationic configurations, as reported in our previous work.28 We investigated the effect of the precursor solution pH on the coloration, crystalline phase, composition, cationic configurations and degrees of polyhedral distortions at tetrahedral and octahedral sites of the products. Moreover, we also discuss the growth process of prepared Co–Ga NPs by comparing the local structure around cobalt atoms in the solid products obtained at 200 °C, 300 °C, and 400 °C.
2. Experimental procedure
2.1. Preparation of cobalt gallate NPs
The supercritical hydrothermal synthesis of Co–Ga NPs was performed as follows. Cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, 99.5%) and gallium(III) nitrate n-hydrate (Ga(NO3)3·nH2O, n = 7–9, 99.9%) were purchased from FUJIFILM Wako Pure Chemical Co. and used without further purification. Co(NO3)2·6H2O and Ga(NO3)3·nH2O were dissolved in pure water at concentrations of 0.025 mol L−1 and 0.10 mol L−1, respectively. For pH control, an aqueous solution of 1.0 mol L−1 NaOH was prepared using sodium hydroxide (NaOH, Kishida Chemical Co., Ltd, purity over 97.0%) and added dropwise to adjust the pH of the reactant solution to 7, 9, and 11. The added volume of NaOH for each condition was recorded, and solid precipitates with a pale pink color were observed at this moment. Prepared precursor solutions (1.5 mL) were poured into a pressure-resistant Hastelloy C-276 reactor (inner volume of 5.0 mL). After being tightly capped, the reactor was placed in an electric furnace with a preheated temperature of 400 °C. After a synthesis time of 10 min, the reaction was terminated by removing the reactor and submerging it into a cold water bath. Temperatures inside the reactor during the heating and cooling process were recorded (see Fig. S1 in the ESI†).2.2. Product collection and purification
After synthesis, solid products were collected from the reactant solutions by centrifugation (4 °C, 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 20 min) and decantation and were freeze-dried (FDS-1000, Tokyo Rikakikai Co., Ltd) for X-ray diffraction (XRD), scanning electron microscopy (SEM), and transmission electron microscopy (TEM) analyses.
000 rpm, 20 min) and decantation and were freeze-dried (FDS-1000, Tokyo Rikakikai Co., Ltd) for X-ray diffraction (XRD), scanning electron microscopy (SEM), and transmission electron microscopy (TEM) analyses.
2.3. Product characterization
The crystalline phase of the freeze-dried solid products was analyzed by AERIS (PANalytical) with Cu-Kα radiation operating at 40 kV and 15 mA. XRD patterns were recorded in the range of 10° < 2θ < 70°. For SEM observation, a JSM-7610F (JEOL Ltd) with an accelerating voltage of 15 kV was used. For TEM observation, a JEM-1400 Plus (JEOL Ltd) was used, and energy-dispersive X-ray spectroscopy (EDS) was conducted to confirm the elemental composition of the products.The Co/Ga molar ratios in the solid products were measured using inductively coupled plasma-atomic emission spectroscopy (ICP-AES) analysis. The solid products were dissolved in a mixed solution of nitric acid and hydrogen peroxide in a volume ratio of 1:1. For solid products that were hard to dissolve, hydrochloric acid was added. Nitric acid (HNO3, weight fraction in the range of 60–61%), hydrochloric acid (HCl, weight fraction in the range of 35.0–37.0%), hydrogen peroxide (H2O2, 30.0–35.5%), and standard solutions for ICP-AES analysis were purchased from FUJIFILM Wako Pure Chemical Co. and used as received without any further purification. Next, an SPS7800 (Seiko Instruments) was used to measure the Co and Ga concentrations in the resultant aqueous solution. Additionally, we performed ICP-AES analysis to measure Ni, Cr and Fe along with Co and Ga in the solid products when taking the issue of contamination from the reactor into consideration. Moreover, the concentrations of unreacted Co and Ga in the supernatant of the reactant solutions after synthesis were also measured by ICP-AES analysis, based on which the conversion of Co and Ga for each pH condition was calculated.
To investigate the main valence states of metal ions and their cationic configurations in the solid products, Ga K-edge and Co K-edge X-ray absorption fine structure (XAFS) spectra were measured at BL-9A and BL-12C, Photon Factory, KEK, Japan, using the transmission method. Data processing of the X-ray absorption near-edge structure (XANES) region and extended X-ray absorption fine structure (EXAFS) region in the XAFS spectra was carried out with a data processing program, Athena.29 In particular, the absorption edges of the products were determined as the photon energies at the maximum of the first derivative of the normalized μ(E) in the XANES region.30 The cationic configurations of the products were determined by using a linear combination of k3-weighted χ(k) oscillations, and they were used as the initial cationic configurations in Rietveld refinement using RIETAN-FP.31 Specifically, the metal occupancy of Co atoms at tetrahedral and octahedral sites for each pH condition was quantitatively evaluated by considering that the measured k3-weighted χ(k) oscillations at the Co K-edge of the products are a linear combination of those of IV(Co)VI[Ga2]O4 (normal spinel) and IV(Ga)VI[CoGa]O4 (inverse spinel).28 The k3-weighted χ(k) oscillations of IV(Co)VI[Ga2]O4 and IV(Ga)VI[CoGa]O4 at the Co K-edge were theoretically calculated by Larch using all possible scattering paths, an amplitude reduction factor of 0.70, and a thermal and static disorder parameter σ2 of 0.01.32
The diffuse reflectance spectra in the UV–Vis region of the solid products were measured by a UV–Vis spectrophotometer (V-650, JASCO Co.) using an integrating sphere (SIV-767). The measurement range was 200–900 nm, and the scanning speed was 100 nm min−1. A color compass MFA (AT System Co., Ltd) with a spectrometer (C12880MA, Hamamatsu Photonics K. K., spectral response range of 340–850 nm) was used to measure the CIE XYZ parameters of the products. Then, the CIE XYZ parameters were converted to sRGB and RGB parameters using a gamma correction of 2.4.
The degree of distortions in the solid products was evaluated by Raman spectroscopy. Raman spectra were collected using inVia Reflex (RENISHAW). The excitation was achieved by using 532 nm green laser emission (J150GS, KYOCERA SOC Co.) The laser power output and irradiation time on the sample were set to 1%–5% and 30–300 s, respectively.
3. Results and discussion
3.1. Effects of precursor solution pH on the coloration, crystalline phase, and composition of solid products
The coloration of the solid products prepared at each pH by the present technique was investigated first. Fig. 2 shows digital images of the solid products synthesized at each pH, together with their corresponding RGB parameters converted from measured XYZ parameters in the CIE 1931 XYZ color space. The colors obtained at pH 7, pH 9, and pH 11 were greenish-blue, blue, and yellowish-green, respectively. Therefore, solid products with tunable coloration were successfully synthesized at a fixed temperature of 400 °C by supercritical hydrothermal synthesis.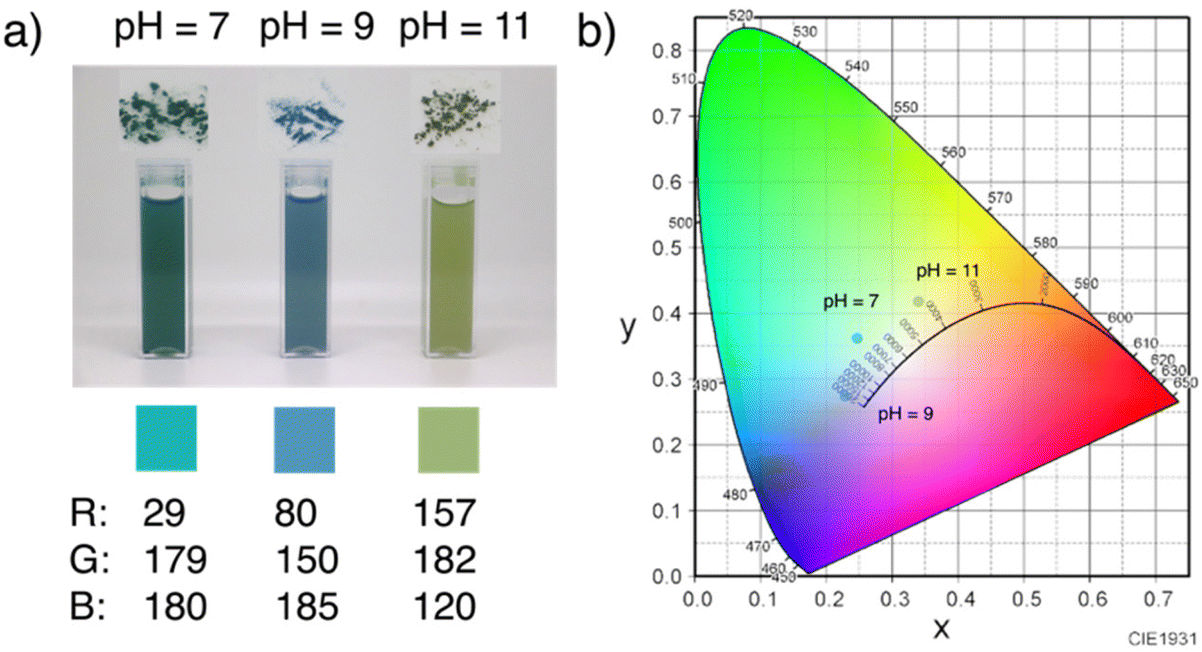 | ||
| Fig. 2 (a) Digital images and calculated corresponding RGB parameters (γ = 2.4) of the solid products produced at 400 °C using precursor solutions with pH 7–11 and (b) measured XYZ parameters based on the CIE 1931 XYZ color space. | ||
To confirm whether the coloration change was due to the difference in the crystal structure, we investigated the crystalline phase of the products. Fig. 3 shows the measured XRD patterns of the products. All of them agreed with that of spinel-type CoGa2O4 (ICSD: 172183, Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), and slight rightward shifts were observed for all three pH conditions. The crystallite sizes were evaluated by the Williamson–Hall equation, as shown in Fig. S2 (ESI†), and the calculated crystallite sizes are summarized in Fig. 3. They were in the range of 10 nm to 53 nm, which corresponded to the obtained narrowed full width at half maximum (FWHM) values of the XRD patterns at pH 11. As shown in Fig. 3, there was a large difference in size between NPs at pH 11 with a green color (53 nm) and those at pH 9 with a blue color (10 nm). To confirm whether the size effect of the NPs could result in the changing color, we compared Co–Ga NPs prepared at pH 9 in this study to those prepared at pH 7 using a precursor solution with a high Co/Ga molar ratio (Co/Ga = 0.50) in our previous study.28 They have comparable crystallite sizes (this study: 10 nm; previous study: 13 nm) but a large difference in coloration. Therefore, we consider that the color difference between NPs prepared at different pH in this study may not be caused by the size effect (see Fig. S3 in ESI†).
m), and slight rightward shifts were observed for all three pH conditions. The crystallite sizes were evaluated by the Williamson–Hall equation, as shown in Fig. S2 (ESI†), and the calculated crystallite sizes are summarized in Fig. 3. They were in the range of 10 nm to 53 nm, which corresponded to the obtained narrowed full width at half maximum (FWHM) values of the XRD patterns at pH 11. As shown in Fig. 3, there was a large difference in size between NPs at pH 11 with a green color (53 nm) and those at pH 9 with a blue color (10 nm). To confirm whether the size effect of the NPs could result in the changing color, we compared Co–Ga NPs prepared at pH 9 in this study to those prepared at pH 7 using a precursor solution with a high Co/Ga molar ratio (Co/Ga = 0.50) in our previous study.28 They have comparable crystallite sizes (this study: 10 nm; previous study: 13 nm) but a large difference in coloration. Therefore, we consider that the color difference between NPs prepared at different pH in this study may not be caused by the size effect (see Fig. S3 in ESI†).
 | ||
| Fig. 3 XRD patterns of the solid products produced at 400 °C using precursor solutions with pH 7–11. | ||
The crystallite sizes of the Co–Ga NPs were further confirmed by SEM and TEM observations. Fig. 4 shows SEM, TEM, and TEM-EDS images of the prepared Co–Ga NPs. The SEM and TEM images indicated particle sizes comparable to the calculated crystallite sizes and an octahedral shape of the Co–Ga NPs prepared at all pH conditions. Meanwhile, the TEM-EDS images showed a uniform element distribution of Ga and Co in the prepared Co–Ga NPs (Fig. 4(c) and Fig. S4 in ESI†). As shown in Fig. 3 and 4, a larger crystallite size was obtained at higher pH conditions, which can be explained by the formation mechanism reported in our previous work.28 As previously reported, the solid precipitates with a pale pink color in the precursor solutions are a layered double hydroxide-like material. In this layered double hydroxide structure, both cobalt and gallium ions are in octahedral coordination. Meanwhile, these solid precipitates could act as a starting material and finally form spinel-type cobalt gallate metal oxides during the temperature increase in supercritical hydrothermal synthesis. Based on the previous discussion, we consider that the solid precipitates could show high stability in a high-pH environment, such as pH 11. Thus, the reaction rate of the formation from the layered double hydroxide-like starting material to the spinel-type cobalt gallate may decrease at pH 11, resulting in an increased crystallite size owing to improved crystal growth.
 | ||
| Fig. 4 (a) SEM images and (b) TEM images of Co–Ga NPs produced at 400 °C using precursor solutions with pH 7–11, and (c) TEM-EDS results showing the element distribution of the NPs produced at pH 11. | ||
ICP-AES analysis was conducted to investigate the composition of the Co–Ga NPs and the possibility of contamination. As shown in Fig. 5, the prepared Co–Ga NPs had nonstoichiometric compositions, and the Co/Ga composition increased from 0.282 to 0.728 with increasing pH. The Co/Ga molar ratios of the NPs prepared at pH 9 and pH 11 greatly surpassed those in the precursor solutions (Co/Ga = 0.25), especially the Co/Ga molar ratio at pH 11, which exceeded that in stoichiometric CoGa2O4 (Co/Ga = 0.50). To explain the increasing nonstoichiometric compositions, we focused on the conversions of cobalt and gallium and their percentages in the aqueous phase of the reactant solutions at room temperature (see Fig. S5 in ESI†). As shown in Fig. S5,† large amounts of gallium were found to dissolve in the aqueous phase of the reactant solutions, especially at pH 11, and the percentage showed the highest value of 0.931. Compared to gallium, cobalt remained in the solid phase of the reactant solutions at room temperature, and the conversions were close to 1.0 at all pH conditions. Therefore, we suppose that gallium may prefer to dissolve in the aqueous phase and attempt to remain unreacted during synthesis at higher pH. As a result, the decreasing conversion of gallium contributed to the increasing Co/Ga composition from pH 7–11. Meanwhile, contamination was barely detected except at pH 7, where a nonnegligible amount of Ni contamination was detected. These Ni2+ ions could exist at the octahedral site and result in an unexpected green hue, as shown in Fig. 2.33
 | ||
| Fig. 5 M/Ga (M = Co, Ni, Cr, and Fe) molar ratios of Co–Ga NPs produced at 400 °C using precursor solutions with pH 7–11 measured by ICP-AES. | ||
To investigate the main valence states of Ga and Co in the prepared nonstoichiometric Co–Ga NPs, normalized μ(E) values were calculated from the XAFS spectra in the XANES region. Fig. 6(a) and (b) show the normalized μ(E), and Fig. 6(c) and (d) show the first derivative of the normalized μ(E) of our products, commercially available references, and CoGa2O4 produced by the solid-state reaction method using a mixed powder of CoO and Ga2O3 at 1300 °C for 24 hours. The maximum peak of the first derivative of the normalized μ(E) is labeled with a black circle, at which the photon energy was determined as the absorption edge, as previously introduced. The Ga–K absorption edges of the Co–Ga NPs prepared at pH 7, 9, and 11 were 10370.7 eV, 10370.4 eV, and 10370.4 eV, respectively, which were almost the same as those of β-Ga2O3 and CoGa2O4 (10370.4 eV) (Fig. 6(c)). Meanwhile, the Co–K absorption edges of the Co–Ga NPs prepared at pH 7, 9, and 11 were 7718.52 eV, 7718.53 eV, and 7718.59 eV, respectively, which were close to those of CoO and CoGa2O4 (7718.66 eV and 7718.53 eV) (Fig. 6(d)). Therefore, Ga3+ and Co2+ are considered the main valence states of the prepared nonstoichiometric Co–Ga NPs. Nevertheless, to further investigate the chemical condition of metal atoms in the products, photon energies at the maximum of the white lines in the normalized μ(E) measured at the Ga and Co–K edge of the products were also compared to corresponding references (Fig. 6(a) and (b)). The maximum value of the white line for each pH condition at the Ga K-edge was close to those of β-Ga2O3 and CoGa2O4 (10375.4 eV), but that at the Co K-edge was slightly larger than those of CoO and CoGa2O4 (7723.80 eV and 7724.47 eV). Therefore, there could be a small amount of Co3+ existing in the Co–Ga NPs produced at pH 7 and pH 9 and more at pH 11. In summary, color-controlled nonstoichiometric Co–Ga NPs with little Co3+ were successfully synthesized by supercritical hydrothermal synthesis at a fixed low temperature of 400 °C without calcination when using precursor solutions with a low Co/Ga molar ratio of 0.25 and pH 7–11.
 | ||
| Fig. 6 The normalized μ(E) at the (a) Ga K-edge and (b) Co K-edge, first derivative of the normalized μ(E) at the (c) Ga K-edge and (d) Co K-edge in the XANES region of nonstoichiometric Co–Ga NPs produced at 400 °C using precursor solutions with pH 7–11 and corresponding references (@KEK PF BL-9A; the maximum of the while line of the normalized μ(E), and that of the first derivative of the normalized μ(E) are labeled with a black circle). | ||
3.2. Investigations of the UV–Vis absorption spectra of prepared nonstoichiometric Co–Ga NPs
Nonstoichiometric spinel-type Co–Ga NPs with Ga3+ and Co2+ as the main valences and greenish-blue, blue, and yellowish-green colors were prepared at pH 7, 9, and 11, respectively. To study the relationship between their nonstoichiometric composition (Co/Ga molar ratio) and coloration, we explored the coloration mechanism based on investigations of the UV–Vis region absorptions and cationic configurations. In this section, we measured the diffuse reflectance spectra of the nonstoichiometric Co–Ga NPs. The Kubelka–Munk absorbance in the UV–Vis region is shown in Fig. 7. | ||
| Fig. 7 UV–Vis absorption spectra converted from diffuse reflectance spectra of nonstoichiometric Co–Ga NPs produced at 400 °C using precursor solutions with pH 7–11. | ||
At pH 7 and pH 9, where products with blue hues were obtained, evident triplet absorption was observed at 560 nm, 600 nm, and 650 nm. We consider that the spin-allowed d–d transition of ν1 [4A2(F) → 4T1(P)] of Co2+–O4 could result in the triplet absorption band based on reports by Llusar et al.34 These three transitions could give rise to blue coloration owing to absorptions at 560 nm (green region), 600 (yellow-orange region), and 650 nm (red region).34 In addition, another broad absorption peak was observed below approximately 400 nm. We consider that the charge transfer between the oxygen anion and Co2+ at the tetrahedral site (O2−–Co2+ (Tet.)) could contribute to this absorption.35 When compared to pH 7 and pH 9, the UV–Vis absorption spectrum at pH 11 showed a more intensive absorption at 400–500 nm and an area with larger absorption at approximately 700 nm. We consider that the intensive absorption at 400–500 nm at pH 11 could also be due to the charge transfer of O2−–Co2+ (Tet.), similar to pH 7 and pH 9 but with a larger intensity. We will discuss our assumptions on this significant increase in the absorbance intensity at 400–500 nm later in section 3.4. In regard to the area with a large absorption at approximately 700 nm, we consider that the charge transfer between the oxygen anion and Co3+ at the octahedral site (O2−–Co3+ (Oct.)) is contributable based on previous studies.35,36 Note that three absorption peaks relating to ν1 [4A2(F) → 4T1(P)] at pH 11 were not evident, which might be because of less Co2+–O4 compared to pH 7 and pH 9. The band gaps of the prepared Co–Ga NPs were evaluated by Tauc plot (see Fig. S6 in the ESI†). The band gaps at pH 7 and pH 9 were approximately 3.31 eV, which could contribute to the absorption at approximately 375 nm. Meanwhile, NPs prepared at pH 11 with the highest Co/Ga molar ratio (Co/Ga = 0.728) showed a much lower band gap of 2.54 eV. This band gap could cause absorption at approximately 488 nm at pH 11.
Based on previous investigations, we assume that there could be more Co2+–O4 in the Co–Ga NPs prepared at pH 7 and pH 9 than at pH 11 because of the exhibition of blue color and the evident triplet absorption of ν1 at 560–650 nm. Meanwhile, there could be larger numbers of Co3+–O6 in the NPs prepared at pH 11 than at pH 7 and pH 9 because of the intensive charge transfer absorption at approximately 700 nm. Generally, the coloration change from pH 7–11 is considered to be due to the change in the cationic configuration. The cationic configuration will be quantitatively determined in section 3.3. Moreover, the effect of the polyhedral distortions at the tetrahedral and octahedral sites on the coloration will be discussed later in section 3.4.
3.3. Investigations of the cationic configurations of prepared nonstoichiometric Co–Ga NPs
To confirm our predictions on the cationic configuration in section 3.2, we quantitatively determined the occupancies of cobalt and gallium atoms at tetrahedral and octahedral sites in the products based on the XAFS measurement and Rietveld refinement, as shown in Fig. 8. Specifically, Fig. 8(a) shows the measured k3-weighted χ(k) oscillations at the Co K-edge of the prepared Co–Ga NPs. The cationic configurations at the tetrahedral and octahedral sites were determined by fitting these k3-weighted χ(k) curves. Fig. 8(b) shows the linear combination when using k3-weighted χ(k) oscillations that were theoretically calculated for IV(Co)VI[Ga2]O4 (normal spinel) and IV(Ga)VI[CoGa]O4 (inverse spinel) arrangements. Fig. 8(b) also shows the corresponding coefficient of determination (R2 factor) for each pH condition. Next, theoretically determined cationic configurations were used as the initial cationic configuration required in the Rietveld refinement to calculate the XRD patterns of the products. The calculated XRD patterns were compared with the observed patterns for each pH condition to examine the reliability of the calculated cationic configurations. As a result, the R2 factor in Fig. 8(b) indicates a good fit of the linear combination for each pH, and the calculated XRD patterns fit the observed patterns in the Rietveld refinement well, as shown in Fig. 8(c) and Fig. S7 (ESI†). In summary, the cationic configurations of the prepared nonstoichiometric Co–Ga NPs were quantitatively determined by linear fitting of XAFS results using the Co K-edge k3-weighted χ(k) oscillations of IV(Co)VI[Ga2]O4 and IV(Ga)VI[CoGa]O4 arrangements and were confirmed to have high reliability by Rietveld refinement using XRD results. | ||
| Fig. 8 (a) Measured k3-weighted χ(k) oscillations in the EXAFS region at the Co K-edge of nonstoichiometric Co–Ga NPs produced at 400 °C using precursor solutions with pH 7–11, (b) linear combination fitting results, and (c) result of Rietveld refinement using RIETAN-FP (pH 9, 400 °C). | ||
The theoretically determined cationic configurations and Rietveld refinement results of the prepared nonstoichiometric Co–Ga NPs and those of the calcined stoichiometric CoGa2O4 with a bright blue color determined by Naka et al.15 are summarized in Table S1 (ESI†). The cationic configurations are depicted in Fig. 9 for a better comparison.
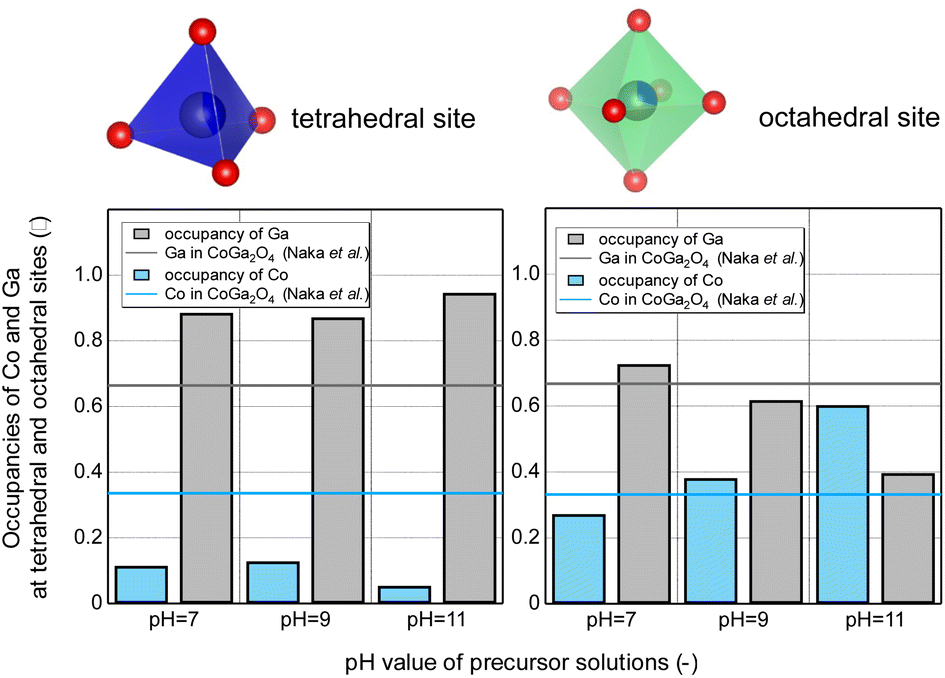 | ||
| Fig. 9 Occupancies of Ga and Co at the tetrahedral and octahedral sites of nonstoichiometric Co–Ga NPs produced at 400 °C using precursor solutions with pH 7–11 derived from the linear combination of k3-weighted χ(k) oscillations at the Co K-edge (black and blue lines show the Ga and Co occupancies in stoichiometric CoGa2O4 produced from a solid-state reaction15). | ||
Unlike what we assumed from the UV–Vis absorption spectra, cobalt ions in the products mainly occupy the octahedral site even at pH 7 and pH 9, at which point products with a blue hue were obtained. These results show that our products were highly inverse spinel compounds. When focusing on the blue and black lines representing the Co and Ga occupancies in the calcined CoGa2O4, our nonstoichiometric products have a completely different cationic configuration. That is, the cobalt occupancies are lower at the tetrahedral site but comparable or much higher at the octahedral site.
As shown in Fig. 9, nonstoichiometric Co–Ga NPs produced at pH 11 with a yellowish-green color were found to have the largest octahedral cobalt occupancy (approximately 0.603). Notably, the octahedral cobalt occupancy at pH 11 greatly surpassed that in inverse CoGa2O4 (0.50). One of the possible reasons is that a part of the Co ions existed as Co3+ at the octahedral site. To verify this, we focused on the photon energies at the maximum of the XAFS white lines in the normalized μ(E) measured at the Co–K edge (see Fig. 6(b)). The photon energy at pH 11 was larger than those at pH 7 and pH 9, indicating the presence of more Co3+ ions. Therefore, we consider that Co3+ ions may replace Ga3+ ions at the octahedral site as Co3+–O6 at pH 11. This resulted in a large octahedral cobalt occupancy of 0.603 at pH 11 and an area with a large absorption at approximately 700 nm owing to the charge transfer of O2−–Co3+ (Oct.). Meanwhile, because the number of octahedral sites is 2 times that of tetrahedral sites in a spinel structure, this large octahedral cobalt occupancy resulted in a high Co/Ga molar ratio for NPs produced at pH 11, as shown in Fig. 5. The products obtained at pH 7 and pH 9, with an octahedral cobalt occupancy comparable to that of the calcined CoGa2O4, exhibited a blue hue. However, due to the lower tetrahedral cobalt occupancy, they showed a Co/Ga molar ratio lower than the stoichiometric value of 0.50.
To explain why the cobalt occupancies of the prepared Co–Ga NPs were lower at the tetrahedral site but comparable or much higher at the octahedral site when compared to the calcined CoGa2O4, we focus again on the gallium ions in the aqueous phase of the reactant solutions, as mentioned in our previous discussion on the increasing nonstoichiometric compositions of the products in section 3.2. We suppose that gallium ions may prefer to dissolve in the aqueous phase during the initial temperature increase and be stable in tetrahedral coordination, such as Ga(OH)4−. With a further temperature increase, some of the Ga(OH)4− species become unstable and may prefer to incorporate into the tetrahedral site with ease. Currently, many cobalt ions are considered to remain in the octahedral coordination because of the rapid reaction from the layered double hydroxide-like material to the spinel phase. Previous assumptions could result in a large cobalt octahedral occupancy (highly inverse spinel compounds) at all pH conditions.
3.4. Investigations of the degree of distortions at tetrahedral and octahedral sites of prepared nonstoichiometric Co–Ga NPs
When transition metal ions exist in distorted polyhedrons with less symmetric geometries compared to regular octahedral or tetrahedral geometries, they may have different absorption in the visible region, i.e., produce different colors.37 In this study, the polyhedral distortions in the prepared NPs may be induced by the Jahn–Teller effect of the transition metal ions (Co2+, Co3+) and may also be due to the synthesis conditions.38 These polyhedral distortions may affect the absorptions of Co2+–O4 and Co2+/3+–O6 in the prepared Co–Ga NPs and further affect the coloration of the NPs. Therefore, we simply evaluated the degrees of the polyhedral distortions at tetrahedral and octahedral sites in the spinel lattice by using Raman spectroscopy.Raman spectra in the range of 150–850 nm of the prepared NPs and reference of calcined CoGa2O4 are shown in Fig. 10(a). Because CoGa2O4 belongs to the Fdm space group, we theoretically considered five Raman active modes (A1g + Eg + 3F2g) and assigned them to the observed intensive peaks of calcined CoGa2O4.39 Notably, the presence of an extra peak marked with an asterisk (A*1g) was considered to be due to the degree of inversion.39,40 Compared to the calcined CoGa2O4, the Raman spectra of the prepared Co–Ga NPs generally show broader peaks, and peaks such as Eg and F2g (2) were not evident. In particular, the increase in the breadth of the observed peaks in our products was considered the result of polyhedral distortions within the spinel lattice.41 In Fig. 10(a), Raman spectra of our NPs also showed peak shifts compared to the calcined CoGa2O4. We consider that polyhedral distortions could affect the bond lengths of Co–O and Ga–O and result in a shift in the peak center. In this study, we focused on the full width at half maximum (FWHM), that is, the breadth of the peaks of F2g (1) and A*1g in the Raman spectra, to investigate tetrahedral distortions and the FWHM of A1g to investigate octahedral distortions of the NPs. Multiple peak fitting was conducted for the calcined CoGa2O4 and each pH condition, as shown in Fig. 10(b–e). The FWHM results as well as the peak location of each fitted peak are summarized in Table S2 (ESI†). Notably, the peak of F2g (3) was also intense in our products. However, this peak is considered to be associated with vibrations of oxygen atoms at both tetrahedral and octahedral sites and was not discussed in this study.42
 | ||
| Fig. 10 (a) Raman spectra of nonstoichiometric Co–Ga NPs produced at 400 °C using precursor solutions with pH 7–11 and calcined CoGa2O4 (1300 °C, 24 h) and their (b–e) peak fitting results. | ||
According to reports by Bouchard and Gambardella, the A1g peak is characteristic of vibrations involving the motion of oxygen atoms at octahedral sites, and the F2g (1) peak is associated with the motion of oxygen atoms at tetrahedral sites.41 Previous reports are on the study of a normal spinel of CoAl2O4, and we referred to those results in our study of CoGa2O4. However, the prepared Co–Ga NPs and the calcined CoGa2O4 are spinel compounds with inversions, and they have the additional characteristic peak of A*1g. We assigned A*1g to stretching vibrations involving oxygen atoms at tetrahedral sites.39 When focusing on the FWHM of the peaks of F2g (1), A1g, and A*1g, NPs prepared at pH 9 showed the largest FWHM value at F2g (1) and A*1g and those at pH 11 showed the largest FWHM value at A1g (see Table S2 in ESI†). Therefore, there could be large polyhedral distortions existing at the tetrahedral site at pH 9 and at the octahedral site at pH 11. Nevertheless, NPs prepared at pH 7 also showed a considerable degree of distortion at the tetrahedral site when focusing on the large FWHM at A*1g.
Next, we correlated the degree of polyhedral distortions to the UV–Vis absorption spectra of the prepared Co–Ga NPs. At pH 7 and pH 9, the large degrees of polyhedral distortions at the tetrahedral site might decrease the crystal field strength (10Dq) and may further intensify the triplet absorption band of Co2+–O4, as reported by Raj and Rao.43 As a result, evident triplet absorption at 560–650 nm and a blue hue were observed from the NPs at pH 7 and pH 9 despite the low tetrahedral cobalt occupancy. At pH 11, where there could be more Co2+/3+–O6, we consider that the large degree of distortions at the octahedral site might alleviate the effect of Laporte–forbidden transitions of the octahedral complexes (Co2+/3+–O6) due to asymmetric octahedrons.17 In our work, octahedral distortions might intensify the transitions of ν2 [1A1g → 1T2g] of Co3+–O6. This ν2 transition was observed at 430 nm in Co3O4 spinel compounds, which have a high content of Co3+–O6 similar to the NPs at pH 11 in this work.44 We also consider that the large absorption at 400–500 nm might also contain intensified absorptions of transitions ν3 [4T1g(F) → 2A1g(G)] and ν4 [4T1g(F) → 4T1g(P)] of Co2+–O6. They were observed at 470 nm and 510 nm in an alumina-supported cobalt system.45 In summary, all those intensified possible absorptions together with the absorption at 488 nm induced by the band gap absorption at pH 11 might finally cause a significant increase in the absorbance intensity at 400–500 nm when compared to pH 7 and pH 9 and result in an intensive green hue in the NPs prepared at pH 11.
In summary, color-controlled nonstoichiometric Co–Ga NPs prepared by the present technique were found to have different cationic configurations at tetrahedral and octahedral sites compared to the calcined stoichiometric CoGa2O4. Prepared NPs were also found to have considerable degrees of polyhedral distortions. Based on previous discussions of the main valences, cationic configurations, and degrees of distortions at the tetrahedral and octahedral sites, the relationship between the prepared Co–Ga NPs and their colorations is summarized in Table 1.
| pH 7 | pH 9 | pH 11 | |
|---|---|---|---|
| Composition (Co/Ga molar ratio) | 0.282 | 0.423 | 0.728 |
| Cobalt valence | Co2+ as the main valence | Co2+ as the main valence | Co2+ and Co3+ |
| Tetrahedral cobalt occupancy (0.336 for calcined CoGa2O415) |
0.115 (<0.336) | 0.129 (<0.336) | 0.0540 (<0.336) |
| Octahedral cobalt occupancy (0.332 for calcined CoGa2O415) |
0.273 (≈0.332) | 0.382 (≈0.332) | 0.603 (>0.332) |
| Number of octahedrally coordinated Co3+ ions (Co3+–O6) | Small | Small | Large |
| Distortions at tetrahedral sites | Large than calcined CoGa2O4 (when focusing on A*1g) | The largest | Smaller than calcined CoGa2O4 (when focusing on A*1g) |
| Distortions at octahedral sites | Larger than calcined CoGa2O4 | Larger than calcined CoGa2O4 | The largest |
| Contamination | Nonnegligible Ni2+ ions | Almost none | Almost none |
| Coloration | Greenish-blue | Blue | Yellowish-green |
3.5. Growth process of prepared nonstoichiometric Co–Ga NPs
Durable inorganic blue color materials have continuously attracted attention from human beings for a long time.46,47 In this work, we successfully prepared nonstoichiometric Co–Ga NPs with high crystallinity and a blue hue at a fixed low temperature of 400 °C without calcination but with less brilliance than calcined stoichiometric CoGa2O4, as shown in Fig. S8 (ESI†). We consider this result to be due to a lower tetrahedral cobalt occupancy that could result in weaker triplet absorption of the previously introduced spin-allowed transition of Co2+–O4. To find synthesis conditions that could allow more Co2+–O4, i.e., realizing a more brilliant blue color, we investigated the growth process in the present technique. Specifically, we conducted synthesis at 200 °C and 300 °C using the same precursor solution (Co/Ga = 0.25) and measured the XAFS spectra at the Co K-edge of the solid particles prepared at each pH condition. Next, Fourier transforms of the k3-weighted χ(k) oscillations in the EXAFS region of the products were derived. We investigated the growth process and discussed the strategy for a more brilliant blue color using the present technique by comparing the local structures (Fourier transforms) of the solid products produced at 200 °C, 300 °C, and 400 °C with that of calcined CoGa2O4.As shown in Fig. 11, the Fourier transforms of the k3-weighted χ(k) oscillations of most solid products and the calcined CoGa2O4 generally showed an intensive peak at 1.5 Å and two intensive peaks ranging from 2.2 to 3.5 Å. These peaks correspond to Co–O interactions and interactions between the metal atoms of MOct.–MOct. (2.2–3.0 Å) and MTet.–MOct./Tet. (3.0–3.5 Å), respectively. When focusing on the calcined CoGa2O4 (dotted line) with a brilliant blue color, there is nearly no difference in height between the two intense peaks ranging from 2.2 to 3.5 Å. Therefore, to prepare Co–Ga NPs exhibiting an intense blue color, the difference between the two peaks from 2.2 to 3.5 Å needs to be as small as possible.
 | ||
| Fig. 11 Fourier transforms of the k3-weighted χ(k) oscillations in the EXAFS region at the Co K-edge (@KEK PF BL-12C) of stoichiometric CoGa2O4 prepared by calcination at 1300 °C for 24 h (dotted line) and solid products prepared at 200 °C, 300 °C, and 400 °C using precursor solutions with (a) pH 7, (b) pH 9, and (c) pH 11. | ||
This difference was smaller at pH 7, larger at pH 9, and much larger at pH 11. This result indicates that cobalt ions show higher selectivity for the octahedral site during the growth process at higher pH. At pH 9, the differences between the two peaks at 2.2–3.5 Å significantly decreased with increasing synthesis temperature. This indicates the possibility of producing Co–Ga NPs with a more brilliant blue color when using a higher synthesis temperature in the present technique, even for an alkaline environment of pH 9. Unlike pH 9, the difference between the two peaks changed only slightly at a high synthesis temperature when using the precursor solution of pH 11. We consider this result to probably be due to an extremely high octahedral site selectivity of cobalt ions at pH 11. Therefore, Co–Ga NPs with a more brilliant blue coloration could be realized in an acidic synthesis environment or in an alkaline synthesis environment (pH < 11) when using a synthesis temperature higher than 400 °C in the present technique.
4. Conclusions
In this research, color-controlled nonstoichiometric spinel-type cobalt gallate nanopigments were realized by a rapid supercritical hydrothermal synthesis at 400 °C for 10 min without calcination when using a precursor solution with a low Co/Ga molar ratio (0.25) and different pH values. To summarize, the prepared Co–Ga NPs exhibited a higher Co/Ga molar ratio (0.282–0.728), a larger amount of Co3+, a larger octahedral cobalt occupancy (0.273–0.603), and a larger degree of octahedral distortions as the precursor solution pH increased. Generally, the color manipulation from greenish-blue to yellowish-green is probably due to an increasing amount of Co2+/3+–O6 and an increasing degree of distortions at the octahedral site in the spinel structure at higher pH. In particular, nonstoichiometric Co–Ga NPs with a blue color (R: 80, G: 150, B: 185) were successfully synthesized without calcination for the first time and were found to have a large degree of tetrahedral distortions. Additionally, investigations of the growth process of Co–Ga NPs during temperature increase indicated the possibility of producing Co–Ga NPs with a more brilliant blue color when using a higher synthesis temperature in the present technique. Our synthesis method has merits such as a lower synthesis temperature, a shorter synthesis time, use of inexpensive metal nitrate as raw materials, and less use of cobalt compared to traditional calcination methods. We believe that the present technique could lead to energy-efficient and environmentally friendly production of coloristic cobalt gallate nanopigments in the future.Author contributions
Bo Xie: Conceptualization, formal analysis, investigation (material production and XRD, SEM, ICP-AES, Raman spectroscopy and XAFS measurements), validation, visualization, writing – original draft, writing – review & editing. Chiya Numako: Formal analysis, investigation (XAFS measurements), resources, validation, writing – review & editing. Takashi Naka: Formal analysis, investigation (material production of calcined CoGa2O4), resources, validation, writing – review & editing. Seiichi Takami: Conceptualization, funding acquisition, investigation (XAFS measurements), formal analysis, project administration, resources, supervision, validation, writing – review & editing.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers 20H02514 and 23H01752 and was partially supported by the World Premier International Research Center Initiative (WPI), MEXT, Japan. XAFS measurements were conducted under the approval of the Photon Factory Program Advisory Committee (Proposal No. 2021G143, 2021G584). We acknowledge the Division for Medical Research Engineering, Graduate School of Medicine, Nagoya University for usage of the JSM-7610F and JEM-1400 Plus and corresponding technical support from Mr Koji Itakura.References
- V. D’Ippolito, G. B. Andreozzi, U. Hålenius, H. Skogby, K. Hametner and D. Günther, Phys. Chem. Miner., 2015, 42, 431 CrossRef.
- B. Serment, M. Gaudon, A. Demourgues, A. Noël, G. Fleury, E. Cloutet, G. Hadziioannou and C. Brochon, ACS Omega, 2020, 5, 18651 CrossRef CAS PubMed.
- Y. Youn, J. Miller, K. Nwe, K.-J. Hwang, C. Choi, Y. Kim and S. Jin, ACS Appl. Energy Mater., 2019, 2, 882 CrossRef CAS.
- G. B. Andreozzi, V. D’Ippolito, H. Skogby, U. Hålenius and F. Bosi, Phys. Chem. Miner., 2019, 46, 343 CrossRef CAS.
- E. Chavarriaga, A. Lopera, C. Bergmann and J. Alarcón, Bol. Soc. Esp. Ceram. Vidrio, 2020, 59, 176 CrossRef CAS.
- H. R. Hedayati, A. A. Sabbagh Alvani, H. Sameie, R. Salimi, S. Moosakhani, F. Tabatabaee and A. Amiri Zarandi, Dyes Pigm., 2015, 113, 588 CrossRef CAS.
- Y. Zhang, Y. Zhao, Z. Wang and L. Shang, Int. J. Appl. Ceram. Technol., 2020, 17, 2368 CrossRef CAS.
- F. T. Şenberber and O. D. Özdemir, Russ. J. Inorg. Chem., 2020, 65, 2020 CrossRef.
- M. Yoneda, K. Gotoh, M. Nakanishi, T. Fujii and T. Nomura, Powder Technol., 2018, 323, 574 CrossRef CAS.
- S. Kimura, Y. Kaneko, K. Marumoto and Y. Suzuki, J. Ceram. Soc. Jpn., 2020, 128, 260 CrossRef CAS.
- Y. H. Hou, Y. J. Zhao, Z. W. Liu, H. Y. Yu, X. C. Zhong, W. Q. Qiu, D. C. Zeng and L. S. Wen, J. Phys. D: Appl. Phys., 2010, 43, 445003 CrossRef.
- P. Porta and A. Anichini, J. Chem. Soc., Faraday Trans. 1, 1980, 76, 2448 RSC.
- J. G. Na, T. D. Lee and S. J. Park, J. Mater. Sci. Lett., 1993, 12, 961 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- T. Naka, T. Nakane, S. Ishii, M. Nakayama, A. Ohmura, F. Ishikawa, A. de Visser, H. Abe and T. Uchikoshi, Phys. Rev. B, 2021, 103, 224408 CrossRef CAS.
- S. Mathur, C. Cavelius and H. Shen, Z. Anorg. Allg. Chem., 2009, 635, 2106 CrossRef CAS.
- S. Yaemphutchong, W. Wattanathana, K. Chansaenpak, S. Singkammo, P. Kanjanaboos, P. Siri-apai, S. Janejobsakonkit, P. Pipattanaporn, N. Suetrong, S. Wannapaiboon and Y. Hanlumyuang, Ceram. Int., 2022, 48, 18490 CrossRef CAS.
- S. K. Biswas, D. Dhak, A. Pathak and P. Pramanik, Mater. Res. Bull., 2008, 43, 665 CrossRef CAS.
- W. J. Stark, P. R. Stoessel, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 5793 RSC.
- P. M. T. Cavalcante, M. Dondi, G. Guarini, M. Raimondo and G. Baldi, Dyes Pigm., 2009, 80, 226 CrossRef CAS.
- D. Rangappa, S. Ohara, S. Takami, T. Naka, A. Kondo, M. Ishii, T. Kobayashi and T. Adschiri, Mater. Res. Innovations, 2012, 16, 30 CrossRef CAS.
- D. Rangappa, T. Naka, A. Kondo, M. Ishii, T. Kobayashi and T. Adschiri, J. Am. Chem. Soc., 2007, 129, 11061 CrossRef CAS PubMed.
- J. Lu, K. Minami, S. Takami and T. Adschiri, Chem. Eng. Sci., 2013, 85, 50 CrossRef CAS.
- M. Zayat and D. Levy, Chem. Mater., 2000, 12, 2763 CrossRef CAS.
- N. El Habra, L. Crociani, C. Sada, P. Zanella, M. Casarin, G. Rossetto, G. Carta and G. Paolucci, Chem. Mater., 2007, 19, 3381 CrossRef CAS.
- C. M. Álvarez-Docio, J. J. Reinosa, A. del Campo and J. F. Fernández, Dyes Pigm., 2017, 137, 1 CrossRef.
- F. Tielens, M. Calatayud, R. Franco, J. M. Recio, J. Pérez-Ramírez and C. Minot, Solid State Ionics, 2009, 180, 1011 CrossRef CAS.
- B. Xie, C. Numako, T. Naka and S. Takami, Cryst. Growth Des., 2023, 23, 2511 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537 CrossRef CAS PubMed.
- A. A. Hummer and A. Rompel, Adv. Protein Chem. Struct. Biol., 2013, 93, 257 CAS.
- F. Izumi and K. Momma, Solid State Phenom., 2007, 130, 15 CAS.
- M. Newville, J. Phys.: Conf. Ser., 2013, 430, 012007 CrossRef CAS.
- M. Gaudon, L. C. Robertson, E. Lataste, M. Duttine, M. Ménétrier and A. Demourgues, Ceram. Int., 2014, 40, 5201 CrossRef CAS.
- M. Llusar, A. Forés, J. A. Badenes, J. Calbo, M. A. Tena and G. Monrós, J. Eur. Ceram. Soc., 2001, 21, 1121 CrossRef CAS.
- M. Taguchi, T. Nakane, K. Hashi, S. Ohki, T. Shimizu, Y. Sakka, A. Matsushita, H. Abe, T. Funazukuri and T. Naka, Dalton Trans., 2013, 42, 7167 RSC.
- B. Serment, C. Brochon, G. Hadziioannou, S. Buffière, A. Demourgues and M. Gaudon, RSC Adv., 2019, 9, 34125 RSC.
- S. Tamilarasan, S. Laha, S. Natarajan and J. Gopalakrishnan, Eur. J. Inorg. Chem., 2016, 288 CrossRef CAS.
- C. L. Bull, C. J. Ridley, N. P. Funnell, C. W. Wilson and S. G. Macleod, Mater. Adv., 2021, 2, 5096 RSC.
- V. D'Ippolito, G. B. Andreozzi, D. Bersani and P. P. Lottici, J. Raman Spectrosc., 2015, 46, 1255 CrossRef.
- X. Lu, W. Bian, Y. Li, H. Zhu, Z. Fu and Q. Zhang, J. Am. Ceram. Soc., 2018, 101, 1646 CrossRef CAS.
- M. Bouchard and A. Gambardella, J. Raman Spectrosc., 2010, 41, 1477 CrossRef.
- C. M. Álvarez-Docio, J. J. Reinosa, A. Del Campo and J. F. Fernández, J. Alloys Compd., 2019, 779, 244 CrossRef.
- A. K. V. Raj and P. P. Rao, ChemistrySelect, 2021, 6, 11344 CrossRef CAS.
- Y. Brik, M. Kacimi, M. Ziyad and F. Bozon-Verduraz, J. Catal., 2001, 202, 118 CrossRef CAS.
- A. Jean-Marie, A. Griboval-Constant, A. Y. Khodakov and F. Diehl, Catal. Today, 2011, 171, 180 CrossRef CAS.
- K. Kupferschmidt, Science, 2019, 364, 424 CrossRef CAS PubMed.
- B. A. Duell, J. Li and M. A. Subramanian, ACS Omega, 2019, 4, 22114 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: (a) Temperature inside the reactor, (b) results of Williamson–Hall plot, (c) discussions on size effect, (d) TEM-EDS results at pH 7 and pH 9, (e) conversions of cobalt and gallium and their percentages in aqueous phase at R.T., (f) band gap calculations, (g) Rietveld refinement results, (h) results of peak fitting of Raman spectra, and (i) digital images of the nonstoichiometric Co–Ga NPs prepared at 400 °C, pH 9 and calcined CoGa2O4. See DOI: https://doi.org/10.1039/d3dt03086e |
| This journal is © The Royal Society of Chemistry 2023 |