
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA robust Zintl cluster for the catalytic reduction of pyridines, imines and nitriles†
Bono
van IJzendoorn
 ,
Jessica B. M.
Whittingham
,
George F. S.
Whitehead
,
Nikolas
Kaltsoyannis
* and
Meera
Mehta
*
,
Jessica B. M.
Whittingham
,
George F. S.
Whitehead
,
Nikolas
Kaltsoyannis
* and
Meera
Mehta
*
Department of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: nikolas.kaltsoyannis@manchester.ac.uk; meera.mehta@manchester.ac.uk
First published on 12th September 2023
Abstract
Despite p-block clusters being known for over a century, their application as catalysts to mediate organic transformations is underexplored. Here, the boron functionalized [P7] cluster [(BBN)P7]2− ([1]2−; BBN = 9-borabicyclo[3.3.1]nonane) is applied in the dearomatized reduction of pyridines, as well as the hydroboration of imines and nitriles. These transformations afford amine products, which are important precursors to pharmaceuticals, agrochemicals, and polymers. Catalyst [1]2− has high stability in these reductions: recycling nine times in quinoline hydroboration led to virtually no loss in catalyst performance. The catalyst can also be recycled between two different organic transformations, again with no loss in catalyst competency. The mechanism for pyridine reduction was probed experimentally using variable time normalization analysis, and computationally using density functional theory. This work demonstrates that Zintl clusters can mediate the reduction of nitrogen containing substrates in a transition metal-free manner.
Introduction
Dihydropyridine derivatives are ubiquitous in naturally occurring materials, biologically active compounds including protonated nicotinamide adenine dinucleotide (NADH), and pharmaceuticals such as the cardiovascular medications nifedipine, pranidipine, nimodipine and amlodipine (Fig. 1).1,2 Thus, methodologies that construct dihydropyridines have garnered significant attention in synthetic chemistry. Usually, this scaffold is constructed by reducing pre-activated pyridines with strong reductants.3–5 This is often a multistep process, which requires an organometallic nucleophile, reducing the atom economy of the reaction, and suffers from poor regioselectivity. To address these limitations, catalyzed protocols relying on transition metal-based catalysts have been developed with mild reductants, including boranes, silanes and H2 gas.6–13 Amongst these protocols, those that employ boranes and silanes are desirable as H2 gas is both flammable and can lead to uncontrolled over reduction.13 The need to develop more environment-friendly processes has driven innovation towards catalysts based on inexpensive and abundant elements, including phosphorus and boron. With respect to facilitating pyridine hydroborations a handful of molecular metal-free catalysts have been reported. Specifically, Kinjo and Speed have reported diazaphosphorus catalysts,14,15 while Park and Chang, Li and Wang, and Wright have employed simple boranes to mediate this transformation.16–18 | ||
| Fig. 1 Example pharmaceuticals that feature the dihydropyridine moiety, previous examples of Zintl-based catalysts, and this work. | ||
Compared to homogenous catalysts, heterogeneous catalysts offer the advantages of greater stability and recyclability,19 as well as easier separations and thus lower purification costs. One inexpensive and abundant material that would be a sustainable alternative to metal-based catalysts is red phosphorus. However, its applications in catalysis are hardly known, with limited examples as a photocatalyst for bacterial deactivation,20 water splitting,21 and CO2 reduction.22 The poor solubility of red phosphorus makes it difficult to study and has stifled the discovery of new reactivity. Zintl clusters occupy the space between soluble molecules and insoluble solids, and can thus be considered stepping stones to uncovering new reactivity with heterogeneous materials.23,24 In particular, [P7]3− can be regarded as a fragment of red phosphorus23,25–29 and, once functionalized, has good solubility in common solvents allowing in situ investigations. Uncovering new reactivity and catalysis with [P7] clusters could be extended to related reactions with heterogeneous phosphorus.
Only a few examples of Zintl-based clusters in catalytic applications have been reported (Fig. 1). In most cases, the cluster is a spectator ligand coordinated to an expensive metal that mediates the transformation. In 2020 and 2022, Goicoechea and Weller coordinated a [Ge9] cluster to a Rh metal centre and affected the hydrogenation of alkenes, as well as H/D exchange reactions.24,30 Scheschkewitz coordinated a silicon cluster to Ir which then facilitated the isomerization of alkenes.31 Sun and Zhang dispersed [Ru@Sn9]6− onto a CeO2 surface to catalyse the reverse water–gas shift reaction, although whether the cluster remains intact after dispersion is not yet fully established.32 In 2022, we began to study [P7] clusters in transition-metal free catalysis. We have reported that boranes can be tethered using aliphatic linkers onto the [P7] cluster and then exploited in Lewis acid catalysis while the cluster remains an innocent platform.33 We have found that in the case of catalyst ([(BBN)P7]2− ([1]2−); BBN = 9-borabicyclo[3.3.1]nonane), where the boron is directly coordinated to [P7], the reduction of carbonyls and CO2 could be mediated with high efficiency compared to other metal-free catalysts.34 Mechanistic studies revealed that the [P7] cluster is non-innocent and substrate activation happens between both the boron and phosphorus centres in a cooperative fashion.
Herein, we report that catalytic amounts of [(BBN)P7]2− ([1]2−) promote the hydroboration of pyridines, imines, and nitriles. In the case of pyridine hydroboration, dihydropyridine products are formed, while the reduction of imines and nitriles gave simple amines. With over 40% of pharmaceuticals and pharmaceutical candidates featuring amine functional groups, it is the second most popular functional group in drug design after aromatic rings.35 Amines are also important components in agrochemicals, polymers, and dyes.36–39 Although the hydroboration of pyridines, imines and nitriles is well known via organometallic, molecular main group chemistry, and occasionally in a catalyst-free manner,40–56 herein the reductions are performed under mild conditions and represent the first of such reactivity where a Zintl cluster is a catalytic participant, establishing a new platform for these transformations.
Results and discussion
Catalytic reduction of N-heteroarenes
We have previously reported that the sodium and potassium salts of catalyst [(BBN)P7]2− ([1]2−) are prepared by dehydrocoupling the 9-borabicyclo[3.3.1]nonane dimer (HBBN dimer) with the respective [M(18-c-6)]2[HP7] (M = Na, K; 18-c-6 = 1,4,7,10,13,16-hexaoxacyclooctadecane) salts. The protonated [HP7]2− precursor is prepared in pyridine. We found that if adventitious pyridine was present in this precursor, and a slight excess of HBBN dimer used during the preparation of [Na(18-c-6)]2[(BBN)P7]2− ([Na(18-c-6)]2[1]), small amounts of clear crystals could be obtained from the reaction mixture. Single crystal X-ray diffraction (XRD) studies revealed two different crystalline products of different morphology. The first contains a BBN 1,4-hydroborated pyridine (2b′) (Scheme 1). The hydroborated pyridine has a C2–C3–C4 bond angle of 109.22(9)°, and hence a tetrahedral geometry at C3, and two hydrogens could be located on this carbon (Fig. 2). The N atom (N1) from the hydroborated pyridine coordinates the boron from the BBN unit with a bond length of 1.5440(13) Å. The B is also bound to an unreduced pyridine, confirmed by the longer B1–N2 bond length of 1.6773(13) Å. Hence, the structure of 2b′ is consistent with 1,4-hydroboration of pyridine with HBBN to give a dihydropyridine followed by coordination of a second pyridine to the boron atom. The second product (2b′′) contains two dihydropyridines and crystallizes as the [Na(THF)2(18-c-6)] salt (Fig. 3). This is presumably formed from 1,4-hydroboration of pyridine followed by reaction with another molecule of itself to form the dihydropyridine adduct. Analytically pure [(BBN)P7]2− ([1]2−) could be prepared by removing any residual pyridine from [HP7]2− under reduced pressures for extended periods of time, and controlling the stoichiometry of [HP7]2−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HBBN to be precisely 1:1. Serendipitous observation of 2b′ and 2b′′ prompted us to investigate the [1]2− cluster as a catalyst for the hydroboration of pyridines.
:HBBN to be precisely 1:1. Serendipitous observation of 2b′ and 2b′′ prompted us to investigate the [1]2− cluster as a catalyst for the hydroboration of pyridines.
 | ||
| Scheme 1 Serendipitous generation of 2b′ and 2b′′. | ||
 | ||
| Fig. 2 Molecular structure of 2b′. Anisotropic displacement ellipsoids pictured at 50% probability. Only selected hydrogen atoms shown for clarity. Boron: green; carbon: black; nitrogen: blue, hydrogen: white. Selected bond length [Å]: B1–N1 1.5440(13), B1–N2 1.6779(13), C1–C2 1.3402(14), C2–C3 1.5044(16); selected bond angles [°]: C2–C3–C4 109.22(9). | ||
 | ||
| Fig. 3 Molecular structure of 2b′′ in the [Na(THF)2(18-c-6)][2b′′] salt. Anisotropic displacement ellipsoids pictured at 50% probability. Only selected hydrogen atoms shown and [Na(THF)2(18-c-6)]+ cation omitted for clarity. Boron: green; carbon: black; nitrogen: blue; hydrogen: white. Selected bond length [Å]: B1–N1 1.581(3), B1–N2 1.582(2), C1–C2 1.345(3), C2–C3 1.501(3); selected bond angles [°]: C2–C3–C4 109.04(18). | ||
First, 10 mol% [Na(18-c-6)]2[1] was investigated with equimolar amounts of pyridine and HBBN in THF-d8 at 50 °C and found to give 66% conversion to the 1,4- and 1,2-hydroborated products in a 99:1 ratio (Table 1). Changing the borane reductant from HBBN to catecholborane (HBcat) gave a similar distribution of products, but with only 6% overall conversion. Meanwhile, when pinacolborane (HBpin) was employed as the reductant complete conversion to the 1,4- and 1,2-hydroborated products was observed, now in a 95:5 ratio. Moving to the less coordinating polar solvent ortho-difluorobenzene (oDFB) increased conversions to the dihydropyridine products, from 66% to 91% when HBBN reductant was employed, and from 6% to 22% in the case of HBcat, while maintaining product selectivity. When the solvent was altered to C6D6 no reduction of pyridine was observed with HBBN or HBcat, and conversion with HBpin declined to 22%. Changing the solvent from THF to oDFB with HBpin as the reductant maintained complete conversion of pyridine to the dihydropyridine products. This diminished reactivity is presumably due to the lower solubility of the dianionic catalyst in the non-polar solvent. Next, in oDFB with 1:1 HBpin and pyridine the catalyst loading of [Na(18-c-6)]2[1] was reduced from 10 mol% to 5 mol% and again complete conversion to the dihydropyridine products was obtained with no change to product selectivity. Decreasing the catalyst loading to 1 mol% maintained complete hydroboration of pyridines, but the product distribution between the 1,4-hydroborated and 1,2-hydroborated isomers was 93:7. Because of the greater solubility of HBpin compared to HBBN the room temperature experiment was also tested. 1 mol% [Na(18-c-6)]2[1], HBpin and pyridine were allowed to react in oDFT at room temperature and found to give 90% conversion to the 1,4- and 1,2-hydroborated products in an 83:17 proportion. The difference in product distribution between the room temperature and analogous 50 °C experiment (83:17 and 93:7 respectively) is consistent with the 1,4-hydroborated pyridine being the thermodynamic product. Heating a reaction mixture containing both the 1,4- and 1,2-hydroborated products in a 86:14 respective ratio to 50 °C for 24 hours in the presence of [Na(18-c-6)]2[1] and HBpin did not change the product distributions, indicating no subsequent conversion between the 1,4- and 1,2-hydroborated products.
|
|
|||||
|---|---|---|---|---|---|
| Catalyst | Loading (mol%) | Solvent | H-[B] | Temp. (°C) | Conv.a (%) (b:c) |
|
a Determined by 1H NMR spectroscopy, based on C–H bond formation. Ratio b:c given in parentheses.
|
|||||
| [Na(18-c-6)]2[1] | 10 | THF-d8 | (HBBN)2 | 50 | 66 (99:1) |
| [Na(18-c-6)]2[1] | 10 | THF-d8 | HBcat | 50 | 6 (99:1) |
| [Na(18-c-6)]2[1] | 10 | THF-d8 | HBpin | 50 | >99 (95:5) |
| [Na(18-c-6)]2[1] | 10 | C6D6 | (HBBN)2 | 50 | 0 |
| [Na(18-c-6)]2[1] | 10 | C6D6 | HBcat | 50 | 0 |
| [Na(18-c-6)]2[1] | 10 | C6D6 | HBpin | 50 | 22 (87:13) |
| [Na(18-c-6)]2[1] | 10 | oDFB | (HBBN)2 | 50 | 91 (99:1) |
| [Na(18-c-6)]2[1] | 10 | oDFB | HBcat | 50 | 22 (99:1) |
| [Na(18-c-6)]2[1] | 10 | oDFB | HBpin | 50 | >99 (96:4) |
| [Na(18-c-6)]2[1] | 5 | oDFB | HBpin | 50 | >99 (94:6) |
| [Na(18-c-6)]2[1] | 1 | oDFB | HBpin | 50 | >99 (93:7) |
| [Na(18-c-6)]2[1] | 0.2 | oDFB | HBpin | 50 | 12 (91:9) |
| [Na(18-c-6)]2[1] | 1 | oDFB | HBpin | RT | 90 (83:17) |

To better understand the role of the cluster catalyst, several control reactions were undertaken (Table 2). First, in the absence of catalyst (entries 1 and 2) in oDFB with HBpin and pyridine at both room temperature and 50 °C no conversion to the hydroborated products was observed. Secondly, the literature-known tris-functionalized (Me3Si)3P7 cluster was prepared57 and found to be catalytically inactive at 5 mol% catalyst loading at 50 °C (entry 3). Next, (HBBN)2 (entry 4) was tested at 5 mol% catalyst loading, 10 mol% borane monomer loading, and again found to be completely inactive towards pyridine hydroboration. As anticipated, when [K(18-c-6)]2[1] (entry 5) was employed at 5 mol% catalyst loading the overall conversion to the 1,4- and 1,2-hydroborated products was >99% in an 88:12 ratio, in line with the reactivity of the sodium salt. When the unfunctionalized trianionic cluster precursor [Na(DME)x]3[P7] was investigated (entry 6) it was found to be catalytically inactive. However, surprisingly when 18-c-6 was added to [Na(DME)x]3[P7] (entry 7), 70% catalytic conversion to the dihydropyridine products was observed with 75% of that converted to the 1,4-hydroborated product and 25% to the 1,2-hydroborated product. The same reactivity was observed with the potassium salt, where the unfunctionalized cluster (entry 8) in the absence of 18-c-6 showed no catalytic activity and with 18-c-6 (entry 9) gave 68% conversion to the pyridine 1,4- and 1,2-hydroborated products. Next, 18-c-6 itself and 18-c-6 with sodium triflate (NaOTf) were each tested (entries 10 and 11) at 10 mol% catalyst loadings, and both found to be inactive. These results are consistent with the 18-c-6 and the s-block cation not being independently catalytically active, meaning that the [P7]3− component must be involved in the catalysis. It is expected that the role of 18-c-6 is in increasing the solubility of the naked trianionic [P7]3− salt, which can then coordinate to the boron of H–Bpin and promote transfer of the hydride to the pyridine substrate, followed by subsequent Bpin transfer to the cluster. 31P NMR (nuclear magnetic resonance) studies on the reaction of [P7]3−, HBpin and pyridine in the presence of 18-c-6 revealed the formation of a functionalized Zintl cluster which is believed to be [(Bpin)P7]2−. We have previously shown that the [HP7]2− catalyst precursor could be activated in situ using HBpin to form [(Bpin)P7]2−.34 The 31P NMR spectrum from this in situ generated [(Bpin)P7]2− and from the reaction mixture when [P7]3− is used as the catalyst were in good agreement (see ESI section 2.4†). Reaction of [P7]3− with an excess of HBpin and 18-c-6 did not show any new resonances in the 31P NMR spectrum. Thus, it was suspected that a substrate must be present to accept the hydride from HBpin and form [(Bpin)P7]2−. This need for a substrate was further confirmed by addition of trityl tetrakis(pentafluorophenyl)borate as a stoichiometric hydride acceptor in place of the substrate, which results in a 31P NMR spectrum in good agreement with the spectra observed from the reactions where [P7]3− and [HP7]2− are used as (pre)catalysts. [Na(18-c-6)]2[HP7] was also tested (entry 12) as a pre-catalyst in the hydroboration of pyridine, and at 5 mol% catalyst loading found to give 72% conversion to the dihydropyridine products with 81% of that converted to the 1,4-hydroborated product (2b) and 19% to the 1,2-hydroborated product (2c). These control reactions are consistent with the [P7] cage being necessary to promote the catalysis, the naked [P7]3− cluster having some (pre)catalytic competency, and also cooperative reactivity with a boron directly on the cluster, catalyst [1]2−, having enhanced catalytic activity.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Loading (mol%) | Temp. (°C) | Conv.a (%) (2b:2c) |
|
a Determined by 1H NMR spectroscopy, based on C–H bond formation. Ratio 2b:2c given in parentheses.
b Pre-catalyst.
|
||||
| 1 | None | None | RT | 0 |
| 2 | None | None | 50 | 0 |
| 3 | (Me3Si)3P7 | 5 | 50 | 0 |
| 4 | (HBBN)2 | 5 | 50 | 0 |
| 5 | [K(18-c-6)]2[1] | 5 | 50 | >99 (88:12) |
| 6 | [Na(DME)]3[P7] | 5 | 50 | 0 |
| 7 | [Na(DME)]3[P7] + 18-c-6b | 5 | 50 | 70 (75:25) |
| 8 | K3P7 | 5 | 50 | 0 |
| 9 | K3P7 + 18-c-6b | 5 | 50 | 68 (75:25) |
| 10 | 18-c-6 | 10 | 50 | 0 |
| 11 | NaOTf + 18-c-6 | 10 | 50 | 0 |
| 12 | [Na(18-c-6)]2[HP7]b | 5 | 50 | 72 (81:19) |

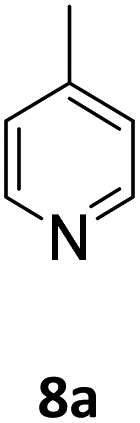
Next, the scope of pyridine hydroboration with 5 mol% [Na(18-c-6)]2[1] and HBpin in oDFB at 50 °C was expanded (Table 3). 2-Methylpyridine (3a) was hydroborated to 53% conversion after 48 h. This lower conversion is believed to be due to the increased steric demand, and in fact only the 1,4-hydroborated product was observed. Similarly, 2-methoxypyridine (4a) is only hydroborated in trace amounts to 4b. Whereas, efforts to hydroborate 2-chloro pyridine and 2-phenyl pyridine were unsuccessful. In the case of meta-substitution, 3-methoxypyridine (5a), 3-chloropyridine (6a), and 3,5-lutidine (7a) are all hydroborated in moderate to low conversions, between 57% and 29%. Hydroboration of the para-substituted 4-methylpyridine (8a) results in 30% conversion to a 50:50 mixture of 8b and 8c.









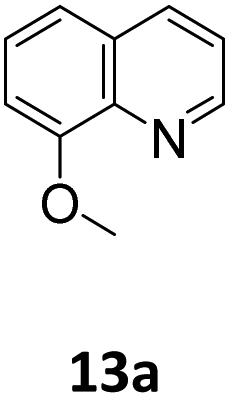
Next, the hydroboration of quinolines and an acridine was explored with 5 mol% [Na(18-c-6)]2[1] and HBpin in oDFB at 50 °C (Table 4). The hydroboration of quinolines and an acridine was found to be more facile than pyridine. For instance, quinoline (9a) was hydroborated in complete conversion to the 1,4- (9b) and 1,2- (9c) products in 88% and 12% product distribution after 18 h rather than the 48 h required for pyridine (2a). Due to the more facile hydroboration of 9a, select control reactions were repeated with 9a. In the absence of catalyst or when employing 5 mol% HBBN dimer, no hydroboration of 9a was observed. In-line with the pyridine hydroboration (Table 2, entry 7), when 5 mol% K3P7 + 18-c-6 was employed, 9a was hydroborated with an overall conversion of 75% and with products 9b:9c in a 4:1 ratio. Further, the planar substitution of the phenyl ring in quinoline meant the decoration at this ring was less obstructive to the catalyst when compared to pyridines. 6-Methylquinoline (10a), 6-methoxyquinoline (11a), and 6-bromoquinoline (12a) are all hydroborated in nearly complete conversion, 97–99%, after 18 hours. Similarly, 8-methoxyquinoline (13a) was hydroborated in high conversion, 89%, with complete selectivity for 13b observed. 6-Bromoquinoline (14a) was hydroborated to complete conversion with a 96:4 1,4-hydroboration: 1,2-hydroboration product ratios. Finally, acridine (15a) was hydroborated to 15b in 93% conversion after 24 hours. In all these cases, as anticipated, the formation of the 1,4-hydroborated product is preferred over the 1,2-product.
|
|
|||
|---|---|---|---|
| Substrate | Product(s) and distribution | Time (h) | Conv.a (%) |
| a Determined by 1H NMR spectroscopy, based on C–H bond formation. Yield after work-up shown in parentheses. | |||

|

|
18 | |
| 9a R = H |
9b:9c: R = H (95:5) |
9b + c: > 9 (89) | |
| 10a R = Me |
10b:10c: R = Me (96:4) |
10b + c: 97 (92) | |
| 11a R = MeO |
11b:11c: R = MeO (96:4) |
11b + c: >99 (91) | |
| 12a R = Br |
12b:12c: R = Br (96:4) |
12b + c: >99 (87) | |
|

|
48 | 89 |

|

|
18 | >99 (89) |

|

|
24 | 93 (86) |

Catalytic reduction of imines and carbonitriles
In an effort to broaden the scope of the study, the hydroboration of other C–N multiple bonded substrates was investigated, specifically imines and nitriles. Beginning with the imines, 1 mol% [1]2− was allowed to react with 1 equivalent of an imine and 1 equivalent of HBpin at RT in oDFB. When N-benzylideneaniline (16a) was employed as the imine, after 48 h 25% conversion to the borylamine (16b) was obtained. However, increasing the catalyst loading to 2.5 mol% now gave 16b in 98% conversion. As expected, changing the solvent from oDFB to THF decreased catalytic reactivity (see ESI Table S1†), consistent with the previously reported hydroboration of carbonyls34 and N-heteroarenes discussed above. Control reactions were undertaken (Table 5) and similar to the hydroboration of the N-heteroarenes no conversion was observed in the absence of a catalyst or when using (Me3Si)3P7 as the catalyst (entries 1 and 2). HBBN dimer (entry 3) was observed to stoichiometrically hydroborate N-benzylideneaniline (16a), but not catalytically. Again 18-c-6, NaOTf and the unfunctionalized clusters [Na(DME)x]3[P7] and K3P7 (entries 4–6 and 8) were found to be catalytically inactive Whereas addition of 18-c-6 to the [P7] salts (entries 7 and 9) resulted in high conversions, 90–91%, of 16b being formed. A similar conversion of 92% was observed when [Na(18-c-6)]2[HP7] was employed as a pre-catalyst (entry 10). Further, as expected 2.5 mol% catalyst loading of [K(18-c-6)]2[1] (entry 11) afforded 16b in complete conversion, in good agreement with the sodium salt [Na(18-c-6)]2[1].|
|
|||
|---|---|---|---|
| Entry | Catalyst | Loading (mol%) | Conv.a (%) |
| a Determined by 1H NMR spectroscopy, based on C–H bond formation. b Pre-catalyst. | |||
| 1 | None | None | 0 |
| 2 | (Me3Si)3P7 | 5 | 0 |
| 3 | (HBBN)2 | 5 | 10 |
| 4 | 18-c-6 | 10 | 0 |
| 5 | NaOTf + 18-c-6 | 10 | 0 |
| 6 | [Na(DME)]3[P7] | 5 | 1 |
| 7 | [Na(DME)]3[P7] + 18-c-6b | 5 | 90 |
| 8 | K3P7 | 5 | 1 |
| 9 | K3P7 + 18-c-6b | 5 | 91 |
| 10 | [Na(18-c-6)]2[HP7]b | 5 | 92 |
| 11 | [K(18-c-6)]2[1] | 2.5 | >99 |

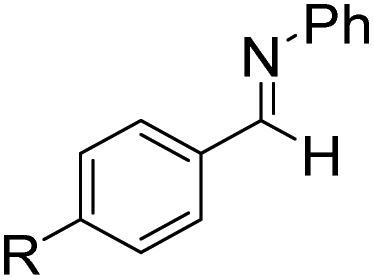
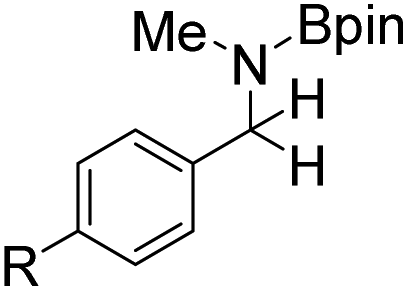
Next, para-substituted N-phenyl-1-(p-tolyl)methanimine (17a), 1-(4-methoxyphenyl)-N-phenylmethanimine (18a), and 1-(4-bromophenyl)-N-phenylmethanimine (19a) were all efficiently hydroborated to the boryl amines 17b, 18b, and 19b, correspondingly, in high yields, between 86% and 97% (Table 6). Imines 17a and 18a bearing the electron donating substituents methyl and methoxy required longer reaction times compared to imine 19a with the electron withdrawing bromine substituent. In a similar fashion, hydroboration of substrate 20a, where the N-substitution is altered from phenyl to methyl, still gave the borylamine product 20b in high yield. N-methyl-1-(p-tolyl)methanimine (21a), N-methyl-1-(4-methoxyphenyl)methanimine (22a), and N-methyl-1-(4-methoxyphenyl)methanimine (23a) were also converted to the corresponding amines 21b, 22b, and 23b in high yields, ranging from 85% to 99%. However, when the steric bulk on the N-substituent was increased, for example in the case of substrates N-t-butyl-1-phenylmethanimine, N-t-butyl-1-cyclohexylmethanimine and N-(2,4,6-trimethylphenyl)-1-phenylmethanimine, no catalytic conversions could be obtained. However, pyridine functionalized imines 24a and 25a were efficiently hydroborated to the amines 24b and 25b in 92% and 97% conversion, respectively. Addition of a second equivalent of HBpin after 24b was formed did not lead to subsequent hydroboration of the pyridine. The alkyl functionalized imine 1-cyclohexyl-N-methylmethanimine (26a) was also hydroborated to 26b in complete conversion, but 48 h were required. Efforts to hydroborate the less reactive ketimine N-(1-phenylethylidene)aniline only resulted in trace amounts of product being observed even at elevated temperatures (50 °C).
|
|
|||
|---|---|---|---|
| Substrate | Product | Time (h) | Conv.a (%) |
| a Determined by 1H NMR spectroscopy, based on C–H bond formation. Yield after work-up shown in parentheses. | |||
|

|
||
| 16a: R = H | 16b: R = H | 16b: 36 | 16b: 98 (86) |
| 17a: R = Me | 17b: R = Me | 17b: 48 | 17b: 86 |
| 18a: R = MeO | 18b: R = MeO | 18b: 48 | 18b: 94 (81) |
| 19a: R = Br | 19b: R = Br | 19b: 36 | 19b: 97 (85) |

|
|
48 | |
| 20a: R = H | 20b: R = H | 20b: 93 (86) | |
| 21a: R = Me | 21b: R = Me | 21b: >99 (87) | |
| 22a: R = MeO | 22b: R = MeO | 22b: 85 (84) | |
| 23a: R = Br | 23b: R = Br | 23b: 97 (91) | |

|

|
36 | 97 (82) |

|

|
36 | 92 (89) |

|

|
48 | >99 (86) |

The successful catalytic hydroboration of imines encouraged us to investigate carbonitriles. When benzonitrile (27a) was allowed to react with one equivalent of HBpin with 5 mol% [1]2− in oDFB at 50 °C only 50% conversion to the double hydroborated product 27b and 50% unreacted 27a could be observed. Thus, two equivalents of HBpin were employed in the nitrile reductions to hydroborate both of the π-bonds.
When 1 mol% of catalyst [Na(18-c-6)]2[1] was allowed to react with 1 equivalent of benzonitrile (27a) and 2 equivalent HBpin at RT the bis(boryl)amine 27b was afforded in only 5% conversion (Table 7). Increasing the catalyst loading to 5 mol% increased the conversion to 18%, while increasing the temperature to 50 °C further increased the conversion to 97% after 48 hours. As observed in the hydroboration of carbonyls,34 N-heteroarenes and imines, using THF as the solvent decreased the catalytic performance.
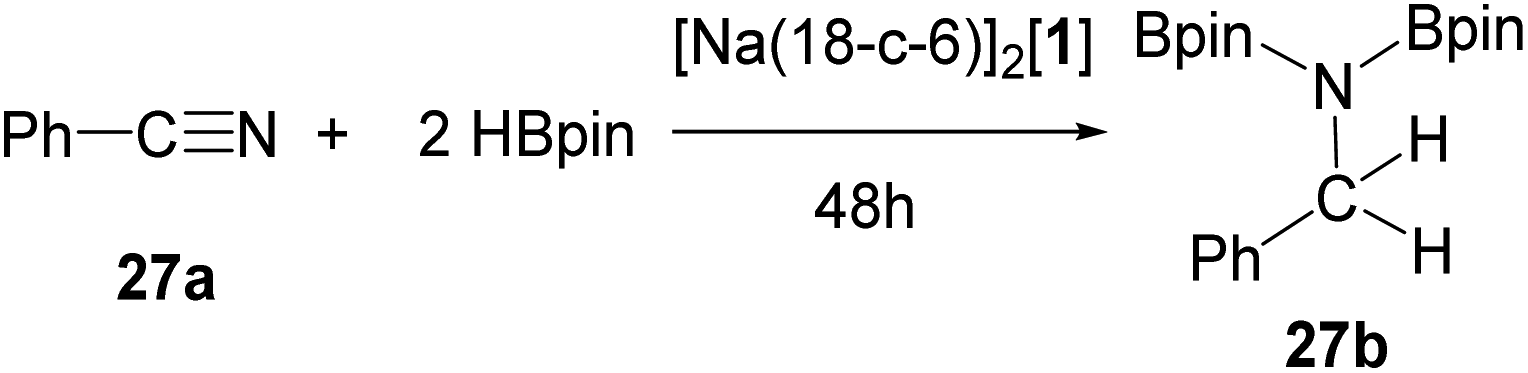
The same control reactions were tested as in the hydroboration of N-heteroarenes and imines (Table 8). Again, no catalytic conversion was observed in the absence of catalyst, with (Me3Si)3P7, HBBN dimer, 18-c-6, NaOTf, or K3P7 (entries 1–5 and 8). However, when [Na(DME)]3[P7] was employed as a catalyst 2% conversion to 27b was observed (entry 6). Addition of 18-c-6 to [Na(DME)]3[P7] and K3P7 (entries 7 and 9) again increased the catalytic performance, giving 68% and 64% of 27b, respectively. Whereas, when the [Na(18-c-6)]2[HP7] precursor (entry 10) was employed as a pre-catalyst only 12% conversion to 27b was observed. Again, changing the s-block cation from sodium to potassium had a minimal impact on the catalytic competency (entry 11), with [K(18-c-6)]2[1] at 5 mol% catalyst loading giving 97% conversion to 27b.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Loading (mol%) | Conv.a (%) |
| a Determined by 1H NMR spectroscopy, based on C–H bond formation. b Pre-catalyst. | |||
| 1 | None | None | 0 |
| 2 | (Me3Si)3P7 | 5 | 0 |
| 3 | (HBBN)2 | 5 | 6 |
| 4 | 18-c-6 | 10 | 0 |
| 5 | NaOTf + 18-c-6 | 10 | 0 |
| 6 | [Na(DME)]3[P7] | 5 | 2 |
| 7 | [Na(DME)]3[P7] + 18-c-6b | 5 | 68 |
| 8 | K3P7 | 5 | 0 |
| 9 | K3P7 + 18-c-6b | 5 | 64 |
| 10 | [Na(18-c-6)]2[HP7]b | 5 | 12 |
| 11 | [K(18-c-6)]2[1] | 5 | 97 |

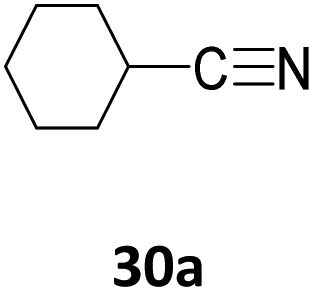
With 5 mol% catalyst loading of [Na(18-c-6)]2[1] at 50 °C, 4-bromobenzonitrile (28a) and 4-methoxybenzonitrile (29a) were both hydroborated to their respective bis(boryl)amine products 28b and 29b in 65% and 82% conversion (Table 9). Both also required longer reaction times compared to benzonitrile 27a. Whereas, hydroboration of the alkyl carbonitriles cyclohexylcarbonitrile (30a) and butylnitrile (31a) only showed moderate conversions to their bis-hydroborated products, giving 43% and 29% conversion correspondingly, while the pyridine substituted carbonitrile 32a afforded 32b in 5% conversion after 48 h. In the hydroboration of 32a no hydroboration of the pyridine moiety was observed.








To probe the chemoselectivity of these hydroborations, a competition reaction was carried out between benzophenone, acridine (15a), and N-benzylideneaniline (16a), with increasing amounts of HBpin (Scheme 2). These substrates were selected as they could be hydroborated in high yields in contrast to pyridines or nitriles. Using one equivalent of HBpin, exclusive hydroboration of benzophenone was observed. Addition of a second equivalent of HBpin resulted in further hydroboration of 15a and 16a to give 15b and 16b in a 59:41 ratio, suggesting that 15a and 16a are almost equally susceptible to hydroboration under these conditions. Addition of a third equivalent of HBpin resulted in complete hydroboration of all substrates.
 | ||
| Scheme 2 Competition reaction between benzophenone, 15a, and 16a. | ||
Catalyst recycling
To investigate [Na(18-c-6)]2[1] catalyst stability, the hydroboration of quinoline (9a) was performed as described above (5 mol% loading, 1 eq. HBpin, 50 °C, 18 h). After completion, the reaction mixture was investigated by 1H and 31P NMR spectroscopy and no evidence of catalyst decomposition was observed (ESI section 5.1†). To test subsequent catalyst recyclability, the reaction sample was reloaded with 1 equivalent of quinoline and 1 equivalent of HBpin. Again, at 50 °C, complete conversion was observed after 18 hours. The catalyst was recycled a total of 9 times without loss of catalyst performance and with only a slight difference in selectivity (Fig. 4). It was also found that the catalyst could be recovered after the catalytic hydroboration of 9a was complete, and then re-used in a separate batch without loss of performance. Further, catalyst [Na(18-c-6)]2[1] was applied in the previously reported reduction of benzaldehyde,34 separated from the reaction mixture, and then recycled in the hydroboration of 15a with no loss in catalyst performance observed, see ESI section 5.2.† This recycling is consistent with living catalysis. Further, for the catalytic hydroboration of 15a and 16a, the reaction was successfully scaled to 5.58 mmol with no loss in catalytic performance, resulting in 1.45 g (85% isolated yield) of 15b and 1.68 g (97% isolated yield) of 16b. | ||
| Fig. 4 Catalyst [Na(18-c-6)]2[1] recycling for the hydroboration of quinoline. | ||
Mechanistic investigations
Thomas and co-workers have previously reported the ‘hidden role’ that BH3 and borohydrides can play in hydroboration catalysis.58 To test for hidden BH3 and borohydride generation, the hydroboration of pyridine (2a), N-benzylideneaniline (16a), and benzonitrile (27a) was performed using the optimized conditions described above, specifically: for pyridine 1 eq. HBpin, 50 °C, 5 mol% [Na(18-c-6)]2[1]; for N-benzylideneaniline 1 eq. HBpin, RT, 2.5 mol% [Na(18-c-6)]2[1]; and for benzonitrile 2 eq. HBpin, 50 °C, 5 mol% [Na(18-c-6)]2[1]. After each reaction was complete, an excess of tetramethylethylenediamine (TMEDA) was added to capture borohydrides and allow for their detection by NMR spectroscopy. No evidence of TMEDA-captured BH3 could be detected in any of these reaction mixtures by 11B NMR spectroscopy (see ESI section 6†). Next [Na(18-c-6)]2[1] was reacted with HBpin under our most vigorous reaction conditions (50 °C, 48 h). The reaction mixture was then investigated by NMR spectroscopy before and after TMEDA was added to the reaction mixture. And again, no evidence of BH3 formation was detected. Further, 5 mol% BH3·SMe2 was employed as a catalyst itself for the hydroboration of pyridine, N-benzylideneaniline, and benzonitrile and was found to perform significantly worse than [Na(18-c-6)]2[1]. Thus, hidden borohydride catalysis is not expected to occur in these transformations.For the hydroboration of nitriles and imines we expect a similar mechanism as previously reported for [Na(18-c-6)]2[1] in the hydroboration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.34 Stoichiometric reactions of [Na(18-c-6)]2[1] with N-benzylideneaniline or with benzonitrile both show formation of a new (asymmetric) functionalized cluster at RT, in-line with activation of the substrate before HBpin, see ESI section 7.† Whereas, when [Na(18-c-6)]2[1] was investigated in pyridine as a solvent, no evidence of coordination could be observed. We have previously reported that stoichiometric reaction of HBpin with [Na(18-c-6)]2[1] at 50 °C gave NMR data consistent with formation of either intermediate I1 or I2 shown in Scheme 4. Further, a mechanism where the pyridine and HBpin form an adduct which then transfers hydride to the boron on [Na(18-c-6)]2[1] could be envisioned, similar to that reported by Li and Wang.18 During this mechanism, because HBpin and pyridine pre-form an adduct prior to interaction with the catalyst, we would expect no scrambling of the boron group on pyridine. In contrast, if pyridine is coordinated to the borane once there has been activation at the catalyst, there is some probability that the BBN unit on the catalyst could transfer to pyridine rather than Bpin. In order to test for this boron scrambling, a stoichiometric reaction between HBpin, pyridine (2a), and [Na(18-c-6)]2[1] was conducted (Scheme 3). Analysis of the reaction mixture by NMR spectroscopy revealed compound 2b to be the major product and the BBN analogue 2b′ as the minor product in a 7:3 ratio. When 2b was allowed to react with HBBN dimer in the presence of [Na(18-c-6)]2[1] at 50 °C, exchange of the boron-moieties and liberation of HBpin was observed. However, no scrambling was observed when [Na(18-c-6)]2[1] was allowed to react with 2b alone. Further, in our previous stoichiometric studies between [Na(18-c-6)]2[1] and HBpin there was no evidence of HBBN formation.34 Therefore, we do not expect 2b′ to formed after catalysis.
O bonds.34 Stoichiometric reactions of [Na(18-c-6)]2[1] with N-benzylideneaniline or with benzonitrile both show formation of a new (asymmetric) functionalized cluster at RT, in-line with activation of the substrate before HBpin, see ESI section 7.† Whereas, when [Na(18-c-6)]2[1] was investigated in pyridine as a solvent, no evidence of coordination could be observed. We have previously reported that stoichiometric reaction of HBpin with [Na(18-c-6)]2[1] at 50 °C gave NMR data consistent with formation of either intermediate I1 or I2 shown in Scheme 4. Further, a mechanism where the pyridine and HBpin form an adduct which then transfers hydride to the boron on [Na(18-c-6)]2[1] could be envisioned, similar to that reported by Li and Wang.18 During this mechanism, because HBpin and pyridine pre-form an adduct prior to interaction with the catalyst, we would expect no scrambling of the boron group on pyridine. In contrast, if pyridine is coordinated to the borane once there has been activation at the catalyst, there is some probability that the BBN unit on the catalyst could transfer to pyridine rather than Bpin. In order to test for this boron scrambling, a stoichiometric reaction between HBpin, pyridine (2a), and [Na(18-c-6)]2[1] was conducted (Scheme 3). Analysis of the reaction mixture by NMR spectroscopy revealed compound 2b to be the major product and the BBN analogue 2b′ as the minor product in a 7:3 ratio. When 2b was allowed to react with HBBN dimer in the presence of [Na(18-c-6)]2[1] at 50 °C, exchange of the boron-moieties and liberation of HBpin was observed. However, no scrambling was observed when [Na(18-c-6)]2[1] was allowed to react with 2b alone. Further, in our previous stoichiometric studies between [Na(18-c-6)]2[1] and HBpin there was no evidence of HBBN formation.34 Therefore, we do not expect 2b′ to formed after catalysis.
 | ||
| Scheme 3 Stoichiometric hydroboration of pyridine. | ||
 | ||
| Scheme 4 Computed mechanism for the [1]2−-catalyzed hydroboration of pyridine. Electronic energies (kcal mol−1) and, in parentheses, Gibbs energies are given for the individual steps. Transition state energies are given in purple. Only key H atoms are shown. Functional: wB97XD; basis set: def2-TZVP. The choice of computational methodology, and the presentation of both electronic and Gibbs energies, follows from our previous work in this area.34 An alternative representation of this scheme, including ball and stick images of all stationary points, is given in the ESI section 9.3.† | ||
To gain further insight into the mechanism of pyridine hydroboration, we turned to computational quantum chemistry in the form of hybrid density functional theory. Details of the computational methodology, and the coordinates and energies of all stationary points, are collected in the ESI section 9.† A computed mechanism is given in Scheme 4.
Following on from our previous computational mechanistic studies in ref. 34, we begin from [1]2−. Addition of H[B] forms intermediate I1. This is mildly exothermic at the electronic energy level, but endothermic at the Gibbs level. Note that the present I1 in Scheme 4 is not the I1 in ref. 34, but is actually slightly more stable. I1 then forms I2 (I1 in ref. 34) via transition state 1 (TS1). Addition of pyridine to I2viaTS2 yields I3, which then undergoes H atom transfer from B to the pyridine ring viaTS3 to give I4. Very exothermic dissociation of the hydroborated pyridine from I4 then returns us to [1]2−. A second isomer related to I4 (I4_iso), where the pyridine is coordinated to the BBN moiety rather than Bpin was also computed and found to be 5.4 kcal mol−1 more stable. This intermediate is expected to be related to the formation of 2b′ from Scheme 3. However, conversion of I4 to this other isomer is expected to be a multi-step process which is currently under investigation. The higher Lewis acidity and pinched back alkyl groups of BBN presumably contribute to the preference for borohydride formation at the BBN moiety over the Bpin for [I1] and [I2], allowing for selective N–Bpin formation rather than N–BBN. For similar reasons, along with the anionic nature of the cluster, a stronger P–B bond for the P–BBN moiety could be expected compared to the P–Bpin moiety. The anionic nature of the cluster is also thought to prevent quenching of the boron in [1]2— by the N-heterocycle.
Next, a variable time normalization analysis method reported by the Burés group was used to determine the order of the reagents using [Na(18-c-6)]2[1] as a catalyst in the hydroboration of pyridine.59,60 The analysis supports a zero order in concentration of pyridine, and first order in the concentration of HBpin and catalyst. Monitoring the catalysis by 31P NMR spectroscopy revealed [1]2− to be present during the whole catalysis, therefore it is proposed to be the catalytic resting state. This observation, together with the variable time normalization analysis, is consistent with the formation of I1 from [1]2− (Scheme 4) being the rate determining step, although despite extensive efforts, the transition state between these true minima could not be located computationally.
Conclusions
In summary, the boron-functionalized Zintl cluster [(BBN)P7]2− ([1]2−) mediates the reduction of a broad range of pyridines, nitriles, and imines, thus affording dihydropyridine and amine products. Interestingly, the naked unfunctionalized [P7]3− with a cation sequestering agent also showed some catalytic competency, albeit lower than that of [1]2−. Further, it was shown that catalyst [1]2− could be recycled several times without loss in performance and two catalytic transformations were scaled to 5.58 mmol. This contribution further affirms the utility of Zintl clusters in the field of catalysis.Author contributions
Synthesis and catalysis was performed by B. v. I. Synthesis of imine substrates was performed by J. B. M. W. Crystal X-ray diffraction data was collected and solved by G. F. S. W. DFT studies were performed by N. K. The manuscript was written through contributions from B. v. I, M. M. and N. K. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for funding (EP/V012061/1) and supporting a DTA studentship (B. v. I.). We also thank Gareth Smith for mass spectrometric analyses, Anne Davies and Martin Jennings for elemental analyses, and Ralph Adams for NMR spectroscopic enquiries. We are also grateful to the University of Manchester for computing resources via its Computational Shared Facility and associated support services.References
- Y. Ling, Z. Y. Hao, D. Liang, C. L. Zhang, Y. F. Liu and Y. Wang, Drug Des., Dev. Ther., 2021, 15, 4289–4338 CrossRef CAS PubMed.
- A. Parthiban and P. Makam, RSC Adv., 2022, 12, 29253–29290 RSC.
- D. M. Stout and A. I. Meyers, Chem. Rev., 1982, 82, 223–243 CrossRef CAS.
- U. Eisner and J. Kuthan, Chem. Rev., 1972, 72, 1–42 CrossRef CAS.
- J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed.
- D. S. Wang, Q. A. Chen, S. M. Lu and Y. G. Zhou, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed.
- Y.-G. Zhou, Acc. Chem. Res., 2007, 40, 1357–1366 CrossRef CAS PubMed.
- F. Glorius, N. Spielkamp, S. Holle, R. Goddard and C. W. Lehmann, Angew. Chem. Int. Ed., 2004, 43, 2850–2852 CrossRef CAS PubMed.
- F. Glorius, Org. Biomol. Chem., 2005, 3, 4171–4175 RSC.
- A. N. Kim and B. M. Stoltz, ACS Catal., 2020, 10, 13834–13851 CrossRef CAS PubMed.
- M. P. Wiesenfeldt, Z. Nairoukh, T. Dalton and F. Glorius, Angew. Chem. Int. Ed., 2019, 58, 10460–10476 CrossRef CAS PubMed.
- X. Wang, Y. Zhang, D. Yuan and Y. Yao, Org. Lett., 2020, 22, 5695–5700 CrossRef CAS PubMed.
- S. Park and S. Chang, Angew. Chem. Int. Ed., 2017, 56, 7720–7738 CrossRef CAS PubMed.
- B. Rao, C. C. Chong and R. Kinjo, J. Am. Chem. Soc., 2018, 140, 652–656 CrossRef CAS PubMed.
- T. Hynes, E. N. Welsh, R. McDonald, M. J. Ferguson and A. W. H. Speed, Organometallics, 2018, 37, 841–844 CrossRef CAS.
- E. Kim, H. J. Jeon, S. Park and S. Chang, Adv. Synth. Catal., 2020, 362, 308–313 CrossRef CAS.
- E. N. Keyzer, S. S. Kang, S. Hanf and D. S. Wright, Chem. Commun., 2017, 53, 9434–9437 RSC.
- X. Fan, J. Zheng, Z. H. Li and H. Wang, J. Am. Chem. Soc., 2015, 137, 4916–4919 CrossRef CAS PubMed.
- G. A. Somorjai and Y. Li, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 917–924 CrossRef CAS PubMed.
- T. K. Athira, M. Roshith, R. Kadrekar, A. Arya, M. S. Kumar, G. Anantharaj, L. Gurrala, V. Saranyan, T. G. Satheesh Babu and V. R. K. Darbha, Mater. Res. Express, 2020, 7, 104002 CrossRef CAS.
- Y. Zhu, J. Ren, X. Zhang and D. Yang, Nanoscale, 2020, 12, 13297–13310 RSC.
- C.-M. Fung, C.-C. Er, L.-L. Tan, A. R. Mohamed and S.-P. Chai, Chem. Rev., 2022, 122, 3879–3965 CrossRef CAS PubMed.
- B. van Ijzendoorn and M. Mehta, Dalton Trans., 2020, 49, 14758–14765 RSC.
- O. P. E. Townrow, C. Chung, S. A. Macgregor, A. S. Weller and J. M. Goicoechea, J. Am. Chem. Soc., 2020, 142, 18330–18335 CrossRef CAS PubMed.
- M. Ruck, D. Hoppe, B. Wahl, P. Simon, Y. Wang and G. Seifert, Angew. Chem. Int. Ed., 2005, 44, 7616–7619 CrossRef CAS PubMed.
- M. Jo, A. Dragulescu-Andrasi, L. Z. Miller, C. Pak and M. Shatruk, Inorg. Chem., 2020, 59, 5483–5489 CrossRef CAS PubMed.
- A. Dragulescu-Andrasi, L. Z. Miller, B. Chen, D. T. McQuade and M. Shatruk, Angew. Chem. Int. Ed., 2016, 55, 3904–3908 CrossRef CAS PubMed.
- L. Du, Y. Zhao, L. Wu, X. Hu, L. Yao, Y. Wang, X. Bai, Y. Dai, J. Qiao, M. G. Uddin, X. Li, J. Lahtinen, X. Bai, G. Zhang, W. Ji and Z. Sun, Nat. Commun., 2021, 12, 4822 CrossRef CAS PubMed.
- Y. Zhou, S. R. Elliott and V. L. Deringer, Angew. Chem., Int. Ed., 2023, 62, e202216658 CrossRef CAS PubMed.
- O. P. E. Townrow, S. B. Duckett, A. S. Weller and J. M. Goicoechea, Chem. Sci., 2022, 13, 7626–7633 RSC.
- N. E. Poitiers, L. Giarrana, V. Huch, M. Zimmer and D. Scheschkewitz, Chem. Sci., 2020, 11, 7782–7788 RSC.
- Y. Wang, C. Zhang, X. Wang, J. Guo, Z.-M. Sun and H. Zhang, ACS Catal., 2020, 10, 7808–7819 CrossRef CAS.
- B. L. L. Réant, B. van Ijzendoorn, G. F. S. Whitehead and M. Mehta, Dalton Trans., 2022, 51, 18329–18336 RSC.
- B. van Ijzendoorn, S. F. Albawardi, I. J. Vitorica-Yrezabal, G. F. S. Whitehead, J. E. McGrady and M. Mehta, J. Am. Chem. Soc., 2022, 144, 21213–21223 CrossRef PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- K. S. Hayes, Appl. Catal., A, 2001, 221, 187–195 CrossRef CAS.
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed.
- V. Froidevaux, C. Negrell, S. Caillol, J.-P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- S. A. Lawrence, Amines: Synthesis, Properties and Applications, Cambridge University Press, 2004 Search PubMed.
- A. R. Bazkiaei, M. Findlater and A. E. V. Gorden, Org. Biomol. Chem., 2022, 20, 3675–3702 RSC.
- A. D. Sadow, Early Main Group Metal Catalysis, 2020, pp. 201–224 Search PubMed.
- S. J. Geier, C. M. Vogels, J. A. Melanson and S. A. Westcott, Chem. Soc. Rev., 2022, 51, 8877–8922 RSC.
- M. Magre, M. Szewczyk and M. Rueping, Chem. Rev., 2022, 122, 8261–8312 CrossRef CAS PubMed.
- M. Arrowsmith, M. S. Hill, T. Hadlington, G. Kociok-Köhn and C. Weetman, Organometallics, 2011, 30, 5556–5559 CrossRef CAS.
- G. S. Kumar, J. Bhattacharjee, K. Kumari, S. Moorthy, A. Bandyopadhyay, S. K. Singh and T. K. Panda, Polyhedron, 2022, 219, 115784 CrossRef.
- N. Sarkar, S. Bera and S. Nembenna, J. Org. Chem., 2020, 85, 4999–5009 CrossRef CAS PubMed.
- D. Bedi, A. Brar and M. Findlater, Green Chem., 2020, 22, 1125–1128 RSC.
- M. Bhandari, M. Kaur, S. Rawat and S. Singh, Inorg. Chem., 2023, 62, 6598–6607 CrossRef CAS PubMed.
- W. Zou, L. Gao, J. Cao, Z. Li, G. Li, G. Wang and S. Li, Chem. – Eur. J., 2022, 28, e202104004 CrossRef CAS PubMed.
- X. Wang and X. Xu, RSC Adv., 2021, 11, 1128–1133 RSC.
- V. K. Pandey, S. N. R. Donthireddy and A. Rit, Chem. - Asian J., 2019, 14, 3255–3258 CrossRef CAS PubMed.
- H. Yang, L. Zhang, F.-Y. Zhou and L. Jiao, Chem. Sci., 2020, 11, 742–747 RSC.
- D. Hayrapetyan and A. Y. Khalimon, Chem. – Asian J., 2020, 15, 2575–2587 CrossRef CAS PubMed.
- E. Jeong, J. Heo, S. Park and S. Chang, Chem. – Eur. J., 2019, 25, 6320–6325 CrossRef CAS PubMed.
- J. Légaré Lavergne, H.-M. To and F.-G. Fontaine, RSC Adv., 2021, 11, 31941–31949 RSC.
- A. D. Bage, T. A. Hunt and S. P. Thomas, Org. Lett., 2020, 22, 4107–4112 CrossRef CAS PubMed.
- M. Cicač-Hudi, J. Bender, S. H. Schlindwein, M. Bispinghoff, M. Nieger, H. Grützmacher and D. Gudat, Eur. J. Inorg. Chem., 2016, 2016, 649–658 CrossRef.
- A. D. Bage, K. Nicholson, T. A. Hunt, T. Langer and S. P. Thomas, ACS Catal., 2020, 10, 13479–13486 CrossRef CAS.
- C. D. T. Nielsen and J. Burés, Chem. Sci., 2019, 10, 348–353 RSC.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028–2031 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2260639 and 2260640. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02896h |
| This journal is © The Royal Society of Chemistry 2023 |