DOI:
10.1039/D3DT02722H
(Perspective)
Dalton Trans., 2023,
52, 14259-14286
Cooperation towards nobility: equipping first-row transition metals with an aluminium sword
Received
21st August 2023
, Accepted 25th August 2023
First published on 8th September 2023
Abstract
The exploration for noble metals substitutes in catalysis has become a highly active area of research, driven by the pursuit of sustainable chemical processes. Although the utilization of base metals holds great potential as an alternative, their successful implementation in predictable catalytic processes necessitates the development of appropriate ligands. Such ligands must be capable of controlling their intricate redox chemistry and promote two-electron events, thus mimicking well-established organometallic processes in noble metal catalysis. While numerous approaches for infusing nobility to base metals have been explored, metal–ligand cooperation has garnered significant attention in recent years. Within this context, aluminium-based ligands offer interesting features to fine-tune the activity of metal centres, but their application in base metal catalysis remains largely unexplored. This perspective seeks to highlight the most recent breakthroughs in the reactivity of heterobimetallic aluminium–base-metal complexes, while also showcasing their potential to develop novel and predictable catalytic transformations. By turning the spotlight on such heterobimetallic species, we aim to inspire chemists to explore aluminium–base-metal species and expand the range of their applications as catalysts.
1. Introduction
Noble metals (NM) are privileged elements that enable straightforward modification of organic molecules, thus establishing themselves as indispensable workhorses in the chemical industry. Their dominance stems from their ability to coordinate substrates and undergo well-understood two-electron organometallic steps, providing controllable and predictable chemical transformations.1–3 Yet, their limited availability, fluctuating costs and huge carbon footprint related to their production (e.g., the production of 1 kg of pure Rh metal generates 32t of CO2) represent major drawbacks to overcome if chemists aim at developing truly sustainable industrial synthetic processes.4 While catalysts based on abundant, inexpensive, and sustainable base metals (BM) have recently emerged as promising alternatives,5–9 their potential to replace noble metals in catalysis is hampered by their intrinsic differences in orbital overlap, bonding, and electronic structure. Contrary to 4d and 5d metals, reactivity on 3d metal centres often proceeds through one-electron redox events, resulting in difficulties in fully understanding, controlling and maintaining the function of the catalyst in chemical transformations.10–12 Fe, Cr, Ni and Co represent attractive alternatives to pension off noble metals in key industrial processes,13–17 yet these metals demand adequate ligands to tame one of their several catalytically active redox couples.18 Indeed, fine-tuning their reactivity will certainly enable novel and challenging reactions that are unachievable with current technologies. Several strategies have been employed to control the chemistry of base metals in catalytic setups, including the use of redox active ligands,19–21 and metal–ligand cooperation.22,23 It is noteworthy that these approaches with Earth-abundant metals have been routinely employed by Nature in enzymes with metallic active centres to perform transformations that have no parallel in synthesis.24
A particularly interesting approach that allows control of redox events in base metal centres is the use of chemical metal–ligand cooperation, in which actor ligands participate in bond-forming and breaking events. Combining base metals with ligands endowed with low-lying LUMOs, spanning from olefins to Lewis acids,25 enables anchoring points for substrates during catalytic cycles, which facilitates their activation in a cooperative, predictable and fundamentally unique manner. In this vein, carbenes are a family of ligands endowed with ideal properties to engage base metals in cooperative two-electron events: they are strong σ-donors and can accept charge through their low-lying empty p-orbital.26,27 Nonetheless, their use in cooperative catalysis is prevented by the stability of the newly formed carbon–element bond after activation.28–30 Pairing base metals with heavier neighbours from group 14 is an approach that has been gaining increased interest recently,31–34 as weaker bonding after activation with such elements promotes small molecule activation in an efficient and synergistic manner.35,36 A powerful alternative that has been recently exploited to overcome the reluctance of carbene–metal systems to undergo cooperative catalysis is the use of elements from group 13.37–39 Such elements possess unoccupied p-orbitals, in both high-valent and low-valent states, and coordination of ligands to such orbitals appears to be more labile, which facilitates bond scission and, subsequently, catalysis. Particularly attractive is the use of low-valent group 13 ligands, which are strong σ-donors.40 Indeed, the use of boron-based anionic ligands (boryl-type ligands),41 which can be embedded in pincer scaffolds,42 has enabled cooperative catalytic processes with base metals,43–47 including Ni, Co and Cu.
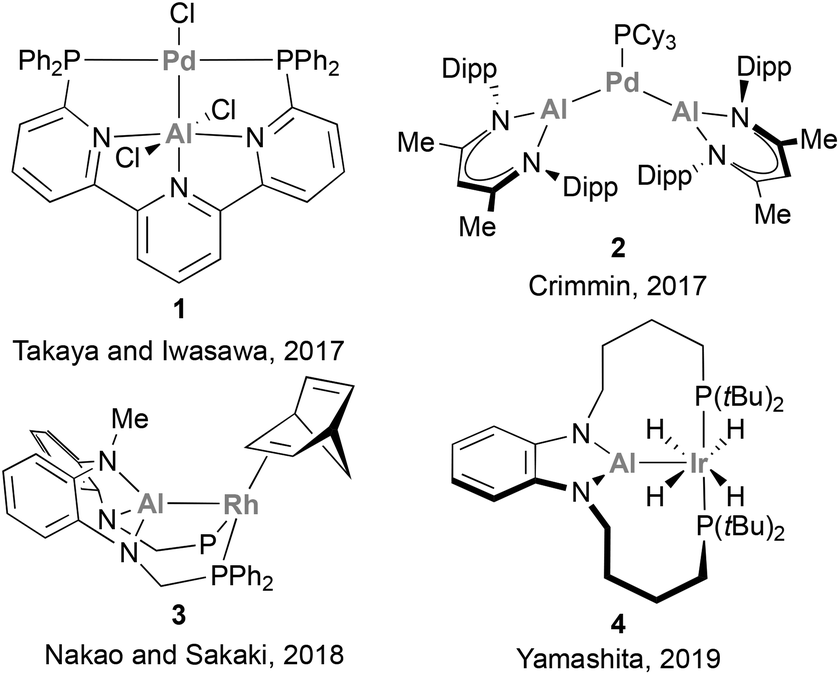
The potential of such low valent group 13 species, combined with recent advances in low-valent aluminium (Al) chemistry,48–53 promoted a shift in interest towards the use of Al in cooperative catalytic transformations.54 Al, the most abundant metal in the Earth's crust, is less electron-negative compared with B (1.47 vs. 2.01),55 which endows Al(I) centres with a better σ-donor ability, resulting in the formation of stable, coordinatively unsaturated electron-rich metal centres. Furthermore, Al species possess highly acidic p-orbitals and a larger coordination number, key features that enable diverse substrate-coordination modes. Importantly, Al centres can be readily installed in a variety of scaffolds, which allows facile modification of their electronic and steric environment. All these attractive features have been recently employed to develop catalysts that pair Al with a noble metal, including Pd,56 Ir,57 and Rh,58–62 (species 1–4, Scheme 1) thus enabling the catalytic functionalization of C–X bonds and small molecules.
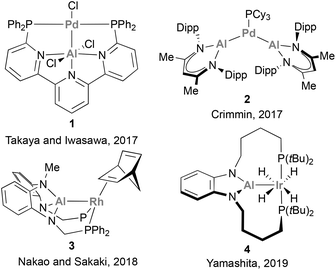 |
| | Scheme 1 Catalytically active aluminium–noble-metal species for the functionalisation of organic molecules. | |
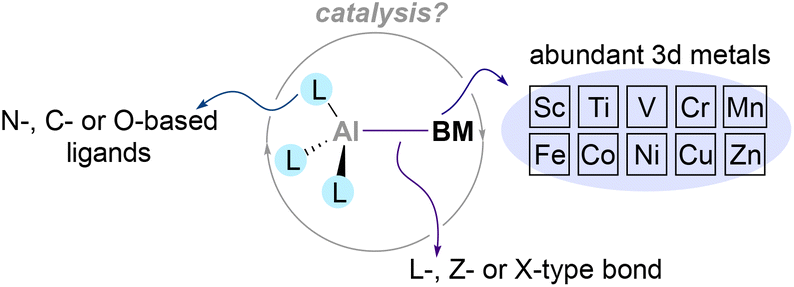
Despite these pioneering studies, heterobimetallic complexes in which Al is paired and directly bonded to a 3d metal remain underexplored, and their use in catalysis remains an uncharted territory. This is surprising considering the existing synthetic methodologies to functionalise organic molecules that leverage from the synergistic interaction of base metals salts with aluminium-based additives.63–65 Thus, in this perspective we will highlight the current state-of-the-art regarding the synthesis and reactivity of main group metalloligands coordinated to base metals,66–68 focusing on aluminium–base-metal species that contain an aluminium–base-metal bond (Scheme 2). By doing so, we believe this perspective will encourage chemists to gain further insight into the reactivity of compounds that pair aluminium with abundant 3d metals. The heterobimetallic complexes described in the following section present an exceptional opportunity to tame and exploit unique cooperative two-electron processes, endowing nobility to base metals and take a step towards a more sustainable future.
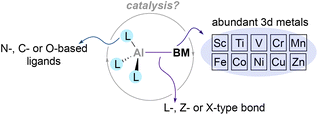 |
| | Scheme 2 Heterobimetallic complexes pairing aluminium with base metals and their potential benefits. | |
2. General synthesis of aluminium–base-metal complexes
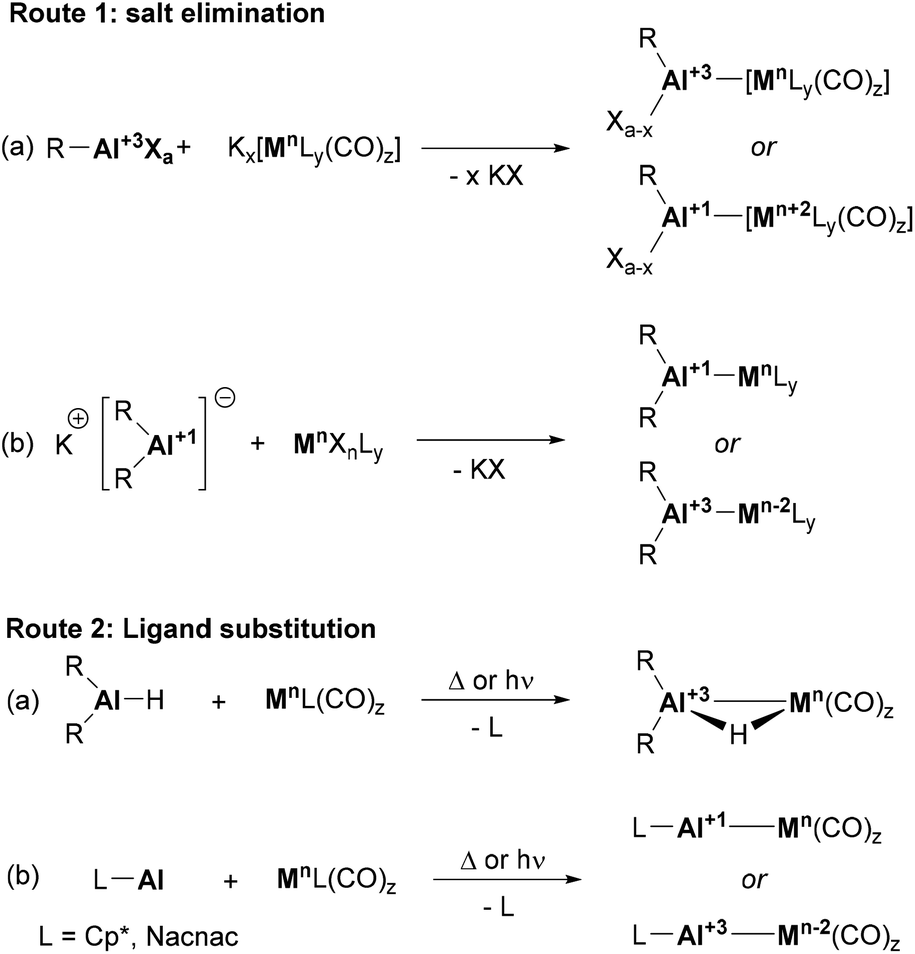
The synthesis of heterobimetallic Al–BM complexes can be achieved from a wide range of precursors via two main transformations: salt elimination and ligand substitution. The former method is usually limited to the use of metal carbonyl salts (e.g., Collman's reagent)69 together with Al(III) halide precursors (Scheme 3, route 1a). However, the recent development of stable Al(I) anions has boosted the applicability of this approach using aluminyl salts together with metal carbonyls, which broadened the scope of BM precursors that can be employed to achieve heterobimetallic Al-supported species (Scheme 3, route 1b). The second methodology, ligand substitution, is particularly effective when the synthesis of Al–BM complexes with bridging H atoms is targeted, and starts from Al(III) hydride precursors (Scheme 3, route 2a). Neutral Al(I) species such as Cp*Al and Nacnac Al(I) complexes are also good L-type ligands capable of undergoing ligand substitution reactions (Scheme 3, route 2b). It is worth mentioning that substitution usually occurs with metal-carbonyl and metal-olefin species under thermal or photochemical conditions.
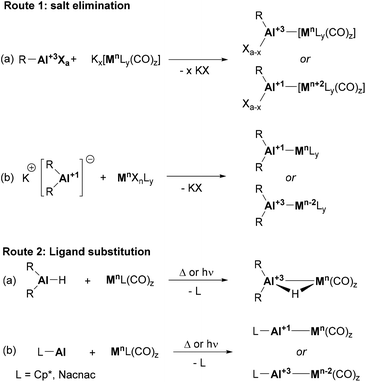 |
| | Scheme 3 Typical procedures for the construction of Al–BM bonds. | |
However, several aluminium–base-metal species have been also obtained following less explored approaches that, despite being rare and specific, are intriguing from a mechanistic point of view. These transformations would include a carboalumination reaction of a Cr–Cr quintuple bond,70 a Ga(I)-to-Al(I) formal exchange,71 insertion reactions of Al(I) species into TM–N bonds,72 and direct coordination of AlX3 species to TM complexes.73 As this perspective is focused on the reactivity of heterobimetallic complexes bearing an aluminium–base–metal bond, the synthesis of these compounds will be briefly addressed in the following section, focusing on complexes whose reactivity has been explored.
Noteworthy, the reported synthetic approaches to forge intermetallic bonds between Al and first-row transition metals (TM) have been also employed with heavier group neighbours. While 4d and 5d metal complexes supported by aluminium-based ligands can be generated through salt elimination with anionic Al(I) species or simple coordination, they also present exclusive routes that are not applicable to 3d metals. These unique pathways to achieve aluminium–TM bonds include oxidative addition to Al–Cl or Al–H bonds, alkane elimination, addition of trialkylaluminium species to vinylidene complexes, and simple coordination followed by reduction.74 It is worth noting that several formal oxidative addition examples have been reported for base metals and Al–Cl species; however, they are labelled as salt elimination (Scheme 3, route 1a, bottom) and limited to the use of supernucleophilic, low oxidation state metallic centres,69,75,76 in contrast to their heavier analogues.
3. Reactivity of aluminium–base-metal complexes
Heterobimetallic aluminium–base-metal complexes exhibit a wide range of reactivity patterns in small molecule activation and catalysis. This section will discuss such reactivity, as well as key structural features of some aluminium–base-metal species. This will be done following the first-row transition metals series and classifying reactivity patterns into specific transformations, including small molecule activation (e.g. N2, CO2, CO, azides, etc.), C–H activation and oxidation reactions. Examples are presented in chronological order, except in cases where subsequent studies complemented earlier ones. It is important to emphasise that oxidation states will be explicitly depicted for those species that have undergone extensive investigation, and authors have unambiguously assigned them in their respective studies.
3.1 Scandium
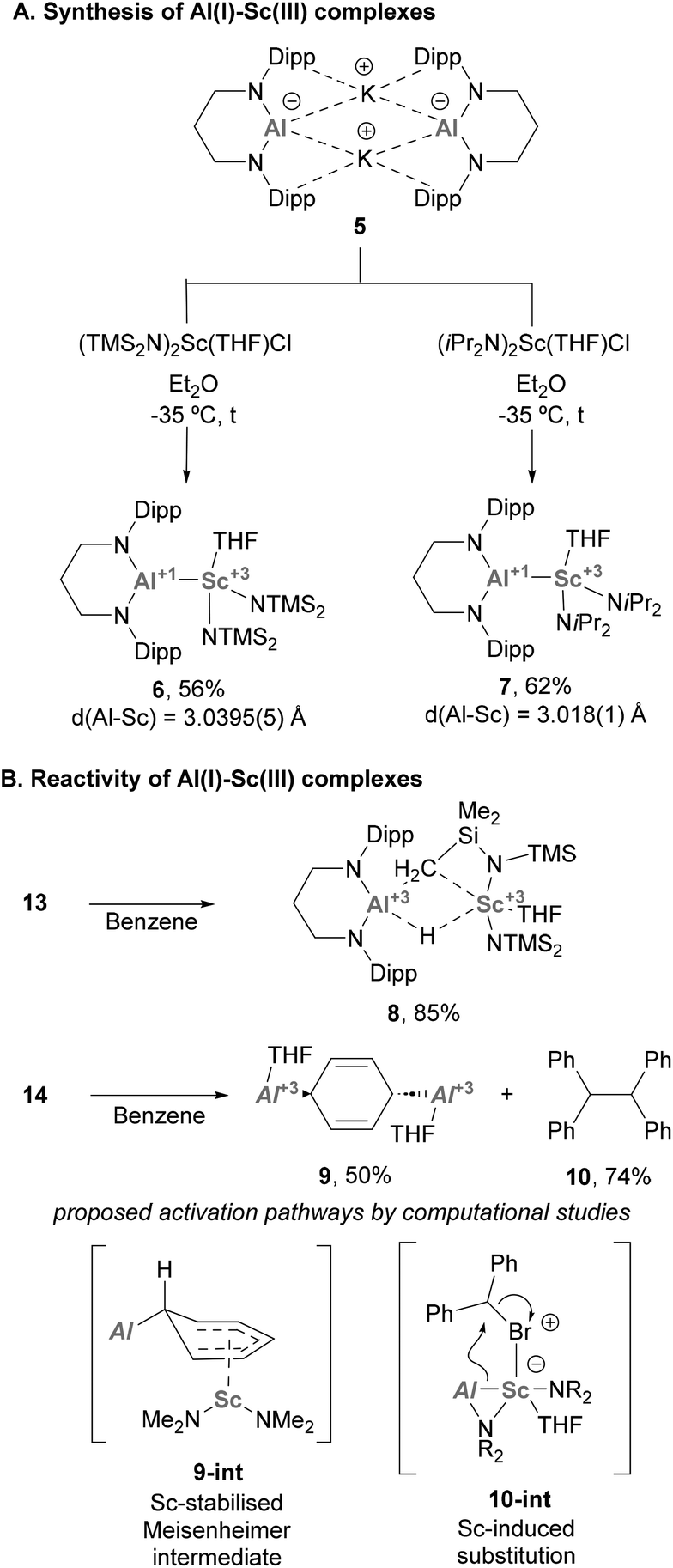
Heterobimetallic complexes bearing an Al–Sc bond are still rare and have received limited attention until a recent report from Yamashita and co-workers.77 In this work, the authors synthesised a novel Al(I) anion (5, Scheme 4A), which was used to furnish two unprecedented complexes featuring Al(I)–Sc(III) bonds via salt elimination. Indeed, reaction of 5 with selected bis-amido Sc(III) chloride species furnished complexes 6 and 7, which were isolated in good yields (Scheme 7A). Single Crystal X-Ray Diffraction (SC-XRD) analysis of such species showed an Al–Sc bond length of 3.0395(5) and 3.018(1) Å for 6 and 7, respectively, thus lying between the sum of Al and Sc covalent (2.74 Å) and van der Waals (3.95 Å) radii.78 This feature, which was previously observed in Al–Y complexes,74 strongly suggests a partial ionic character of the intermetallic bond. Additionally, the Al–N distances in 6 and 7 are essentially identical compared with aluminium anion 5 (Al–N = 1.849(3) and 1.854(4) Å), which supports the +1-oxidation state on the Al centre.
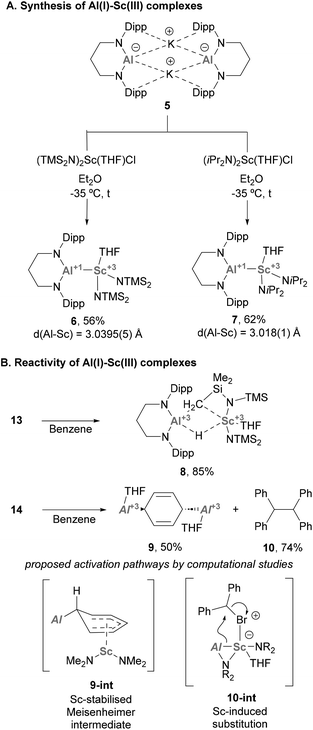 |
| | Scheme 4 Synthesis and reactivity of Al(I)–Sc(III) species. | |
Regarding the reactivity of such heterobimetallic complexes, 6 was found to be stable in the solid state. However, when dissolved in benzene, it slowly decomposed to complex 8 (Scheme 4B, top) via C–H bond activation of the methyl group of the btsa ligand. This transformation was proposed to proceed through oxidative addition at the Al(I) centre. Heterobimetallic Al–Sc systems were also tested towards SN2-type reactivity. Complex 7, which is stable in solution, was reacted with alkyl electrophiles (e.g. bromodiphenylmethane, Ph2CHBr), resulting in the formation of species 9, which exhibits a 1,4-dialuminiumcyclohexadiene motif. In addition, homocoupling product 10 was obtained in 74% yield (Scheme 4B, bottom). Interestingly, formation of species 9 does not occur in the absence of alkyl halide or when using toluene as solvent. Moreover, homocoupling product 10 was observed when reactions were run both in benzene and toluene, suggesting that they are not involved in its formation. Computational studies suggest complex 9 is formed via exchange of the coordinated THF in 7 for benzene, followed by a nucleophilic attack of the Al(I) lone pair involved in Al–Sc bonding. This would yield an Sc-stabilised Meisenheimer complex (9-int) as the key intermediate of this transformation. As for 10, computational studies indicate a synergistic activation of the C–Br bond through a 2e− process. First, the halide coordinates to the Sc centre, which triggers a nucleophilic attack of the Al(I) atom that cleaves the intermetallic bond to furnish a new Al–C bond (10-int). This new organometallic bond can act as nucleophile and attack a Ph2CHBr molecule, leading to the formation of 10.
As observed, the chemistry of Al–Sc heterobimetallic complexes is still in its early stages of exploration and requires further development towards the development of novel small molecule activation pathways. Nevertheless, the aforementioned findings reported by Yamashita suggest that combining Al with Sc holds promise for the development of efficient systems in cooperative C–H and C–X bond activation and catalysis.
3.2 Titanium
Since the discovery of the Ziegler–Natta process,79,80 Ti complexes have been broadly applied in polymer and synthetic organic chemistry.81 Due to the significance of such polymerization processes and the practicality of Tebbe's reagent,82 the combination of Ti systems with aluminium species has been an intense area of research.83–110 Although Ti and Al have been demonstrated to effectively work in synergistic processes, heterobimetallic systems featuring a Ti–Al bond remain largely underexplored,111 and their application to catalytic transformations is a feature yet to be discovered.
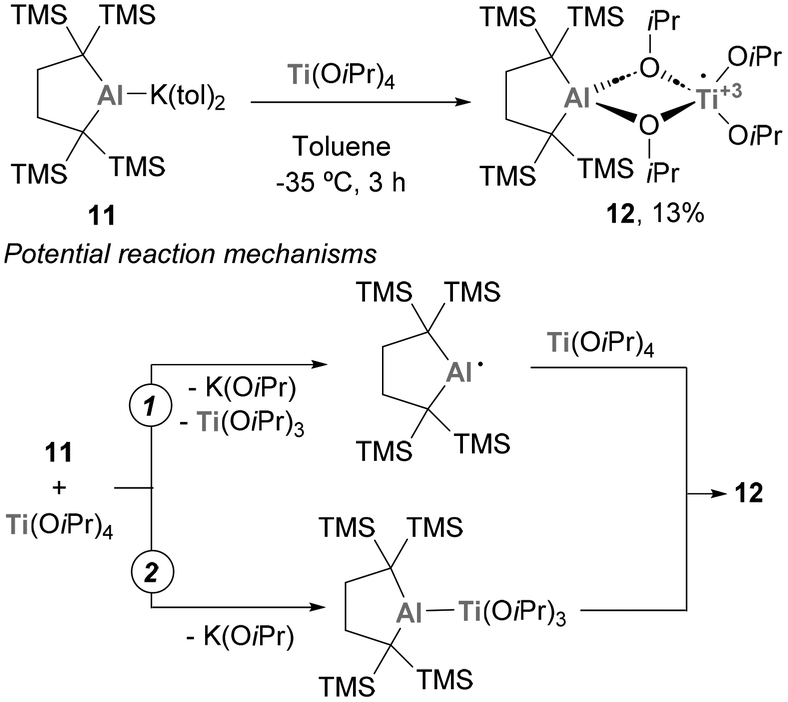
The chemistry of systems featuring a Ti–Al bond has also received attention from Yamashita and co-workers.112 In this study, an anionic Al(I) complex (11) was reacted with Ti(OiPr)4, leading to the formation of a system containing a Ti and an Al atom with no direct Ti–Al bond (Scheme 5). It is worth mentioning that, as a possible mechanism for the formation of 12, two different mechanistic routes were proposed. Pathway 1 involves the formation of an open-shell Al(II) centre, with subsequent release of Ti(OiPr)3, while pathway 2 entails the intermediacy of a heterobimetallic species containing a Ti–Al bond. In both cases, the reaction results in a concomitant redox step that leads to the formation of species 12. Unfortunately, the reactivity of such species was not studied in further detail.
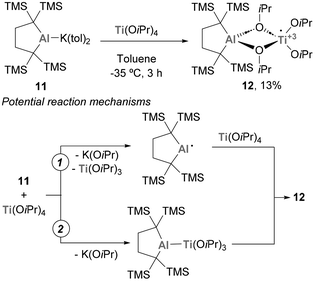 |
| | Scheme 5 Reaction between a low valent Al(I) and Ti(OiPr)4. | |
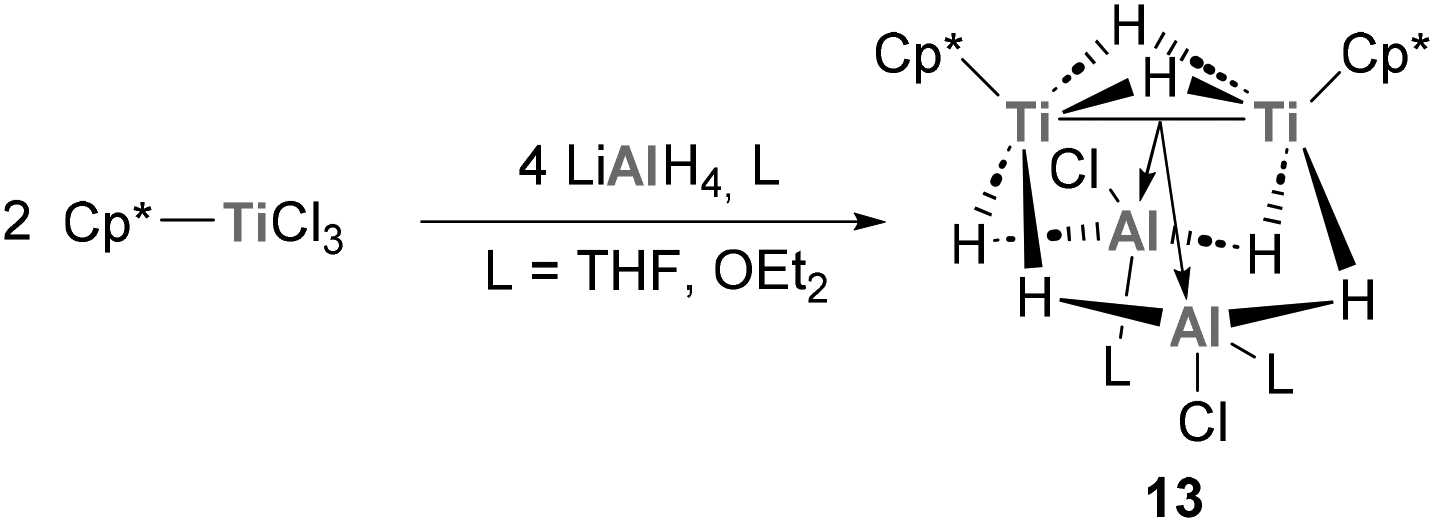
Jover and Yélamos have recently reported the synthesis of low-oxidation state Ti species with an intriguing Ti–Al bonding situation.113 Reaction of Cp*TiCl3 with LiAlH4 in the presence of donor solvents such as THF or diethyl ether results in the formation of [{TiCp*(μ-H)}2{(μ-H)2AlCl(L)}2] (13, Scheme 6). Interestingly, this family of mixed Ti–Al heterobimetallic clusters presents average titanium–aluminium distances of 2.644(10) Å, which are smaller than the sum of covalent radii (2.81 Å) and shorter than those reported for heterometallic complexes with Ti(III)–H–Al units. Indeed, this short distance is explained by two strong interactions between the Ti–Ti bond and the empty s orbitals of Al, leading to unprecedented Ti–Ti → Al interactions in which the Lewis base donor is the pair of electrons involved in the Ti–Ti bond. The reactivity of such bonds is yet to be investigated, but holds the promise of unique transformations exploiting the rich redox ability of heterobimetallic Ti–Al species.
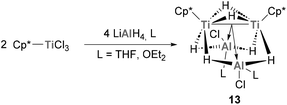 |
| | Scheme 6 Synthesis of heterobimetallic Ti–Al clusters featuring Ti–Ti → Al interactions. | |
3.3 Vanadium
Similarly to its 3d neighbour from group 4, V salts have found broad application in polymer chemistry when combined with Al(III) organometallic additives.114–118 However, there is a lack of examples of heterobimetallic complexes featuring a V–Al bond, and the study of the reactivity at the interface of such species is yet to be meticulously explored.
3.4 Chromium
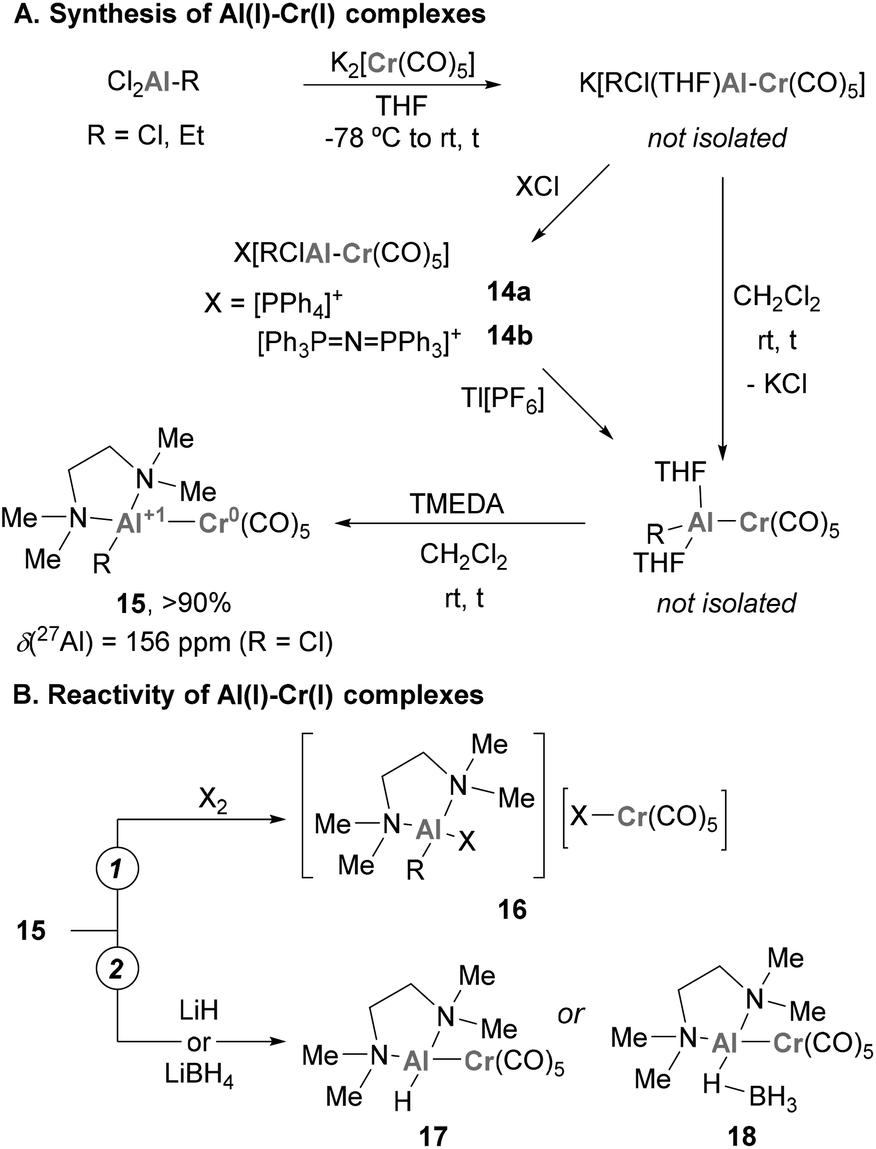
Pioneering examples of heterobimetallic complexes containing Cr–Al bonds were reported in 1996 by Fischer and co-workers.119–121 As shown in Scheme 7A, reacting common Al(III) halide precursors with K2[Cr(CO)5] in THF, the authors synthesised a collection of anionic species K[RCl(THF)Al–Cr(CO)5] through salt elimination. These compounds can be isolated as solvent-free X[RClAl–Cr(CO)5] species (14a–b) through cation exchange (X = PPh4 or Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N+PPh3). When DCM is added to these species together with a bidentate amine (TMEDA), a second elimination of KCl takes place, yielding [(TMEDA)RAl–Cr(CO)5] species 15. Unfortunately, the authors were not able to analyse the structure of such species by SC-XRD, but their main features were assigned by comparison with its heavier Ga(I) analogue (R = Me). Spectroscopically, a significant shift from 210 to 156 ppm was observed by 27Al-NMR when 15 was compared with Al(III) species, which indicates the presence of an Al(I) centre in 15. The reactivity of heterobimetallic Cr–Al complex 15 was scrutinised towards the oxidative addition of halogens X2, resulting in the oxidative cleavage of the Cr–Al bond (16, Scheme 7B, pathway 1). Additionally, ligand exchange with hydride sources such as LiH and LiBH4 produced hydride and borohydride analogues of complex 17 (Scheme 7B, pathway 2).
N+PPh3). When DCM is added to these species together with a bidentate amine (TMEDA), a second elimination of KCl takes place, yielding [(TMEDA)RAl–Cr(CO)5] species 15. Unfortunately, the authors were not able to analyse the structure of such species by SC-XRD, but their main features were assigned by comparison with its heavier Ga(I) analogue (R = Me). Spectroscopically, a significant shift from 210 to 156 ppm was observed by 27Al-NMR when 15 was compared with Al(III) species, which indicates the presence of an Al(I) centre in 15. The reactivity of heterobimetallic Cr–Al complex 15 was scrutinised towards the oxidative addition of halogens X2, resulting in the oxidative cleavage of the Cr–Al bond (16, Scheme 7B, pathway 1). Additionally, ligand exchange with hydride sources such as LiH and LiBH4 produced hydride and borohydride analogues of complex 17 (Scheme 7B, pathway 2).
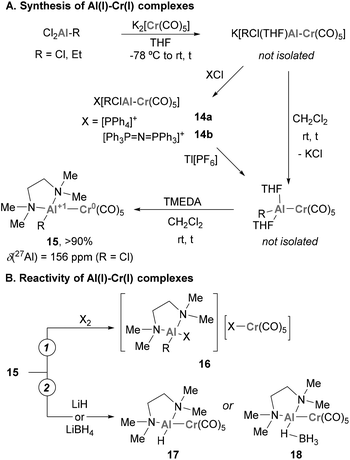 |
| | Scheme 7 First examples of Al(I)–Cr(0) complexes synthesised by salt metathesis. | |
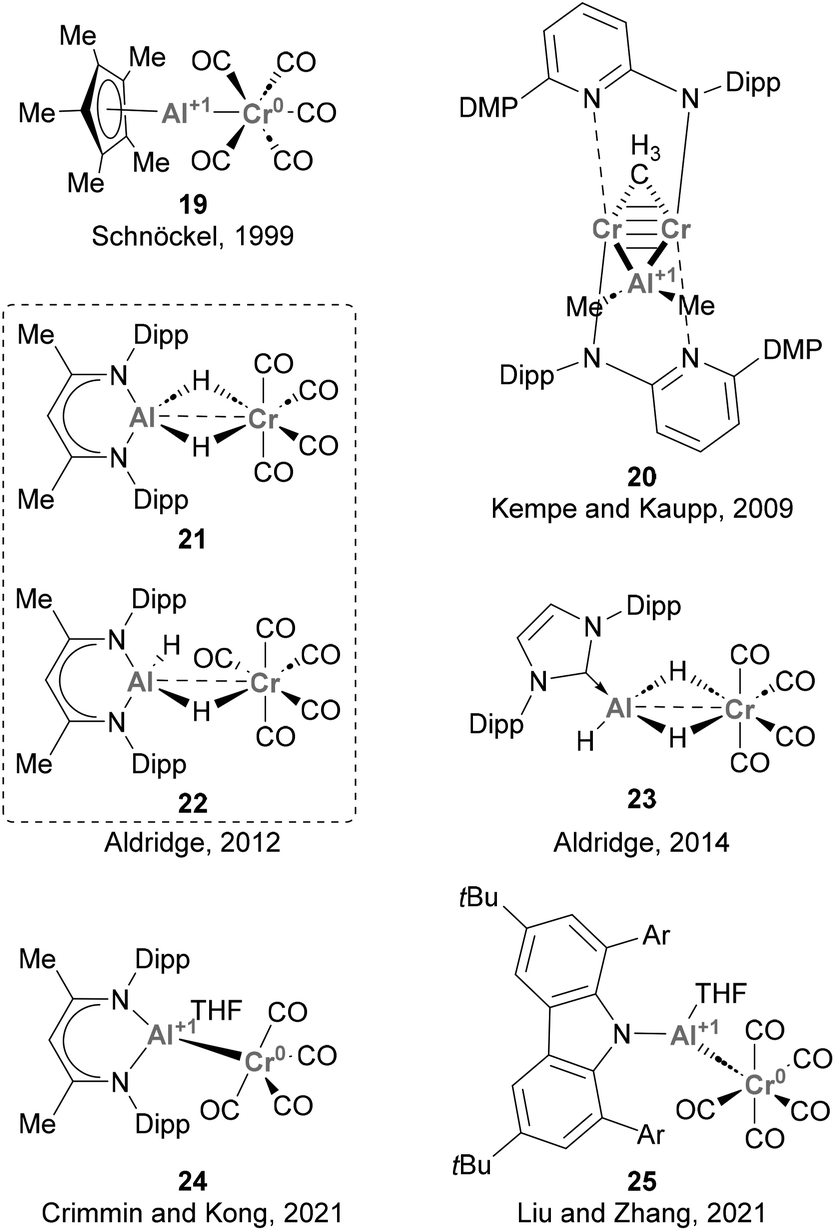
Since then, various synthetic approaches for accessing heterobimetallic species featuring Cr–Al bonds have been reported (Scheme 8). Studies from Schnöckel (19),122 Aldridge (21–23),123,124 Crimmin and Kong (24),125 and Liu and Zhang (25)126 achieved such heterobimetallic species by ligand substitution using low oxidation state Cr carbonyl species. In contrast, Kempe and Kaupp synthesised species 20via carboalumination.70 These works provided key insights into the synthesis and electronic structure of species featuring Cr–Al bonds, as well as the nature of such intermetallic bond. These studies have also provided information about the formal oxidation states of Cr and Al, which was unambiguously assigned to be Cr(0) and Al(I) in species 20, 19, 24 and 25. Noteworthy, and as far as we know, the ability of species depicted in Scheme 8 towards small molecule activation and catalysis has not been explored so far. Hence, the chemical characteristics and reactivity of the Cr–Al bond are yet to be thoroughly investigated. This aspect is particularly intriguing considering the remarkable capacity of low oxidation state Cr species to activate a diverse range of chemical bonds,127,128 which ensures a rich redox reactivity for species featuring Cr–Al bonds and offers the opportunity for future investigations in this field.
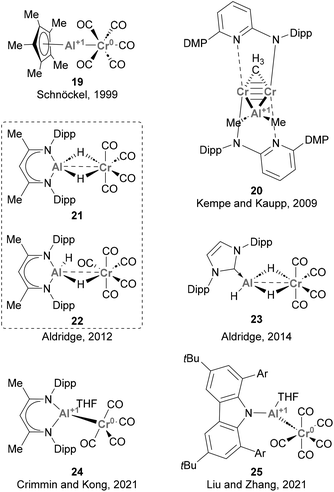 |
| | Scheme 8 Cr–Al complexes structurally characterised. | |
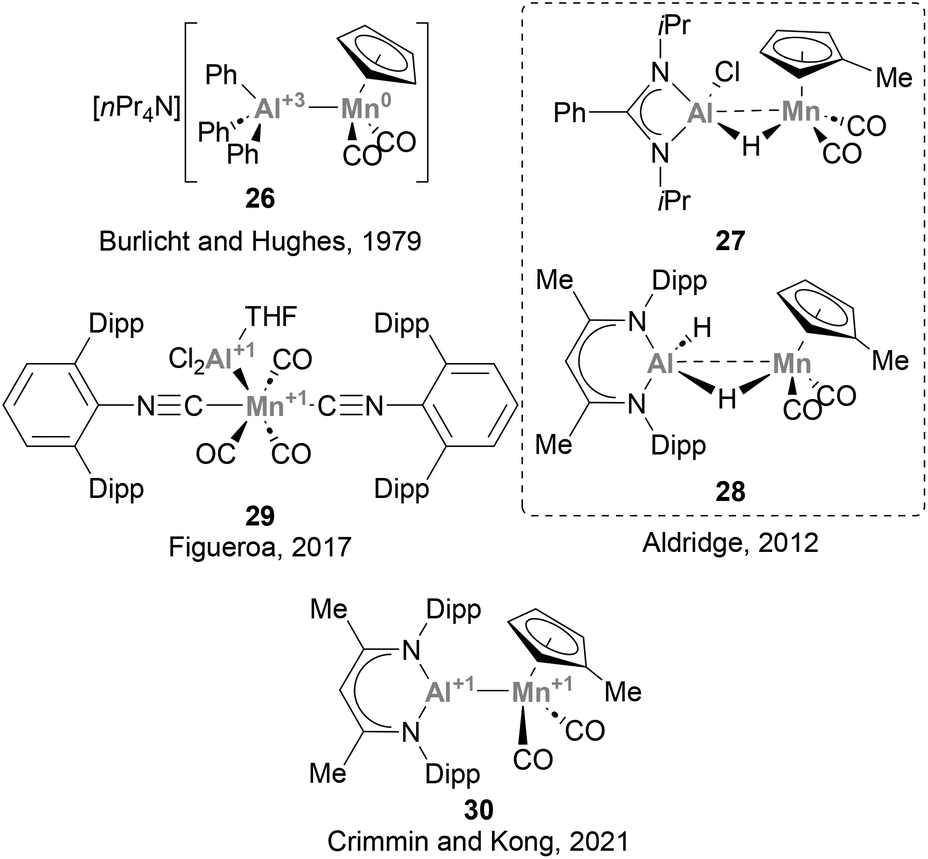
3.5 Manganese
The lightest element of group 7, Mn, has been largely underexplored in heterobimetallic complexes bearing a direct Mn–Al bond, and only a handful of examples have been described so far.73,123,125,129,130 Burlitch, Hughes and co-workers pioneered the field, reporting the first complex featuring a direct Mn–Al bond (26, Scheme 9) in 1979.73 In this species, AlPh3 acts as a Z-type ligand, accepting charge from the anionic [CpMn(CO)2]− fragment. Complexes 27 and 29 were synthesised following salt elimination routes with Mn(I) and Mn(-I) precursors, respectively, while 28 and 30 were accessed via decarbonylative ligand substitution. While the intermetallic bond in 27 and 28 was not discussed in detail, species 29 and 30 were unambiguously assigned as low oxidation state Al(I)–Mn(I) heterobimetallic systems by means of computational and experimental studies. Unfortunately, and similarly to its group 6 neighbour, the reactivity of Mn–Al species towards the activation of small molecules and strong bonds has not received attention from the chemical community. Given the abundance of Mn, its potential to catalyse a wide range of organic reactions, including C–H activation and oxidation,131–133 as well as its ability to engage in visible-light-induced transformations,134 the use of heterobimetallic Mn–Al species in small molecule activation and catalysis represents a promising opportunity towards more sustainable organic synthesis.
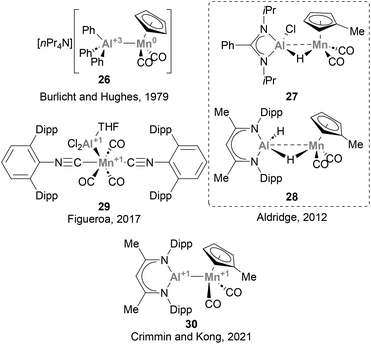 |
| | Scheme 9 Mn–Al species structurally characterised. | |
3.6 Iron
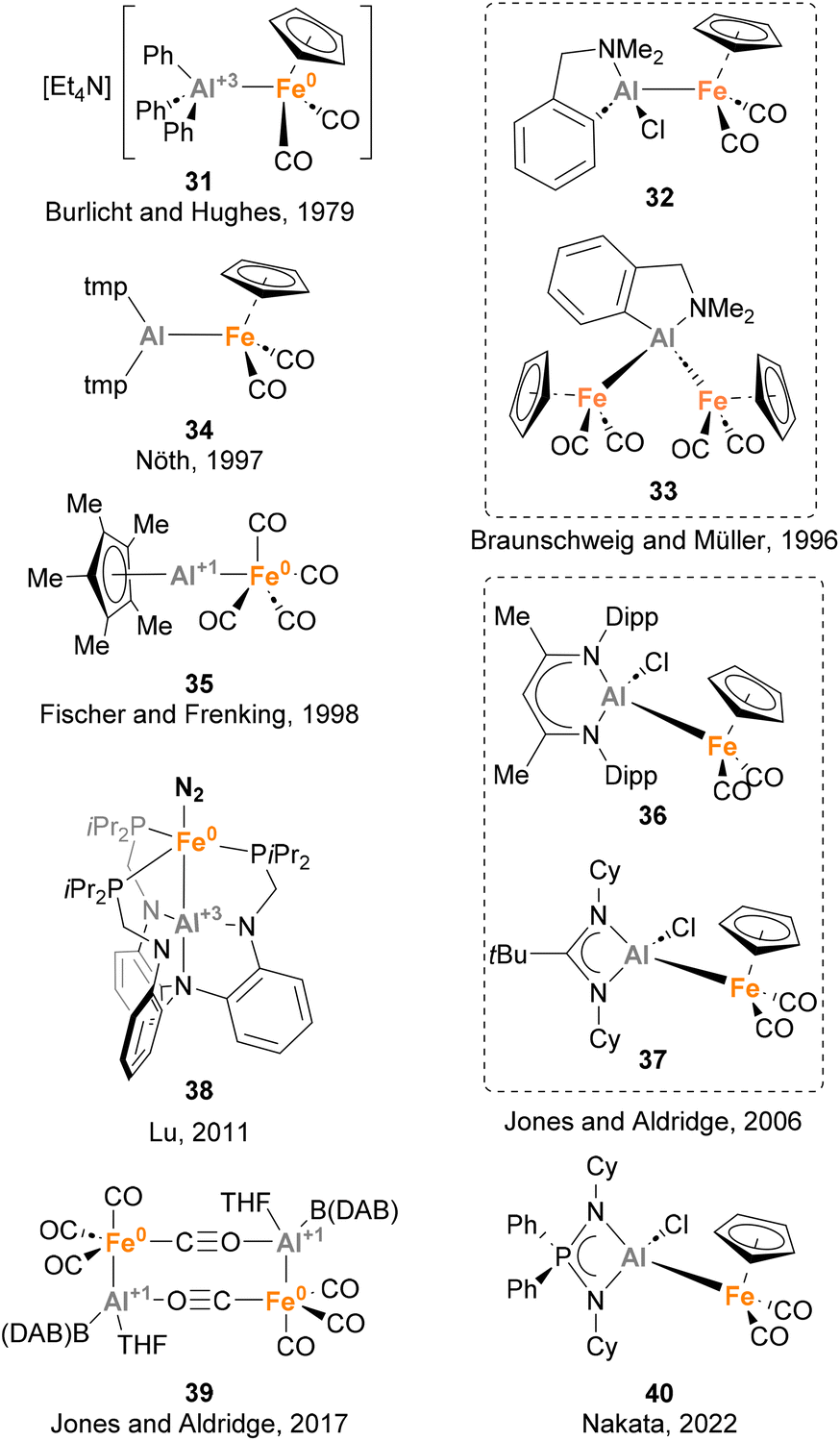
Iron is the 3d metal that has been more studied in heterobimetallic complexes that feature an Fe–Al bond, thus forming metal complexes containing the most abundant metals in the Earth's crust. Nowadays, a plethora of heterobimetallic Fe–Al complexes have been described (Scheme 10).73,129,135–151 These complexes attracted the interest of the inorganic community due to their particular bonding situation, as well as their reactivity in cooperative small molecule activation. To the best of our knowledge, the first heterobimetallic complex bearing an Fe–Al (31) bond was reported in 1979 by Burlicht, Hughes and co-workers.73 Similarly to Mn species 26, this Fe–Al species features a Z-type coordination of AlPh3, which acts as acceptor. The most exploited route to synthesise such complexes is the salt metathesis approach, which was used by Braunschweig and Müller (32 and 33),136 Nöth (34),138 Jones and Aldridge (36, 37 and 39),140,145 and Nakata (40),148 while ligand substitution has been reported by Fischer and Frenking137 and Lu142 for the synthesis of 35 and 38, respectively. While the reactivity of complexes depicted in Scheme 10 was not investigated, the potential of Fe–Al bonds in small molecule activation and catalysis has been studied in detail using other heterobimetallic complexes, including serendipitous transformations. These reactions, which are detailed in the section below, include C–H bond cleavage and small molecule activation processes, as well as catalytic reduction of N2 to NH3.
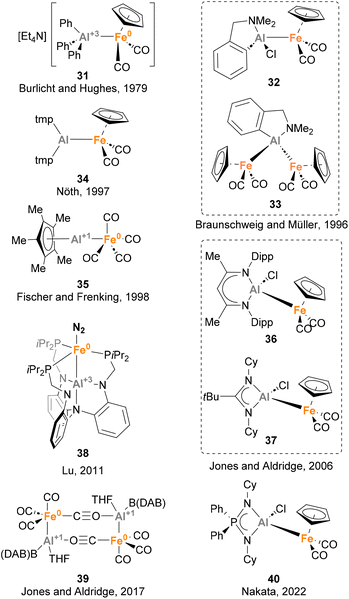 |
| | Scheme 10 Structurally characterised Fe–Al complexes. | |
3.6.1 C–H bond activation.
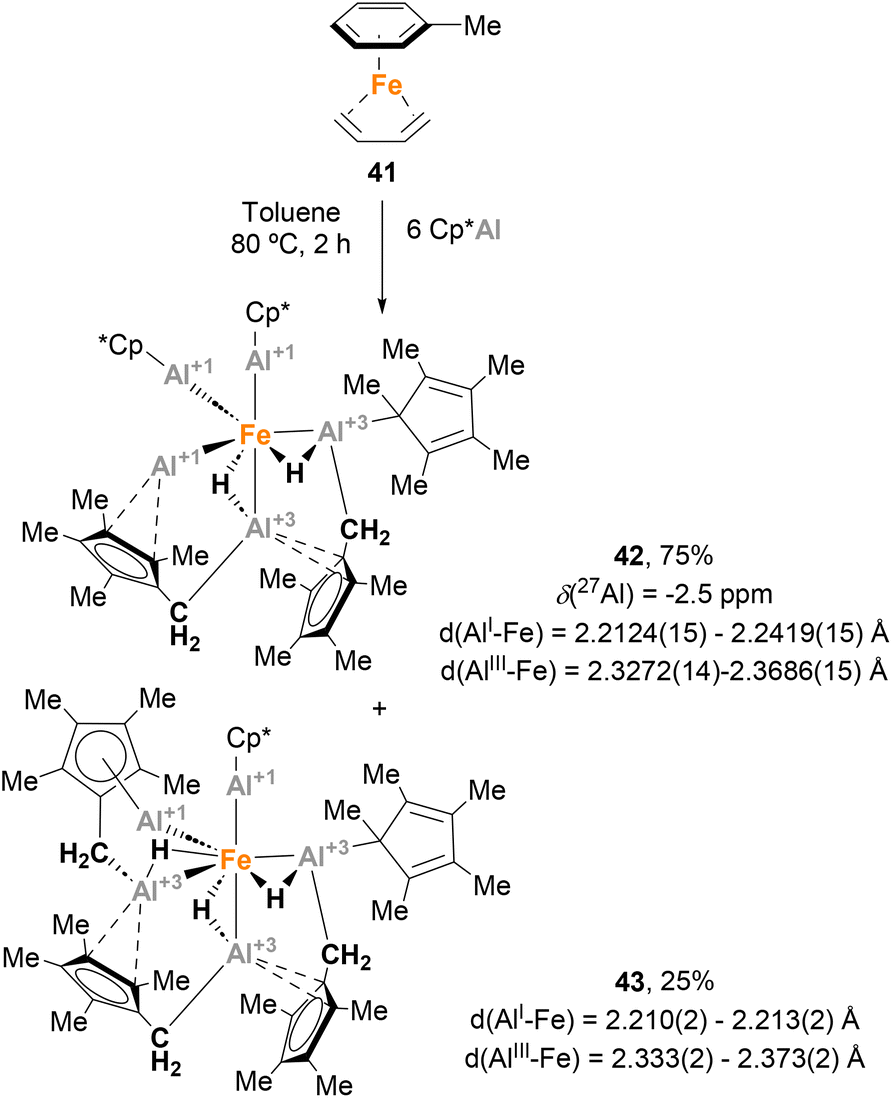
Trying to test the limits of Fe(0) complexes in terms of number of Cp*Al coordinated to the metal, Fischer and co-workers focused their efforts on the synthesis of [Fe(AlCp*)5] complexes, which were predicted to be stable by DFT calculations.37,152 With this goal in mind, [Fe(η6-toluene)(η4-1,3-butadiene)] 41 was reacted with a large excess of Cp*Al (Scheme 11). However, instead of [Fe(AlCp*)5], the reaction resulted in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of two isomers, 42 and 43, respectively.139 SC-XRD analysis of 42 showed Fe–Al distances in agreement with Fe(0)–Al(I) oxidation states for the AlCp* moieties (2.2124(15)–2.2419(15) Å)120 and Fe(0)–Al(III) oxidation states for the [HAl(CH2C5Me4)] fragments (2.3272(14)–2.3686(15) Å). SC-XRD analysis of 43 exhibited similar structural parameters. The formation of these structures can be explained through ligand substitution to generate [Fe(AlCp*)n] species, followed by a C–H bond activation step involving methyl groups of the Cp* ligands. As far as we know, this study involving the serendipitous Csp3–H activation of Cp* ligands represents the first example of the ability of heterobimetallic Fe–Al species to perform C–H bond activation.
:1 mixture of two isomers, 42 and 43, respectively.139 SC-XRD analysis of 42 showed Fe–Al distances in agreement with Fe(0)–Al(I) oxidation states for the AlCp* moieties (2.2124(15)–2.2419(15) Å)120 and Fe(0)–Al(III) oxidation states for the [HAl(CH2C5Me4)] fragments (2.3272(14)–2.3686(15) Å). SC-XRD analysis of 43 exhibited similar structural parameters. The formation of these structures can be explained through ligand substitution to generate [Fe(AlCp*)n] species, followed by a C–H bond activation step involving methyl groups of the Cp* ligands. As far as we know, this study involving the serendipitous Csp3–H activation of Cp* ligands represents the first example of the ability of heterobimetallic Fe–Al species to perform C–H bond activation.
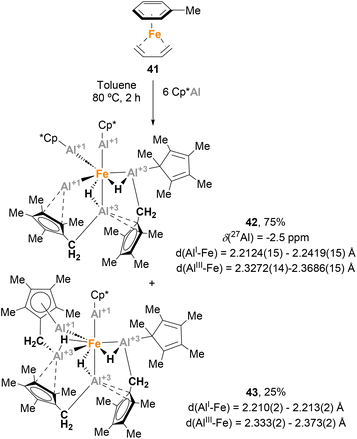 |
| | Scheme 11 First example of C–H bond activation mediated by an Fe–Al complex. | |
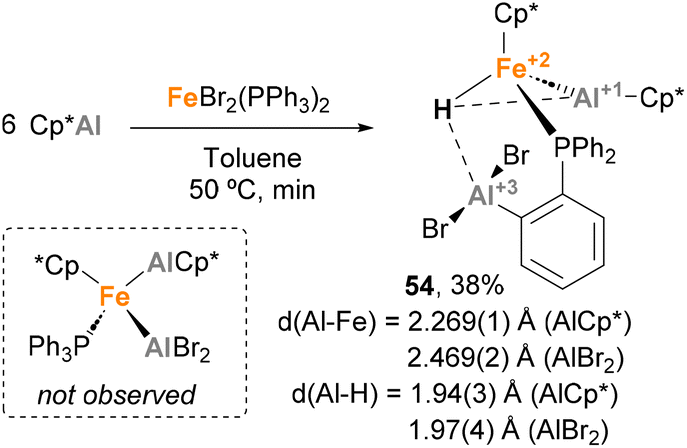
Fischer and co-workers observed a similar transformation while studying the insertion of Cp*Al into Fe–Br bonds.141 While reacting ECp* (E = Ga, In) with [FeBr2(PPh3)2] (X = Cl, Br) resulted in a Cp* transfer process, when [FeBr2(PPh3)2] was treated with Cp*Al, complex 44 was obtained in 38% yield (Scheme 12). In this case, SC-XRD analysis also shows different Fe–Al bond lengths of 2.269(1) Å (Fe–AlCp*) and 2.469(2) Å (Fe–AlBr2–Ph), in agreement with the previously reported values for Fe(0)–Al(I) and Fe(0)–Al(III) bonds, respectively.73,120,139 In this example, formation of 44 can be rationalised by a Csp2–H bond activation of the phenyl residue from the phosphine ligand after formation of putative [FeCp*(AlCp*)(AlBr2)(PPh3)] species, which were not observed. This unique reactivity of Fe–Al bonds towards C–H activation, which was not observed when using Ga/In Cp* analogues, can be attributed to the higher acidity of Al, which leads to more reactive ambiphilic systems.
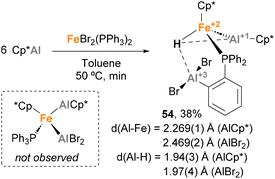 |
| | Scheme 12 C–H bond insertion in the synthesis of a Fe–Al complex reported by Fischer. | |
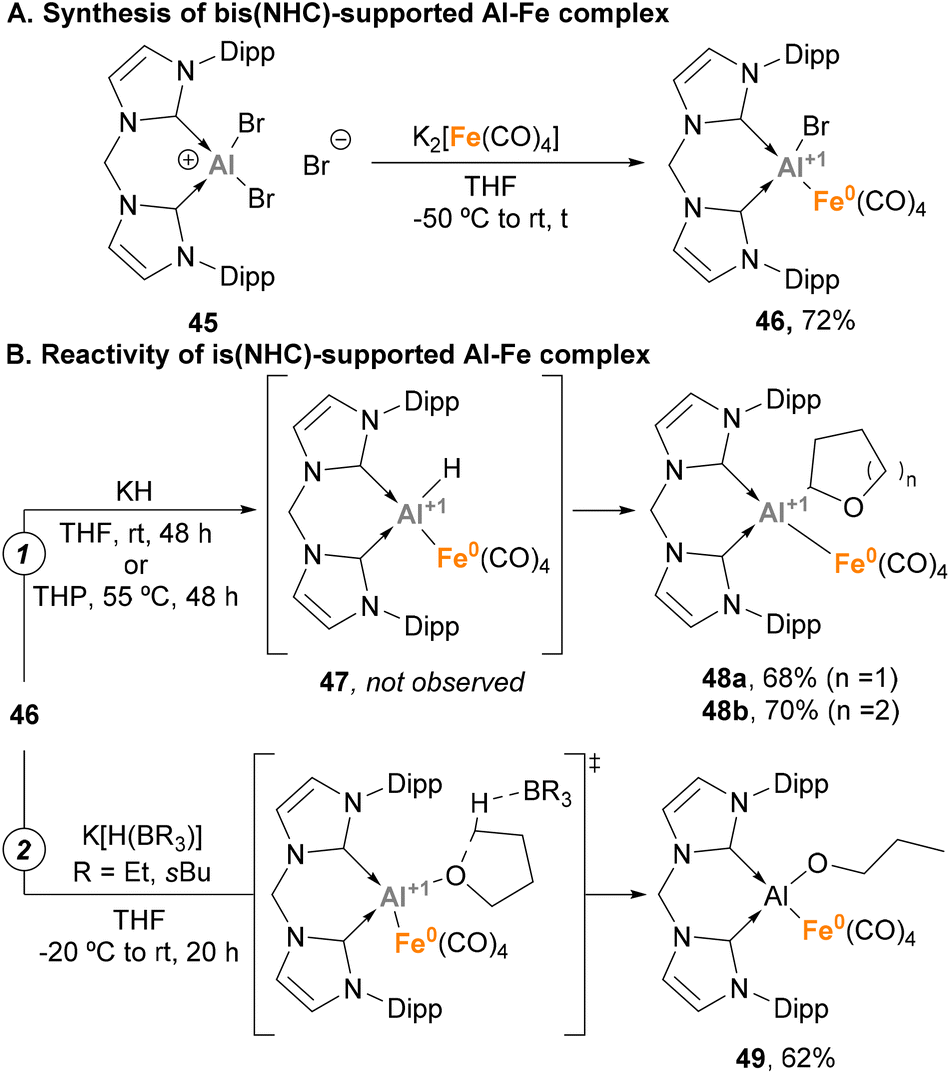
Taking advantage of the steric protection that bis-NHC ligands provide, Driess and co-workers focused on the synthesis of low oxidation state Al(I)–H species embedded in a Fe–Al unit.143 To this end, a salt elimination-reduction strategy was employed starting from CNCDippAlBr3 (45) and Collman's reagent analogue K2[Fe(CO)4], resulting in a heterobimetallic Fe(0)–Al(I) complex 46 (Scheme 13A). With the goal of achieving Al–H species, different hydride sources were employed (Scheme 13B). Surprisingly, when KH was added to 46 in tetrahydrofuran (THF) or tetrahydropyran (THP), Al(I)–H species 47 were not detected (Scheme 13B, pathway 1). However, complexes 48a–b were formed in high yields. This transformation is a good example of α-metalation of O-heterocycles with concomitant H2 extrusion, as evidenced by spectroscopic and computational studies. Usually, α-metalation processes with organolithium or organopotassium reagents lead to the cleavage and polymerisation of the heterocycle,153,154 which makes this reaction particularly interesting. On the contrary, when K[H(BR3)] species were employed as hydride sources a ring-opening reaction of THF was observed, yielding complex 55 (Scheme 13B, pathway 2). This transformation, which represents a pioneering example of C–O cleavage with heterobimetallic Fe–Al species, is proposed to start by THF coordination to the Al(I) LUMO, followed by hydride-mediated ring-opening of THF.
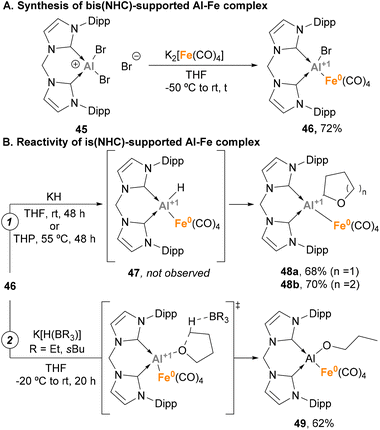 |
| | Scheme 13 C–H metalation and C–O bond cleavage mediated by Fe–Al species. | |
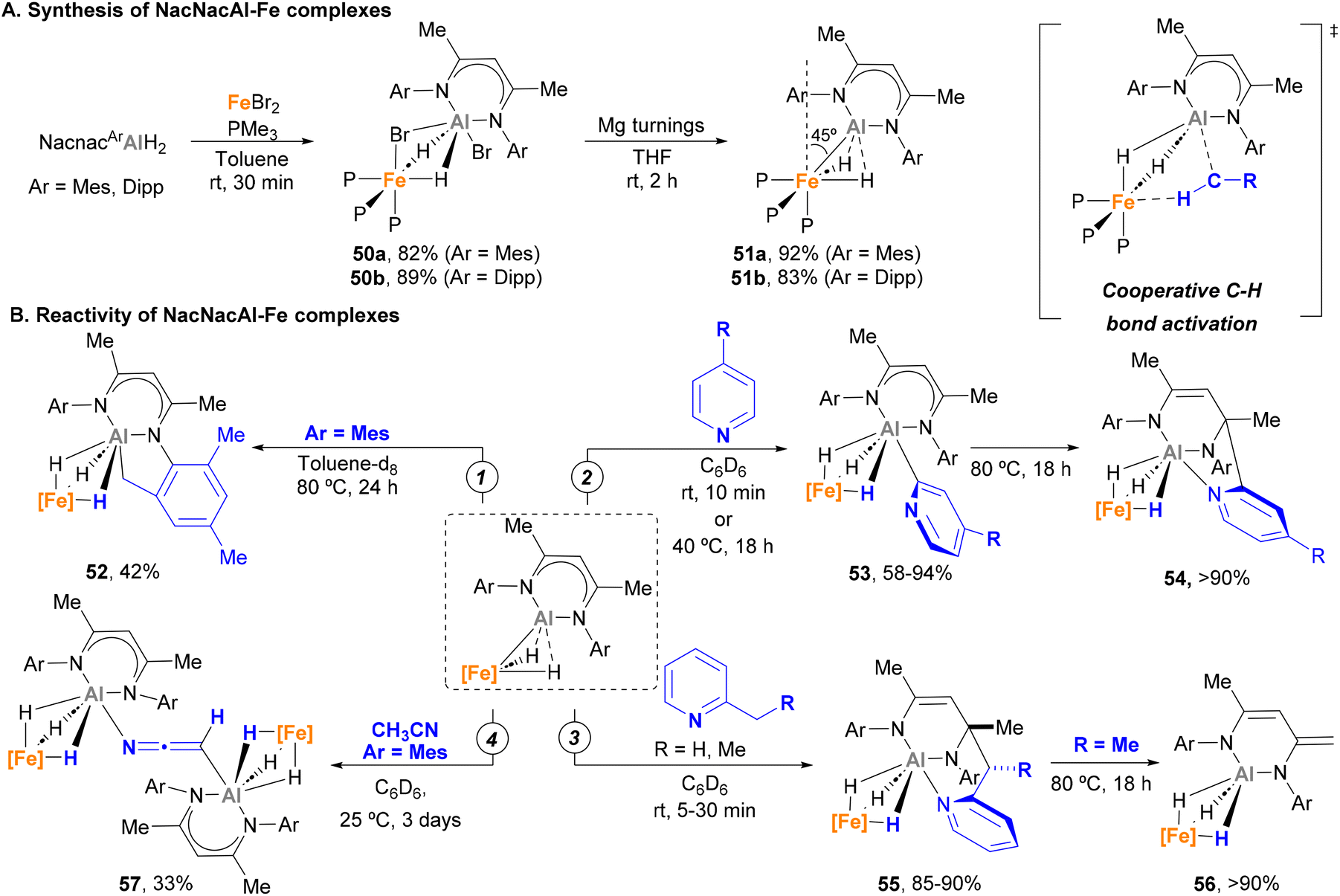
Over the past year, Crimmin and co-workers made important contributions to the development of heterobimetallic Fe–Al species and their application in C–H activation. In 2022, they successfully synthesised and studied the reactivity of novel low-spin κ2-hydride Fe(II)–Al(I) complexes.150 These species are readily accessed in excellent yields via a two-step procedure (Scheme 14A) that involves a first ligand exchange with FeBr2 with NacnacAlH2 and phosphine ligands (50a–b) followed by reduction with Mg turnings (51a–b). SC-XRD analysis unveiled Fe–Al bond distances close to the sum of their covalent radii for 50a–b (2.453(1)–2.459(1) Å vs. a covalent radii of 2.43 Å),78 prompting the authors to define these systems as σ-alane iron(II) complexes. When reduced with Mg, Fe–Al bond significantly shortened in species 51a–b (2.2176(13)–2.194(1) Å), which indicates a reduction of the Al centre to a +1 oxidation state. Computational studies of species 51a–b show these entities reside in an intermediate state between an aluminylene–Fe(II) complex and a highly polarized system comprising a cationic Al unit and an anionic Fe dihydride fragment. Additionally, SC-XRD analysis revealed that the Fe centre possesses a distorted octahedral geometry, in which the aluminyl ligand bends away from the axial position by 45°. This results in an unusual ground state destabilisation that strongly impacts their chemical behaviour.
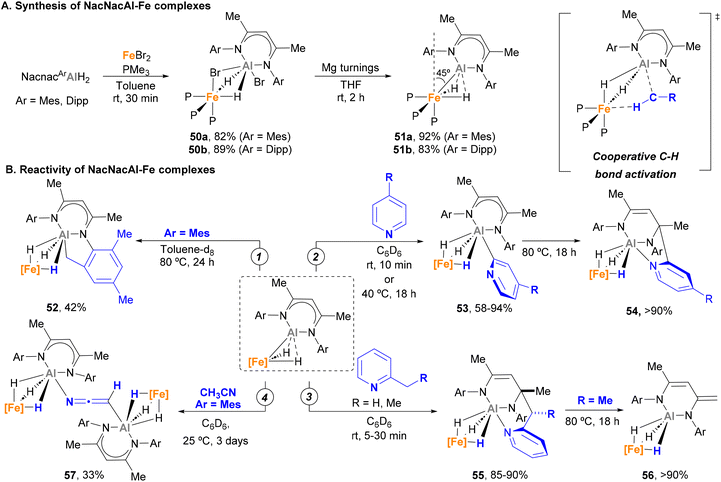 |
| | Scheme 14 C–H bond activation mediated by a low-spin d6 Al–Fe complex. | |
In contrast to traditional, fully saturated d6 low spin Fe complexes, which tend to be stable and unreactive, it was found that complexes 51a–b were capable of performing C–H bond activation. Indeed, when 51a is heated in toluene solution, cyclometalated product 52 is formed via Csp3–H activation (Scheme 14B, pathway 1). More remarkably, complexes 51a–b were able to selectively activate the C2–H bonds of pyridines with a variety of para-substituents, leading to κ3-trihydride Fe–Al species 53 (Scheme 14B, pathway 2). Computational studies indicate that the key step for such C–H bond activation pathways involves a cooperative reductive deprotonation reaction in which the Fe–Al bond behaves as a frustrated Lewis-pair (FLP), where the HOMO (Fe–Al σ-bonding orbital) acts as donor and the LUMO (p-orbital on Al) as acceptor.
In a subsequent study, Crimmin and co-workers further tested the reactivity of species 51 towards meta- and ortho-substituted pyridines.155 This investigation unveiled that meta substituents hamper C2–H bond activation, leading to C6-aluminated heterocycles. Interestingly, these complexes are not stable upon heating, which leads to a nucleophilic attack of the organometallic Al–Cpyridine bond to the Nacnac ligand and the formation of a tricoordinated Al fragment (54). On the other hand, when 2-alkyl pyridines were used (Scheme 14B, pathway 3), Csp3–H activation occurs under mild conditions, leading to species 55 after nucleophilic attack on the ligand. The thermal stability of complex 55 was also investigated. Interestingly, these studies revealed a series of rearrangements (when R = H) and deprotonations (when R = Me) involving the Nacnac ligand, with concomitant extrusion of the initial 2-alkyl pyridine and formation of the corresponding κ3-hydride Fe–Al complex 56. This mechanism was supported by computational studies, showing a feasible energy activation barrier of 25.3 kcal mol−1 for the process depicted in pathway 3. More recently, Gorgas and Crimmin reported a double Csp3–H activation of CH3CN using 57 (Scheme 14B, pathway 4).156 SC-XRD analysis exhibited a μ2-κC-κN-[CHCN]2− bis-anion as a bridging unit through the Al atoms (57). Computational studies indicate that the monodeprotonated acetonitrile complex acts as intermediate, while the distorted octahedral geometry of the Fe species contributes towards making possible this unique reactivity.
3.6.2 Small molecule activation.
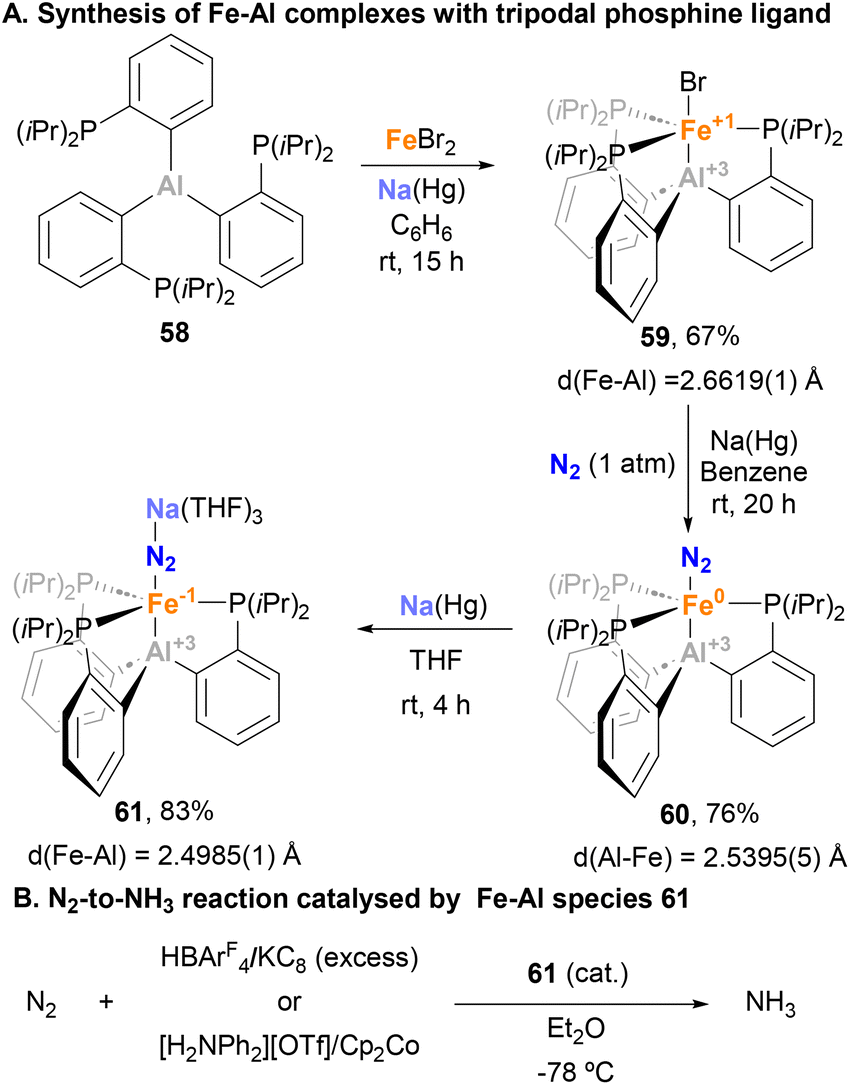
Given the ability of tripodal, tetradentate Fe–B species to catalyse N2-to-NH3 reduction,157 Peters and Fajardo decided to investigate the impact of heavier Al congeners in N2 activation reactions.147 Thus, starting from FeBr2 and ligand 58 scaffold in the presence of Na(Hg) amalgam, complex 59 was achieved in 67% yield (Scheme 15A). Remarkably, the Fe–Br remained intact in 59, in contrast to metal–aluminium complexes in which halogen abstraction by the Al centre takes place.158 In 59, SC-XRD analysis unveiled an Fe–Al bond length of 2.6619(1) Å, slightly longer than the sum of covalent radii. The reduction of 59 using Na(Hg) under N2 atmosphere led to the formation of complex 60, which shows an Fe–Al bond length 2.5393(5) Å, suggesting that 59 induces a closer proximity between the two metal atoms compared with AltraPhos (depicted in Scheme 10, complex 38).142 The Fe–Al bonding in 59 and 60 was described as a dative Fe → Al(III) interaction, where aluminium acts as a Z-type ligand that stabilises the σ-(Fe–Br/N2) orbital of Fe dz2 parentage. For this reason, further reduction of 60 with Na(Hg) leads to a well-defined Fe(-I)–Al(III) species 61, which shows an even shorter Fe–Al bond length (2.485(1) Å). This clearly suggests a stronger stabilising donor–acceptor interaction with the Al centre, as aforementioned. After characterisation of complex 61, its ability to catalytically activate N2 was studied (Scheme 15B). Cyclic voltammetry and FT-IR experiments were conducted to identify the extent of N2 activation in species 61 and its corresponding analogues containing group 13 elements B and Ga. The results revealed a comparable degree of N2 activation among these species, suggesting that the presence of the group 13 element bonded to the Fe centre has minimal impact. However, the efficiency of 61 towards N2 reduction was found to be lower compared with the B analogue with two different sets of proton source and reductant combinations. The authors attributed this lower activity to a higher preference of species 61 for competing Hydrogen Evolution Reaction (HER), particularly when the reductant employed was milder (Cp2Co vs. KC8), preventing the regeneration of the active species. It is noteworthy to mention that, to the best of our knowledge, this process represents the sole reported reaction wherein an Fe–Al system functions as a catalyst.
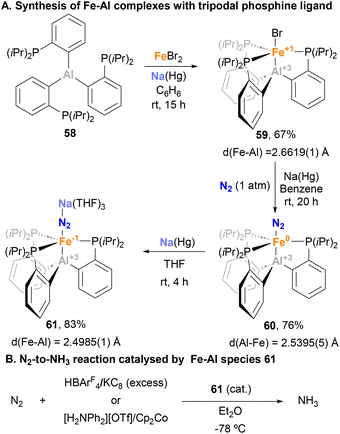 |
| | Scheme 15 Al–Fe complexes employed in catalytic N2-to-NH3 reduction. | |
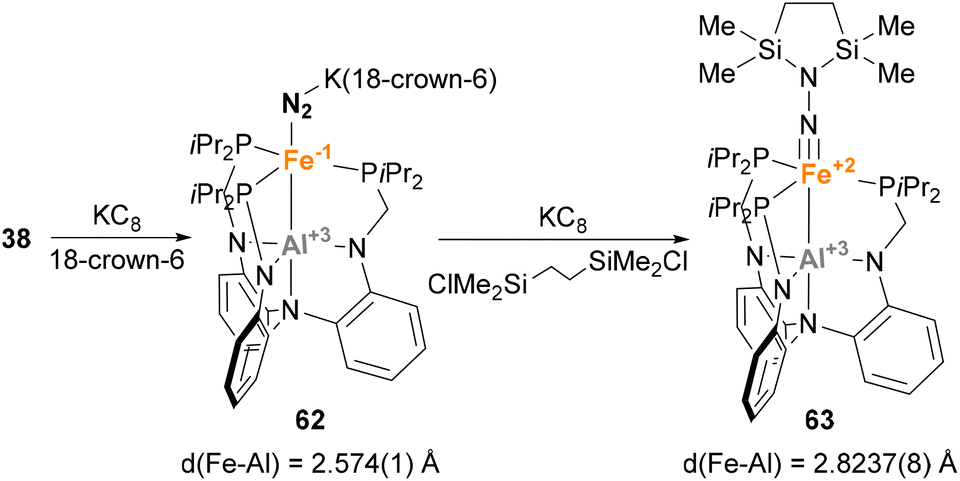
Similar reactivity towards N2 activation was described by Lu and co-workers using AltraPhos-supported Fe–Al species 38 (Scheme 16).159 After one-electron reduction in THF, species 62 is readily prepared. As expected, spectroscopic and SC-XRD studies indicate that species 62 presents a weaker N–N bond compared with 38 due to stronger π-back donation. In addition, sizeable changes also occur in the Fe–Al bond, which shortens 0.23 Å upon reduction. These features suggest that reduction occurs at the Fe centre, which adopts a formal oxidation state value of −1. Species 62 allowed functionalisation of the bound dinitrogen by reacting it with 1,2-bis(chlorodimethylsilyl)ethane in the presence of KC8. The reaction occurs smoothly, and generates diamagnetic Fe(II) species 63 in a transformation that involves double substitution of the distal nitrogen atom as well as four-electron reduction of the N–N bond. Contrary to the previous example, catalytic functionalisation was not explored, but 63 represents an exceptional opportunity to explore complete N–N bond scission with release of amines.
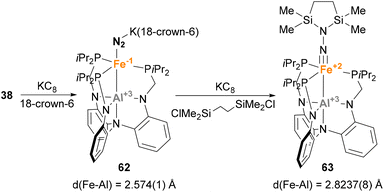 |
| | Scheme 16 Al–Fe complexes employed in catalytic N2-to-NH3 reduction. | |
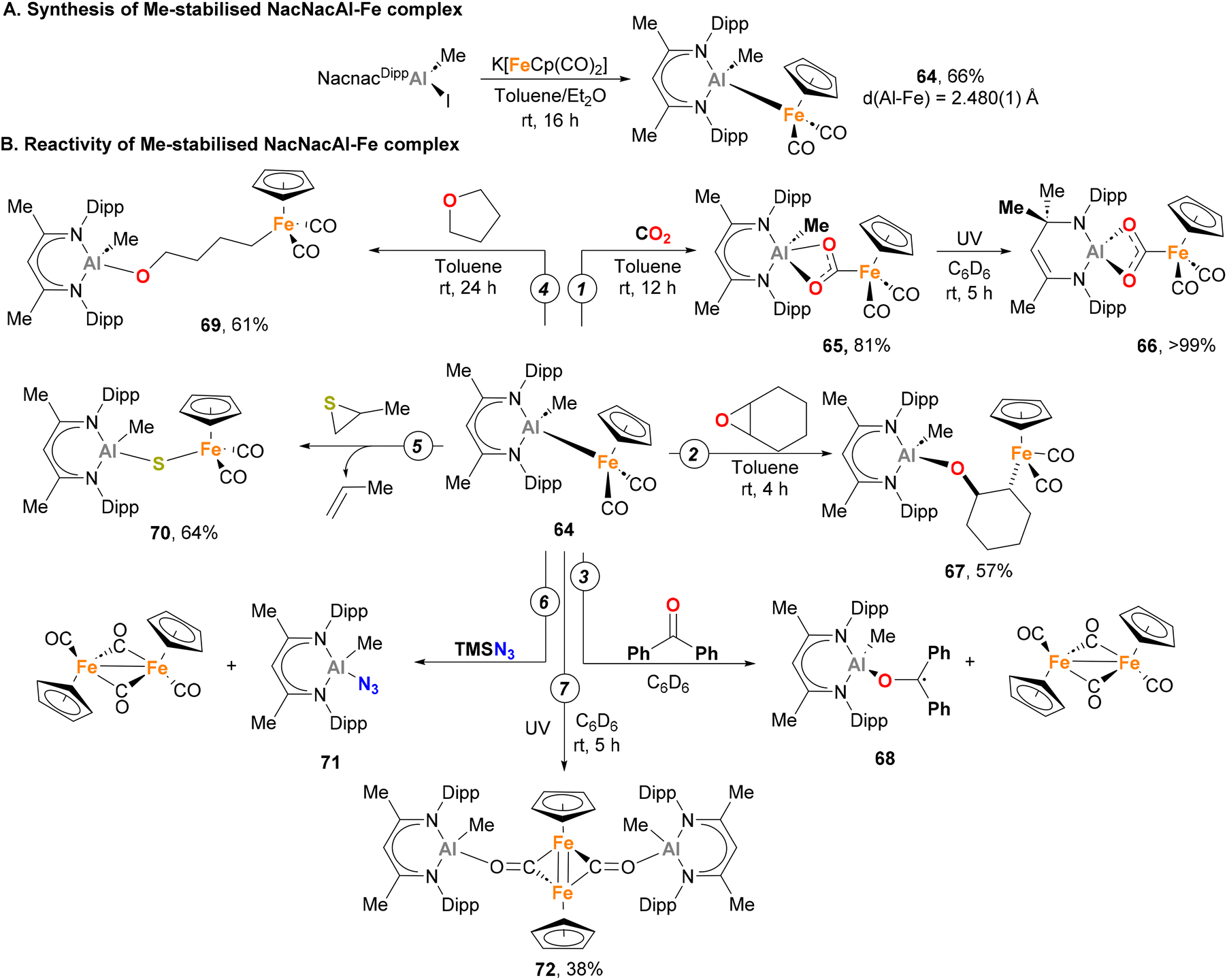
Recently, Mankad and co-workers described the synthesis of a novel Fe–Al heterobimetallic complex, and extensively investigated its reactivity.149 By reacting a Nacnac Al(III) iodide complex with K[FeCp(CO)2], complex 64 could be obtained through a salt elimination route (Scheme 17A). SC-XRD analysis exhibited a tetracoordinated Al centre with distorted tetrahedral geometry, and the Fe–Al bond length was comparable to the Fe–Al species reported by Aldridge (Scheme 10, species 36 and 37).129 Computational insight into the atomic charge and bonding nature of species 64 unambiguously determined a +3 oxidation state for Al and a zero oxidation state for Fe. Regarding its reactivity, 64 generates adduct 65 upon reaction with CO2 under mild reaction conditions (Scheme 17B, pathway 1), representing the first example of Al(III)–base-metal-mediated CO2 activation. Interestingly, irradiation of complex 65 with UV light triggered a formal 1,3-methyl shift, leading to the formation of species 66. Species 64 is also able to ring-open epoxides, such as cyclohexene oxide, yielding product 67 (Scheme 17B, pathway 2). Such reactivity studies were complemented with a computational analysis of the reaction profile, aiming at the elucidation of the cooperative pathways governing CO2 and C–O activation. Surprisingly, computational studies revealed a low energetic barrier for the homolytic dissociation of the Fe–Al bond in complex 64, which at the same time is similar to the experimental barrier obtained. Based on this, a mechanism in which the Fe–Al bond dissociates to generate a highly reactive metalloradical Fe(I)–Al(II) pair was proposed. Further evidence of the intermediacy of a metalloradical Fe(I)–Al(II) pair was gathered when 64 was mixed with benzophenone (Scheme 17B, pathway 3), leading to the formation of the NMR-silent species 68. It is worth noting that the origin of the low energetic barrier for homolytic Fe–Al cleavage is attributed to the significant steric hindrance of the NacnacDipp ligand. In contrast, the less bulky [NacnacPhAl(Me)-FeCp(CO)2] displays an absence of reactivity towards CO2 activation, further underscoring the influence of the NacnacDipp ligand's steric hindrance.
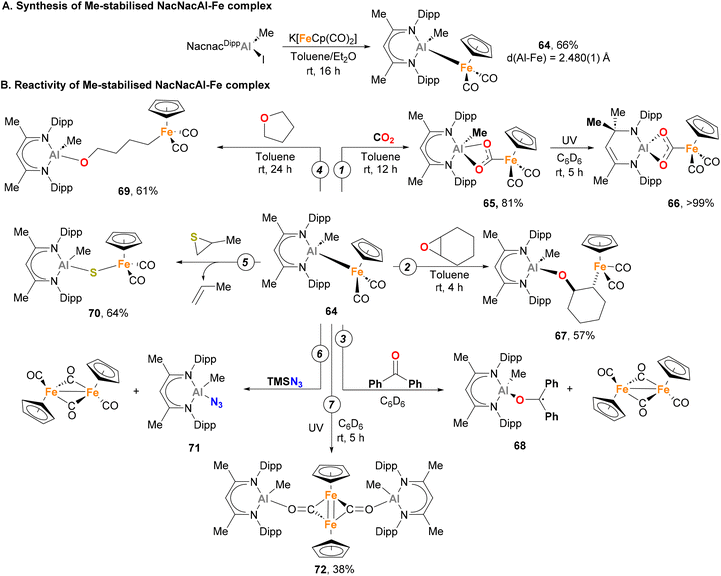 |
| | Scheme 17 Stoichiometric small molecule activation by a heterobimetallic Fe–Al complex via metalorradical pairs. | |
This first study prompted Mankad and co-workers to further explore the reactivity of complex 64 under thermal and photochemical conditions.151 The more intricate cooperative ring-opening reaction of THF was subjected to testing (Scheme 17B, pathway 4), yielding species 69 where Fe and Al moieties were found to be connected by a linear four-carbon chain. This transformation represents an uncommon radical cleavage of C–O bonds of THF, as typical mechanisms with other Al or Fe complexes involve cationic intermediates.160,161 When an episulfide was reacted with 64, rather than forming ring-opening product, the reaction resulted in a sulfur atom transfer into the Fe–Al bond (70, Scheme 17B, pathway 5). Species with weak and labile bonds, such as TMS–N3, were also activated, yielding 71 with concomitant dimerization of the [FeCp(CO)2] metalloradical fragment (Scheme 17B, pathway 6). Finally, dimerization reaction and CO insertion into the Fe–Al bond was also observed when 64 was irradiated with UV light, forming species 72, which can be formulated as having a FeFe double bond (Scheme 17B, pathway 7).
3.6.3 Miscellaneous reactivity.
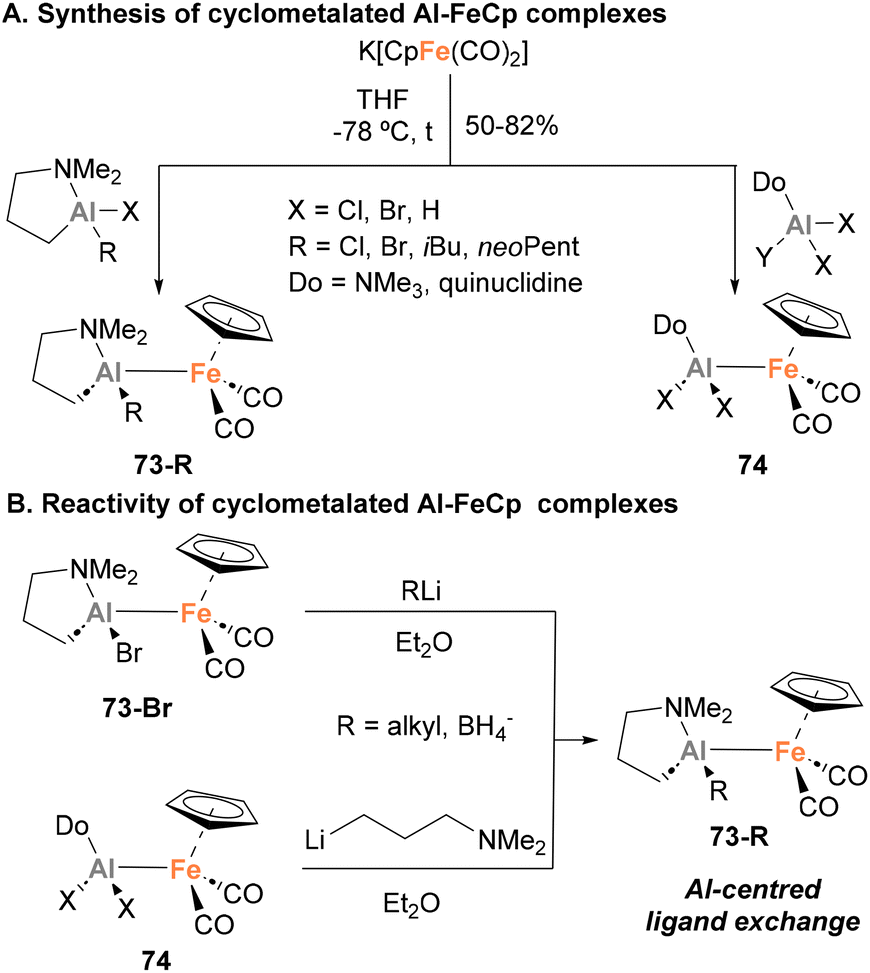
In 1994, Fischer and Priermeier reported a collection of heterobimetallic complexes featuring an Fe–Al bond, aiming at the synthesis of Al-containing thin films for microelectronics.135 Using a salt metathesis approach, distinctly substituted haloalumanes were reacted with K[CpFe(CO)2], obtaining species 73-R and 74 in moderate to excellent yields (Scheme 18A). The structure of such heterobimetallic complexes was unambiguously assigned by SC-XRD analysis (73-iBu), showing an unsupported Fe–Al unsupported bond. The Fe–Al distance (2.456(1) Å) in 73-iBu was similar to the distance reported for the adduct [Ph3Al–FeCp(CO)2]−31,73 which led the authors to suggest a Z-type donor–acceptor intermetallic bond. Nonetheless, in this case no formal oxidation states were unambiguously assigned to the metal centres. Albeit these species were not tested towards small molecule activation, complex 73-Br can undergo ligand exchange at the Al centre with alkyllithium reagents or LiBH4, resulting in the formation of the corresponding alkyl or borohydride derivative (Scheme 18B). Additionally, species 74 can also undergo Al-centred ligand exchange transformations to form alumacycles 73-R when mixed with Li–(CH2)3–NMe2. As mentioned above, these heterobimetallic Fe–Al species were employed to obtain thin films based on Al and Fe, and their reactivity towards small molecule activation and cooperative cleavage of strong bonds was not explored.
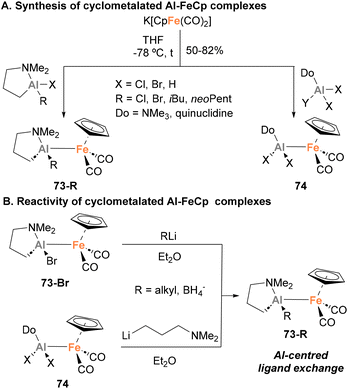 |
| | Scheme 18 Salt-metathesis-based synthesis and derivatisation of Al–Fe systems. | |
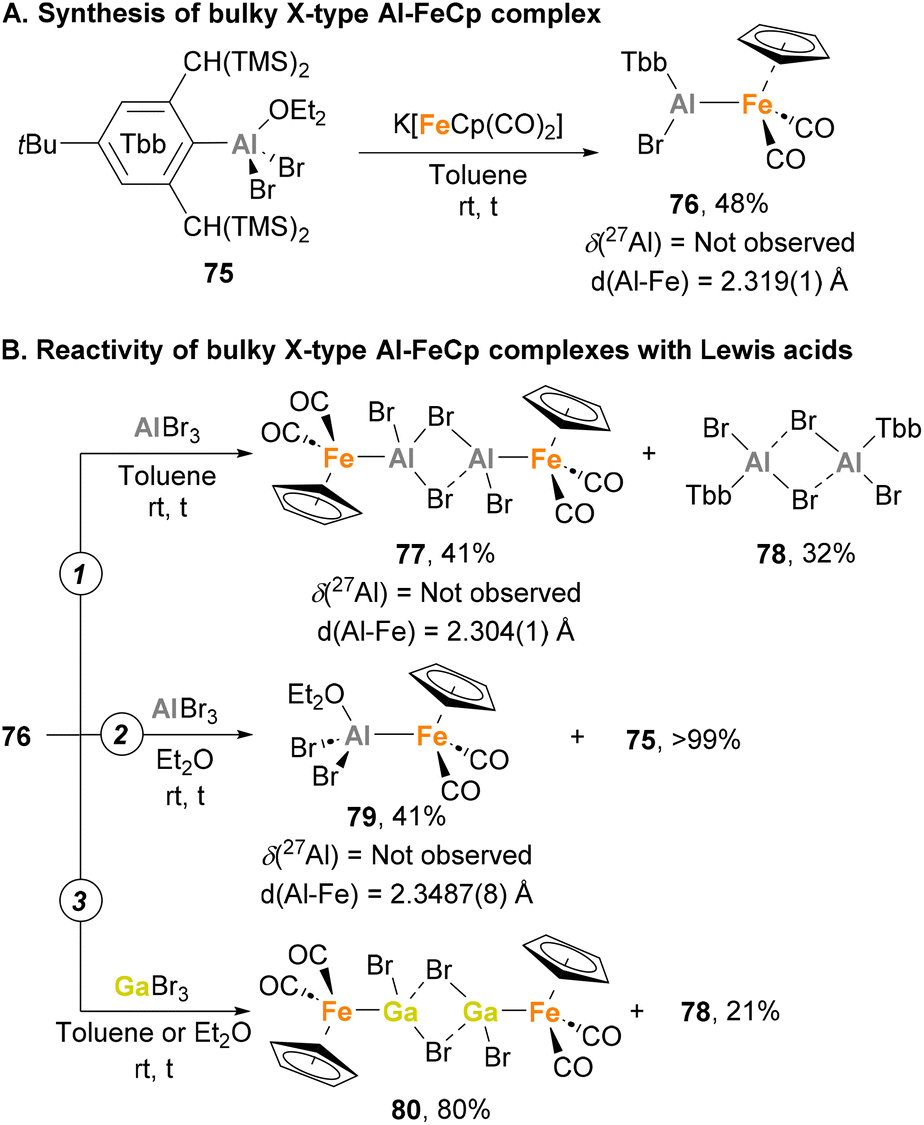
All the reported heterobimetallic Fe–Al species described so far, with exemption of Nöth's138 and Crimmin's150 examples, include a four-coordinated, fully saturated Al centre. An example of unsaturated Fe–Al species was recently introduced by Tokitoh and co-workers.144,146 To do so, Tbb–AlBr2·OEt275 was employed as precursor (Tbb = 2,6-bis(bis(trimethylsilyl)methyl)-4-(tert-butyl)phenyl), which was treated with the anionic carbonyl salt K[FeCp(CO)2] to obtain complex 76 in 48% yield after salt elimination (Scheme 19A). Analysis of this complex after reaction completion by 27Al NMR revealed no signal, suggesting a tricoordinated structure of the Al centre that was unambiguously confirmed by SC-XRD analysis.162 Although the nature of the Fe–Al bond was studied by means of computational techniques, no discussion about the formal oxidation states of each metal was provided. Regarding its reactivity, when 76 was mixed with EBr3 (E = Al, Ga) in toluene, a formal exchange of the aluminyl moiety occurred, generating Br2Al–Fe complex 77 (Scheme 19B, pathway 1) and Br2Ga–Fe complex 80 as dimeric species (Scheme 19B, pathway 3), in both cases with subsequent formation of Al(III) species 78. When the reaction with AlBr3 was performed in a coordinating solvent such as diethyl ether, monomeric complex 79 was generated (Scheme 19B, pathway 2). Contrarily, reaction of 76 with GaBr3 in diethyl ether led to dimeric complex 80, which can be explained invoking the higher Lewis acidity of the Al centre, making a better acceptor for weakly coordinating ligands. It is worth mentioning that 77 and 79 were analysed in depth, including Al27 NMR and characterisation by SC-XRD. Fe–Al bonds (77: 2.304(1) Å, 79: 2.3487(8) Å) were revealed to be similar to parent Fe–Al species 76 (2.319(1) Å), and both complexes were completely NMR silent. Computational studies of the intermetallic bond in 76 were performed using Natural Bond Order (NBO) analysis, and although formal oxidation states were not explicitly assigned, they strongly suggest the existence of an Fe(II)–Al(I) bond, where aluminium functions as an X-type donor ligand.
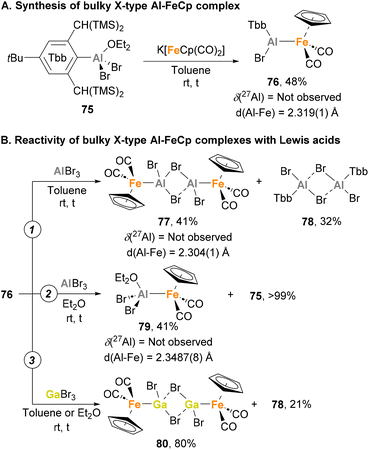 |
| | Scheme 19 Synthesis of bulky Al–Fe system and its reactivity with Lewis acids. | |
Overall, heterobimetallic species containing an Fe–Al bond have undergone extensive study since their discovery in the late 1970s. Initially considered structural curiosities, their reactivity did not attract significant attention until recent years. Notably, Driess, Crimmin, Mankad, Peters, and others have demonstrated the potential of these species in small molecule activation, including the cleavage of strong C–H bonds, leading to their application in crucial processes such as catalytic N2-fixation reactions. Nonetheless, taking into account the importance of Fe in organic synthesis,163 chemists have just scratched the surface of the actual potential of heterobimetallic Fe–Al species in small molecule activation. Considering the abundance and environmentally benign properties of both Fe and Al, the combination of these elements in heterobimetallic species capable of performing cooperative catalytic transformations represents a promising avenue that is likely to garner increasing attention in the future.
3.7 Cobalt
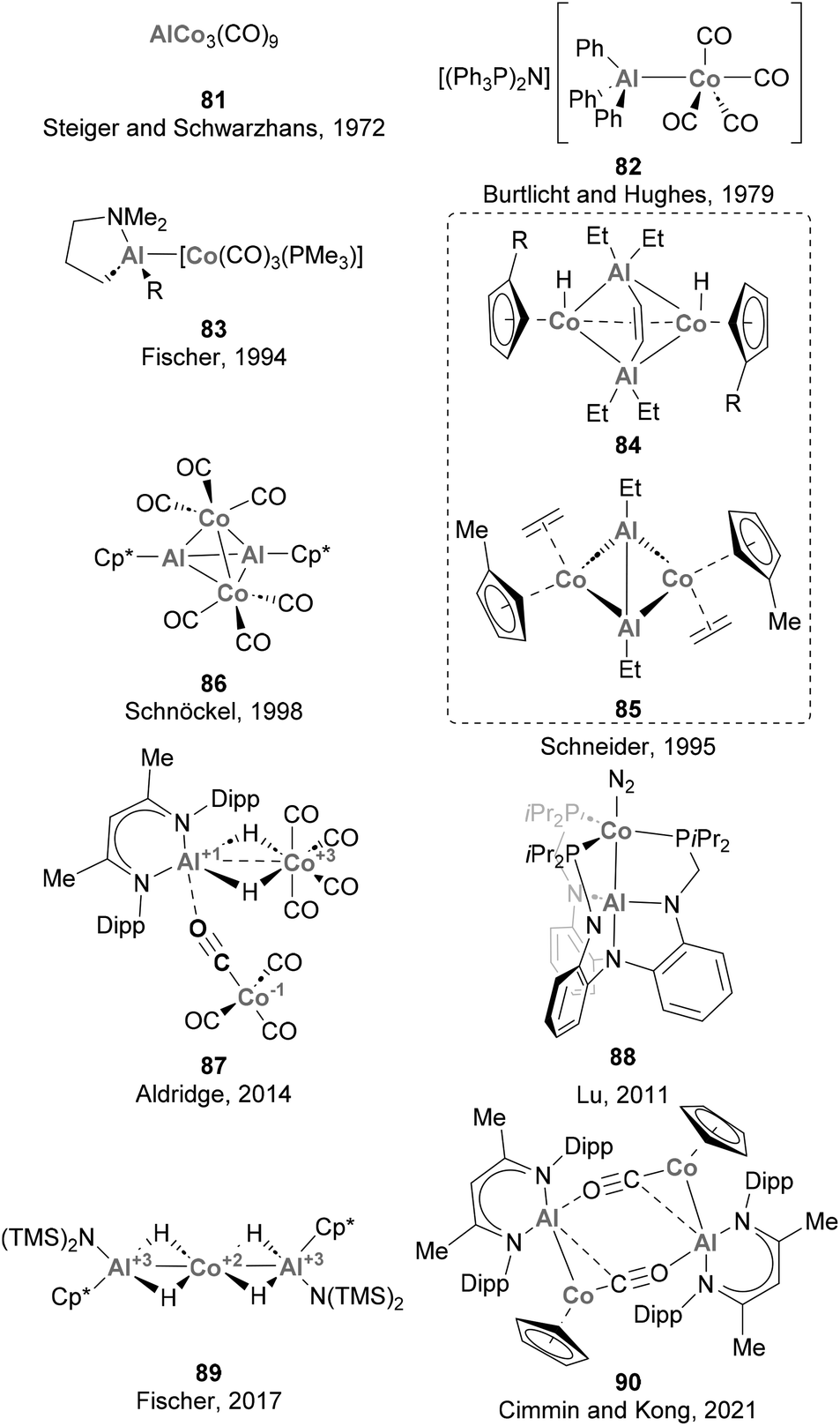
Steiger and Schwarzhans pioneered the field of heterobimetallic aluminium–base-metal complexes in 1972 with the first example of a Co complex containing an Al-based ligand (81, Scheme 20).164 While this represented a milestone in the field, the full characterisation of a heterobimetallic species featuring a Co–Al had to wait more than 20 years, when Schneider and co-workers reported a SC-XRD analysis of species 84 and 85.165 Furthermore, it is worth noting that the majority of the Co–Al systems reported subsequent to these pioneering examples,72,73,124,125,135,142,164–166 depicted in Scheme 20, have predominantly remained as structural curiosities without significant exploration in the realms of small molecule activation and catalysis. Among these complexes, only 81 and 83 were synthesised through salt elimination, prevailing the use of direct coordination of low-valent Al species and ligand substitution reactions.
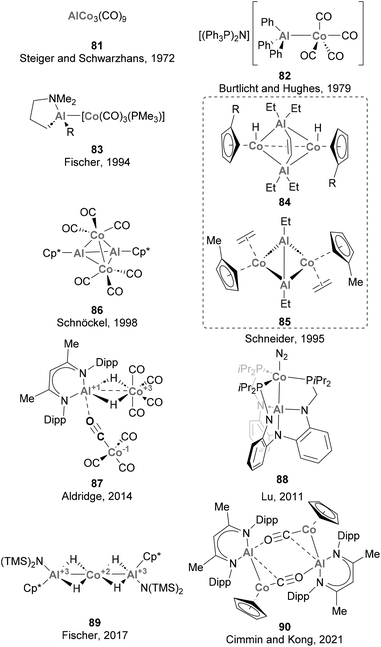 |
| | Scheme 20 Structurally characterised Co–Al complexes. | |
Recently, heterobimetallic species featuring a Co–Al bond have received attention from the organometallic community, and their reactivity has been explored in C–H bond and small molecule activation. However, it is worth noting that the catalytic application of such species remains very limited, as Co–Al complexes find application as catalysts in CO2 hydrogenation and oxidation of alcohols (see below).
3.7.1 Oxidation of alcohols.
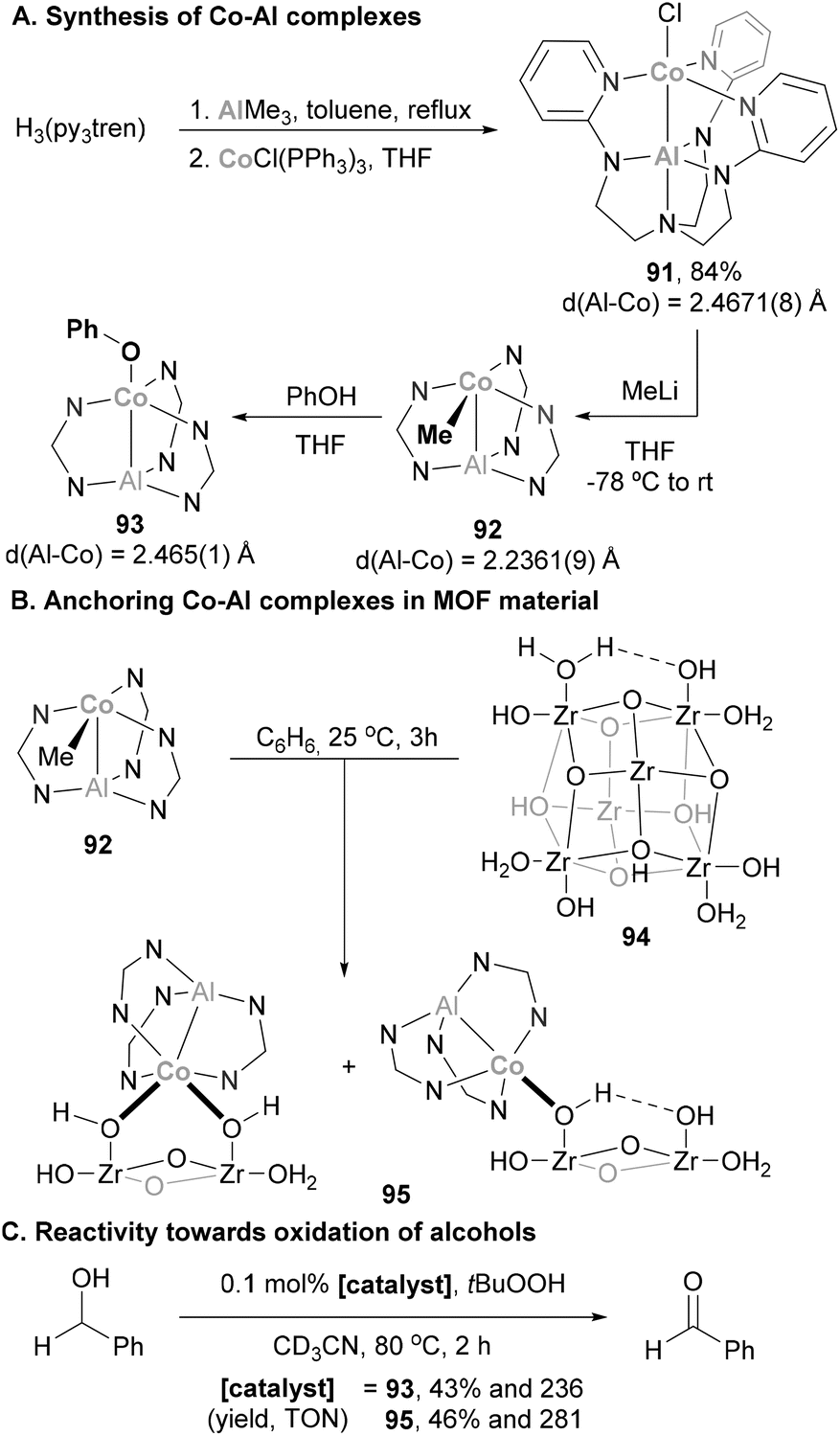
Taking advantage of a double-decker ligand, Lu and co-workers synthesised a heterobimetallic Al(III)–Co(I) complex for MOF functionalisation.167 Starting from H3(py3tren), the Al–Co complex 91 was readily obtained in a 2-step procedure (Scheme 21A). The structure of 91 was unambiguously confirmed by SC-XRD experiments, featuring a Co–Al bond length of 2.4671(8) Å, 5% shorter than the sum of Al and Co covalent radii (2.60 Å),78 indicating a donor–acceptor interaction. Transmetallation reaction with MeLi results in the formation of organometallic Co–Me species 92. This complex was anticipated to be a good precursor for the functionalisation of metal organic frameworks (MOF), since its reaction with –OH residues on the MOF surface (94) was anticipated to be straightforward. In this process, organometallic species 92 was reacted with phenol, producing phenoxide complex 93 under mild conditions. Encouraged by these results, 92 was mixed with MOF 94, obtaining Co–Al single sites on a MOF (Scheme 21B, 95, simplified structures). Species 93 and 95 were tested in alcohol oxidation reactions, and albeit they present similar activity towards the formation of benzaldehyde, species 94 decomposes to an ill-defined structure and is not amenable for catalyst recycling.
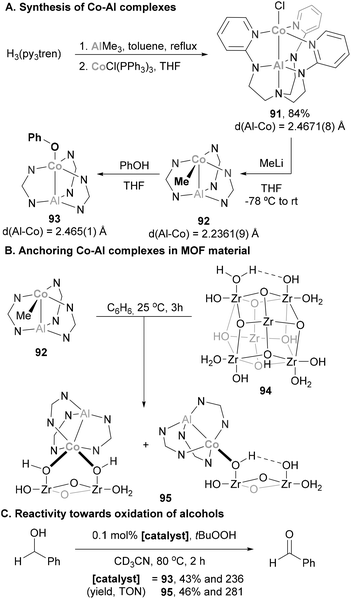 |
| | Scheme 21 Catalytic oxidation of benzyl alcohol performed by Co–Al systems anchored in MOF material (simplified structure drawn for clarity). | |
3.7.2 C–H activation.
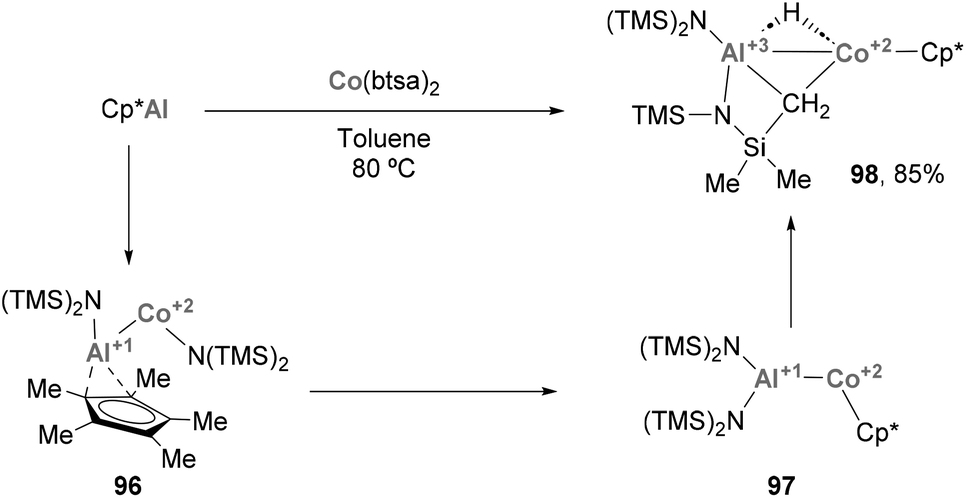
Heterobimetallic species featuring a Co–Al bond have also been involved in the activation of relatively inert C–H bonds. Fischer and co-workers recently reported how low valent ECp* (E = Al, Ga) species trigger Csp3–H activation of –Me residues when mixed with Co(btsa)2 salts (Scheme 22), resulting in complex 98.72 SC-XRD analysis of this heterobimetallic complex presents a Co–Al distance of 2.402(2) Å, slightly longer than the previously described examples124,165,166 but shorter than reported AlCo alloys,168 while the Co–Cp* distance (1.709 Å) is in agreement with a Co(II) oxidation state (1.714 Å for Cp*2Co vs. 1.623 Å for [Cp*2Co][PF6]).169,170 Formation of species 98 can be rationalised by a sequence of events, which is illustrated in Scheme 22. The reaction starts with the insertion of Cp*Al into a Co–N bond (96), followed by Cp* migration from Al to Co leading to 97 and a subsequent C–H activation of the one of the methyl groups of the btsa ligands, resulting in the formation of 98 in 85%. This example illustrates the still underexplored ability of systems featuring a Co–Al bonds to cleave and functionalise C–H bonds.64 Taking into account the ability of Co to perform such transformations,171–174 together with precedents with heavier group 9 elements,58,60 intensive investigation of Co–Al systems will be crucial to develop novel methods for functionalizing C–H bonds.
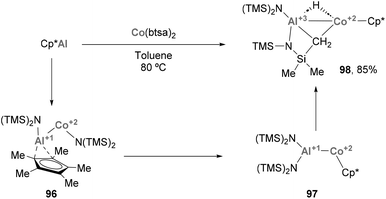 |
| | Scheme 22 Formation of Co–Al species via a C–H activation mechanism. | |
3.7.3 Small molecule activation.
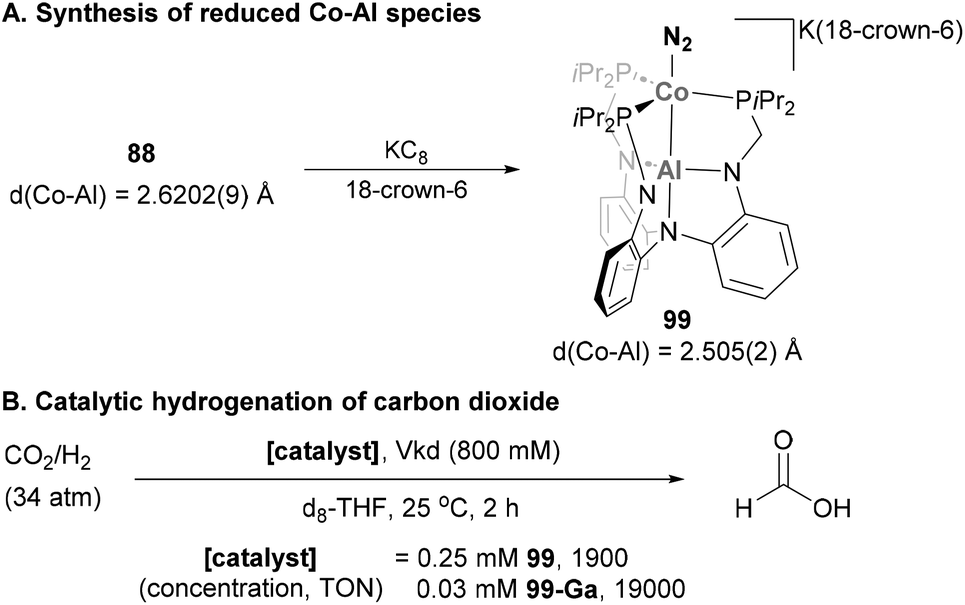
Lu and co-workers exploited the use of AltraPhos in heterobimetallic Co–Al species containing a Z-type Co → Al interaction (Scheme 23A).142 Indeed, such interaction allowed the synthesis of stable Co–Al species 99 with a formal Co(-I) centre after one-electron reduction of complex 88 with KC8.159 Similarly to its Fe analogue (see Scheme 16), the bonded dinitrogen molecule in 99 also presents an enlarged N–N bond compared with its precursor as a result of stronger π-back donation. In addition, the Co–Al bonds in 99 is 0.12 Å shorter than in 88, thus indicating a Co-centred reduction event that yields a reduced Co atom in a formal oxidation state value of −1. In this case, and in contrast to highly reduced Fe species 62, the bound dinitrogen molecule could not be functionalised with 1,2-bis(chlorodimethylsilyl)ethane as this reaction led to an intractable mixture of products. Nonetheless, the highly reduced Co–Al species 99 proved to be excellent at binding dihydrogen, a property that was exploited in catalysis to hydrogenate CO2 (Scheme 23B).175 The mechanism of CO2 hydrogenation was only studied in depth for the Ga analogue 99-Ga, resulting in an unprecedented Co(-I)/Co(I) catalytic cycle with a key oxidative addition of Co(-I) into the H–H bond. Hydrogenation with species 99 is proposed to proceed through an analogous mechanism, representing a unique catalytic hydrogenation example of a heterobimetallic Co–Al species.
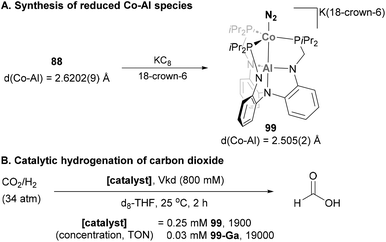 |
| | Scheme 23 Synthesis of reduced Co–Al species and their use in catalytic reduction of CO2. | |
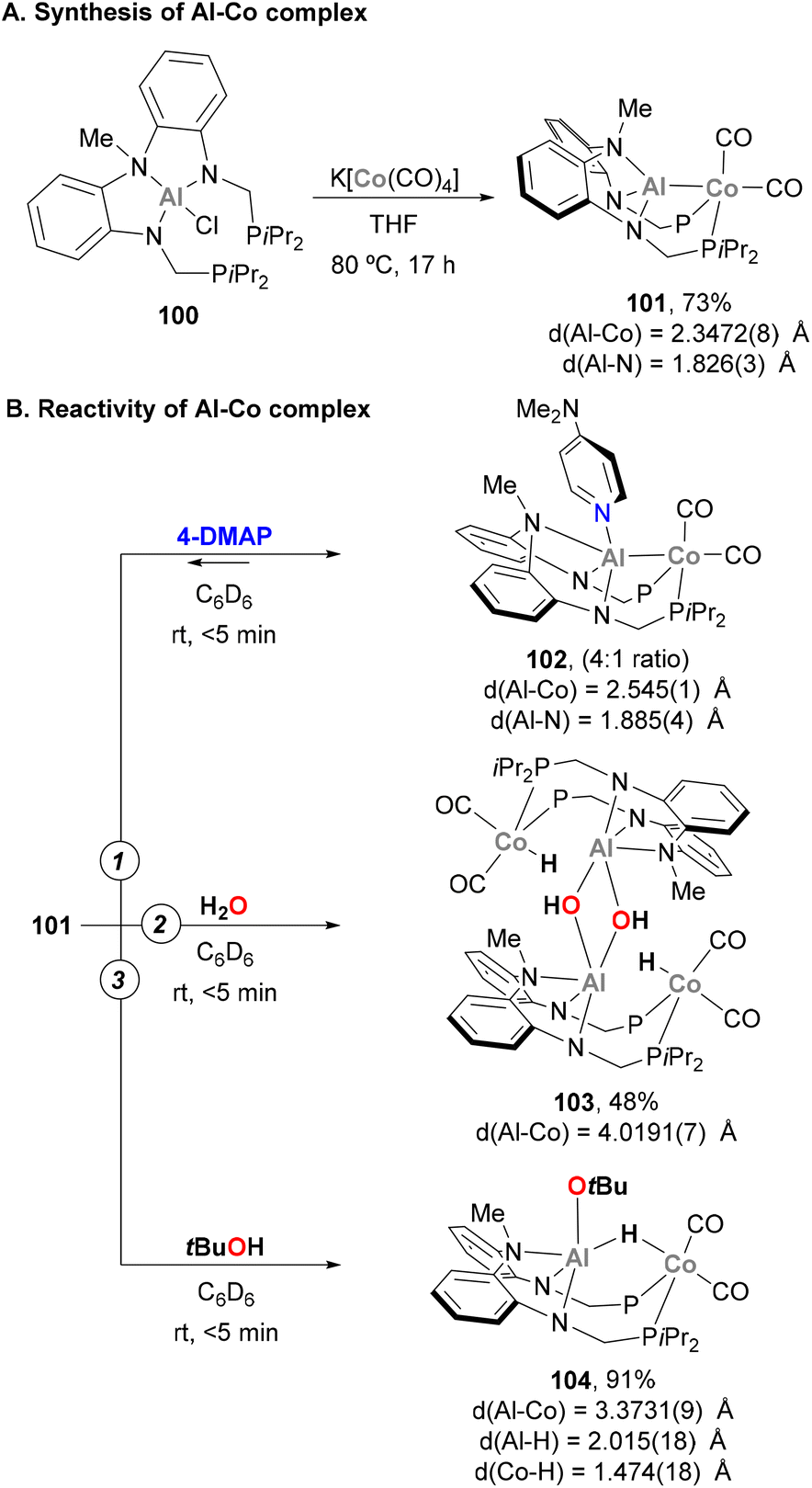
The activation of E–H bonds using Co–Al species has also been explored by Nakao and co-workers.176 Using a PAlP pincer and following a salt metathesis approach, Al(III) chloride precursor 100 was reacted with K[Co(CO)4], yielding heterobimetallic complex 101 in 73% yield (Scheme 24A). SC-XRD analysis indicates that the Al and Co centres adopt tetrahedral and bipyramidal configurations, respectively. In addition, Co–Al (2.3472(8) Å) and Al–Namide (1.826(3) Å) distances in 101 are similar to previously reported Co(I)–Al(I) complexes, suggesting a low oxidation state Co centre. Two main transformations were studied with heterobimetallic complex 101. Firstly, the coordination ability of 4-dimethylaminopyridine (4-DMAP) was found to be reversible, illustrating the high Lewis acidity of the Al centre (102, Scheme 24B, pathway 1). SC-XRD analysis of 102 shows an expected elongation of Co–Al (2.545(1) Å) and Al–Namide (1.885(4) Å) distances after coordination of DMAP, as well as a trigonal bypiramidal Al centre. Inspired by a recent report from Ozerov,177 the ability of 101 for O–H bond activation was tested with H2O and tBuOH. This resulted in the isolation of dimeric complex 103 (Scheme 24B, pathway 2) and complex 104 in 91% yield (Scheme 24B, pathway 3). It is important to note that in both cases, the integrity of the Co–Al bond was lost, as evidenced by the long Co–Al distances after O–H bond activation (4.0191(7) Å for 103 and 3.3731(9) Å for 104), generating an Al alkoxide and a Co hydride. In complex 104, Al–H and Co–H bond lengths suggest that the μ-H ligand is a Co hydride, with partial coordination to the Al centre. It is important to note that treatment of complex 101 with PhNH2 or n-BuNH2, in contrast to Ozerov's work, did not afford N–H bond activation products. This fact indicates that the O–H activation by complex 101 does not proceed via a cooperative mechanism and can be described as a protonation of the low oxidation state Co centre.
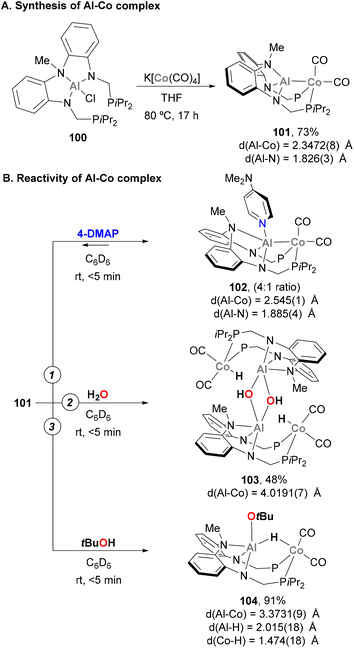 |
| | Scheme 24 O–H bond activation by a PAlP Co–Al complex. | |
Contrary to heavier heterobimetallic M–Al (M = Rh and Ir) group 9 analogues,57–59,61,178 cooperative small molecule activation with heterobimetallic Co–Al species needs further investigation if these species are to be applied to catalytic transformations. This is an exceptional opportunity to unlock the full potential of the field and pave the way for the development of predictable and efficient transformations using inexpensive and benign elements of the d and p blocks.
3.8 Nickel
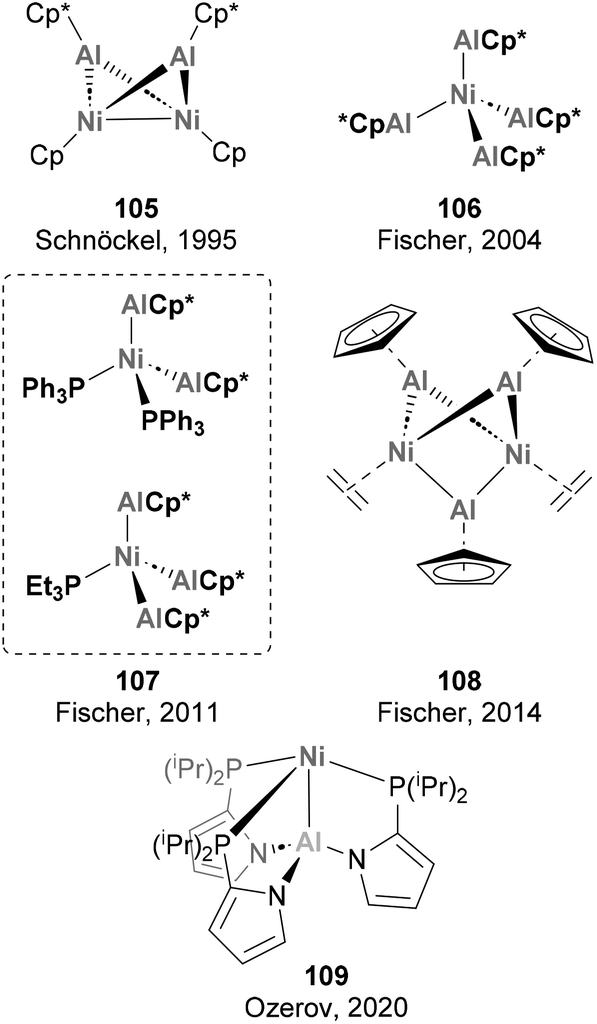
A long story of success is shared by Ni and Al when combined together, starting with their role in polymerisation reactions and the so-called “Nickel effect”,179 which almost one century ago set the basis for the discovery of the Ziegler catalysts in Mülheim an der Ruhr.79,180 Despite this early discovery and the potential intermediacy of Ni–Al species in hydroalumination reactions and polymerisation,181,182 well-defined heterobimetallic species with Ni–Al bonds remained elusive until 1995, when Schnöckel and co-workers described species 105 (Scheme 25).183 While Ni has also been extensively used in combination with Al additives in catalytic processes, the synthesis and reactivity of heterobimetallic species with Ni–Al bonds has not received the same attention. Pioneering examples of heterobimetallic species with Ni–Al bonds whose reactivity has not been studied are depicted in Scheme 25. As shown, Ni centres coordinated to AlCp* fragments have dominated this realm from 1995 to 2014 (105–109),71,183–187 with major contributions by Fischer and co-workers.
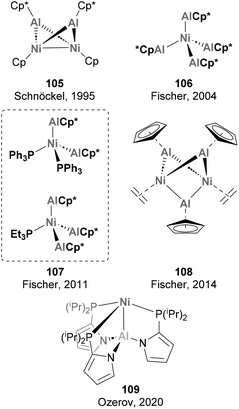 |
| | Scheme 25 Structurally characterised Ni–Al complexes. | |
The reactivity of Ni–Al bonds has been studied in a more systematic manner over the past five years, and relevant works are exemplified below. Among the transformations investigated, stoichiometric small molecule activation has been the most extensively reported. Notably, a recent report highlights a unique example in which a heterobimetallic complex featuring two Ni–Al bonds is used as catalyst for the dearomatisation of N-heterocycles.
3.8.1 C–H activation.
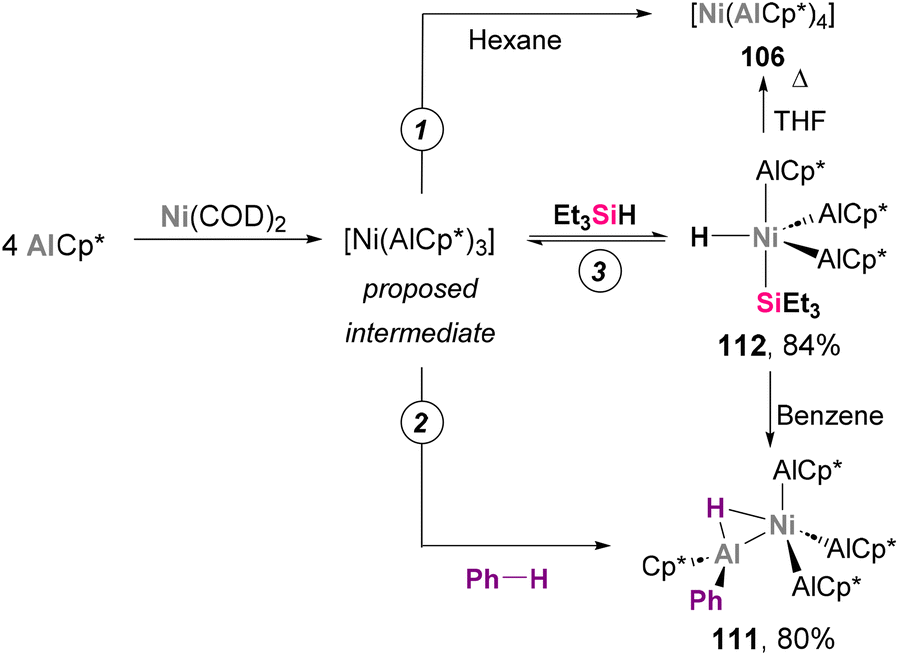
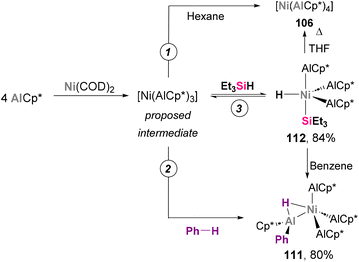
The cleavage of C–H bonds has been an extensive area of research in heterobimetallic aluminium–base-metal complexes. In 2004, Fischer and co-workers discovered several E–H (E = C, Si) activation transformations aiming to synthesise species 106, which represented innovative reactivity examples of Ni–Al complexes.184 As previously reported, treatment of Ni(COD)2 with 4 equivalents of AlCp* in hexane results in the formation of [Ni(AlCp*)4] 106 (Scheme 26, pathway 1). In an attempt to improve the yield of this transformation, the authors employed benzene as solvent, observing no traces of 106 but quantitative formation of 111 (Scheme 26, pathway 2). This product results from C–H bond cleavage of the benzene, which illustrates the ability of Ni–Al bonds to perform this transformation. SC-XRD of 111 exhibits a Ni–Al bond distance of 2.2105(11) Å (AlCp* fragment) and 2.2912(11) Å (for HAlPhCp* fragment), which suggest a +3 and +1 oxidation state for the Al centres in the H-bridged Al atom and the AlCp* ligands, respectively. Alternative control experiments indicate a 16-electron complex with chemical formula [Ni(AlCp*)3] as intermediate, as tetracoordinated Ni species 106 was found to be chemically inert. Interestingly, when 3 equivalents of Et3SiH were added to the mixture of Ni(COD)2 and AlCp*, complex 112 was obtained in high yield after a reversible Ni-centred Si–H bond insertion (Scheme 26, pathway 3). SC-XRD analysis of 112 reveals a distorted trigonal bypiramidal geometry, with Ni–Al distances between 2.180(7) and 2.208(10) Å. In a later wok by the same authors, this reversibility was employed for the selective synthesis of [Ni(PEt3)(AlCp*)3] 107, which cannot be accessed through conventional one-pot procedures.186
 |
| | Scheme 26 Reactivity of Ni–Al complexes towards E–H (E = C, Si) bonds. | |
3.8.2 Small molecule activation.
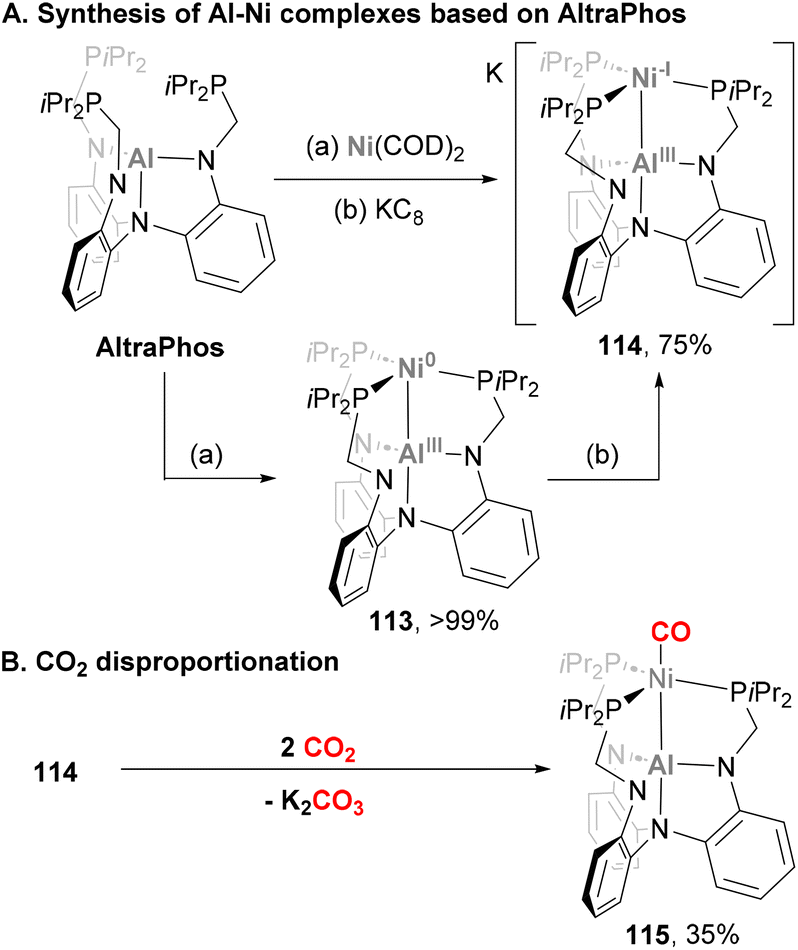
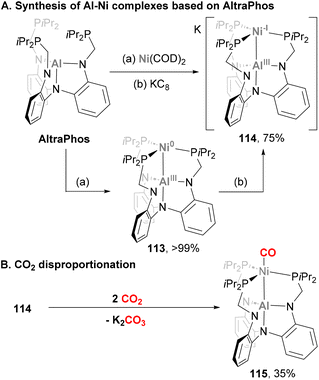
Complexes bearing a Ni–Al bond have also been applied to small molecule activation, albeit most of their reactivity so far involves ligand exchange studies. An example of such processes was reported by Lu and co-workers using AltraPhos as ligand (Scheme 27),142 similarly to Fe and Co complexes 38 and 88, respectively. Reaction of Ni(COD)2 with AltraPhos yields a diamagnetic, formally Ni(0)–Al(III) complex 113.188 Interestingly, 27Al NMR studies together with a Ni–Al distance of 2.450(1) Å suggests direct Ni–Al bonding, presumably via a Ni → Al dative bond. A complex recently reported by Ozerov and co-workers (109, Scheme 25) displays similar features compared with 113, although in this work a tris(N-pyrrolyl)aluminium ligand was used.187 Contrary to its Ga and In analogues, 113 was shown to be inactive in catalytic olefin hydrogenation. In a subsequent study from Lu and co-workers, reduction of Ni-alane species 113 with KC8 resulted in a dark red solution containing radical anion 114,189 which was crystallised in the presence of cryptand crypt-222. SC-XRD studies revealed that the Ni–Al bond distance in 114 contracts to 2.389(1) Å as a result of a Ni-centred reduction and a subsequent stronger Ni → Al donation. Furthermore, combined experimental and computational studies of such species support its formulation as formally Ni(-I)–Al(III), with an unexpected stabilisation arising from a one-electron polar σ-(Ni–Al) and a two-electron (3dz2) → Al dative bond. The reactivity of this open-shell Ni–Al species towards CO2 activation was then tested.190 Stirring 114 under a CO2 atmosphere yielded K2CO3 and a 1:1 mixture of 113 and CO adduct 115via a disproportionation reaction. Formation of such species was rationalised via a nucleophilic attack of metalloradical 114 to CO2, followed by a reduction mediated by another molecule of species 114 to generate a formally dianionic nickel diolate. The latter species is proposed to insert a second CO2 molecule to generate a Ni di-CO2 intermediate, which decays to CO32− and heterobimetallic complex 115. This species was independently synthesized by treatment of 114 with ethyl formate, confirming its structural features. SC-XRD analysis resulted in a Ni–Al bond distance of 2.626(1) Å, which suggests the Ni–Al bond in 115 is weaker compared with 114, probably due to the strong π-acceptor ability of CO as ligand.
 |
| | Scheme 27 Synthesis and reactivity of Ni-alane species based on AltraPhos. | |
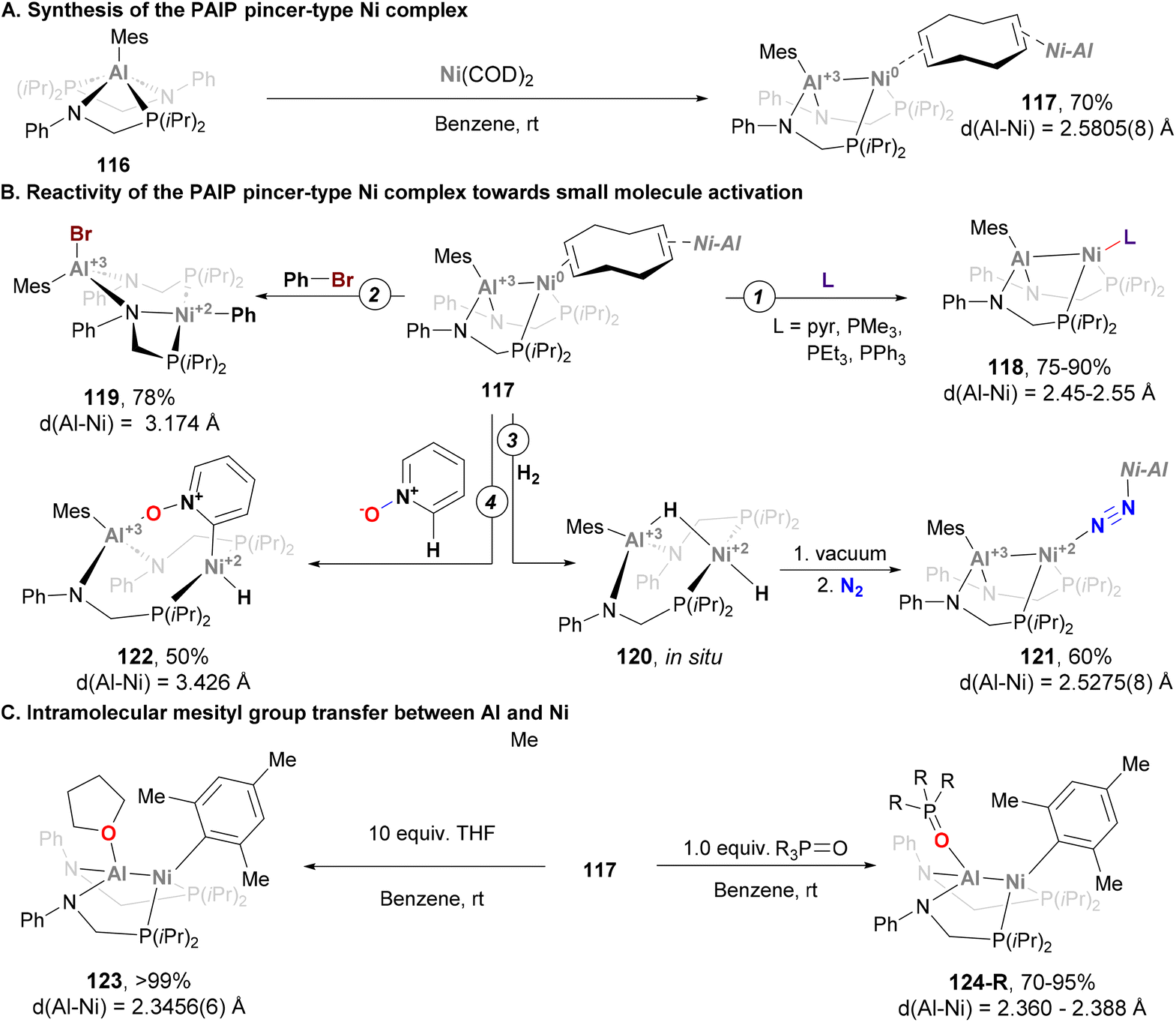
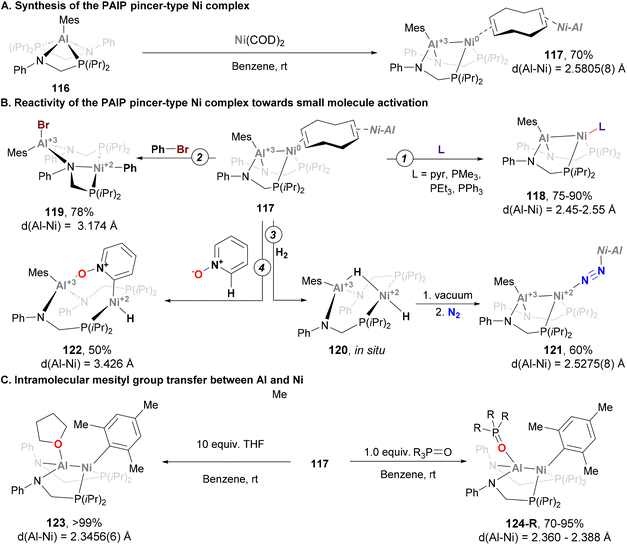
In the example depicted in Scheme 27, the Al atom is buried, and its role is limited to indirectly adjusting the reactivity of the Ni centre. In a recent example, Lu and co-workers designed a Ni–Al system featuring a ligand platform that enables substrates to interact with both Ni and Al centres (Scheme 28), and promote cooperative activation of substrates.191 Treatment of the corresponding Al-based metalloligand 116 with Ni(COD)2 yields a dimer heterobimetallic pincer species (117) bridged by cyclooctadiene (Scheme 28A). Complex 117 has been fully characterised, and its SC-XRD analysis revealed an intriguing Ni–Al distance of 2.5805(8) Å, which is longer than the sum of covalent radii. This bond length, together with the observed pyramidalization of the Al centre suggest a weak Z-type Ni → Al interaction.78,192 After characterisation of heterobimetallic species 117, its reactivity towards ligand substitution with pyridine and phosphines was tested, resulting in the formation of species 118 (Scheme 28B, pathway 1). This species was also fully characterised, revealing that ligand substitution occurs exclusively at the Ni centre, showing virtually identical Ni–Al bond lengths compared with parent complex 117. More interestingly, cooperative activation of small molecules was investigated taking advantage of the open coordination environment of the bimetallic Ni–Al unit. As the authors point out in their study, it is important to note that the Ni → Al interaction breaks after substrate activation as a consequence of Ni(0) being oxidised to Ni(II). As an example of C–X activation, Ph–Br was reacted with 117, furnishing species 119 (Scheme 28B, pathway 2). This example, which represents the first Ph–Br oxidative addition into an aluminium–base-metal bond, shows ligation of the halide to the Al centre, while the phenyl group coordinates the Ni atom. Addition of H2 to 117 generates species 120, which features a bridged Al–H–Ni and a terminal hydride Ni–H moieties, similarly to previously reported Ni → B species.193 Interestingly, complex 120 releases H2 upon exposure to vacuum or Ar and converts to 121 when stored under a N2 atmosphere (Scheme 28B, pathway 3). This reactivity shows that the Ni → Al bond interaction not only facilitates cooperative activation of substrates, but also promotes reductive elimination. The open coordination environment of the bimetallic Ni–Al unit also allows the Al centre to direct substrates for selective C–H activation at the Ni site. Indeed, this reactivity was demonstrated by the reaction of 117 with pyridine N-oxide, which resulted in the ortho C–H oxidative addition product 122 in 50% yield (Scheme 28B, pathway 3). Spectroscopic evidence and SC-XRD analysis is consistent with a Ni centre coordinated by a terminal hydride ligand, as well as a C2-cyclometalated pyridine N-oxide moiety. Importantly, the Al centre engages in a strong O → Al interaction (1.814(4) Å) with the pyridine N-oxide ligand, which indicates a coordination prior to the C–H oxidative addition event. This transformation is highly relevant, as it represents a unique example of selective ortho C–H activation of pyridine N-oxides with well-defined heterobimetallic complexes pairing a base metal with Al. An intriguing reaction occurred when species 117 was reacted with weak donors such as THF or phosphine oxides (123 and 124, respectively; Scheme 28C). SC-XRD revealed a Ni–Al complex where the mesityl group shifted from Al to Ni, with a significant contraction of the Ni–Al distance from 2.5805(8) Å to 2.3456(6) Å. These features suggest a ligand-responsive conversion of a Ni-alane to a Ni–aluminyl complex, which is reversible upon addition of more COD ligand. Computational analysis shows that the Ni–Al in 123 and 124 lies in between a Ni(0) → Al(III) interaction and a polarised covalent Ni(I)–Al(II) bond. Overall, the rich reactivity in cooperative activation of substrates, together with its reversible redox processes, represent key features that make species 117 a good candidate to be applied in catalytic transformations.
 |
| | Scheme 28 Synthesis and reactivity of PAlP Ni–Al complex. | |
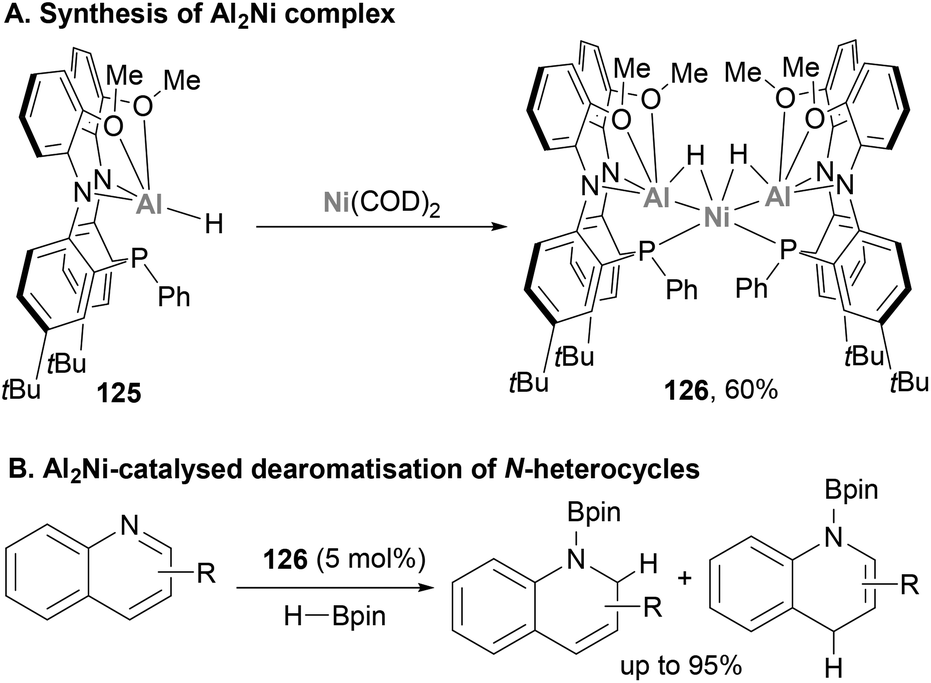
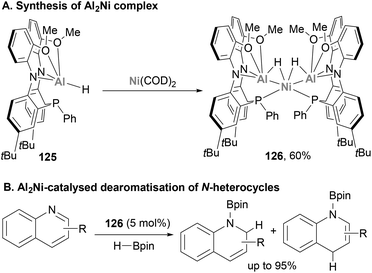
Very recently, Shoshani and co-workers developed a well-defined heterobimetallic Ni–Al complex (126) with catalytic application,194 which was synthesised through metalation of the Al(III)–H precursor 125 with Ni(COD)2 (Scheme 29A). SC-XRD analysis unveiled a trimetallic NiAl2(μ2-H)2, in which both Al atoms are bridged to the Ni centre by a hydride ligand. In addition, the Ni centre adopts a distorted tetrahedral geometry consistent with an 18 electron Ni(0) centre. In addition, Ni–Al distances were found to be short (2.3629(7) and 2.3658(6) Å), indicating a strong interaction between both atoms. Indeed, these distances are consistent with previously reported Ni–alane species,142,187 and are shorter than the sum of Ni and Al covalent radii.78 Based on these structural features, the oxidation state of the metal centres was assigned as +3 for Al and 0 for Ni. Considering the importance of hydride species in hydrogenation and hydroboration of heterocyclic compounds, the ability of 126 towards the catalytic hydroboration of quinoline was tested (Scheme 29B). Using HBpin as hydride source and catalytic quantities of 126 (5 mol%), quinoline was hydrogenated quantitatively. Catalytic amounts of starting Al(III)–H 125 species or Ni(COD)2 resulted in lower yields and slower reaction rates, which highlights the synergistic effect of the Ni–Al bond and its potential in catalysis. Nonetheless, the authors highlight that further studies are needed to identify the responsible catalytically active species and to determine the true nature of the Ni–Al bond. Albeit the field is still in its infancy, this pioneering study opens the door to a new horizon of catalytic reactivity and the application of heterobimetallic Ni–Al complexes in organic synthesis and catalytic small molecule functionalisation.
 |
| | Scheme 29 Synthesis and reactivity of Ni-alane species featuring two Ni–Al bonds. | |
3.8.3 Miscellaneous reactivity.
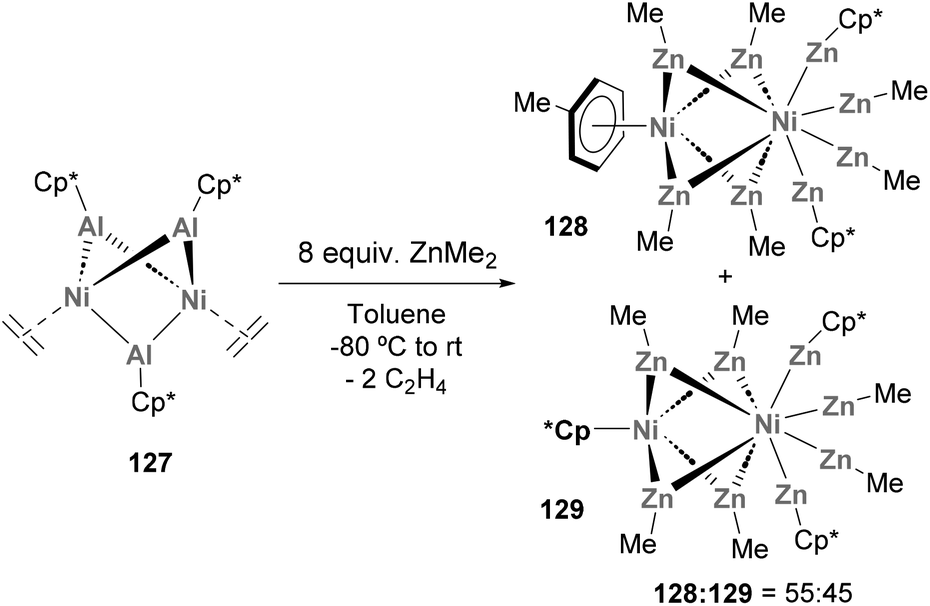
Heterobimetallic Ni–Al species have also been used to produce metallic clusters. In 2014, Fischer and co-workers described the synthesis of novel all-hydrocarbon ligand-stabilised binuclear clusters of metal-core composition Ni2Zn7Al (Scheme 30).71 To do so, species 127, which is synthesised through an unconventional pathway involving transmetallation, was reacted with 8.0 equiv. of ZnMe2. After Al/Zn ligand exchange, a 55:45 mixture of organometallic 128 and 129 species was obtained and products were fully characterised by computational studies, spectroscopic techniques and SC-XRD, revealing short Ni–Zn distances. Furthermore, an open-shell configuration was assigned to species 129, which makes it an exceptional example within mononuclear Zn-rich molecules that strictly follow closed-shell 18 valence electron counting. Furthermore, the discovery of such Ni2Zn7Al species from heterobimetallic Ni–Al complexes opens the door to new strategies for the synthesis of larger heterometallic aggregates.
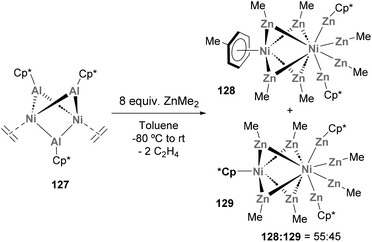 |
| | Scheme 30 Reactivity of Ni2Al3 cluster towards transmetallation with ZnMe2. | |
In summary, the synthesis and reactivity of heterobimetallic species featuring a Ni–Al bond have opened new avenues in C–H functionalization, small molecule activation, production of new metallic clusters, and, importantly, catalytic processes for the functionalization of organic molecules. It is worth mentioning that, although not emphasized in this review, Ni–Al species have also shown promise as efficient catalysts for polymerization.195 The pioneering studies reported in this section have laid the foundation for further exploration. In fact, we believe future research in this area will likely focus on developing catalytic systems and exploring the potential of heterobimetallic Ni–Al species in synthetic transformations.
3.9 Copper
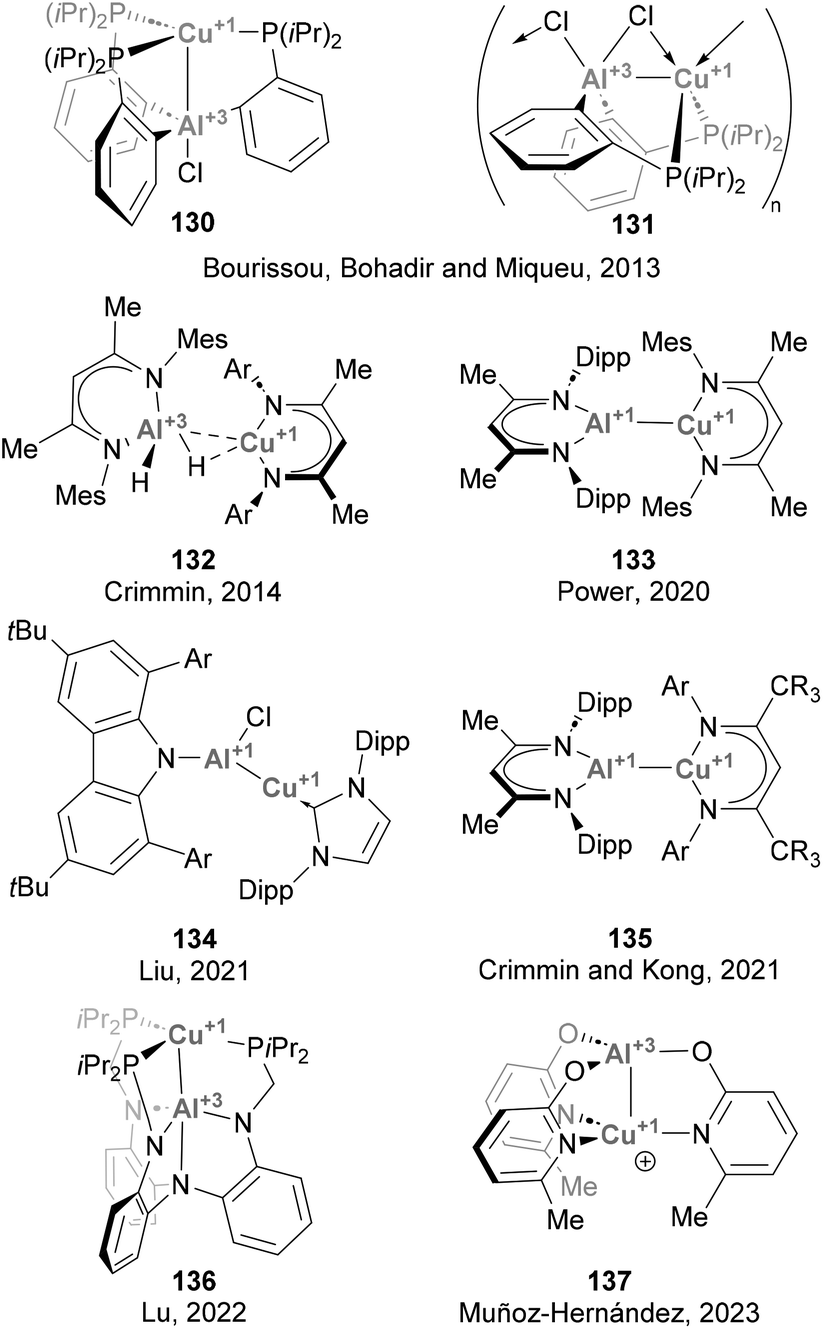
Forging Cu–Al bonds has attracted increasing attention in recent years, particularly since the discovery of aluminyl anions.48,49,51 To date, more than a dozen of examples have been reported, although most of them only contributed to expanding the collection of structures featuring a Cu–Al intermetallic bond, and their reactivity was not further investigated (Scheme 31). The synthesis of compounds featuring Cu–Al bonds can be tracked down to a study from 2013 by Bourissou, Bouhadir, Miqueu and co-workers,158 who reported two complexes featuring Z-type Cu(I)–Al(III) interactions, 130 and 131. After this groundbreaking example, a series of complexes featuring Cu–Al bonds was recently synthesised and fully characterised by Crimmin (132 and 135),125,196 Power (133),197 Liu (134),126 Lu (135)198 and Muñoz-Hernández (137).199 Synthesis of such heterobimetallic Cu–Al species was achieved through conventional methods, such as ligand substitution and direct complexation of neutral low-valent Al(I) species, while salt metathesis has been more extensively utilised when Cu was paired to Al starting from Al(I) anions, as reported by Hill and McMullin and Aldridge.200,201 While the species depicted in Scheme 31 were not tested towards small molecule activation, the reactivity of Cu–Al species with X-type Al(I) ligands has seen significant advances during the last couple of years (see below), demonstrating promising potential for catalysis.
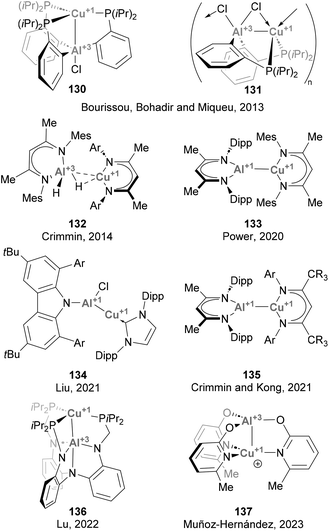 |
| | Scheme 31 Structurally characterised Cu–Al complexes. | |
3.9.1 Small molecule activation.
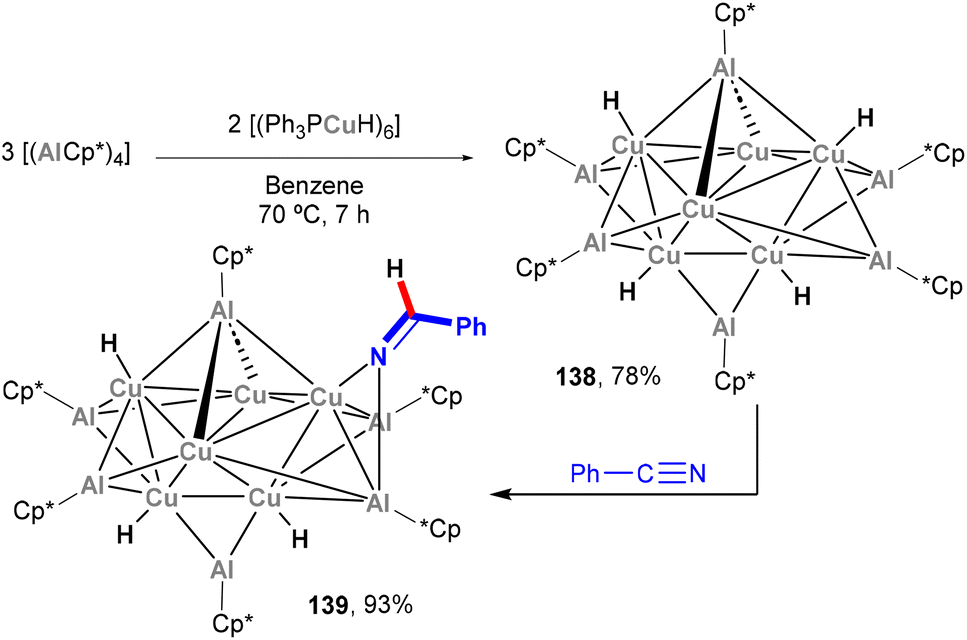
In 2014, a report on the synthesis and reactivity of the heterobimetallic Cu6Al6 cluster was published by Fischer and co-workers (Scheme 32).202 Species 138, which is synthesised by treating [(Ph3PCu)6H6] with Cp*Al in a 2:3 ratio, was revealed to contain a Cu6 unit inside an octahedral Al6 moiety, with the resulting Cu6Al6 core being wrapped in a shell of Cp* and H ligands. Interestingly, species 138 offers the opportunity to investigate the surface reactivity of the (Cu6Al6)H4 unit towards hydrogenation of unsaturated organic functional groups. Indeed, 138 was shown to react with benzonitrile, as depicted in Scheme 32, furnishing 139 in quantitative yield as the 1:1 insertion product. The reactivity of these clusters was not studied in further detail, but their hydrogenation ability exemplifies the potential of complexes bearing Cu–Al bonds in small molecule activation and functionalisation of chemical feedstock.
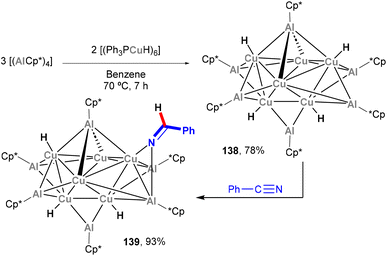 |
| | Scheme 32 Synthesis of Cu6Al6 clusters and their surface reactivity with PhCN. | |
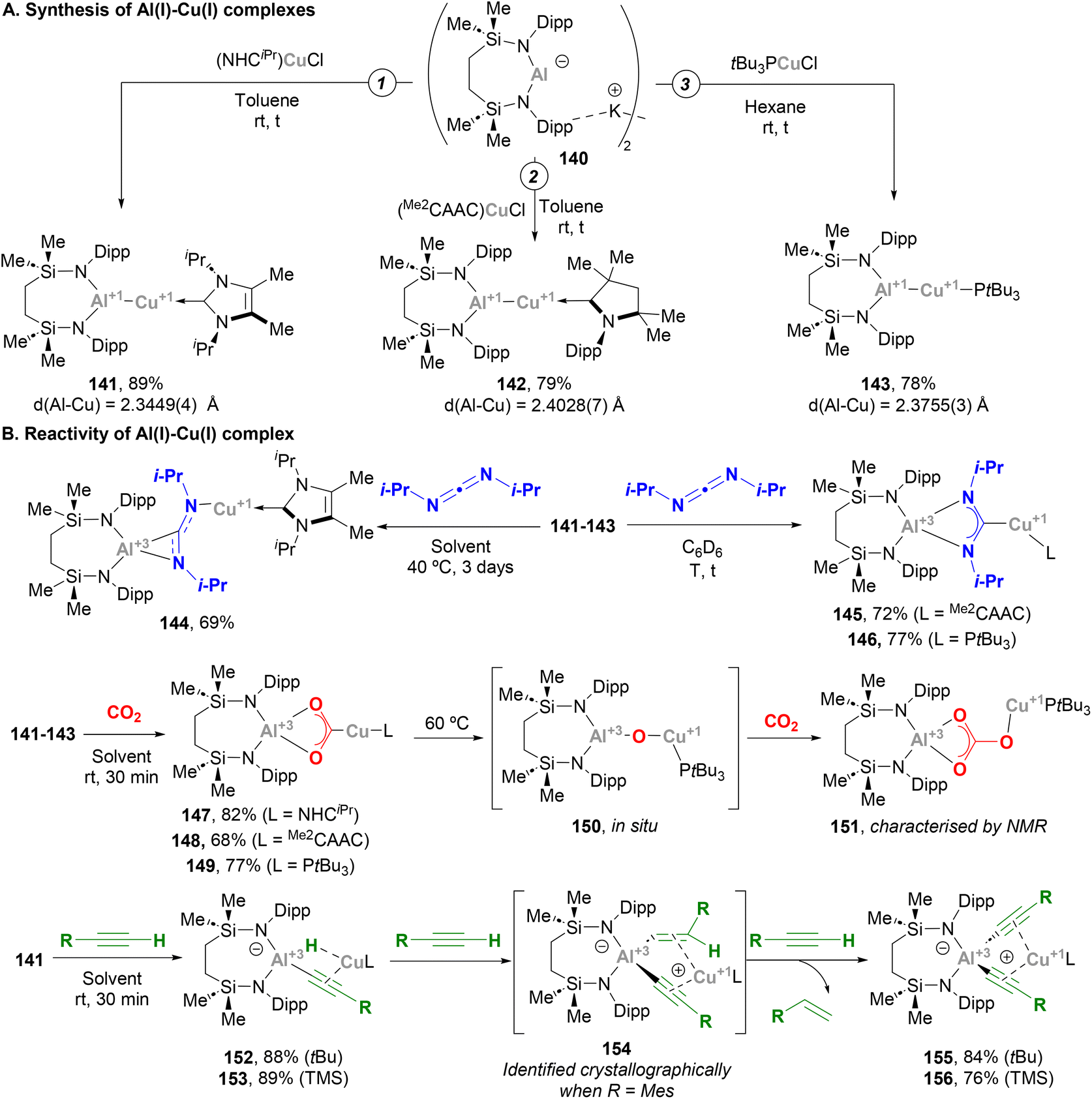
McMullin, Hill and co-workers have largely contributed to this area during the last three years. Inspired by Aldridge's breakthrough in the field of anionic Al(I) species,48 the authors designed a novel seven-membered heterocyclic potassium diamidoalumanyl salt (140, Scheme 33) with formula [K{Al(SiNDipp)}]2.203 Through a salt elimination route, anionic Al(I) species 140 was treated with Cu(I) chloride carbene complexes, [(NHCiPr)CuCl] and [(Me2CAAC)CuCl],200 which acted as precursors to synthesise heterobimetallic Cu–Al species 141 and 142 in 89% and 79% yield, respectively (Scheme 33A, pathways 1 and 2). In a later work by Hill, Mahon and McMullin, the same methodology was employed for the synthesis of Cu phosphine complex 143 in 78% yield.204 The three complexes were isolated and characterised through SC-XRD, unveiling Cu–Al (141: 2.3449(4) Å, 142: 2.4028(7) Å, 143: 2.3755(3) Å) and Al–N (141: 1.8464(10) Å, 142: 1.8607(18) Å, 143: 1.848(1) Å) distances in agreement with previously reported complexes (Scheme 31), suggesting a dative X-type Al(I) → Cu(I) bond. The nature of the Cu–Al intermetallic bond was further studied by testing the reaction of Cu–Al complexes with heteroallenes. Thus, when 141–143 were treated with N,N′-diisopropylcarbodiimide, complexes 144–146 were obtained in excellent yields (Scheme 33B, in blue). Noteworthy, as shown in Scheme 36B, the insertion mode of N,N′-diisopropylcarbodiimide into the Cu–Al bond is modulated by the ligand attached to the Cu centre, furnishing a Cu–C bond (145 and 146) or a Cu–N bond (144). Reaction of heterobimetallic species featuring a Cu–Al bond with CO2 resulted in 147–149 in good yields (Scheme 33B, in red). Interestingly, CO2 inserts into the Cu–Al bond, which cleaves after activation with concomitant oxidation of Al(I) to Al(III). These results highlight the ambiphilic character of the aluminyl–Cu species. Indeed, computational mechanistic insight into these transformations indicates a polarised Cuδ−–Alδ+ bond, in which the Al centre acts as an electrophilic site and the Cu atom as a nucleophile. Decomposition of species 149 results in oxo-bridged species 150, which reacts with another molecule of CO2 to eventually furnish carbonate-bridged species 151. Further computational studies by Lin, Sheong and co-workers205 concluded that the rate-determining insertion of CO2 and carbodiimide is followed by several rearrangements, supporting McMullin and Hill's work. The insertion, however, is proposed to be initiated by the nucleophilic attack of the σ-(Cu–Al) bond on the heteroallene coupling partner, providing valuable insight for developing future Cu–Al-mediated reactivity.
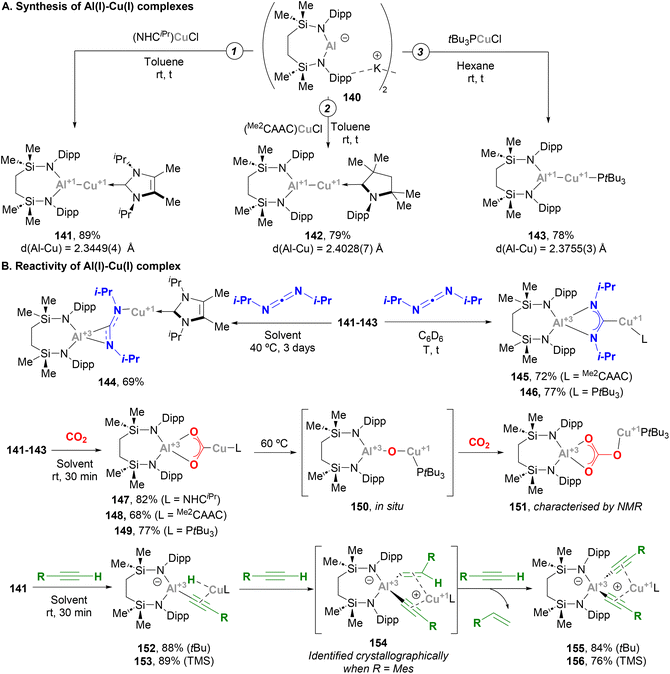 |
| | Scheme 33 Synthesis and reactivity of Cu–Al species reported by Mohan, McMullin and Hill. | |
Indeed, Hill and McMullin recently investigated the reactivity of Cu–Al species towards terminal alkynes.206 When complex 141 was treated with three equivalents of a terminal alkyne RCCH, bis(alkynyl)aluminate complex 155 or 156 were obtained with concomitant extrusion of the corresponding alkene (Scheme 33B, in green). An in-depth study allowed the authors to determine the sequence of intermediates involved in this transformation. Thus, after a first insertion of the Cu–Al bond into the alkyne C–H bond, cuprous (hydrido)(alkynyl)aluminates 152 and 153 are formed, in a transformation that could be classified as a Csp–H activation event. This step is followed by a hydroalumination reaction of a second molecule of alkyne, which furnishes (E-alkenyl)(alkynyl)aluminates. These intermediates could be isolated and characterised crystallographically only when alkynes bearing bulky aryl substituents were used (154). Finally, the last RCCH equivalent undergoes σ-bond metathesis with the alkenyl fragment of the (E-alkenyl)(alkynyl)aluminate via a Cu-acetylide intermediate, releasing the corresponding alkene and bis(alkynyl)aluminates 155 and 156 after Cu-to-Al alkyne migration. It is important to note that the experimental study was fully supported by DFT calculations, which provided further insight into the mechanism of this unprecedented reaction. The overall process represented a distinctive scenario of alkyne transfer semi-hydrogenation, in which the combination of the protic acetylenic C–H bond and the reducing ability of the σ-(Cu–Al) bond furnishes the necessary hydride source.
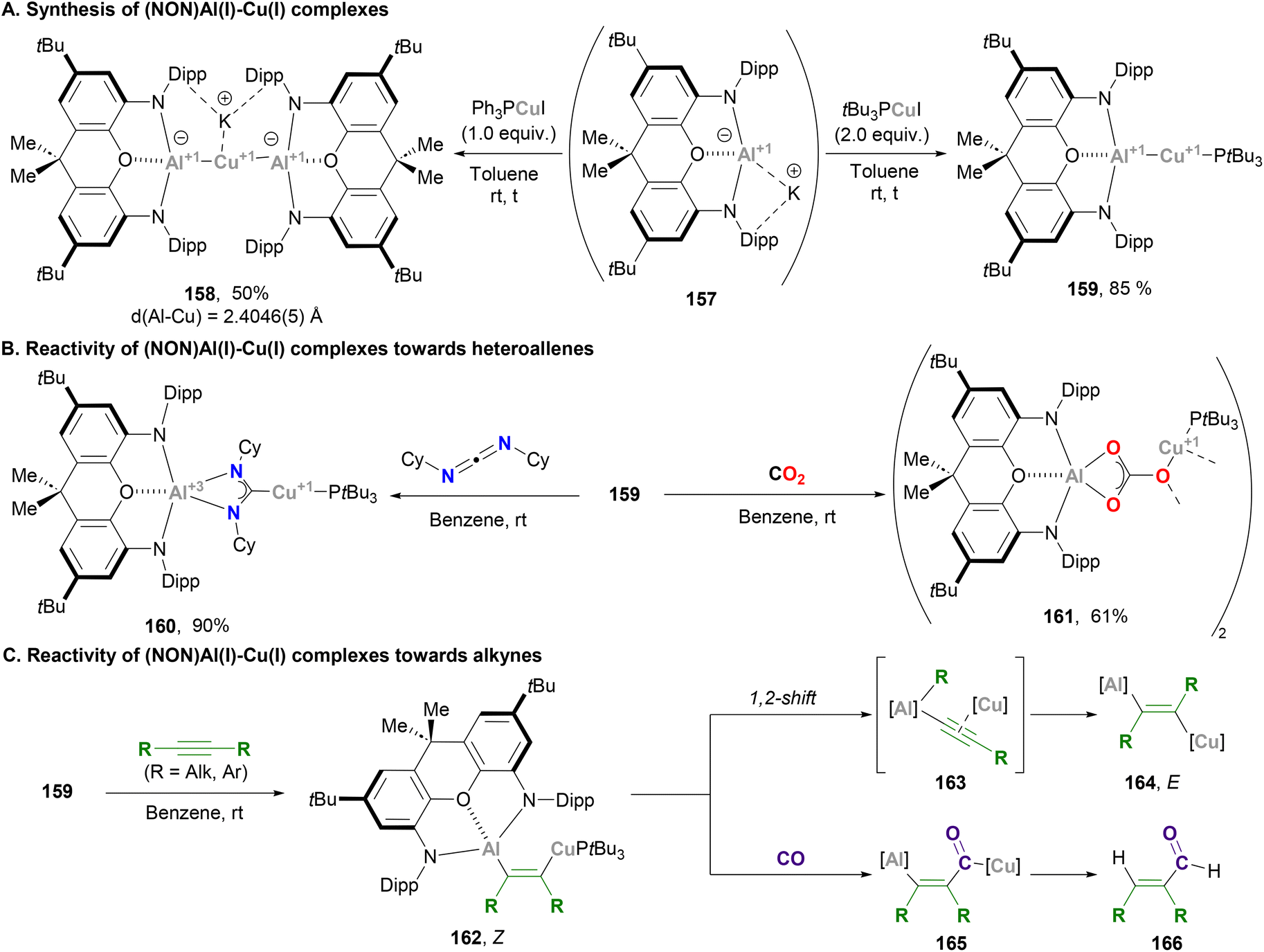
Aldridge and Goicoechea took advantage of their ground-breaking nucleophilic aluminium anion reported in 2018,48 and together with Zhao and Frenking contributed to the development of heterobimetallic complexes pairing aluminium with Cu. Following McMullin and Hill's work on Cu–Al species,200 Aldridge reported the synthesis of xanthene-supported Al(I)-coinage metal bimetallics and their reactivity towards CO2 insertion reactions (Scheme 34). The xanthene-supported Al(I) anion 157 was treated with equimolar amounts of copper–phosphine complex Ph3PCuI, affording the bisaluminyl cuprate complex 158 in 50% yield after salt elimination and ligand exchange (Scheme 34A, left). The solid-state structure of 158 features a Cu–Al bond average distance of 2.4046(5) Å, which is shorter than the sum of their covalent radii (2.53 Å) and in agreement with previously reported Cu(I)–Al(I) species (vide supra). With the goal of synthesising 1:1 bimetallic systems with Cu salts that allow retention of the Cu–P linkage, phosphines with stronger σ-donor abilities were explored. Indeed, when the reaction was performed with 2.0 equivalents of tBu3PCuI, species 159 was obtained (Scheme 34A, right). Albeit there was no SC-XRD of this complex, its structure was unambiguously assigned by means of spectroscopic experiments and comparison with Ag and Ga analogues. After structural characterisation studies, the reactivity of Cu–Al bonds was interrogated with heteroallenes, including CO2. Thus, reaction of complex 159 with N,N′-diisopropylcarbodiimide led to the formation of species 160 (Scheme 34B, pathway 1), whereas stirring 159 under a CO2 atmosphere led to carbonate 161 (Scheme 34B, pathway 2). Mechanistic studies unveiled that, similarly to previous examples,200 these products arise from an initial nucleophilic attack of the σ-(Cu–Al) bond on the heteroallene, which yields species featuring μ-κ1(C):κ2(E,E′) bridging units and Cu–C/Al–E bonds. Formation of carbonate species 161 was rationalised via a similar mechanism compared with species 151 (Scheme 33), which involves a rate-limiting CO extrusion and the intermediacy of a reactive μ-oxide Cu–O–Al species.
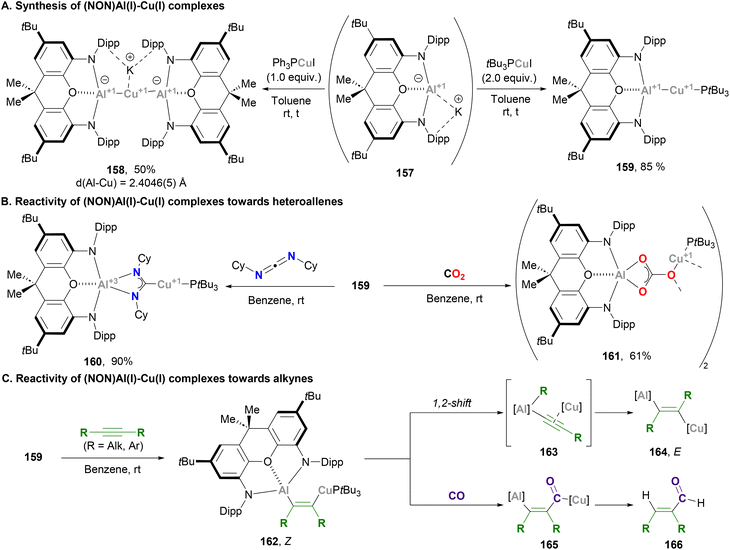 |
| | Scheme 34 Synthesis and reactivity of Cu–Al species reported by Frenking, Goicoechea and Aldridge. | |
A follow-up study from Aldridge and co-workers focused on the reactivity of xanthene-based heterobimetallic Cu–Al systems towards C–C triple bonds (Scheme 34C).207 Contrary to the Cu–Al system reported by Hill and McMullin,206 when alkynes are reacted with 159, spectroscopic features of the product suggest the formation of Z-alkene 162. Indeed, SC-XRD analysis of the product with R = Et shows addition of the Cu–Al unit across the alkyne triple bond in syn fashion, which represents the first example of an (aluminylalkenyl)copper complex and the first structurally characterised 1,2-hetero-dimetallation by insertion of an alkyne into a heterobimetallic M–M′ bond. Interestingly, if the reaction times are extended complete consumption of 162 is observed, and spectroscopic analysis of the crude reaction indicates formation of the corresponding anti dimetallated alkene 164. The reaction mechanism of this isomerisation from Z to E is reminiscent of a recent report by Yamashita and co-workers describing the chemistry of B–Au species.208 Based on this mechanism, isomeration from 162 is proposed to occur through Cu-alkynyl intermediate 163, which can produce formal anti or syn dimetallation products after a 1,2-shift of the R group. Evidence of this mechanism was obtained by isolating the corresponding (NON)Al–Et complex, whose formation unambiguously demonstrates that C-to-Al migration is viable. Computational studies from Sorbelli, Belpassi and Belanzoni provided further insight into this mechanism and supported the idea of a cooperative radical-like reactivity of the Cu–Al bond.209 Reactivity of 162, which contains a Cu–C and an Al–C bond, was also explored in the presence of CO. This resulted in an unprecedented copper acyl complex (165) after insertion of CO into the Cu–C bond, while the Al–C bond remained intact. It is important to note that this reaction was observed exclusively with the Z-(aluminylalkenyl)copper complex, which highlights the role played by Al, which offers additional stabilisation of the acyl unit in 165via an O → Al interaction. In addition, hydrolysis of such carbonylated species results in the formation of α,β-unsaturated aldehydes 166via the net hydroformylation of an alkyne.
Overall, the recent eruption of heterobimetallic species featuring a Cu–Al bond has the potential to revolutionise the field of small molecule activation, as they present unprecedented reactivity modes. Based on the examples reported in this section, together with the breadth of application of boryl-based Cu species in organic synthesis,43 the field of aluminyl–Cu species is expected to grow in the future, providing new and unexplored reactivity avenues for the most abundant, benign and inexpensive coinage metal.
3.10 Zinc
Heterobimetallic complexes bearing a covalent Zn–Al bond were reported a few years ago by Fischer and co-workers while studying the reactivity of 1st-row transition metals complexes bearing btsa ligands towards ECp* (E = Al, Ga) units.72 Insertion of AlCp* into the Zn–N bond of Zn(btsa)2 resulted in complex 167, with concomitant reduction of the Zn centre and formation of the first example of a heterobimetallic complex featuring a Zn–Al bond. Small molecule activation pathways of this early example, together with a recent Zn–Al species reported by Paparo and Jones (168), were not further investigated (Scheme 35).
 |
| | Scheme 35 Structurally characterised Zn–Al complexes. | |
3.10.1 Small molecule activation.
The reactivity of heterobimetallic complexes featuring a Zn–Al bond remained unexplored until 2018, when Crimmin and co-workers synthesised a collection of heterobimetallic complexes that included an aluminium–base-metal complex featuring an L-type Zn–Al interaction.210 To do so, neutral Al(I) precursor NacnacDippAl (169) was treated with NacnacMesZnEt (170), affording complex 171 (Scheme 36A) after a formal insertion of the Al(I) reagent into a Zn–C bond. SC-XRD analysis of such species revealed a Zn–Al bond length of 2.4877(10) Å, which was found to be similar to the Zn–Al distances observed by Fischer (167, 2.448(2) Å).72 The reactivity of these complexes was tested for C–F bond activation. Contrary to heterobimetallic species featuring a Zn–Mg interaction, which proved to be active in C–F bond cleavage, complex 171 remained unreacted when mixed with a variety of fluorinated (hetero)arenes (Scheme 36B). This inactivity towards C–F bond activation was attributed to two main factors: the tetrahedral geometry of the Al centre, which diminished its Lewis acidity; and the steric hindrance of both Nacnac ligands, which disfavours the required disposition in the C–F bond activation TS (172). Additionally, computational studies are in agreement with the experimental results, revealing a large energetic barrier for this transformation with a value of ΔG‡ = 65.1 kcal mol−1.
 |
| | Scheme 36 Synthesis and attempted C–F bond activation with Zn–Al species. | |
In 2021, Harder and co-workers successfully achieved C–F bond activation using a heterobimetallic Zn–Al system.211 In this work, the synthesis of Zn–Al complexes from 169 and 173 was attempted using fluorobenzene as solvent (Scheme 37). Instead of the corresponding heterobimetallic species bearing a Zn–Al bond, the reaction resulted in mixture of a variety of species, from which Zn and Al complexes 174 and 175 were identified. These species are the result of a fast C–F bond activation of fluorobenzene, an unprecedented result with heterobimetallic Zn–Al systems that does not occur with Zn/Ga, Mg/Al or Ca/Al metal pairs.59,212–215 The authors speculated about the possibility of an intermediate species featuring a Zn–Al bond as they were able to isolate and characterise the corresponding Zn–Ga analogue. Nonetheless, computational studies indicate that C–F bond activation does not proceed through a previously formed heterobimetallic complex. Instead, the transformation is proposed to proceed via an FLP-type SNAr reaction in which Al(I) species 169 acts as nucleophile while Zn cation 173 activates fluorobenzene, making it more electrophilic.
 |
| | Scheme 37 Successful C–F bond activation with Zn–Al species via FLP-type mechanism. | |
The synthesis of an unambiguously characterised low valent Zn(II)–Al(I) complex was recently reported by Aldridge, Goicoechea and co-workers taking advantage of the practicality of the salt elimination approach with anionic Al(I) species 158.216 Indeed, the reaction between 158 and NacnacMesZnI (176) results in the formation of a well-defined complex featuring a Zn–Al bond in 85% yield (177, Scheme 38A), the structure of which was unambiguously confirmed by SC-XRD analysis. Furthermore, such analysis revealed a Zn–Al bond length of 2.468(1) Å, which falls within the range of previously reported distances for heterobimetallic species with a Zn–Al bond (vide supra). Complementary computational studies suggest a high degree of Zn–Al covalency, which was further tested by means of reactivity towards heteroallenes such as diisopropylcarbodiimide and CO2 (Scheme 38B, pathways 1 and 2, respectively). Although in both cases addition into the Zn–Al bond was observed, furnishing species 178 and 179, only the CO2 adduct could be isolated and fully characterised. As shown in Scheme 38B, insertion of CO2 generates new Zn–C and Al–O bonds, unveiling the nucleophilic character of the Zn centre, the electrophilic role played by the Al atom and the ambiphilic character of the heterobimetallic complex.
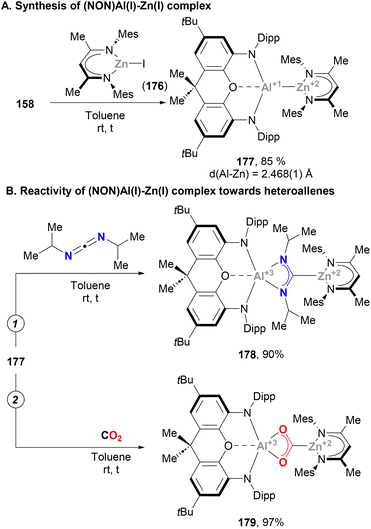 |
| | Scheme 38 Synthesis and reactivity of (NON)Al–Zn species towards heteroallenes. | |
A similar heterobimetallic Zn(II)–Al(I) complex (180) featuring a SIONOSi backbone has been recently reported by McMullin and Coles (Scheme 39).217 Complex 180 presents a Zn–Al bond length of 2.4860(5) Å, almost identical to species 177, and shows the same reactivity pattern with CO2. Interestingly, McMullin and Coles have been able to isolate and fully characterise the corresponding μ-oxo bridge Zn–O–Al species (181), which has been previously proposed to participate as intermediate in the conversion of CO2 to carbonate in bimetallic systems.201 Indeed, when species 181 was treated with CO2, a carbonate species analogous to 162 was obtained, demonstrating the intermediacy of μ-oxo M–O–Al species.
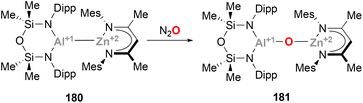 |
| | Scheme 39 Reactivity of (SiNONSi)Al–Zn towards nitrous oxide. | |
4. Conclusion and prospect
In summary, heterobimetallic complexes pairing abundant base metals with aluminium are emerging as promising alternatives to noble metals in small molecule activation, mainly due to the synergy of aluminium with 3d centres. The studies showcased in this review demonstrate the potential of aluminium-based ligands to confer nobility to base metals and their potential to unveil cooperative catalytic methods for the functionalisation of organic molecules. Paralleling recent applications with noble metals (vide supra), implementation of heterobimetallic Al–base-metal complexes in C–X cleavage (X = hydrogen, halogen, ether, amine, etc.) and small molecule activation has gained significant attention, particularly with the development of X-type aluminyl anions. Such species have proved to be exceptional ligands not only for transition metal centres but also for various elements within the main group.203,216,218 We believe that this field will see further advancements combining forces through a multi-disciplinary approach, where the expertise of synthetic and computational chemists will be key to enhance the understanding of fundamental organometallic cooperative events involving heterobimetallic Al–BM complexes. Additionally, efforts should be directed towards expanding the catalytic applications beyond hydrogenation, exploring the integration of heterobimetallic complexes with popular sustainable technologies such as photochemical,219 electrochemical and mechanochemical transformations. Furthermore, it is essential to extend the scope of heterobimetallic sustainable chemistry to encompass the main group elements and explore the potential applications of Al–MG (MG = main group) species in small molecule activation. This expansion would open new avenues for utilizing abundant resources in catalysis and contribute to the development of more sustainable chemical processes. By pursuing these directions, it will be possible to unlock the full potential of aluminium-based heterobimetallic complexes and drive innovation towards a greener and more efficient future in synthetic and coordination chemistry.
Author contributions
All authors participated in the writing, reviewing, and editing of the manuscript. O. P. supervised the project. Additionally, all authors have given their approval for the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support for this work was provided by Queen Mary University of London. This project has received funding from the Royal Society (Research Grant RGS/R1/221326) and the Engineering and Physical Sciences Research Council (EP/X019306/1). S. Fernández thanks the “Fundación Ramón Areces” for a Postdoctoral Fellowship (BEVP34A6835). We thank Dr S. Arseniyadis for insightful discussions and generous support.
References
-
R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, John Wiley & Sons, Hoboken, NJ, 2005 Search PubMed.
-
J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Mill Valley, CA, 2010 Search PubMed.
-
A. de Meijere and F. Diederich, Metal-Catalyzed Cross-Coupling Reactions, Wiley–VCH Verlag GmbH & Co. KGaA., Mörlenbach, Germany, 2004 Search PubMed.
- P. Nuss and M. J. Eckelman, PLoS One, 2014, 9, e101298 CrossRef PubMed.
- R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Science, 2020, 369, eabc3183 CrossRef CAS PubMed.
- P. J. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495–2495 CrossRef CAS PubMed.
- Y. Feng, S. Long, X. Tang, Y. Sun, R. Luque, X. Zeng and L. Lin, Chem. Soc. Rev., 2021, 50, 6042–6093 RSC.
- T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed.
- L. Rocard, D. Chen, A. Stadler, H. Zhang, R. Gil, S. Bezzenine and J. Hannedouche, Catalysts, 2021, 11, 674 CrossRef CAS.
- R. Arevalo and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 9106–9123 CrossRef CAS PubMed.
- T. Zell and R. Langer, ChemCatChem, 2018, 10, 1930–1940 CrossRef CAS.
- P. L. Holland, Acc. Chem. Res., 2015, 48, 1696–1702 CrossRef CAS PubMed.
- P. J. Chirik, K. M. Engle, E. M. Simmons and S. R. Wisniewski, Org. Process Res. Dev., 2023, 27(7), 1160–1184 CrossRef CAS.
- B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed.
- C. B. Larsen and O. S. Wenger, Chem. – Eur. J., 2018, 24, 2039–2058 CrossRef CAS PubMed.
- J. E. Zweig, D. E. Kim and T. R. Newhouse, Chem. Rev., 2017, 117, 11680–11752 CrossRef CAS PubMed.
- B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed.
-
R. J. M. K. Gebbink and M.-E. Moret, Non–Noble Metal Catalysis: Molecular Approaches and Reactions, Wiley–VCH Verlag GmbH & Co. KGaA., Weinheim, Germany, 2019 Search PubMed.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- A. Nakada, T. Matsumoto and H.-C. Chang, Coord. Chem. Rev., 2022, 473, 214804 CrossRef CAS.
- J. I. van der Vlugt, Chem. – Eur. J., 2019, 25, 2651–2662 CrossRef CAS PubMed.
- M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC.
- J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 2012, 363–375 CrossRef CAS.
- M. D. Wodrich and X. Hu, Nat. Rev. Chem., 2017, 2, 1–7 Search PubMed.
- D. G. A. Verhoeven and M.-E. Moret, Dalton Trans., 2016, 45, 15762–15778 RSC.
- K.-S. Feichtner and V. H. Gessner, Chem. Commun., 2018, 54, 6540–6553 RSC.
- A. Singh and D. Gelman, ACS Catal., 2020, 10, 1246–1255 CrossRef CAS.
- D. V. Gutsulyak, W. E. Piers, J. Borau-Garcia and M. Parvez, J. Am. Chem. Soc., 2013, 135, 11776–11779 CrossRef CAS PubMed.
- R. A. Manzano and R. D. Young, Coord. Chem. Rev., 2021, 449, 214215 CrossRef CAS.
- A. Singh and D. Gelman, ACS Catal., 2020, 10, 1246–1255 CrossRef CAS.
- P. M. Keil, T. Szilvási and T. J. Hadlington, Chem. Sci., 2021, 12, 5582–5590 RSC.
- P. M. Keil and T. J. Hadlington, Angew. Chem., Int. Ed., 2022, 61, e202114143 CrossRef CAS PubMed.
- P. M. Keil, A. Soyemi, K. Weisser, T. Szilvási, C. Limberg and T. J. Hadlington, Angew. Chem., Int. Ed., 2023, 62, e202218141 CrossRef CAS PubMed.
- J. Kim, Y.-E. Kim, K. Park and Y. Lee, Inorg. Chem., 2019, 58, 11534–11545 CrossRef CAS PubMed.
- J. Kim, Bull. Korean Chem. Soc., 2022, 43, 538–548 CrossRef CAS.
- J. A. Cabeza and P. García-Álvarez, Chem. – Eur. J., 2023, 29, e202203096 CrossRef CAS PubMed.
- S. González-Gallardo, T. Bollermann, R. A. Fischer and R. Murugavel, Chem. Rev., 2012, 112, 3136–3170 CrossRef PubMed.
- C. Gemel, T. Steinke, M. Cokoja, A. Kempter and R. A. Fischer, Eur. J. Inorg. Chem., 2004, 2004, 4161–4176 CrossRef.
- G. Linti and H. Schnöckel, Coord. Chem. Rev., 2000, 206, 285–319 CrossRef.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef CAS PubMed.
- Y. Segawa, M. Yamashita and K. Nozaki, Organometallics, 2009, 28, 6234–6242 CrossRef CAS.
- D. Hemming, R. Fritzemeier, S. A. Westcott, W. L. Santos and P. G. Steel, Chem. Soc. Rev., 2018, 47, 7477–7494 RSC.
- F. Kong, P. Ríos, C. Hauck, F. J. Fernández-de-Córdova, D. A. Dickie, L. G. Habgood, A. Rodríguez and T. B. Gunnoe, J. Am. Chem. Soc., 2023, 145, 179–193 CrossRef CAS PubMed.
- T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 13672–13683 CrossRef CAS PubMed.
- P. Ríos, A. Rodríguez and J. López-Serrano, ACS Catal., 2016, 6, 5715–5723 CrossRef.
- F. W. Seidel and K. Nozaki, Angew. Chem., Int. Ed., 2022, 61, e202111691 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef CAS PubMed.
- M. P. Coles and M. J. Evans, Chem. Commun., 2023, 59, 503–519 RSC.
- K. Hobson, C. J. Carmalt and C. Bakewell, Chem. Sci., 2020, 11, 6942–6956 RSC.
- M. Zhong, S. Sinhababu and H. W. Roesky, Dalton Trans., 2020, 49, 1351–1364 RSC.
- Y. Liu, J. Li, X. Ma, Z. Yang and H. W. Roesky, Coord. Chem. Rev., 2018, 374, 387–415 CrossRef CAS.
- N. Hara, K. Semba and Y. Nakao, ACS Catal., 2022, 12, 1626–1638 CrossRef CAS.
- A. L. Allred and E. G. Rochow, J. Inorg. Nucl. Chem., 1958, 5, 264–268 CrossRef CAS.
- J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2017, 139, 6074–6077 CrossRef CAS PubMed.
- S. Morisako, S. Watanabe, S. Ikemoto, S. Muratsugu, M. Tada and M. Yamashita, Angew. Chem., Int. Ed., 2019, 58, 15031–15035 CrossRef CAS PubMed.
- N. Hara, T. Saito, K. Semba, N. Kuriakose, H. Zheng, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2018, 140, 7070–7073 CrossRef CAS PubMed.
- I. Fujii, K. Semba, Q.-Z. Li, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2020, 142, 11647–11652 CrossRef CAS PubMed.
- N. Hara, K. Aso, Q.-Z. Li, S. Sakaki and Y. Nakao, Tetrahedron, 2021, 95, 132339 CrossRef CAS.
- R. Seki, N. Hara, T. Saito and Y. Nakao, J. Am. Chem. Soc., 2021, 143, 6388–6394 CrossRef CAS PubMed.
- I. Fujii, K. Semba and Y. Nakao, Org. Lett., 2022, 24, 3075–3079 CrossRef CAS PubMed.
- S. Ogoshi, M. Ueta, T. Aral and H. Kurosawa, J. Am. Chem. Soc., 2005, 127, 12810–12811 CrossRef CAS PubMed.
- C.-S. Wang, S. Di Monaco, A. N. Thai, M. S. Rahman, B. P. Pang, C. Wang and N. Yoshikai, J. Am. Chem. Soc., 2020, 142, 12878–12889 CrossRef CAS PubMed.
- T. Zhang, Y.-X. Luan, N. Y. S. Lam, J.-F. Li, Y. Li, M. Ye and J.-Q. Yu, Nat. Chem., 2021, 13, 1207–1213 CrossRef CAS PubMed.
- R. A. Fischer and J. Weiß, Angew. Chem., Int. Ed., 1999, 38, 2830–2850 CrossRef PubMed.
- C. Gemel, T. Steinke, M. Cokoja, A. Kempter and R. A. Fischer, Eur. J. Inorg. Chem., 2004, 2004, 4161–4176 CrossRef.
- M. J. Butler and M. R. Crimmin, Chem. Commun., 2017, 53, 1348–1365 RSC.
- J. P. Collman, Acc. Chem. Res., 1975, 8, 342–347 CrossRef CAS.
- A. Noor, G. Glatz, R. Müller, M. Kaupp, S. Demeshko and R. Kempe, Nat. Chem., 2009, 1, 322–325 CrossRef CAS PubMed.
- M. Molon, C. Gemel, P. Jerabek, L. Trombach, G. Frenking and R. A. Fischer, Inorg. Chem., 2014, 53, 10403–10411 CrossRef CAS PubMed.
- J. Weßing, C. Göbel, B. Weber, C. Gemel and R. A. Fischer, Inorg. Chem., 2017, 56, 3517–3525 CrossRef PubMed.
- J. M. Burlitch, M. E. Leonowicz, R. B. Petersen and R. E. Hughes, Inorg. Chem., 1979, 18, 1097–1105 CrossRef CAS.
- K. Sugita and M. Yamashita, Chem. – Eur. J., 2020, 26, 4520–4523 CrossRef CAS PubMed , and references within.
- J. P. Collman, R. G. Finke, J. M. Cawse and J. I. Brauman, J. Am. Chem. Soc., 1977, 99, 2515–2526 CrossRef CAS.
- S. M. Rummelt, P. O. Peterson, H. Zhong and P. J. Chirik, J. Am. Chem. Soc., 2021, 143, 5928–5936 CrossRef CAS PubMed.
- G. Feng, K. L. Chan, Z. Lin and M. Yamashita, J. Am. Chem. Soc., 2022, 144, 22662–22668 CrossRef CAS PubMed.
- L. Pauling, J. Am. Chem. Soc., 1947, 69, 542–553 CrossRef CAS.
- K. Ziegler, E. Holzkamp, H. Breil and H. Martin, Angew. Chem., 1955, 67, 541–547 CrossRef CAS.
- G. Natta, J. Polym. Sci., 1955, 16, 143–154 CrossRef CAS.
- E. P. Beaumier, A. J. Pearce, X. Y. See and I. A. Tonks, Nat. Rev. Chem., 2019, 3, 15–34 CrossRef PubMed.
- L. Tebben, C. Mück-Lichtenfeld, G. Fernández, S. Grimme and A. Studer, Chem. – Eur. J., 2016, 23, 5864–5873 CrossRef PubMed.
- M. L. Cooper and J. B. Rose, J. Chem. Soc., 1959, 795–802 RSC.
- E. H. Adema, H. Bos and C. H. Vrinssen, Recl. Trav. Chim. Pays-Bas, 1960, 79, 1282–1288 CrossRef CAS.
- P. E. M. Allen, J. K. Brown and R. M. S. Obaid, Trans. Faraday Soc., 1963, 59, 1808–1814 RSC.
-
G. Natta and F. Danusso, Stereoregular Polymers and Stereospecific Polymerizations, Pergamon Press Ltd, Oxford, United Kingdom, 1967 Search PubMed.
- A. J. Amass, J. N. Hay and J. C. Robb, Br. Polym. J., 1969, 1, 277–281 CrossRef CAS.
- J. N. Hay and R. M. S. Obaid, Eur. Polym. J., 1978, 14, 965–969 CrossRef CAS.
- J. Holton, M. F. Lappert, D. G. H. Ballard, R. Pearce, J. L. Atwood and W. E. Hunter, J. Chem. Soc., Dalton Trans., 1979, 45–53 RSC.
- B. M. Bulychev, S. E. Tokareva, G. L. Soloveichick and E. V. Evdokimova, J. Organomet. Chem., 1979, 179, 263–273 CrossRef CAS.
- V. E. Lvovsky, E. A. Fushman and F. S. Dyachkovsky, J. Mol. Catal., 1980, 10, 43–56 CrossRef CAS.
- B. M. Bulichev, E. V. Evdokimova, A. I. Sizov and G. L. Soloveichik, J. Organomet. Chem., 1982, 239, 313–320 CrossRef CAS.
- E. B. Lobkovskii, G. L. Soloveichik, A. I. Sisov, B. M. Bulychev, A. I. Gusev and N. I. Kirillova, J. Organomet. Chem., 1984, 265, 167–173 CrossRef CAS.
- E. B. Lobkovskii, G. L. Soloveichik, A. I. Sizov and B. M. Bulychev, J. Organomet. Chem., 1985, 280, 53–66 CrossRef CAS.
- J. Poláček, H. Antropiusová, V. Hanuš, L. Petrusová and K. Mach, J. Mol. Catal., 1985, 29, 165–180 CrossRef.
- U. M. Dzhemilev and O. S. Vostrikova, J. Organomet. Chem., 1985, 285, 43–51 CrossRef CAS.
- E. B. Lobkovskii, A. I. Sizov, B. M. Bulychev, I. V. Sokolova and G. L. Soloveichik, J. Organomet. Chem., 1987, 319, 69–75 CrossRef CAS.
- A. I. Sizov, I. V. Molodnitskaya, B. M. Bulychev, E. V. Evdokimova, G. L. Soloveichik, A. I. Gusev, E. B. Chuklanova and V. I. Andrianov, J. Organomet. Chem., 1987, 335, 323–330 CrossRef CAS.
- A. I. Sizov, I. V. Molodnitskaya, B. M. Bulychev, V. K. Bel'skii and G. L. Soloveichik, J. Organomet. Chem., 1988, 344, 185–193 CrossRef CAS.
- T. Y. Sokolova, A. I. Sizov, B. M. Bulychev, E. A. Rozova, V. K. Belsky and G. L. Soloveichik, J. Organomet. Chem., 1990, 388, 11–19 CrossRef CAS.
- M. Bochmann, J. Chem. Soc., Dalton Trans., 1996, 255–270 RSC.
- D. G. Kelly, A. J. Toner, N. M. Walker, S. J. Coles and M. B. Hursthouse, Polyhedron, 1996, 15, 4307–4310 CrossRef CAS.
- J. P. Corden, W. Errington, P. Moore and M. G. H. Wallbridge, Chem. Commun., 1999, 323–324 RSC.
- P. Arndt, A. Spannenberg, W. Baumann, S. Becke and U. Rosenthal, Eur. J. Inorg. Chem., 2001, 2001, 2885–2890 CrossRef.
- M. P. Coles and P. B. Hitchcock, J. Chem. Soc., Dalton Trans., 2001, 1169–1171 RSC.
- V. Tabernero and T. Cuenca, Eur. J. Inorg. Chem., 2005, 2005, 338–346 CrossRef.
- G. B. Nikiforov, H. W. Roesky, P. G. Jones, R. B. Oswald and M. Noltemeyer, Dalton Trans., 2007, 4149–4159 RSC.
- G. K. P. Dathara and D. S. Mainardi, Mol. Simul., 2008, 34, 201–210 CrossRef CAS.
- A. Hernán-Gómez, A. Martín, M. Mena and C. Santamaría, Dalton Trans., 2013, 42, 5076–5084 RSC.
- T. Oswald, M. Diekmann, A. Frey, M. Schmidtmann and R. Beckhaus, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2017, 73, 691–693 CrossRef CAS PubMed.
- P. Corradini and A. Sirigu, Inorg. Chem., 1967, 6, 601–605 CrossRef CAS.
- K. Sugita and M. Yamashita, Organometallics, 2020, 39, 2125–2129 CrossRef CAS.
- E. del Horno, J. Jover, M. Mena, A. Pérez-Redondo and C. Yélamos, Chem. – Eur. J., 2022, 28, e202103085 CrossRef CAS PubMed.
- H. J. de Liefde Meijer, J. W. G. van den Hurk and G. J. M. van der Kerk, Recl. Trav. Chim. Pays-Bas, 1966, 85, 1025–1038 CrossRef CAS.
- A. G. Evans, J. C. Evans and E. H. Moon, J. Chem. Soc., Dalton Trans., 1974, 2390–2395 RSC.
- A. Anundskås and H. A. Øye, J. Inorg. Nucl. Chem., 1975, 37, 1609–1619 CrossRef.
- H. Hagen, J. Boersma and G. van Koten, Chem. Soc. Rev., 2002, 31, 357–364 RSC.
- I. E. Soshnikov, N. V. Semikolenova, A. A. Shubin, K. P. Bryliakov, V. A. Zakharov, C. Redshaw and E. P. Talsi, Organometallics, 2009, 28, 6714–6720 CrossRef CAS.
- M. M. Schulte, E. Herdtweck, G. Raudaschl-Sieber and R. A. Fischer, Angew. Chem., Int. Ed. Engl., 1996, 35, 424–426 CrossRef CAS.
- R. A. Fischer, M. M. Schulte, J. Weiss, L. Zsolnai, A. Jacobi, G. Huttner, G. Frenking, C. Boehme and S. F. Vyboishchikov, J. Am. Chem. Soc., 1998, 120, 1237–1248 CrossRef CAS.
- H. Fölsing, O. Segnitz, U. Bossek, K. Merz, M. Winter and R. A. Fischer, J. Organomet. Chem., 2000, 606, 132–140 CrossRef.
- Q. Yu, A. Purath, A. Donchev and H. Schnöckel, J. Organomet. Chem., 1999, 584, 94–97 CrossRef CAS.
- I. M. Riddlestone, S. Edmonds, P. A. Kaufman, J. Urbano, J. I. Bates, M. J. Kelly, A. L. Thompson, R. Taylor and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 2551–2554 CrossRef CAS PubMed.
- J. A. B. Abdalla, I. M. Riddlestone, J. Turner, P. A. Kaufman, R. Tirfoin, N. Phillips and S. Aldridge, Chem. – Eur. J., 2014, 20, 17624–17634 CrossRef CAS PubMed.
- R. Y. Kong and M. R. Crimmin, Dalton Trans., 2021, 50, 7810–7817 RSC.
- X. Zhang and L. L. Liu, Angew. Chem., Int. Ed., 2021, 60, 27062–27069 CrossRef CAS PubMed.
- X. Cong and X. Zeng, Synlett, 2021, 32, 1343–1353 CrossRef CAS.
- X. Cong and X. Zeng, Acc. Chem. Res., 2021, 54, 2014–2026 CrossRef CAS PubMed.
- I. M. Riddlestone, J. Urbano, N. Phillips, M. J. Kelly, D. Vidovic, J. I. Bates, R. Taylor and S. Aldridge, Dalton Trans., 2012, 42, 249–258 RSC.
- D. W. Agnew, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Dalton Trans., 2017, 46, 6700–6707 RSC.
- R. M. Philip, S. Radhika, C. M. A. Abdulla and G. Anilkumar, Adv. Synth. Catal., 2021, 363, 1272–1289 CrossRef CAS.
- W. Liu and L. Ackermann, ACS Catal., 2016, 6, 3743–3752 CrossRef CAS.
- W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef CAS PubMed.
- R. Jamatia, A. Mondal and D. Srimani, Adv. Synth. Catal., 2021, 363, 2969–2995 CrossRef CAS.
- R. A. Fischer and T. Priermeier, Organometallics, 1994, 13, 4306–4314 CrossRef CAS.
- H. Braunschweig, J. Müller and B. Ganter, Inorg. Chem., 1996, 35, 7443–7444 CrossRef CAS PubMed.
- J. Weiss, D. Stetzkamp, B. Nuber, R. A. Fischer, C. Boehme and G. Frenking, Angew. Chem., Int. Ed. Engl., 1997, 36, 70–72 CrossRef CAS.
- B. N. Anand, I. Krossing and H. Nöth, Inorg. Chem., 1997, 36, 1979–1981 CrossRef CAS PubMed.
- T. Steinke, M. Cokoja, C. Gemel, A. Kempter, A. Krapp, G. Frenking, U. Zenneck and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 2943–2946 CrossRef CAS PubMed.
- C. Jones, S. Aldridge, T. Gans-Eichler and A. Stasch, Dalton Trans., 2006, 5357–5361 RSC.
- B. Buchin, C. Gemel, A. Kempter, T. Cadenbach and R. A. Fischer, Inorg. Chim. Acta, 2006, 359, 4833–4839 CrossRef CAS.
- P. A. Rudd, S. Liu, L. Gagliardi, V. G. Young Jr. and C. C. Lu, J. Am. Chem. Soc., 2011, 133, 20724–20727 CrossRef CAS PubMed.
- G. Tan, T. Szilvási, S. Inoue, B. Blom and M. Driess, J. Am. Chem. Soc., 2014, 136, 9732–9742 CrossRef CAS PubMed.
- T. Agou, T. Yanagisawa, T. Sasamori and N. Tokitoh, Bull. Chem. Soc. Jpn., 2016, 89, 1184–1186 CrossRef CAS.
- D. Dange, C. P. Sindlinger, S. Aldridge and C. Jones, Chem. Commun., 2016, 53, 149–152 RSC.
- T. Yanagisawa, Y. Mizuhata and N. Tokitoh, Heteroat. Chem., 2018, 29, e21465 CrossRef.
- J. Fajardo Jr. and J. C. Peters, Inorg. Chem., 2021, 60, 1220–1227 CrossRef CAS PubMed.
- K. Nakaya, A. Ishii and N. Nakata, Mendeleev Commun., 2022, 32, 71–73 CrossRef CAS.
- S. Sinhababu, M. R. Radzhabov, J. Telser and N. P. Mankad, J. Am. Chem. Soc., 2022, 144, 3210–3221 CrossRef CAS PubMed.
- N. Gorgas, A. J. P. White and M. R. Crimmin, J. Am. Chem. Soc., 2022, 144, 8770–8777 CrossRef CAS PubMed.
- S. Sinhababu and N. P. Mankad, Organometallics, 2022, 41, 1917–1921 CrossRef CAS.
- J. Uddin and G. Frenking, J. Am. Chem. Soc., 2001, 123, 1683–1693 CrossRef CAS PubMed.
- R. Lehmann and M. Schlosser, Tetrahedron Lett., 1984, 25, 745–748 CrossRef CAS.
- R. B. Bates, L. M. Kroposki and D. E. Potter, J. Org. Chem., 1972, 37, 560–562 CrossRef CAS.
- N. Gorgas, A. J. P. White and M. R. Crimmin, Chem. Commun., 2022, 58, 10849–10852 RSC.
- B. Stadler, N. Gorgas, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2023, 62, e202219212 CrossRef CAS PubMed.
- M.-E. Moret and J. C. Peters, J. Am. Chem. Soc., 2011, 133, 18118–18121 CrossRef CAS PubMed.
- M. Sircoglou, N. Saffon, K. Miqueu, G. Bouhadir and D. Bourissou, Organometallics, 2013, 32, 6780–6784 CrossRef CAS.
- P. A. Rudd, N. Planas, E. Bill, L. Gagliardi and C. C. Lu, Eur. J. Inorg. Chem., 2013, 2013, 3898–3906 CrossRef CAS.
- J. P. Campbell and W. L. Gladfelter, Inorg. Chem., 1997, 36, 4094–4098 CrossRef CAS.
- R. F. Moreira, E. Y. Tshuva and S. J. Lippard, Inorg. Chem., 2004, 43, 4427–4434 CrossRef CAS PubMed.
-
T. P. Hanusa, Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, 2015, pp. 1–25 Search PubMed.
- I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- K. E. Schwarzhans and H. Steiger, Angew. Chem., Int. Ed. Engl., 1972, 11, 535–535 CrossRef CAS.
- J. J. Schneider, C. Krüger, M. Nolte, I. Abraham, T. S. Ertel and H. Bertagnolli, Angew. Chem., Int. Ed. Engl., 1995, 33, 2435–2437 CrossRef.
- C. Üffing, A. Ecker, R. Köppe and H. Schnöckel, Organometallics, 1998, 17, 2373–2375 CrossRef.
- A. B. Thompson, D. R. Pahls, V. Bernales, L. C. Gallington, C. D. Malonzo, T. Webber, S. J. Tereniak, T. C. Wang, S. P. Desai, Z. Li, I. S. Kim, L. Gagliardi, R. L. Penn, K. W. Chapman, A. Stein, O. K. Farha, J. T. Hupp, A. B. F. Martinson and C. C. Lu, Chem. Mater., 2016, 28, 6753–6762 CrossRef CAS.
- P. J. Black, Acta Metall., 1956, 4, 172–179 CrossRef CAS.
- M. M. Clark, W. W. Brennessel and P. L. Holland, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, 391–391 CrossRef PubMed.
- M. A. Laffey and P. Thornton, J. Chem. Soc., Dalton Trans., 1982, 313–318 RSC.
- L. Lukasevics, A. Cizikovs and L. Grigorjeva, Chem. Commun., 2021, 57, 10827–10841 RSC.
- M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS.
- S. M. Ujwaldev, N. A. Harry, M. A. Divakar and G. Anilkumar, Catal. Sci. Technol., 2018, 8, 5983–6018 RSC.
- K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208–1219 CrossRef CAS PubMed.
- M. V. Vollmer, J. Ye, J. C. Linehan, B. J. Graziano, A. Preston, E. S. Wiedner and C. C. Lu, ACS Catal., 2020, 10, 2459–2470 CrossRef CAS.
- K. Semba, F. Shimoura and Y. Nakao, Chem. Lett., 2022, 51, 455–457 CrossRef CAS.
- Y. Cao, W.-C. Shih and O. V. Ozerov, Organometallics, 2019, 38, 4076–4081 CrossRef CAS.
- L. Escomel, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2021, 143, 4844–4856 CrossRef CAS PubMed.
- K. Fischer, K. Jonas, P. Misbach, R. Stabba and G. Wilke, Angew. Chem., Int. Ed. Engl., 1973, 12, 943–953 CrossRef.
- K. Ziegler, E. Holzkamp, H. Breil and H. Martin, Angew. Chem., 1955, 67, 426–426 CrossRef CAS.
- J. J. Eisch, X. Ma, M. Singh and G. Wilke, J. Organomet. Chem., 1997, 527, 301–304 CrossRef CAS.
- J. J. Eisch, S. R. Sexsmith and K. C. Fichter, J. Organomet. Chem., 1990, 382, 273–293 CrossRef CAS.
- C. Dohmeier, H. Krautscheid and H. Schnöckel, Angew. Chem., Int. Ed. Engl., 1995, 33, 2482–2483 CrossRef.
- T. Steinke, C. Gemel, M. Cokoja, M. Winter and R. A. Fischer, Angew. Chem., Int. Ed., 2004, 43, 2299–2302 CrossRef CAS PubMed.
- B. Buchin, T. Steinke, C. Gemel, T. Cadenbach and R. A. Fischer, Z. Anorg. Allg. Chem., 2005, 631, 2756–2762 CrossRef CAS.
- M. Molon, T. Bollermann, C. Gemel, J. Schaumann and R. A. Fischer, Dalton Trans., 2011, 40, 10769–10774 RSC.
- Q. Lai, M. N. Cosio and O. V. Ozerov, Chem. Commun., 2020, 56, 14845–14848 RSC.
- R. C. Cammarota and C. C. Lu, J. Am. Chem. Soc., 2015, 137, 12486–12489 CrossRef CAS PubMed.
- M. V. Vollmer, J. Xie, R. C. Cammarota, V. G. Young Jr., E. Bill, L. Gagliardi and C. C. Lu, Angew. Chem., Int. Ed., 2018, 57, 7815–7819 CrossRef CAS PubMed.
- M. V. Vollmer, R. C. Cammarota and C. C. Lu, Eur. J. Inorg. Chem., 2019, 2019, 2140–2145 CrossRef CAS.
- B. J. Graziano, M. V. Vollmer and C. C. Lu, Angew. Chem., Int. Ed., 2021, 60, 15087–15094 CrossRef CAS PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- W. H. Harman and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 5080–5082 CrossRef CAS PubMed.
- E. De Leon, F. Gonzalez, P. Bauskar, S. Gonzalez-Eymard, D. De Los Santos and M. M. Shoshani, Organometallics, 2023, 42, 435–440 CrossRef CAS.
- Z. Weng, S. Teo, L. L. Koh and T. S. A. Hor, Chem. Commun., 2006, 1319 RSC.
- A. E. Nako, Q. W. Tan, A. J. P. White and M. R. Crimmin, Organometallics, 2014, 33, 2685–2688 CrossRef CAS.
- K. L. Mears, C. R. Stennett, E. K. Taskinen, C. E. Knapp, C. J. Carmalt, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2020, 142, 19874–19878 CrossRef CAS PubMed.
- B. J. Graziano, T. R. Scott, M. V. Vollmer, M. J. Dorantes, V. G. Young, E. Bill, L. Gagliardi and C. C. Lu, Chem. Sci., 2022, 13, 6525–6531 RSC.
- O. J. García-de-Jesus, A. Mondragón-Díaz, B. Donnadieu and M.-Á. Muñoz-Hernández, Inorg. Chem., 2023, 62, 2518–2529 CrossRef PubMed.
- H.-Y. Liu, R. J. Schwamm, M. S. Hill, M. F. Mahon, C. L. McMullin and N. A. Rajabi, Angew. Chem., Int. Ed., 2021, 60, 14390–14393 CrossRef CAS PubMed.
- C. McManus, J. Hicks, X. Cui, L. Zhao, G. Frenking, J. M. Goicoechea and S. Aldridge, Chem. Sci., 2021, 12, 13458–13468 RSC.
- C. Ganesamoorthy, J. Weßing, C. Kroll, R. W. Seidel, C. Gemel and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 7943–7947 CrossRef CAS PubMed.
- R. J. Schwamm, M. P. Coles, M. S. Hill, M. F. Mahon, C. L. McMullin, N. A. Rajabi and A. S. S. Wilson, Angew. Chem., Int. Ed., 2020, 59, 3928–3932 CrossRef CAS PubMed.
- H.-Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon and C. L. McMullin, Dalton Trans., 2022, 51, 3913–3924 RSC.
- X. Guo, T. Yang, Y. Zhang, F. K. Sheong and Z. Lin, Inorg. Chem., 2022, 61, 10255–10262 CrossRef CAS PubMed.
- H.-Y. Liu, S. E. Neale, M. S. Hill, M. F. Mahon and C. L. McMullin, Chem. Sci., 2023, 14, 2866–2876 RSC.
- C. McManus, A. E. Crumpton and S. Aldridge, Chem. Commun., 2022, 58, 8274–8277 RSC.
- A. Suzuki, L. Wu, Z. Lin and M. Yamashita, Angew. Chem., Int. Ed., 2021, 60, 21007–21013 CrossRef CAS PubMed.
- D. Sorbelli, L. Belpassi and P. Belanzoni, Inorg. Chem., 2022, 61, 21095–21106 CrossRef CAS PubMed.
- C. Bakewell, B. J. Ward, A. J. P. White and M. R. Crimmin, Chem. Sci., 2018, 9, 2348–2356 RSC.
- A. Friedrich, J. Eyselein, J. Langer, C. Färber and S. Harder, Angew. Chem., Int. Ed., 2021, 60, 16492–16499 CrossRef CAS PubMed.
- D. D. L. Jones, I. Douair, L. Maron and C. Jones, Angew. Chem., 2021, 133, 7163–7168 CrossRef.
- T. X. Gentner, B. Rösch, G. Ballmann, J. Langer, H. Elsen and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 607–611 CrossRef CAS PubMed.
- F. Rekhroukh, W. Chen, R. K. Brown, A. J. P. White and M. R. Crimmin, Chem. Sci., 2020, 11, 7842–7849 RSC.
- A. S. S. Wilson, M. S. Hill, M. F. Mahon, C. Dinoi and L. Maron, Tetrahedron, 2021, 82, 131931 CrossRef CAS.
- M. M. D. Roy, J. Hicks, P. Vasko, A. Heilmann, A.-M. Baston, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 22301–22306 CrossRef CAS PubMed.
- M. J. Evans, G. H. Iliffe, S. E. Neale, C. L. McMullin, J. R. Fulton, M. D. Anker and M. P. Coles, Chem. Commun., 2022, 58, 10091–10094 RSC.
- J. T. Boronski, L. R. Thomas-Hargreaves, M. A. Ellwanger, A. E. Crumpton, J. Hicks, D. F. Bekiş, S. Aldridge and M. R. Buchner, J. Am. Chem. Soc., 2023, 145, 4408–4413 CrossRef CAS PubMed.
- J. T. Moore, M. J. Dorantes, Z. Pengmei, T. M. Schwartz, J. Schaffner, S. L. Apps, C. A. Gaggioli, U. Das, L. Gagliardi, D. A. Blank and C. C. Lu, Angew. Chem., Int. Ed., 2022, 61, e202205575 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Selwin
Fernando
,
Selwin
Fernando