
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron(II) complexes of 2,6-bis(imidazo[1,2-a]pyridin-2-yl)pyridine and related ligands with annelated distal heterocyclic donors†
Rafal
Kulmaczewski
and
Malcolm A.
Halcrow
 *
*
School of Chemistry, University of Leeds, Woodhouse Lane, Leeds, UK LS2 9JT. E-mail: m.a.halcrow@leeds.ac.uk
First published on 2nd October 2023
Abstract
Following a published synthesis of 2,6-bis(imidazo[1,2-a]pyridin-2-yl)pyridine (L1), treatment of α,α′-dibromo-2,6-diacetylpyridine with 2 equiv. 2-aminopyrimidine or 2-aminoquinoline in refluxing acetonitrile respectively gives 2,6-bis(imidazo[1,2-a]pyrimidin-2-yl)pyridine (L2) and 2,6-bis(imidazo[1,2-a]quinolin-2-yl)pyridine (L3). Solvated crystals of [Fe(L1)2][BF4]2 (1[BF4]2) and [Fe(L2)2][BF4]2 (2[BF4]2) are mostly high-spin, although one solvate of 1[BF4]2 undergoes thermal spin-crossover on cooling. The iron coordination geometry is consistently distorted in crystals of 2[BF4]2 which may reflect the influence of intramolecular, inter-ligand N⋯π interactions on the molecular conformation. Only 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Fe:L3 complexes were observed in solution, or isolated in the solid state; a crystal structure of [FeBr(py)2L3]Br·0.5H2O (py = pyridine) is presented. A solvate crystal structure of high-spin [Fe(L4)2][BF4]2 (L4 = 2,6-di{quinolin-2-yl}pyridine; 4[BF4]2) is also described, which exhibits a highly distorted six-coordinate geometry with a helical ligand conformation. The iron(II) complexes are high-spin in solution at room temperature, but 1[BF4]2 and 2[BF4]2 undergo thermal spin-crossover equilibria on cooling. All the compounds exhibit a ligand-based emission in solution at room temperature. Gas phase DFT calculations mostly reproduce the spin state properties of the complexes, but show small anomalies attributed to intramolecular, inter-ligand dispersion interactions in the sterically crowded molecules.
:1 Fe:L3 complexes were observed in solution, or isolated in the solid state; a crystal structure of [FeBr(py)2L3]Br·0.5H2O (py = pyridine) is presented. A solvate crystal structure of high-spin [Fe(L4)2][BF4]2 (L4 = 2,6-di{quinolin-2-yl}pyridine; 4[BF4]2) is also described, which exhibits a highly distorted six-coordinate geometry with a helical ligand conformation. The iron(II) complexes are high-spin in solution at room temperature, but 1[BF4]2 and 2[BF4]2 undergo thermal spin-crossover equilibria on cooling. All the compounds exhibit a ligand-based emission in solution at room temperature. Gas phase DFT calculations mostly reproduce the spin state properties of the complexes, but show small anomalies attributed to intramolecular, inter-ligand dispersion interactions in the sterically crowded molecules.
Introduction
Spin-crossover (SCO) complexes continue to be heavily studied.1–5 Controlling spin state properties is vital for the design of new catalysts6–10 or photosensitisers11,12 from base metal centres.13 Moreover, solid SCO materials are useful testbeds for the crystal engineering of phase transitions in molecular crystals,14,15 while time-resolved spectroscopic and diffraction methods are shedding new light on the atomistic mechanisms of SCO-induced phase transitions.16–18 SCO switching in thin films,19,20 nanoparticles,21,22 monolayers and single molecules19,23–25 is well-established, leading to the use of SCO complexes in nanomolecular electronics.26,27 Lastly, SCO molecules are useful switching components in functional materials which exploit their thermochromism,28,29 or other materials properties associated with the spin transition.30–35 Alternatively, SCO auxiliaries can control fluorescence,36,37 conductivity,38 magnetic39–41 or ferroelectric42 properties in multifunctional composite crystals or soft materials.43Iron(II) complexes of tridentate tris-heterocyclic ligands are particularly fruitful for SCO chemistry.44,45 Iron(II) complexes of ligands derived from trispyrazolylborate,46 trispyrazolyl-methane,46,47 2,6-di(1H-benzimidazol-2-yl)pyridine (bimpy)48 and regioisomers of 2,6-di(pyrazolyl)pyridine (bpp)49–52 can all exhibit SCO at accessible temperatures. These ligand classes can be functionalised using appropriate synthetic starting materials or through functional group manipulations, giving steric and electronic control over the spin states of their iron complexes.48,49,52–56 It also allows functional substituents, long alkyl chains or tether groups to be appended to SCO complexes, producing multifunctional materials or nanostructures based on SCO switches. Complexes of the bimpy57,58 and bpp59–64 ligand families have been particularly useful for the latter goal.
Notwithstanding its multifunctional derivatives, [Fe(bimpy)2]2+ was an early example of a fluorescent SCO complex,65 which have been increasingly developed during the last ten years.36,37 With that in mind, we investigated [Fe(1-bip)2]2+ (1-bip = 2,6-bis{indazol-1-yl}pyridine) and [Fe(2-bip)2]2+ (2-bip = 2,6-bis{indazol-2-yl}pyridine), with the latter being an isosteric analogue of the bimpy complex.66 These also exhibit a ligand-based fluorescence in solution at room temperature, but whose wavelength was unaffected by the spin state of the complexes.66,67
Ruthenium complexes of 2,6-bis(imidazo-[1,2-a]pyridin-2-yl)pyridine (L1; Scheme 1) and its derivatives have been studied by Song et al., as catalysts for a number of reactions.68–72 Following our study of bip ligands, which are constitutional isomers of L1, we now report the iron(II) complex chemistry of L1 and two new analogues, 2,6-bis(imidazo-[1,2-a]pyrimidin-2-yl)pyridine (L2) and 2,6-bis(imidazo-[1,2-a]quinolin-2-yl)pyridine (L3; Scheme 1). We also include a re-investigation of previously reported [Fe(L4)2]2+ (L4 = 2,6-di{quinol-2-yl}pyridine, dqp).73
 | ||
| Scheme 1 The ligands investigated in this study. | ||
Results and discussion
Reaction of 2,6-diacetylpyridine with N-bromosuccinimide in refluxing acetonitrile affords α,α′-dibromo-2,6-diacetylpyridine, in moderate yield. Treatment of that intermediate with 2 equiv. 2-aminopyridine in refluxing acetonitrile gives L1 (Scheme 1), in ca. 60% yield after purification.68 The latter reaction is sensitive to the purity of the α,α′-dibromo-2,6-diacetylpyridine starting material, which should be synthesised in situ and used immediately for the best results. We also screened other 2-aminoazine reagents in this protocol, which afforded L2 (from 2-aminopyrimidine) and L3 (from 2-aminoquinoline) in NMR purity. An attempted synthesis of 2,6-bis(imidazo[1,2-a]pyrazinyl)pyridine from aminopyrazine by this method gave a product that was too insoluble to characterise, however (ESI†). Ligand L4 was synthesised for this study by a literature procedure.73,74Treatment of Fe[BF4]2·6H2O with 2 equiv. L1 or L2 in acetonitrile affords [Fe(L1)2][BF4]2 (1[BF4]2) and [Fe(L2)2][BF4]2 (2[BF4]2) as brown polycrystalline solids after the usual work up. Both complexes form solvated single crystals and, while 2[BF4]2 was obtained as a solvent-free material after drying in vacuo, samples of 1[BF4]2 always contained residual solvent or lattice water by microanalysis. Since the spin state properties of some solvated iron complexes depend significantly on the lattice solvent,75–81 multiple solvates of 1[BF4]2 and 2[BF4]2 were isolated for characterisation.
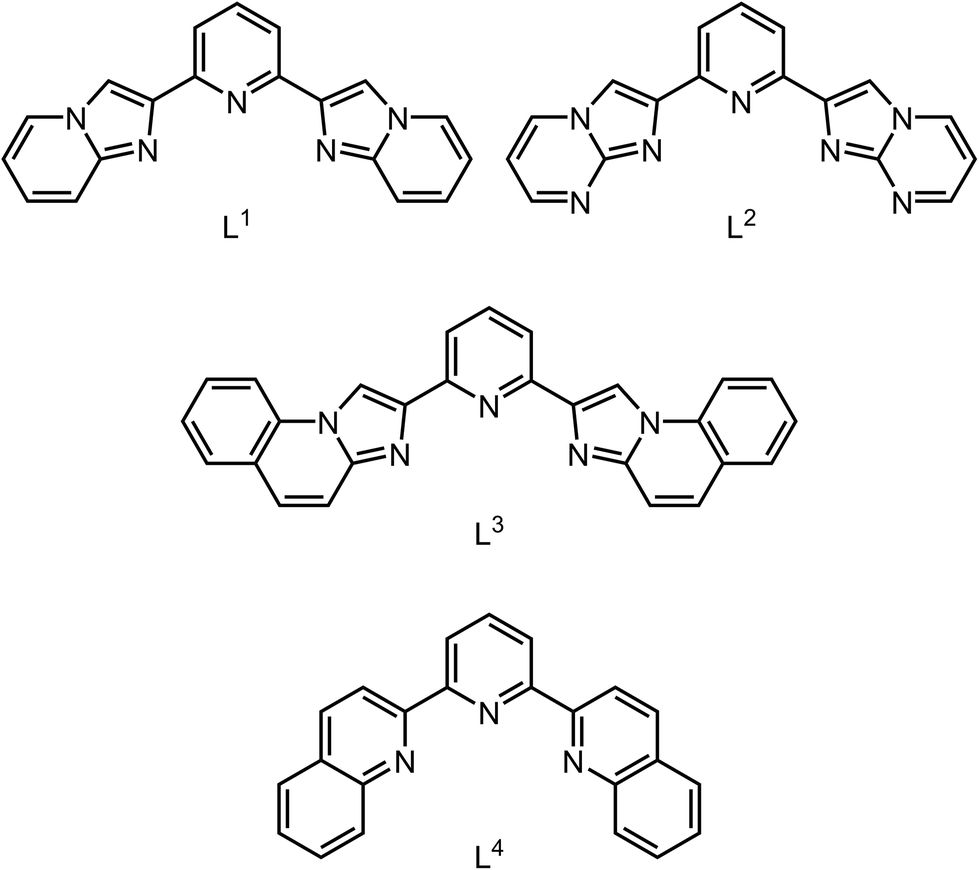
Four crystalline solvates of 1[BF4]2 were characterised by X-ray diffraction, three of which can be considered together. The asymmetric unit of 1[BF4]2·MeCN and 1[BF4]2·mMeNO2 (m ≈ 0.8; both orthorhombic, space group Pccn, Z = 4) contains half a formula unit. Their iron atom lies on a crystallographic C2 axis, so the complex molecule has perfect C2 symmetry (Fig. 1). The solvate 1[BF4]2·1.5MeOH (orthorhombic, Pna21, Z = 4) has similar unit cell dimensions and crystal packing as the first two solvates, but lacks their crystallographic inversion symmetry. All three solvates contain high-spin complex molecules at 120 K, with near-regular coordination geometries showing minor deviations from idealised D2d symmetry (Table 1). The cations associate into zig-zag chains through strong face-to-face π⋯π interactions, involving one imidazopyridyl arm of each ligand. The chains alternate down the c axis, along the [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] and [10] crystal vectors in the Pccn solvates (the unit cell axes are exchanged in the Pna21 setting).
0] and [10] crystal vectors in the Pccn solvates (the unit cell axes are exchanged in the Pna21 setting).
 | ||
| Fig. 1 Two [Fe(L1)2]2+ molecules in 1[BF4]2·MeCN, related by 1 − x, ½ + y, ½ − z. The intermolecular C–H⋯π contact that may inhibit thermal SCO in the three near-isomorphous solvates of 1[BF4]2 is shown in grey, which is positioned to inhibit displacement of those imidazopyridyl groups in the direction shown which would accompany SCO. A space-filling view of this interaction is in Fig. S10.† Colour code: C, white; H, pale grey; Fe, green; N, blue. | ||
| 1[BF4]2·MeCNb | 1[BF4]2·mMeNO2b | 1[BF4]1.6[SiF6]0.2·1.7MeNO2·0.3Et2O | 1[BF4]2·MeOH | |
|---|---|---|---|---|
| a V Oh, Σ and Θ are indices showing the spin state of the complex,82,83 while ϕ and θ measure the orientations of the two tridentate ligands in the molecule.84,85 Typical values for these parameters in complexes related to 1[BF4]2 are given in ref. 51 and 52. b The complex cations in these crystals have crystallographic C2 symmetry. | ||||
| Fe–N{pyridyl} | 2.175(3) | 2.168(3) | 1.920(3), 1.924(3) | 2.171(4), 2.171(4) |
| Fe–N{imidazopyridyl} | 2.191(3), 2.199(4) | 2.195(3), 2.199(3) | 1.979(3)–2.002(3) | 2.193(4)–2.201(4) |
| V Oh | 12.901(15) | 12.873(11) | 9.896(8) | 12.908(15) |
| Σ | 144.4(5) | 145.2(4) | 81.5(4) | 143.5(5) |
| Θ | 473 | 476 | 268 | 470 |
| ϕ | 176.42(18) | 175.81(14) | 178.78(11) | 175.96(14) |
| θ | 89.66(3) | 89.87(2) | 88.89(2) | 89.93(3) |
As discussed below, there is nothing in the molecular geometry of these three solvates that should prevent thermal SCO on cooling. Rather, we attribute their high-spin nature to an intermolecular C–H⋯π contact between molecules in neighbouring cation chains (Fig. 1). While the interaction is not notably short, it is positioned to inhibit the displacement of two of the imidazo-[1,2-a]pyridin-2-yl groups that would accompany contraction of those Fe–N bonds during SCO.
While samples of 1[BF4]2·MeCN and 1[BF4]2·1.5MeOH were homogeneous, red needles of 1[BF4]2·mMeNO2 were often mixed with small black prisms which had the crystallographic composition 1[BF4]1.6[SiF6]0.2·1.7MeNO2·0.3Et2O (triclinic, P, Z = 2). The fractional SiF62− content of these crystals should arise from reaction of the silica crystallisation vials with adventitious F−, produced by hydrolysis of BF4− during the crystallisation process.86 The complex in this crystal is low-spin at 120 K from its metric parameters, which confirms the low-spin state of 1[BF4]2 is thermodynamically accessible (Table 1).
1[BF
4
]
1.6
[SiF
6
]
0.2
·1.7MeNO2·0.3Et2O differs from the other solvates of that complex, in adopting a ‘terpyridine embrace’ packing motif.87 The complex cations interdigitate into four-fold layers in the (10) crystal plane, through edge-to-face and face-to-face π⋯π interactions involving all their imidazopyridyl residues. Nearest neighbour molecules within the layers are related by crystallographic inversion centres, while the layers propagate in 3D by translation along a and b. While they are not perfectly isomorphous, the crystal packing in 1[BF4]1.6[SiF6]0.2·1.7MeNO2·0.3Et2O resembles its regioisomer [Fe(2-bip)2][BF4]2·2MeNO2, which adopts a closely related terpyridine embrace packing motif.66
The solvates of 1[BF4]2 retain crystallinity by powder diffraction upon exposure to air (Fig. S15†). Magnetic susceptibility data show 1[BF4]2·MeCN and 1[BF4]2·1.5MeOH are both high-spin between 5–300 K, which is consistent with their crystal structures at 120 K. Another sample crystallised in small quantities from acetone/diethyl ether was also high-spin (Fig. S16†). The small crystal size of the MeNO2 solvates made it impossible to separate them manually for magnetic characterisation. However, a mixed-phase sample was high-spin at room temperature with an abrupt, partial spin-transition at T½ = 178 K occurring in ca. 45% of the material. That is consistent with a mixture of high-spin and SCO-active phases, as observed crystallographically. Iron(II) complexes related to 1[BF4]1.6[SiF6]0.2·1.7MeNO2·0.3Et2O with terpyridine embrace lattices often show similarly abrupt thermal spin-transitions.66,88,89
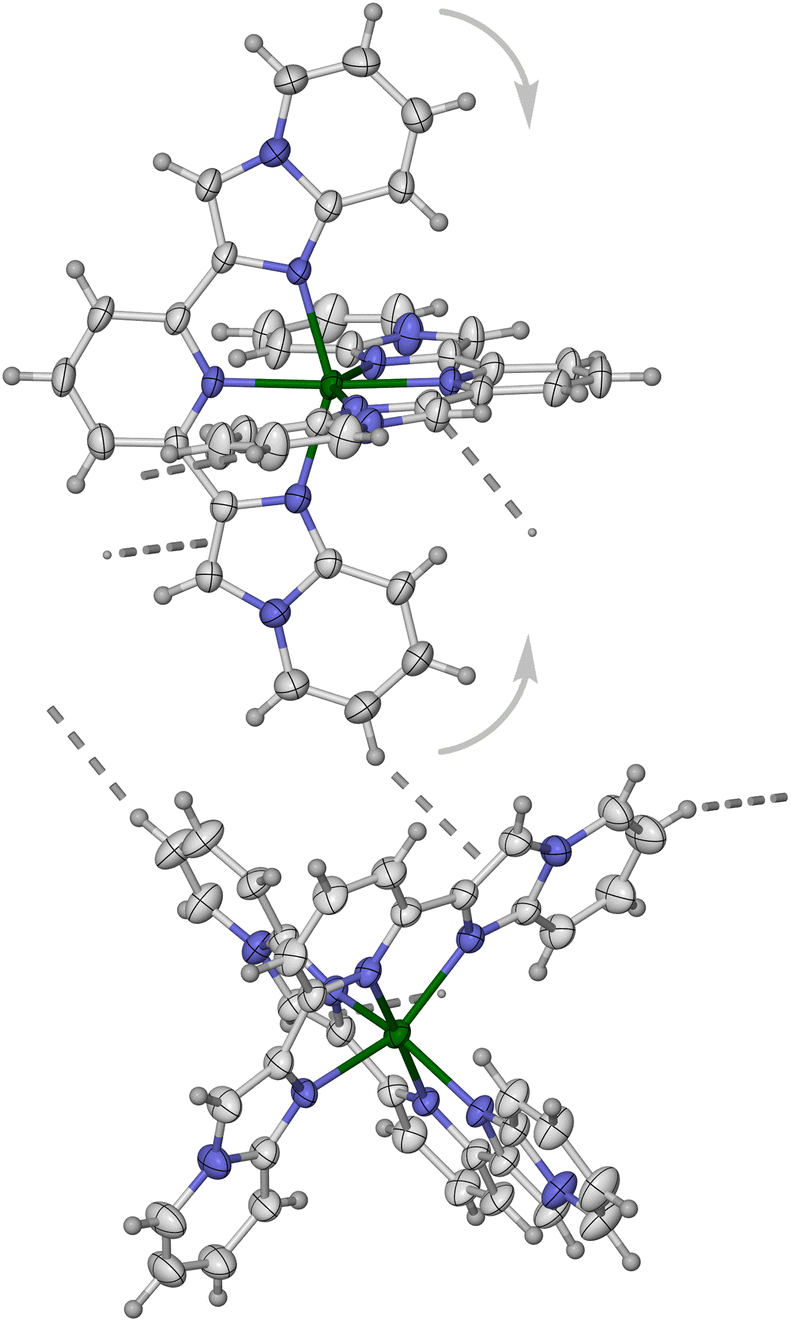
Three solvates of 2[BF4]2 were also crystallised: isomorphous 2[BF4]2·1.5MeCN and 2[BF4]2·Me2CO (both triclinic, P, Z = 2) and 2[BF4]2·3.5MeNO2·0.5Et2O (also triclinic, P, Z = 2 but not isomorphous with the other solvates). These are all crystallographically high-spin at 120 K. However, in contrast to 1[BF4]2, the complex coordination geometry in the 2[BF4]2 solvates is significantly distorted from its idealised D2d symmetry (Fig. 2). This is particularly reflected in the trans-N{pyridyl}–Fe–N{pyridyl} angle (ϕ, Table 2) which lies between 156.26(12)–164.72(7)°, significantly lower than its ideal value of 180°. This type of distortion is well-known in high-spin [Fe(bpp)2]2+ derivatives, and reflects an angular Jahn–Teller distortion of the complex along the Oh–D3d coordinate.51,52 The distortion has a shallow energy profile,85,89,90 and [Fe(bpp)2]2+ derivatives with 148 ≤ ϕ ≤ 180° have been reported.91,92
 | ||
| Fig. 2 A centrosymmetric pair of [Fe(L2)2]2+ molecules in 2[BF4]2·1.5MeCN, showing the intramolecular n⋯π, intermolecular π⋯π and intermolecular C–H⋯N contacts involving the L2 ligands. Colour code: C, white; H, pale grey; Fe, green; N, blue. | ||
| 2[BF4]2·1.5MeCN | 2[BF4]2·Me2CO | 2[BF4]2·3.5MeNO2·0.5Et2O | |
|---|---|---|---|
| Fe–N{pyridyl} | 2.148(2), 2.154(2) | 2.1499(16), 2.1538(16) | 2.160(3), 2.162(3) |
| Fe–N{imidazopyrimidyl} | 2.183(2)–2.201(2) | 2.1849(18)–2.2076(17) | 2.196(3)–2.212(3) |
| V Oh | 12.536(10) | 12.586(6) | 12.430(12) |
| Σ | 140.3(3) | 140.4(2) | 144.8(4) |
| Θ | 467 | 463 | 482 |
| ϕ | 163.52(9) | 164.72(7) | 156.26(12) |
| θ | 87.59(3) | 84.65(1) | 88.37(2) |
Since the distortion is a property of the high-spin state, SCO in a distorted complex requires a significant structural rearrangement to the more regular coordination geometry preferred by the low-spin form. Hence, SCO becomes more disfavoured as ϕ (and θ, Table 2) deviate increasingly from their ideal values in crystalline complexes.85,90 The distortions in 2[BF4]2 lie in the range where SCO is rarely observed (Fig. 3).91,92 Consistent with that, all the solvates of 2[BF4]2 remain high-spin between 5–300 K from magnetic susceptibility data (Fig. S25†).
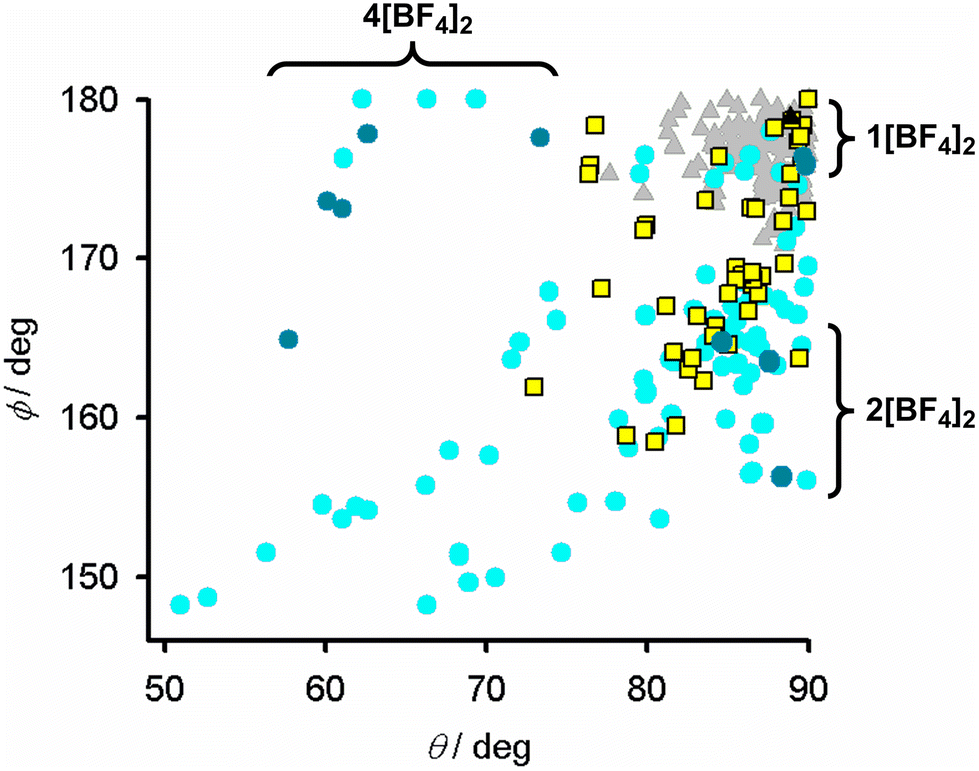 | ||
| Fig. 3 Distortion parameters for [Fe(1-bpp)2]2+ (1-bpp = 2,6-di{pyrazol-1-yl}pyridine) derivatives which are low-spin (grey triangles); high-spin and SCO-active (yellow squares); or remain high-spin on cooling (cyan circles).84,85 Compounds in this work are plotted with the same symbols in darker colouration. The graph is adapted from ref. 92. | ||
These distorted coordination geometries facilitate two different supramolecular interactions involving the L2 ligands (Fig. 2). First, are intramolecular n⋯π contacts between the imidazopyrimidinyl N8 atom of one ligand, and a pyridyl C–N bond of the other ligand.93 Second, is intermolecular π⋯π overlap between nearest neighbour molecules in the lattice. This is always accompanied by a weak intermolecular C–H⋯N contact to the other imidazopyrimidinyl N8 atom acceptor, from the same ligand that forms the intramolecular n⋯π interaction. One ligand in each molecule in isomorphous 2[BF4]2·1.5MeCN and 2[BF4]2·MeCO takes part in these interactions, associating the cations into centrosymmetric pairs. In contrast, both ligands in each molecule of 2[BF4]2·3.5MeNO2·0.5Et2O form the same set of pairwise interactions, affording zig-zag chains of strongly linked cations (Fig. S20 and S21†). The larger ϕ distortion in this solvate facilitates close approach of the neighbour molecule forming its second intramolecular n⋯π interaction.
Samples of 2[BF4]2·1.5MeCN and 2[BF4]2·Me2CO retain crystallinity on exposure to air, but 2[BF4]2·3.5MeNO2·0.5Et2O decomposes through solvent loss to a poorly crystalline red powder. All these materials are high-spin above 5 K, as expected from their crystallographic coordination geometries (Fig. 3).
In contrast to L1 and L2, treatment of Fe[BF4]2·6H2O with 2 equiv. L3 yields brown solids whose microanalyses are consistent with the formulae [FeL3][BF4]2·y(solvent); that is, with a 1:1 Fe:L3 stoichiometry. Consistent with that, freshly prepared solutions of a 1:2 molar mixture of Fe[BF4]2·6H2O and L3 contain a significant amount of uncoordinated ligand by NMR, which is not observed in solutions of 1[BF4]2 and 2[BF4]2. Single crystals were ultimately obtained from a sample of formula [FeBr(py)2L3]Br·0.5H2O (py = pyridine; Fig. 4). The tridentate L3 ligand in that complex displays no obvious steric hindrance to explain its reluctance to form a homoleptic iron complex. Other physical characterisation was performed on a sample with the analytical formula [FeL3][BF4]2·H2O, labelled 3′[BF4]2·H2O.
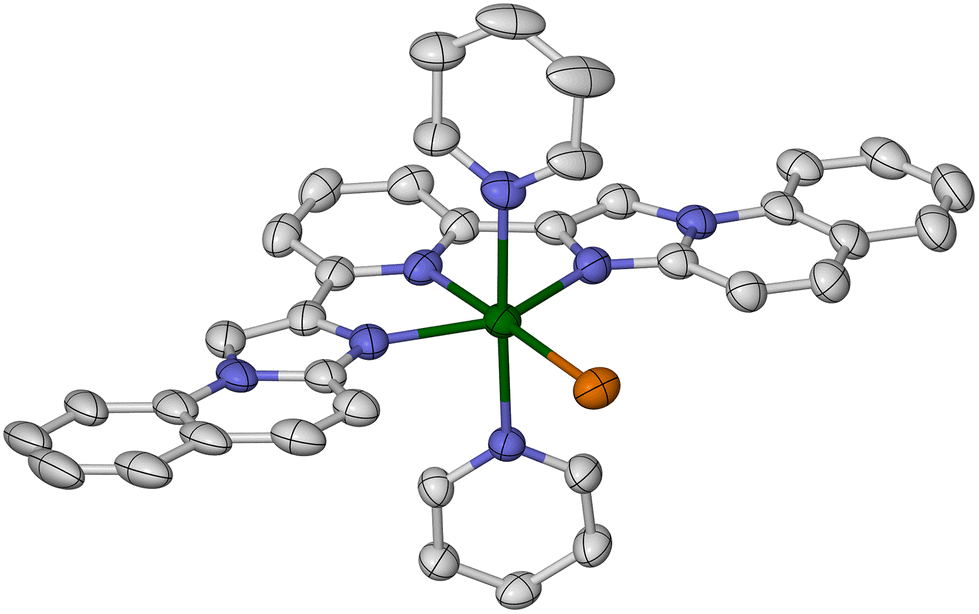 | ||
| Fig. 4 The complex cation in [FeBr(py)2L3]Br·0.5H2O. Displacement ellipsoids are at the 50% probability level, and H atoms are omitted for clarity. Colour code: C, white; Br, orange; Fe, green; N, blue. | ||
Lastly, in 1969 Sinn et al. reported that [Fe(L4)2][ClO4]2 (4[ClO4]2) is high-spin above 85 K in the solid state.73 We have re-investigated this complex as its BF4− salt, 4[BF4]2. Single crystals of 4[BF4]2·1.39MeCN·0.125Et2O·0.25H2O (monoclinic, P21/c, Z = 16, i.e. Z′ = 4) contain four crystallographically unique complex molecules in their asymmetric unit. The four molecules are distinguishable by their molecular conformations, which are strongly flattened with a helical twist (Fig. 5). This is manifested by the dihedral angle between the least squares planes of the L4 ligands in each molecule (θ, Table 3 and Fig. 3).84 Molecules A–D exhibit 57.75(5) ≤ θ ≤ 73.38(4) which is strongly reduced from the idealised value of 90°, while molecule A also has a significant ϕ distortion. The resultant helical ligand conformations give each metal centre a Λ or Δ chirality; the centrosymmetric crystal lattice contains equal numbers of Λ- and Δ-helical molecules. Space-filling models imply the helical distortion is induced by steric repulsion between the quinolyl C8–H groups on each ligand,93 and the pyridyl ring of the other ligand in the molecule. One other homoleptic complex of L4 has also been crystallographically characterised, namely a solvate of [Co(L4)2][PF6]2. That complex has a much smaller helical conformational distortion, with ϕ = 180 and θ = 79.5–80.9°.94
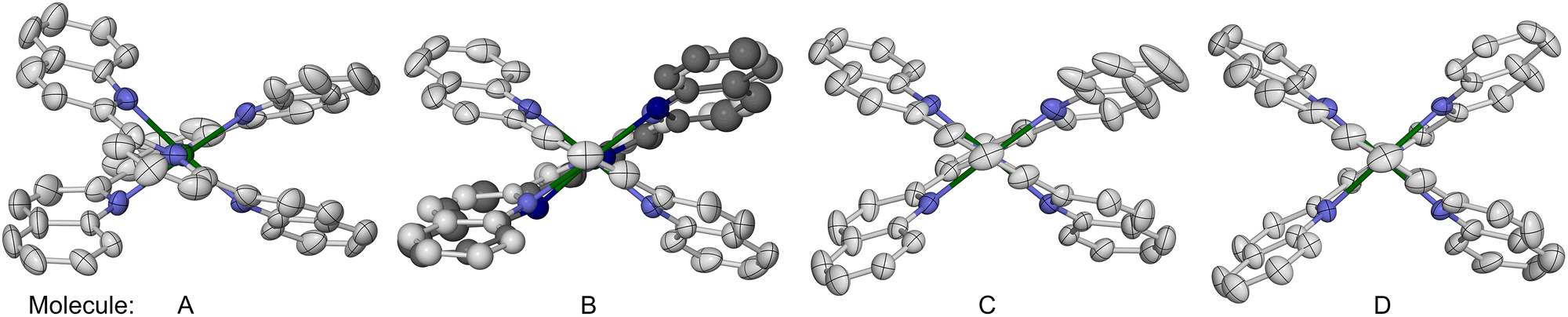 | ||
| Fig. 5 The four unique [Fe(L4)2]2+ molecules in 4[BF4]2·1.39MeCN·0.125Et2O·0.25H2O, viewed along their N{pyridyl}–Fe–N{pyridyl} vectors. Both ligand disorder sites in molecule B are included and distinguished with pale and dark colouration. The crystallographic views of molecules C and D have been inverted, to give them the same handedness as molecules A and B. Displacement ellipsoids are at the 50% probability level, and H atoms are omitted for clarity. Colour code: C, white or dark grey; H, pale grey; Fe, green; N, pale or dark blue. | ||
| Molecule A | Molecule Ba | Molecule C | Molecule D | |
|---|---|---|---|---|
|
a Ligand N(28B)–C(53B) is disordered over two sites, with refined occupancies of 0.61:0.39. Values involving both ligand disorder sites are given in the table.
|
||||
| Fe–N{pyridyl} | 2.073(6), 2.102(6) | 2.100(5), 2.148(9)/2.049(15) | 2.088(5), 2.088(5) | 2.114(5), 2.099(4) |
| Fe–N{quinolinyl} | 2.221(5)–2.319(6) | 2.235(10)–2.335(8) | 2.252(5)–2.298(6) | 2.249(5)–2.286(5) |
| V Oh | 12.73(2) | 12.85(3)/12.99(4) | 12.94(2) | 13.149(18) |
| Σ | 174.6(7) | 195.5(11)/172.9(13) | 177.3(7) | 153.6(7) |
| Θ | 486 | 529/482 | 476 | 476 |
| ϕ | 164.9(2) | 173.6(3)/173.1(5) | 177.8(2) | 177.6(2) |
| θ | 57.75(5) | 60.15(7)/61.04(9) | 62.64(5) | 73.38(4) |
The [Fe(L4)2]2+ cations associate into homochiral helical columns in the crystal lattice, through weak intermolecular π⋯π interactions between their quinolyl groups. The molecules stack as ⋯A⋯B⋯D⋯C⋯ down the [10] crystal vector (Fig. S33–S35†), and left and right-handed columns are arranged into strips whose helicity alternates along c.
As discussed above, the twisted coordination geometries in 4[BF4]2 should strongly favour its high-spin state and inhibit SCO. Consistent with that, the solid complex is fully high-spin between 5–300 K (Fig. S36†).
Solutions of 1[BF4]2, 2[BF4]2 and 4[BF4]2 are paramagnetic, with just one C2 or m-symmetric ligand environment by 1H NMR (Fig. S37–S39†). The NMR spectrum of 4[BF4]2 in CD3CN also contains ca. 0.1-equiv of uncoordinated L4, implying a minor degree of ligand dissociation in solution for that complex. No free ligand was detected in solutions of 1[BF4]2 and 2[BF4]2, however.
The spin states of the complexes were monitored by variable temperature Evans method measurements (Fig. 6). Each compound is fully, or predominantly, high-spin at 300 K which is consistent with their paramagnetic NMR spectra. Solutions of 1[BF4]2 and 2[BF4]2 clearly show the onset of SCO equilibria on cooling, which were fitted to the following parameters: for 1[BF4]2, T½ = 237 ± 3 K, ΔH = 19.3 ± 1.3 kJ mol−1, ΔS = 81 ± 6 J mol−1 K−1; for 2[BF4]2, T½ = 251 ± 1 K, ΔH = 22.4 ± 0.7 kJ mol−1, ΔS = 89 ± 3 J mol−1 K−1. The fitted thermodynamic data are typical for iron(II) complexes of N-donor ligands,55,95,96 with no apparent contributions from other solution equilibria.97,98 In contrast, 3′[BF4]2·H2O and 4[BF4]2 remain high-spin over the liquid range of the solvent.
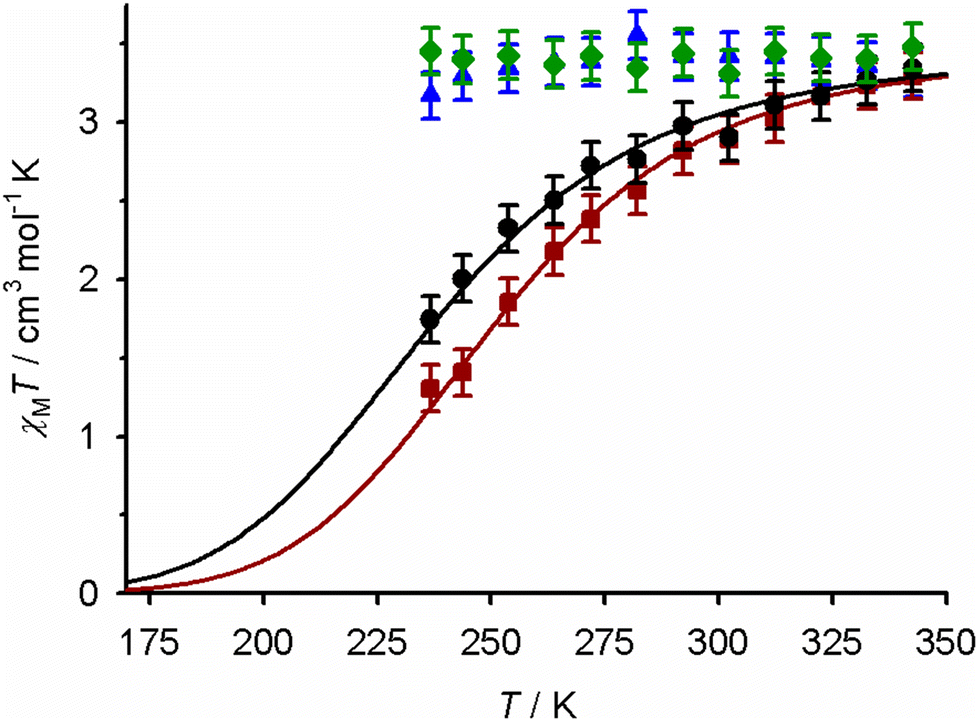 | ||
| Fig. 6 Magnetic susceptibility data in CD3CN solution for: 1[BF4]2 (black ●), 2[BF4]2 (red ■), 3′[BF4]2·H2O (blue ▲), and 4[BF4]2 (green ◆). The lines show the best fit of the data for 1[BF4]2 and 2[BF4]2 to a thermodynamic SCO equilibrium (eqn (1) and (2), Experimental section). | ||
Ligands L1–L4 have a blue fluorescence in acetonitrile solution at room temperature (Fig. 7). Comparison with analogous model compounds implies the near-UV absorptions and emission maxima for L1–L3 should be centred on their imidazo[1,2-a]azine residues.99–101 The L2 emission [λemmax = 457 nm] lies at longer wavelength than for L1 [423 nm], L3 [434 nm] and L4 [435 nm], which might reflect additional dipolar interactions between L2 and the solvent via its imidazopyrimidinyl N8 atoms.93,102 Measurements in different solvents to probe that suggestion were hampered by the poor solubility of L2, however. The absorption and emission spectra for L4 in Fig. 7 are consistent with literature data.94,103,104
 | ||
| Fig. 7 Absorption spectra (left) and normalised emission spectra (right) from the compounds in this work, in acetonitrile solution at 298 K. The UV-vis extinction coefficients for L1–L3 have larger errors, because of their poor solubility. Data from these spectra are listed in Table S10.† | ||
The complexes have a weak blue emission under the same conditions, at 445 ≤ λemmax ≤ 453 nm for 1[BF4]2–3′[BF4]2, and λemmax = 499 nm for 4[BF4]2 (Fig. 7). In most cases, these emissions are red-shifted compared to the corresponding free ligand. The exception is 2[BF4]2, whose emission maximum [445 nm] is essentially identical to 1[BF4]2 [446 nm], but is slightly blue-shifted compared to L2. That supports the involvement of solvation interactions in the longer wavelength emission of L2 (see above). Those interactions would be quenched in 2[BF4]2 whose imidazopyrimidinyl N8 lone pairs93 are oriented towards the interior of the complex (Fig. 2).
Gas phase DFT calculations were performed to confirm the spin state properties of the complexes, using the B86PW91 functional and def2-SVP basis set. Similar protocols performed well in benchmark studies comparing the spin states in different iron(II) complexes,105–107 and we have used this method to elucidate structure:function relationships for SCO in several iron(II) complex families with heterocyclic ligands.108–112 The hypothetical homoleptic complex [Fe(L3)2]2+ (32+) was included in the calculations, for comparison with [Fe(L1)2]2+ (12+) and [Fe(L2)2]2+ (22+). The spin states of [Fe(bimpy)2]2+ and [Fe(2-bip)2]2+, which are isosteric constitutional isomers of 12+, were also examined (Table 4). The corresponding metal-free ligands were minimised by the same protocol, in conformations consistent with tridentate coordination to a metal ion.
| T ½, K | E(HS), Ha | E(LS), Ha | ΔE{HS–LS}, kcal mol−1 | ΔErel{HS–LS},a kcal mol−1 | Free ligand Eav{LP}, eV | |
|---|---|---|---|---|---|---|
| a A positive ΔErel{HS–LS} means the low-spin state is computed to be more stable than for 12+, and vice versa. b Not available. c In acetone solution.48,116 d From ref. 112. e From ref. 66. This value has a larger error because of the limited temperature range of the measurement. | ||||||
| [Fe(L1)2]2+ (12+) | 237 ± 3 | −3273.967089 | −3273.987838 | +13.0 | 0 | −5.66 |
| [Fe(L2)2]2+ (22+) | 251 ± 1 | −3338.084900 | −3338.111325 | +16.6 | +3.6 | −5.73 |
| [Fe(L3)2]2+ (32+) | —b | −3888.377301 | −3888.398581 | +13.6 | +0.3 | −5.69 |
| [Fe(L4)2]2+ (42+) | HS | −3362.198522 | −3362.213259 | +9.3 | −3.7 | −5.43 |
| [Fe(bimpy)2]2+ | 331 ± 5c | −3274.011814 | −3274.038130 | +16.8 | +3.5 | −5.93d |
| [Fe(2-bip)2]2+ | 212 ± 10e | −3273.839226 | −3273.859677 | +12.8 | −0.2 | −6.47 |
The computed Fe–N bond lengths lie within 2.4% of the available crystallographic data, in both spin states (Table S12†). That is a reasonable level of agreement for a calculation of this type. While the complexes mostly minimised to geometries with minor deviations from D2d symmetry, [Fe(L4)2]2+ (42+) was computed with a helical conformation that closely resembles its crystal structure (Fig. S47†). Hence, the crystallographic geometry in 4[BF4]2 is not a crystal packing artefact, but is an intrinsic property of the molecule. It probably arises from intramolecular steric repulsion involving the quinolyl C8–H groups, as discussed above.93
As a pure GGA functional, calculations using B86PW91 consistently overstabilise the low-spin states of metal complexes.113,114 Hence, the spin state energies in Table 4 (ΔErel{HS–LS}) are expressed relative to 12+.66 A positive ΔErel{HS–LS} means the low-spin state is computed to be more stable than for 12+ implying a higher T½, and vice versa. The ΔErel{HS–LS} values follow the trend:
| 42+ < [Fe(2-bip)2]2+ < 12+ ≈ 32+ < 22+ ≈ [Fe(bimpy)2]2+ |
This agrees with the available experimental data, except that the computed low-spin state of 22+ appears overstabilised compared to the other molecules (Fig. 8). With reference to our previous studies, the positive ΔErel{HS–LS} for 22+ seems ca. 5× larger than expected when comparing two compounds whose T½ values differ by just 14 K.108–112
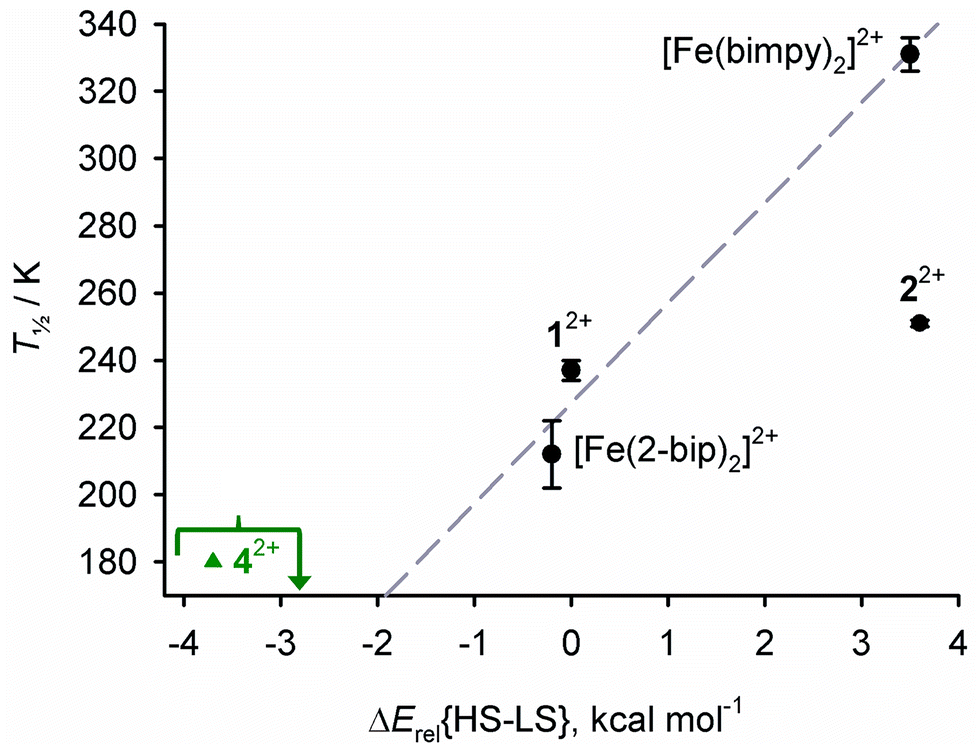 | ||
| Fig. 8 Correlation between measured solution T½ values, and the computed spin state energies in Table 4. The line shows the best fit linear regression of the data points for 12+, [Fe(bimpy)2]2+ and [Fe(2-bip)2]2+.116 Compounds that experimentally show SCO are black circles, and high-spin 42+ (T½ ≤ 180 K) is a green triangle. | ||
Gas phase minimisations of sterically crowded molecules using our protocol can overstabilise the high-spin state, with respect to analogous molecules lacking bulky substituents. We attributed this to intramolecular dispersion interactions between atoms from different residues which are in close contact in the molecule.110,111 Intramolecular dispersion is not included in our protocol, and is difficult to model accurately in single-point DFT calculations.11512+, 32+, 42+, [Fe(bimpy)2]2+ and [Fe(2-bip)2]2+ experience weak inter-ligand C–H⋯π contacts involving their distal benzo groups but 22+ does not, since the relevant C–H groups are replaced by N atoms in that molecule. Hence, since 22+ should experience less intramolecular dispersion, its ΔErel{HS–LS} may not be strictly comparable with the other molecules in the table.
The ligand Brønsted basicities were compared from the average energies of their three lone pair combination MOs (Eav{LP}, Table 4). These give the following trend, where the most basic ligand has the highest average lone pair energy:
| 2-bip < bimpy < L2 ≈ L3 ≈ L1 < L4 |
That is, 2-bip is the least σ-donating ligand in the series and L4 is the strongest. That ordering does not correlate perfectly with the spin states of their complexes, implying the ligand σ-bonding character is not the sole contributor to their different spin state properties. The high-spin nature of 42+, containing the most σ-basic ligand L4, reflects the intramolecular steric hindrance in that molecule.111 The spin states of the other complexes in the study should be less affected by steric factors, however.
Comparison of their frontier orbital energies shows L2 and 2-bip are the most π-acidic of the remaining ligands, and that 22+ and [Fe(2-bip)2]2+ exhibit greater M–L π-back-bonding than the other complexes in the table. That explains the higher T½ shown experimentally by 2[BF4]2, although this is over-estimated by the calculation. Similarly the stronger π-ligand field in [Fe(2-bip)2]2+ partly offsets its weaker Fe–N σ-bonding, leading to just slightly lower ΔErel{HS–LS} and T½ values than 1[BF4]2.
Unlike the other complexes in Fig. 8, the stabilised low-spin state in [Fe(bimpy)2]2+ does not clearly relate to the electronic or steric properties of the bimpy ligand. Thus, no single factor contributing to its larger ΔErel{HS–LS} could be identified from its low-spin d-orbital energies. The π-orbitals of 1H-(benz)imidazolyl groups significantly rehybridise upon dative bond formation, which complicates the analysis of the bonding in that complex.117
The minimisations of 22+ deviate only slightly from idealised D2d symmetry, which contrasts with the distorted coordination geometry found in crystals of 2[BF4]2 (Fig. 2, Table 2). This was investigated by additional minimisations of high-spin 12+ and 22+, with ϕ fixed at values between 165 and 155°.84 This range of distortions carries an energy penalty of <3 kcal mol−1 for 12+, and ≤2 kcal mol−1 for 22+ (ΔE{dist}, Table 5). Since these values lie above kT (0.6 kcal mol−1 at 298 K), the population of distorted molecules in solution should be relatively small at room temperature. We conclude that the distorted geometries observed for 2[BF4]2 are probably induced by crystal packing effects.
| ϕ, deg | E, Ha | ΔE{dist},a kcal mol−1 | |
|---|---|---|---|
| a ΔE(dist) is the energy relative to the corresponding undistorted molecule with ϕ = 180°. b Table 4. c Fixed during the calculation. | |||
| [Fe(L1)2]2+ (12+) | 180 | −3273.967089b | 0 |
| 165c | −3273.965652 | +0.9 | |
| 160c | −3273.964194 | +1.8 | |
| 155c | −3273.962500 | +2.9 | |
| [Fe(L2)2]2+ (22+) | 180 | −3338.084900b | 0 |
| 165c | −3338.083875 | +0.6 | |
| 160c | −3338.083074 | +1.2 | |
| 155c | −3338.081760 | +2.0 | |
ΔE{dist} for 12+ and 22+ is 2–3× larger than for [Fe(1-bpp)2]2+ (1-bpp = 2,6-di{pyrazol-1-yl}pyridine) at each value of ϕ, by the same computational protocol.108 That indicates the steric influence of the annelated distal ligand donor groups on the distorted coordination geometries of 12+ and 22+.
Conclusions
We have surveyed the iron chemistry of 2,6-bis(imidazo[1,2-a]pyridin-2-yl)pyridine (L1), and two new derivatives L2 and L3. While they undergo thermal SCO equilibria in solution, the homoleptic complexes 1[BF4]2 and 2[BF4]2 mostly form high-spin solvates in the crystalline phase. SCO may be inhibited by an intermolecular contact between complex molecules in the 1[BF4]2 solvates (Fig. 1), while 2[BF4]2 adopts a distorted coordination geometry in the crystal that favours a high-spin state (Fig. 2). In the latter case, the geometric distortion is coupled to a pattern of intramolecular n⋯π and intermolecular π⋯π and C–H⋯N interactions that is consistently found in all the solvates examined. However, crystalline 1[BF4]1.6[SiF6]0.2·1.7MeNO2·0.3Et2O is low-spin at 120 K, confirming that the low-spin state of that complex is thermally accessible.Unexpectedly the homoleptic iron(II) complex of L3 was not isolated and is less stable in solution than 1[BF4]2 and 2[BF4]2 by 1H NMR. A 1:1 Fe:L3 complex was structurally characterised, which shows no unexpected features (Fig. 4).
A crystal structure of a salt of [Fe(L4)2]2+, 4[BF4]2, is also reported. That molecule adopts a strongly flattened six-coordinate geometry with a helical ligand conformation, which is imposed by an inter-ligand steric clash involving its quinolyl C8–H groups.93 The molecular helicity in 4[BF4]2 is more pronounced than for the only other structurally characterised [M(L4)2]n+ complex (Mn+ = Co2+)93 but was reproduced by a gas phase DFT minimisation, showing it is an intrinsic property of the molecule.
All the complexes in this work are high-spin in solution at room temperature and show a blue ligand-based emission under ambient conditions (Fig. 8). In most cases, this occurs at longer wavelength than the corresponding metal-free ligand emission. An exception is 2[BF4]2, whose emission maximum is similar to 1[BF4]2 but is slightly blue-shifted compared to L2. That may reflect the influence of dipolar interactions between the solvent molecules and the L2 imidazopyrimidinyl N8 atoms93 on that ligand emission.102 Such interactions should be significant in the free ligand but less so in 2[BF4]2, where those N atoms become sterically inaccessible to the environment.
Gas phase DFT calculations mostly reproduce the spin state properties of 12+–42+, and two constitutional isomers of 12+ from the literature (Table 4). However they overstabilise the low-spin state of 22+ compared to the other molecules, which could reflect weaker intramolecular dispersion interactions in 22+ arising from its reduced steric crowding. The high-spin state and helical molecular geometry of 42+ are reproduced well by the calculation. The ΔErel{HS–LS} spin state energies of the other molecules do not correlate with the ligand σ-basicities, but can be understood as a combination of the metal–ligand σ-bonding and π-bonding character of each ligand.
As well as continuing our synthetic studies, we are seeking a more accurate computational protocol for the spin states of sterically hindered molecules, so the effect of steric crowding in such compounds can be accurately determined. That would have wider value for the design of base metal catalysts for oxidation reactions or other organic transformations, whose mechanisms and effectiveness depend on the spin state of the metal centre.118,119
Experimental
Ligands L1 (ref. 68) and L4 (Scheme 1)74 were synthesised by the literature procedures. Other reagents were purchased commercially and used as supplied. Synthetic protocols for the new ligands L2 and L3 and characterisation data for all the ligands used are given in the ESI.†Synthesis of [Fe(L1)2][BF4]2 (1[BF4]2)
Reaction of Fe[BF4]2·6H2O (0.054 g, 0.16 mmol) with L1 (0.10 g, 0.32 mmol) in nitromethane (10 cm3) rapidly yielded a dark red solution. This was filtered to remove a small amount of brown precipitate, and the filtrate was concentrated to ca. 5 cm3 volume. Slow diffusion of diethyl ether vapour into the solution yielded a red powder, which was recrystallised from different solvents to afford the solvated crystals described below. Yield 0.065 g, 48%. Found C, 51.3; H, 3.39; N, 15.2%. Calcd for C38H26B2F8FeN10·2H2O C, 51.4; H, 3.40; N, 15.8%. 1H NMR (CD3CN) δ 2.7 (4H), 4.0 (4H), 5.8 (4H), 23.1 (2H), 23.4 (4H), 41.2 (4H), 63.6 (4H).Synthesis of [Fe(L2)2][BF4]2 (2[BF4]2)
Method as above, using L2 (0.10 g, 0.32 mmol). The product was initially obtained as a cherry red powder, which was recrystallised from different solvents as below. Yield 0.060 g, 44%. Found C, 47.3; H, 2.51; N, 22.8%. Calcd for C34H22B2F8FeN14 C, 47.7; H, 2.59; N, 22.9%. 1H NMR (CD3CN) δ 4.1 (4H), 9.9 (4H), 15.2 (2H), 25.0 (4H), 36.0 (4H), 53.3 (4H).Synthesis of [FeL3][BF4]2·H2O (3′[BF4]2·H2O)
Method as above, using L3 (0.13 g, 0.32 mmol). Recrystallisation of the product from common organic solvent combinations consistently yielded a brown powder, which analysed as [FeL3][BF4]2·y(solvent). The sample reported here was recrystallised from undried nitromethane/diethyl ether. Yield 0.060 g, 44%. Found C, 49.9; H, 2.97; N, 10.3%. Calcd for C27H17B2F8FeN5·H2O C, 49.2; H, 2.91; N, 10.6%.Synthesis of [FeBr2(py)xL3]·yH2O (py = pyridine)
Addition of hydrated FeBr2 (0.054 g, 0.16 mmol) to a solution of L3 (0.05 g, 0.12 mmol) in 2,2,2-trifluoroethanol (5 cm3) yielded a cloudy brown solution. The mixture was stored in a freezer for 1 h to coagulate the precipitate, then filtered. Layering the dark brown filtrate with pyridine at room temperature yielded dark brown prisms at the solvent interface. The crystals have the crystallographic formulation [FeBr(py)2L3]Br·0.5H2O. However, microanalysis was more consistent with the formula [FeBr(py)L3]Br·2H2O, suggesting some of the pyridine content of the crystals may have been exchanged by atmospheric moisture on exposure to air. Yield 0.060 g, 44%. Found C, 51.5; H, 3.19; N, 11.0%. Calcd for C32H22Br2FeN6·2H2O C, 51.8; H, 3.53; N, 11.3%.Synthesis of [Fe(L4)2][BF4]2 (4[BF4]2)
Method as for 1[BF4]2, using L4 (0.10 g, 0.32 mmol). The product crystallised as small red platelets, which rapidly collapsed to an amorphous pink powder upon isolation. Yield 0.060 g, 44%. Found C, 60.8; H, 3.47; N, 8.69%. Calcd for C46H30B2F8FeN6·H2O C, 60.4; H, 3.53; N, 9.19%. 1H NMR (CD3CN) δ −12.1 (2H), 2.5 (4H), 9.0 (4H), 9.5 (4H), 10.5 (4H), 30.3 (4H), 61.8 (4H), 73.6 (4H).The perchlorate salt of this complex has been reported previously.73
Single crystal structure analyses
Solvate crystals of 1[BF4]2, 2[BF4]2 and 4[BF4]2 were grown by slow diffusion of diethyl ether antisolvent vapour, into solutions of each complex in the appropriate solvent. The crystals of [FeBr(py)2L3]Br·0.5H2O were obtained from a solvent layering experiment, as described above.Experimental details and refinement protocols for the structure determinations are given in the ESI.† All the structures were solved by direct methods (SHELX-TL120), and developed by full least-squares refinement on F2 (SHELXL2018121). Crystallographic figures were produced using XSEED,122 and other publication materials were prepared with OLEX2.123 Unless otherwise stated, the following procedures were applied to the refinements.
Other measurements
Elemental microanalyses were performed by the microanalytical services at the University of Leeds and London Metropolitan University. Diamagnetic NMR spectra employed a Bruker AV3HD spectrometer operating at 400.1 MHz (1H) or 100.6 MHz (13C), while paramagnetic 1H NMR spectra were obtained with a Bruker AV3 spectrometer operating at 300.1 MHz. X-ray powder diffraction data were obtained with a Bruker D8 Advance A25 diffractometer using Cu-Kα radiation (λ = 1.5418 Å).Solid state magnetic susceptibility measurements were performed on a Quantum Design MPMS-3 magnetometer, with an applied field of 5000 G and a scan rate of 5 K min−1. A diamagnetic correction for the sample was estimated from Pascal's constants;124 a diamagnetic correction for the sample holder was also applied. Solvated samples were protected against solvent loss by saturating the (tightly sealed) sample holder capsules with a drop of diethyl ether. Magnetic measurements in solution were obtained by Evans method using a JEOL ECA600ii or a Bruker AV500 spectrometer, operating at 600.13 and 500.05 MHz (1H) respectively.125,126 A diamagnetic correction for the sample,124 and a correction for the variation of the density of the solvent with temperature,127 were applied to these data. The parameters in Table 2 were derived by fitting these data to eqn (1) and (2):
| ln[(1 − nHS(T))/nHS(T)] = ΔH/RT − ΔS/R | (1) |
| ΔS = ΔH/T½ | (2) |
UV-vis spectra were measured using a PerkinElmer Lambda 900 spectrophotometer. Fluorescence measurements under ambient conditions were obtained using a Horiba Fluoromax 3 fluorimeter with constant slit widths of 2 mm. A range of excitation wavelengths were sampled, and the data quoted are for the excitation wavelength that led to the most intense emission for each compound. The sample concentrations for the fluorescence spectra were between 1–4 × 10−5 mol dm−3.
DFT calculations were carried out using SPARTAN'20 for Windows,128 with the B86PW91 functional and the def2-SVP basis set. Low-spin systems were treated as spin-restricted, and high-spin systems were treated as spin-unrestricted. The calculations were carried out in the gas phase, since a solvent gradient for iron is not implemented in SPARTAN'20. The molecules were constructed de novo in the program, then subjected to a preliminary molecular mechanics minimisation before the full DFT energy minimisation was undertaken.
Data availability
Data supporting this study are available in the ESI,† or at https://doi.org/10.5518/1413.Author contributions
RK did all the experimental work in the publication. MAH conceived and supervised the study, assisted with the crystal structure refinements, performed the DFT calculations and wrote the manuscript draft. Both authors approved the final version of the publication.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the EPSRC (EP/K012576/1). The authors thank Simon Barrett (School of Chemistry, University of Leeds) for the Evans method study, and Dr Oscar Cespedes (School of Physics and Astronomy, University of Leeds) for help with the solid state magnetic measurements.References
- Spin Crossover in Transition Metal Compounds I–III: Topics in Current Chemistry, ed. P. Gütlich and H. A. Goodwin, Springer-Verlag, Berlin, 2004, vol. 233–235 Search PubMed.
- Spin-crossover materials – properties and applications, ed. M. A. Halcrow, John Wiley & Sons, Chichester, UK, 2013, p. 568 Search PubMed.
- J. Zarembowitch, F. Varret, A. Hauser, J. A. Real and K. Boukheddaden, C. R. Chim., 2018, 21, 1056 CrossRef CAS.
- K. Senthil Kumar, Y. Bayeh, T. Gebretsadik, F. Elemo, M. Gebrezgiabher, M. Thomas and M. Ruben, Dalton Trans., 2019, 48, 15321 RSC.
- W. Huang, X. Ma, O. Sato and D. Wu, Chem. Soc. Rev., 2021, 50, 6832 RSC.
- M. Puri and L. Que Jr., Acc. Chem. Res., 2015, 48, 2443–2452 CrossRef CAS PubMed.
- M. Milan, M. Salamone, M. Costas and M. Bietti, Acc. Chem. Res., 2018, 51, 1984–1995 CrossRef CAS PubMed.
- M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS PubMed.
- P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495 CrossRef CAS PubMed.
- M. Beller, Chem. Rev., 2019, 119, 2089 CrossRef CAS PubMed.
- C. Förster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057–1070 RSC.
- C. E. Housecroft and E. C. Constable, Chem. Sci., 2022, 13, 1225–1262 RSC.
- M. A. Halcrow, Dalton Trans., 2020, 49, 15560–15567 RSC.
- P. Guionneau, M. Marchivie and G. Chastanet, Chem. – Eur. J., 2021, 27, 1483–1486 CrossRef CAS PubMed.
- S. Xue, Y. Guo and Y. Garcia, CrystEngComm, 2021, 23, 7899–7915 RSC.
- M. Chergui and E. Collet, Chem. Rev., 2017, 117, 11025–11065 CrossRef CAS PubMed.
- K. J. Gaffney, Chem. Sci., 2021, 12, 8010–8025 RSC.
- S. Pillet, J. Appl. Phys., 2021, 129, 181101 CrossRef CAS.
- K. Senthil Kumar and M. Ruben, Angew. Chem., Int. Ed., 2021, 60, 7502–7521 CrossRef PubMed.
- L. Kipgen, M. Bernien, F. Tuczek and W. Kuch, Adv. Mater., 2021 Search PubMed , 33, 2008141 and 2021, 33, 2170354 [correction].
- V. Tangoulis, C. D. Polyzou, P. Gkolfi, N. Lalioti, O. Malina and M. Polaskova, Dalton Trans., 2021, 50, 3109–3115 RSC.
- J. M. Cain, W. He, I. Maurin, M. W. Meisel and D. R. Talham, J. Appl. Phys., 2021, 129, 160903 CrossRef CAS.
- G. D. Harzmann, R. Frisenda, H. S. J. van der Zant and M. Mayor, Angew. Chem., Int. Ed., 2015, 54, 13425–13430 CrossRef CAS PubMed.
- S. K. Karuppannan, A. Martín-Rodríguez, E. Ruiz, P. Harding, D. J. Harding, X. Yu, A. Tadich, B. Cowie, D. Qi and C. A. Nijhuis, Chem. Sci., 2021, 12, 2381–2388 RSC.
- S. Johannsen, S. Ossinger, J. Grunwald, A. Herman, H. Wende, F. Tuczek, M. Gruber and R. Berndt, Angew. Chem., Int. Ed., 2022, 61, e202115892 CrossRef CAS PubMed.
- G. Molnár, S. Rat, L. Salmon, W. Nicolazzi and A. Bousseksou, Adv. Mater., 2018, 30, 1703862 CrossRef PubMed.
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- O. Kahn, J. Krober and C. Jay, Adv. Mater., 1992, 4, 718–728 CrossRef CAS.
- E. Resines-Urien, E. Fernandez-Bartolome, A. Martinez-Martinez, A. Gamonal, L. Piñeiro-López and J. S. Costa, Chem. Soc. Rev., 2023, 52, 705–727 RSC.
- M. Mikolasek, M. D. Manrique-Juarez, H. J. Shepherd, K. Ridier, S. Rat, V. Shalabaeva, A.-C. Bas, I. E. Collings, F. Mathieu, J. Cacheux, T. Leichle, L. Nicu, W. Nicolazzi, L. Salmon, G. Molnár and A. Bousseksou, J. Am. Chem. Soc., 2018, 140, 8970–8979 CrossRef CAS PubMed.
- M. D. Manrique-Juárez, F. Mathieu, A. Laborde, S. Rat, V. Shalabaeva, P. Demont, O. Thomas, L. Salmon, T. Leichle, L. Nicu, G. Molnár and A. Bousseksou, Adv. Funct. Mater., 2018, 28, 1801970 CrossRef.
- S. P. Vallone, A. N. Tantillo, A. M. dos Santos, J. Molaison, R. Kulmaczewski, A. Chapoy, P. Ahmadi, M. A. Halcrow and K. G. Sandeman, Adv. Mater., 2019, 31, 1807334 CrossRef PubMed.
- K. Ridier, Y. Zhang, M. Piedrahita-Bello, C. M. Quintero, L. Salmon, G. Molnár, C. Bergaud and A. Bousseksou, Adv. Mater., 2020, 32, 2000987 CrossRef CAS PubMed.
- M. Romanini, Y. Wang, K. Gürpinar, G. Ornelas, P. Lloveras, Y. Zhang, W. Zheng, M. Barrio, A. Aznar, A. Gràcia-Condal, B. Emre, O. Atakol, C. Popescu, H. Zhang, Y. Long, L. Balicas, J. L. Tamarit, A. Planes, M. Shatruk and L. Mañosa, Adv. Mater., 2021, 33, 2008076 CrossRef CAS PubMed.
- J. Seo, J. D. Braun, V. M. Dev and J. A. Mason, J. Am. Chem. Soc., 2022, 144, 6493–6503 CrossRef CAS PubMed.
- M. K. Javed, A. Sulaiman, M. Yamashita and Z.-Y. Li, Coord. Chem. Rev., 2022, 467, 214625 CrossRef CAS.
- K. Sun, J.-P. Xue, Z.-S. Yao and J. Tao, Dalton Trans., 2022, 51, 16044–16054 RSC.
- M. Wang, Z.-Y. Li, R. Ishikawa and M. Yamashita, Coord. Chem. Rev., 2021, 435, 213819 CrossRef CAS.
- R. Ababei, C. Pichon, O. Roubeau, Y.-G. Li, N. Bréfuel, L. Buisson, P. Guionneau, C. Mathoniére and R. Clérac, J. Am. Chem. Soc., 2013, 135, 14840–14853 CrossRef CAS PubMed.
- D. Pinkowicz, M. Rams, M. Mišek, K. V. Kamenev, H. Tomkowiak, A. Katrusiak and B. Sieklucka, J. Am. Chem. Soc., 2015, 137, 8795–8802 CrossRef CAS PubMed.
- M. Darawsheh, L. A. Barrios, O. Roubeau, S. J. Teat and G. Aromí, Angew. Chem., Int. Ed., 2018, 57, 13509–13513 CrossRef CAS PubMed.
- Y. Sekine, R. Akiyoshi and S. Hayami, Coord. Chem. Rev., 2022, 469, 214663 CrossRef CAS.
- A. Enriquez-Cabrera, A. Rapakousiou, M. Piedrahita Bello, G. Molnár, L. Salmon and A. Bousseksou, Coord. Chem. Rev., 2020, 419, 213396 CrossRef CAS.
- H. A. Goodwin, Top. Curr. Chem., 2004, 233, 59–90 CrossRef CAS.
- M. A. Halcrow, Polyhedron, 2007, 26, 3523–3576 CrossRef CAS.
- G. J. Long, F. Grandjean and D. L. Reger, Top. Curr. Chem., 2004, 233, 91–122 CrossRef CAS.
- O. G. Shakirova and L. G. Lavrenova, Crystals, 2020, 10, 843 CrossRef CAS.
- M. Boča, R. F. Jameson and W. Linert, Coord. Chem. Rev., 2011, 255, 290–317 CrossRef.
- M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493–2514 CrossRef CAS.
- J. Olguín and S. Brooker, Coord. Chem. Rev., 2011, 255, 203–240 CrossRef.
- L. J. Kershaw Cook, R. Mohammed, G. Sherborne, T. D. Roberts, S. Alvarez and M. A. Halcrow, Coord. Chem. Rev., 2015, 289–290, 2–12 CrossRef CAS.
- G. A. Craig, O. Roubeau and G. Aromí, Coord. Chem. Rev., 2014, 269, 13–31 CrossRef CAS.
- T. Buchen and P. Gütlich, Inorg. Chim. Acta, 1995, 231, 221–223 CrossRef CAS.
- J. M. Holland, S. A. Barrett, C. A. Kilner and M. A. Halcrow, Inorg. Chem. Commun., 2002, 5, 328–332 CrossRef CAS.
- L. J. Kershaw Cook, R. Kulmaczewski, R. Mohammed, S. Dudley, S. A. Barrett, M. A. Little, R. J. Deeth and M. A. Halcrow, Angew. Chem., Int. Ed., 2016, 55, 4327–4331 CrossRef CAS PubMed.
- I. Nikovskiy, A. Polezhaev, V. Novikov, D. Aleshin, A. Pavlov, E. Saffiulina, R. Aysin, P. Dorovatovskii, L. Nodaraki, F. Tuna and Y. Nelyubina, Chem. – Eur. J., 2020, 26, 5629–5638 CrossRef CAS PubMed.
- R. Akiyoshi, Y. Hirota, D. Kosumi, M. Tsutsumi, M. Nakamura, L. F. Lindoy and S. Hayami, Chem. Sci., 2019, 10, 5843–5848 RSC.
- Ö. Üngör, E. S. Choi and M. Shatruk, Chem. Sci., 2021, 12, 10765–10779 RSC.
- E. J. Devid, P. N. Martinho, M. V. Kamalakar, I. Šalitroš, U. Prendergast, J.-F. Dayen, V. Meded, T. Lemma, R. González-Prieto, F. Evers, T. E. Keyes, M. Ruben, B. Doudin and S. J. van der Molen, ACS Nano, 2015, 9, 4496–4507 CrossRef CAS PubMed.
- A. Abhervé, M. Palacios-Corella, J. M. Clemente-Juan, R. Marx, P. Neugebauer, J. van Slageren, M. Clemente-León and E. Coronado, J. Mater. Chem. C, 2015, 3, 7936–7945 RSC.
- B. Schäfer, T. Bauer, I. Faus, J. A. Wolny, F. Dahms, O. Fuhr, S. Lebedkin, H.-C. Wille, K. Schlage, K. Chevalier, F. Rupp, R. Diller, V. Schünemann, M. M. Kappes and M. Ruben, Dalton Trans., 2017, 46, 2289–2302 RSC.
- I. Šalitroš, R. Herchel, O. Fuhr, R. González-Prieto and M. Ruben, Inorg. Chem., 2019, 58, 4310–4319 CrossRef PubMed.
- M. Palacios-Corella, J. Ramos-Soriano, M. Souto, D. Ananias, J. Calbo, E. Ortí, B. M. Illescas, M. Clemente-León, N. Martín and E. Coronado, Chem. Sci., 2021, 12, 757–766 RSC.
- I. Galadzhun, R. Kulmaczewski, N. Shahid, O. Cespedes, M. J. Howard and M. A. Halcrow, Chem. Commun., 2021, 57, 4039–4042 RSC.
- M. Hasegawa, F. Renz, T. Hara, Y. Kikuchia, Y. Fukuda, J. Okubo, T. Hoshi and W. Linert, Chem. Phys., 2002, 277, 21–30 CrossRef CAS.
- A. Santoro, L. J. Kershaw Cook, R. Kulmaczewski, S. A. Barrett, O. Cespedes and M. A. Halcrow, Inorg. Chem., 2015, 54, 682–693 CrossRef CAS PubMed.
- S. Vela, C. Gourlaouen, M. Fumanal and J. Ribas-Arino, Magnetochemistry, 2016, 2, 6 CrossRef.
- K. Li, J.-L. Niu, M.-Z. Yang, Z. Li, L.-Y. Wu, X.-Q. Hao and M.-P. Song, Organometallics, 2015, 34, 1170–1176 CrossRef CAS.
- F.-L. Yang, X. Zhu, D.-K. Rao, X.-N. Cao, K. Li, Y. Xu, X.-Q. Hao and M.-P. Song, RSC Adv., 2016, 6, 37093–37098 RSC.
- F.-L. Yang, Y.-H. Wang, Y.-F. Ni, X. Gao, B. Song, X. Zhu and X.-Q. Hao, Eur. J. Org. Chem., 2017, 3481–3486 CrossRef CAS.
- X.-N. Cao, X.-M. Wan, F.-L. Yang, K. Li, X.-Q. Hao, T. Shao, X. Zhu and M.-P. Song, J. Org. Chem., 2018, 83, 3657–3668 CrossRef CAS PubMed.
- X.-M. Wan, Z.-L. Liu, W.-Q. Liu, X.-N. Cao, X. Zhu, X.-M. Zhao, B. Song, X.-Q. Hao and G. Liu, Tetrahedron, 2019, 75, 2697–2705 CrossRef CAS.
- C. M. Harris, H. R. H. Patil and E. Sinn, Inorg. Chem., 1969, 8, 101–104 CrossRef CAS.
- E. Largy, F. Hamon, F. Rosu, V. Gabelica, E. De Pauw, A. Guédin, J.-L. Mergny and M.-P. Teulade-Fichou, Chem. – Eur. J., 2011, 17, 13274–13283 CrossRef CAS PubMed.
- M. Hostettler, K. W. Törnroos, D. Chernyshov, B. Vangdal and H.-B. Bürgi, Angew. Chem., Int. Ed., 2004, 43, 4589–4594 CrossRef CAS PubMed.
- I. Nemec, R. Herchel and Z. Trávníček, Dalton Trans., 2015, 44, 4474–4484 RSC.
- L. J. Kershaw Cook, R. Kulmaczewski, O. Cespedes and M. A. Halcrow, Chem. – Eur. J., 2016, 22, 1789–1799 CrossRef PubMed.
- W. Phonsri, P. Harding, L. Liu, S. G. Telfer, K. S. Murray, B. Moubaraki, T. M. Ross, G. N. L. Jameson and D. J. Harding, Chem. Sci., 2017, 8, 3949–3959 RSC.
- X.-P. Sun, R.-J. Wei, Z.-S. Yao and J. Tao, Cryst. Growth Des., 2018, 18, 6853–6862 CrossRef CAS.
- I. Capel Berdiell, R. Kulmaczewski, N. Shahid, O. Cespedes and M. A. Halcrow, Chem. Commun., 2021, 57, 6566–6569 RSC.
- L. T. Birchall, A. T. Raja, L. Jackson and H. J. Shepherd, Cryst. Growth Des., 2023, 23, 1768–1774 CrossRef CAS.
- V Oh is the volume of the octahedron defined by the FeN6 coordination sphere.83Σ is a general measure of the deviation of a metal ion from an ideal octahedral geometry, while Θ more specifically indicates its distortion towards a trigonal prismatic structure. Σ and Θ are usually larger in the high-spin than in the low-spin state, especially for complexes of chelating ligands with restricted bite angles like L1 and L2; a perfectly octahedral complex gives Σ = Θ = 0.83 Full definitions of Σ and Θ are in the ESI.†.
- P. Guionneau, M. Marchivie, G. Bravic, J.-F. Létard and D. Chasseau, Top. Curr. Chem., 2004, 234, 97–128 CrossRef CAS.
- θ is the dihedral angle between the least squares planes of the two L ligands, and ϕ is the trans-N{pyridyl}–Fe–N{pyridyl} bond angle.85 An ideal D2d-symmetric geometry for a [FeL2]2+ centre (L = a meridional tridentate ligand) gives θ = 90 and ϕ = 180°, but many high-spin complexes of this type exhibit significantly reduced values for these parameters. See the ESI† for more details.
- J. M. Holland, J. A. McAllister, C. A. Kilner, M. Thornton-Pett, A. J. Bridgeman and M. A. Halcrow, J. Chem. Soc., Dalton Trans., 2002, 548–554 RSC.
- P. J. van Koningsbruggen, J. G. Haasnoot, R. A. G. de Graaff and J. Reedijk, J. Chem. Soc., Dalton Trans., 1993, 483–484 RSC.
- I. Dance and M. Scudder, CrystEngComm, 2009, 11, 2233–2247 RSC.
- R. Pritchard, C. A. Kilner and M. A. Halcrow, Chem. Commun., 2007, 577–579 RSC.
- E. Michaels, C. M. Pask, I. Capel Berdiell, H. B. Vasili, M. J. Howard, O. Cespedes and M. A. Halcrow, Cryst. Growth Des., 2022, 22, 6809–6817 CrossRef CAS.
- S. Vela, J. J. Novoa and J. Ribas-Arino, Phys. Chem. Chem. Phys., 2014, 16, 27012–27024 RSC.
- L. J. Kershaw Cook, F. L. Thorp-Greenwood, T. P. Comyn, O. Cespedes, G. Chastanet and M. A. Halcrow, Inorg. Chem., 2015, 54, 6319–6330 CrossRef CAS PubMed.
- R. Kulmaczewski, I. T. Armstrong, P. Catchpole, E. S. J. Ratcliffe, H. B. Vasili, S. L. Warriner, O. Cespedes and M. A. Halcrow, Chem. – Eur. J., 2023, 29, e202202578 CrossRef CAS PubMed.
- This atom label corresponds to the IUPAC atom numbering for imidazo[1,2-a]pyridyl residues, ed. J. Rigaudy and S. P. Klesney, Nomenclature of Organic Chemistry, Sections A, B, C, D, E, F, and H, Pergamon Press, Oxford, 1979, sect. B3 Search PubMed.
- S. Aroua, T. K. Todorova, P. Hommes, L.-M. Chamoreau, H.-U. Reissig, V. Mougel and M. Fontecave, Inorg. Chem., 2017, 56, 5930–5940 CrossRef CAS PubMed.
- H. Toftlund, Monatsh. Chem., 2001, 132, 1269–1277 CrossRef CAS.
- K. P. Kepp, Inorg. Chem., 2016, 55, 2717–2727 CrossRef CAS PubMed.
- N. Hassan, A. B. Koudriavtsev and W. Linert, Pure Appl. Chem., 2008, 80, 1281–1292 CrossRef CAS.
- S. Sundaresan and S. Brooker, Inorg. Chem., 2023, 62, 12192–12202 CrossRef CAS PubMed.
- J. Catalan, E. Mena, F. Fabero and F. Amat-Guerri, J. Chem. Phys., 1992, 96, 2005–2016 CrossRef CAS.
- H. Tomoda, T. Hirano, S. Saito, T. Mutai and K. Araki, Bull. Chem. Soc. Jpn., 1999, 72, 1327–1334 CrossRef CAS.
- A. J. Stasyuk, M. Banasiewicz, M. K. Cyrański and D. T. Gryko, J. Org. Chem., 2012, 77, 5552–5558 CrossRef CAS PubMed.
- D. A. Lerner, P. M. Horowitz and E. M. Evleth, The absorption and emission spectra of imidazo[1,2-a]pyrimidine are solvent-dependent, J. Phys. Chem., 1977, 81, 12–17 CrossRef CAS.
- D. M. Klassen, C. W. Hudson and E. L. Shaddix, Inorg. Chem., 1975, 14, 2733–2736 CrossRef CAS.
- M. L. Stone and G. A. Crosby, Chem. Phys. Lett., 1981, 79, 169–173 CrossRef CAS.
- B. J. Houghton and R. J. Deeth, Eur. J. Inorg. Chem., 2014, 4573–4580 CrossRef CAS.
- S. R. Mortensen and K. P. Kepp, J. Phys. Chem. A, 2015, 119, 4041–4050 CrossRef CAS PubMed.
- O. S. Siig and K. P. Kepp, J. Phys. Chem. A, 2018, 122, 4208–4217 CrossRef CAS PubMed.
- I. Capel Berdiell, R. Kulmaczewski and M. A. Halcrow, Inorg. Chem., 2017, 56, 8817–8828 CrossRef CAS PubMed.
- R. Kulmaczewski, M. J. Howard and M. A. Halcrow, Dalton Trans., 2021, 50, 3464–3467 RSC.
- I. Capel Berdiell, D. J. Davies, J. Woodworth, R. Kulmaczewski, O. Cespedes and M. A. Halcrow, Inorg. Chem., 2021, 60, 14988–15000 CrossRef CAS PubMed.
- N. Shahid, K. E. Burrows, C. M. Pask, O. Cespedes, M. J. Howard, P. C. McGowan and M. A. Halcrow, Inorg. Chem., 2021, 60, 14336–14348 CrossRef CAS PubMed.
- N. Shahid, K. E. Burrows, C. M. Pask, O. Cespedes, M. J. Howard, P. C. McGowan and M. A. Halcrow, Dalton Trans., 2022, 51, 4262–4274 RSC.
- M. Reiher, O. Salomon and B. A. Hess, Theor. Chem. Acc., 2001, 107, 48–55 Search PubMed.
- S. Zein, S. A. Borshch, P. Fleurat-Lessard, M. E. Casida and H. Chermette, J. Chem. Phys., 2007, 126, 014105 CrossRef PubMed.
- D. C. Ashley and E. Jakubikova, Coord. Chem. Rev., 2017, 337, 97–111 CrossRef CAS.
- T ½ for [Fe(bimpy)2]2+ in solution is solvent-dependent, which reflects the influence of hydrogen bonding between its peripheral N–H groups and the solvent on the metal ion ligand field. It shows T½ = 330–335 K in weakly interacting solvents, which should approximate its environment in the gas phase (ref. 48).
- Ł. Boda, M. Boczar, M. J. Wójcik and T. Nakajima, J. Phys. Chem. A, 2021, 125, 6902–6912 CrossRef PubMed.
- A. Fürstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef PubMed.
- M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Barbour, J. Appl. Crystallogr., 2020, 53, 1141–1146 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. J. O'Connor, Prog. Inorg. Chem., 1982, 29, 203–283 CrossRef.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- E. M. Schubert, J. Chem. Educ., 1992, 69, 62 CrossRef CAS.
- B. García and J. C. Ortega, J. Chem. Eng. Data, 1988, 33, 200–204 CrossRef.
- Spartan'20, Wavefunction Inc., Irvine CA, USA, 2020 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols and characterisation data for the ligands in this work; experimental data, refinement details, figures and tables for the crystal structure determinations; additional solid and solution phase characterisation data; and details of the minimised structures from the DFT calculations. CCDC 2288873–2288881. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02747c |
| This journal is © The Royal Society of Chemistry 2023 |