
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and reactivity of Pd(II) imidoyl complexes obtained by insertion of isocyanoferrocene into the Pd–C bonds of orthopalladated precursors†
Michal
Franc
 ,
Petr
Vosáhlo
,
Jiří
Schulz
,
Ivana
Císařová
and
Petr
Štěpnička
*
,
Petr
Vosáhlo
,
Jiří
Schulz
,
Ivana
Císařová
and
Petr
Štěpnička
*
Department of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030, 128 40, Prague, Czech Republic. E-mail: stepnic@natur.cuni.cz
First published on 6th October 2023
Abstract
While the multifaceted reactivity of organic isocyanides has been extensively demonstrated, that of their organometallic analogue, isocyanoferrocene (FcNC; Fc = ferrocenyl), has not yet been adequately explored. This contribution describes the syntheses of novel chelating Pd(II) imidoyl complexes, [(YCH2C6H4C(NFc)-κ2Y,C)PdCl(PR3)], by insertion of FcNC into the Pd–C bond of cyclopalladated precursors [(YCH2C6H4-κ2Y,C)PdCl(PR3)] (Y = Me2N, MeS, R = Ph, Me). The imidoyl complexes underwent facile alkylation with [Me3O][BF4] to produce the cationic aminocarbene complexes [{YCH2C6H4C(N(Me)Fc)-κ2Y,C}PdCl(PR3)][BF4]. All compounds were fully characterised using a combination of spectroscopic methods (NMR, FTIR and ESI MS), cyclic voltammetry and single-crystal X-ray crystallography. In addition, DFT calculations were used to describe the bonding in the two compound families. Analyses with intrinsic bond orbitals (IBOs) and the quantum theory of atoms in molecules (QTAIM) consistently pointed to the transformation of an X-type imidoyl C-ligand (σ-organyl) into an L-type carbene donor upon alkylation, which has only a minor structural consequence. Also reported is the unexpected conversion of the imidoyl complex [(Me2NCH2C6H4C(NFc)-κ2N,C)PdCl(PPh3)] into (Z)-2,2-dimethyl-1-(ferrocenylimino)isoindolin-2-ium tetrafluoroborate as a reductive elimination product, which was induced by Lewis and Brønsted acids.
Introduction
Specific electron properties including partial carbene character1 endow isocyanides with unique reactivity2 and the ability to coordinate transition metals. In addition to being specific σ-donor and π-acceptor ligands,3 coordinated isocyanides readily add alcohols and amines to produce heteroatom-stabilised carbene complexes (reaction A in Scheme 1)4,5 and insert into metal–carbon bonds to afford imidoyl complexes that can be further transformed into aminocarbene complexes by protonation or alkylation (reaction B in Scheme 1).6 | ||
| Scheme 1 Reactions of isocyanides relevant to this work. | ||
We have shown that A and B-type reactions of 1′-(diphenylphosphino)-1-isocyanoferrocene7 afford structurally unique P-chelated aminocarbene complexes (Scheme 2).8 Now, we focus on similar reactions of isocyanoferrocene (FcNC, Fc = ferrocenyl) with cyclometallated Pd(II) precursors. While isocyanide insertions into Pd–C bonds in complexes with orthopalladated ligands are well documented,9,10 subsequent transformations of the resulting imidoyl complexes into carbenes have seldom been reported.6
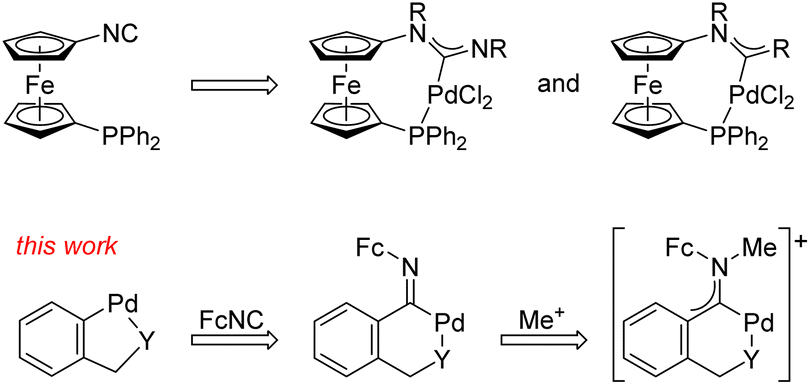 | ||
| Scheme 2 Examples of P-chelated Pd(II) carbene complexes accessible from 1′-(diphenylphosphino)-1-isocyanoferrocene and the aims of the present work focused on isocyanoferrocene (FcNC; Fc = ferrocenyl, Y = NMe2 or SMe). | ||
In this contribution, we describe our study of the reactions between FcNC11,12 and Pd(II) complexes containing orthopalladated benzylamine and benzyl thioether ligands that produced chelating imidoyl complexes, which were subsequently converted into ferrocene-substituted13 Fischer-type aminocarbene complexes with redox-active ferrocenyl pendants by alkylation (Scheme 2, bottom). In particular, we report the syntheses of two series of Pd(II) complexes (imidoyl and aminocarbene) and detailed structural characterisation using a combination of spectroscopic methods, X-ray diffraction analysis, cyclic voltammetry, and DFT calculations. The results of reactivity studies on the imidoyl complexes, which led to the isolation of a structurally uncommon, reductive elimination product, are also reported.
Results and discussion
Syntheses and structural characterisation
The orthopalladated complexes 1 and 2 were synthesised by cleavage of the respective dimeric precursors [(LCY)PdCl]2 with trimethylphosphine and triphenylphosphine (Scheme 3). The reactions with the more stable and easier-to-dose PPh3 were performed using stoichiometric amounts of the starting materials and afforded pure 1a and 2a in practically quantitative yields. The analogous reactions with PMe3 required a slight excess of this air-sensitive phosphine to avoid incomplete conversion of the dimeric Pd(II) precursors. While cleavage of the “benzylamine” complex [(LCN)PdCl]2 (LCN = 2-Me2NCH2C6H4-κ2N,C1) proceeded selectively and produced pure 1b in virtually quantitative yield after simple chromatographic purification, the reaction with the thioether analogue, [(LCS)PdCl]2 (LCS = 2-MeSCH2C6H4-κ2S,C1), gave 2b contaminated with the bis-phosphine complex trans-[(2-MeSCH2C6H4-κC1)PdCl(PMe3)2] (trans-3), arising via decomplexation of the thioether arm by excess PMe3.14 The unwanted formation of trans-3 was suppressed by lowering the amount of PMe3 to a strictly stoichiometric amount (Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PMe3 = 1:1). Conversely, increasing the amount of PMe3 to 2 equiv. per Pd resulted in the selective formation of trans-3, which was isolated and fully characterised, including structure determination (see ESI, Fig. S1†).
:PMe3 = 1:1). Conversely, increasing the amount of PMe3 to 2 equiv. per Pd resulted in the selective formation of trans-3, which was isolated and fully characterised, including structure determination (see ESI, Fig. S1†).
 | ||
| Scheme 3 Syntheses of imidoyl complexes 4 and 5 and their subsequent conversion into carbene complexes 6 and 7 by methylation (all reactions were performed in dichloromethane). | ||
In the next step, compounds 1 and 2 were treated with isocyanoferrocene (1 equiv.) in dichloromethane to produce the respective imidoyl complexes 4 and 5 (Scheme 3). Subsequent methylation with the Meerwein salt, [Me3O][BF4], in CH2Cl2 converted these insertion products into the cationic aminocarbene complexes 6 and 7. Both the imidoyl and carbene complexes were purified by column chromatography over silica gel and isolated in generally good yields (75–90%).
Imidoyl complexes 4 and 5 displayed ions due to [M − Cl]+ in the ESI MS spectra. Their 1H and 13C NMR spectra showed four separate signals for the CH groups of the ferrocene C5H4 ring, which suggested fixed conformation that rendered these groups diastereotopic (at room temperature). The LCS complexes were inherently chiral due to the presence of a stereogenic sulfur atom, which itself made the protons at the ferrocene C5H4 ring diastereotopic. One of the C5H4 signals in the 1H NMR spectrum was always observed at a markedly lower field (δH 6.1–6.4) than the remaining three signals, very likely due to shielding anisotropy. The diastereotopic methyl groups (only for LCN) and methylene protons also gave rise to separate signals. The 3JPC and 4JPH coupling constants observed for the CH2NMe2 groups in 4a and 4b were consistent with the trans-P,N arrangement; the resonances due to the CH2SMe arms were typically observed as broad signals.
The 13C NMR signals of the Pd-bound imidoyl carbons in 4 and 5 were observed near 190 ppm (d, JPC = 2–3 Hz), and the spectra also showed characteristic, low-field signals for the ferrocene Cipso–N carbons, which were split into doublets via interactions with phosphorus (δP 104–106, JPC = 4–6 Hz; cf. δC 105.6 for FcNH215). The 31P NMR signals of 4 and 5 were detected upfield of those for 1 and 2. However, the magnitude of the shift was considerably larger for the triphenylphosphine complexes (ΔP = −16.5 and −13.5 ppm for 4a and 5a, respectively) than for their PMe3 analogues (ΔP = −1.3 ppm for 4b, and −1.4 ppm for 5b). The IR bands attributable to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching modes were observed at approximately 1600 cm−1.
N stretching modes were observed at approximately 1600 cm−1.
Methylation of the imidoyl complex to give Fischer carbenes 6 and 7 altered the NMR spectra rather marginally. The salient change was a downfield shift of the 13C NMR signal due to the Pd-bound carbon atom, as is typical for carbene complexes (δC 227–230 ppm), and the appearance of additional signals due to the introduced methyl group (δH ≈ 3.4–4.0, δC ≈ 48). The presence of the BF4− anion was corroborated by intense and structured ν3 bands at approximately 1060 cm−1 in the IR spectra.16 Consistent with their ionic nature, the carbene complexes displayed strong peaks due to [M − BF4]+ in their ESI MS. Another notable feature was a colour change from orange to red noted visually during the synthesis and illustrated by UV-vis measurements for the pair of compounds 4b and 6b (Fig. 1). In the visible region, imidoyl complex 4b displayed an absorption band at 449 nm (ε = 710 M−1 cm−1 in CH2Cl2), which was bathochromically shifted with respect to that of ferrocene itself (440 nm in ethanol)17 due to the extended conjugated system. After alkylation to give 6b, the absorption band shifted further to lower energies (485 nm), and its intensity increased (ε = 1555 M−1 cm−1).
 | ||
| Fig. 1 UV-vis spectra of 4b, 6b and 8 (c = 0.50 mM in CH2Cl2; optical path: 1 cm). | ||
The structures of all imidoyl and carbene complexes were determined by single-crystal X-ray diffraction analysis. The structures of the representative imidoyl complexes 4a and 5b are shown in Fig. 2 (complete structure diagrams are available in the ESI†). Selected geometric parameters are summarised in Table 1. All compounds crystallised with one structurally independent molecule except for 4b·0.35H2O, which contained two almost identical complex molecules in the asymmetric unit and a water molecule with partial occupation.
 | ||
| Fig. 2 Molecular structures of 4a and 5b (for displacement ellipsoid plots and additional structure diagrams, see the ESI†). | ||
| Parameter | 4a | 4b·0.35H2Ob | 5a | 5b |
|---|---|---|---|---|
| a φ is the torsion angle C11–N1–C1–C2; C2 is the pivotal carbon atom from the C(2-7) phenyl ring bonding to C1. b Data for molecule 1/molecule 2. c The Pd-bound SMe group was disordered and was refined over two positions; parameters for the dominant orientation are given. | ||||
| Y | N2 | N2/N4 | S1 | S1 |
| Pd1–C1 | 1.986(3) | 1.979(2)/1.972(2) | 1.994(1) | 1.994(3) |
| Pd1–Y | 2.201(2) | 2.210(1)/2.202(1) | 2.386(2) | 2.3858(7) |
| Pd1–P1 | 2.2657(8) | 2.2513(5)/2.2495(5) | 2.2916(6) | 2.2705(7) |
| Pd1–Cl1 | 2.4156(7) | 2.4122(5)/2.4156(6) | 2.4091(5) | 2.4115(7) |
| C1–Pd1–Y | 85.64(9) | 87.46(6)/87.26(6) | 86.80(7) | 88.34(7) |
| C1–Pd1–P1 | 94.39(7) | 92.16(5)/90.60(5) | 94.58(4) | 90.87(7) |
| Cl1–Pd1–Y | 89.51(6) | 92.50(4)/93.45(4) | 85.80(7) | 90.84(2) |
| Cl1–Pd1–P1 | 90.39(3) | 87.88(2)/88.82(2) | 92.77(2) | 89.87(2) |
| C1–N1 | 1.265(3) | 1.274(2)/1.278(2) | 1.271(2) | 1.272(3) |
| N1–C11 | 1.410(4) | 1.412(2)/1.409(2) | 1.404(2) | 1.409(3) |
| N1–C1–C2 | 117.6(2) | 118.4(1)/119.2(1) | 117.6(1) | 119.0(2) |
| C1–N1–C11 | 123.3(2) | 122.7(1)/122.1(1) | 123.7(1) | 122.1(2) |
| φ | −176.2(2) | 176.3(1)/−177.3(1) | −178.5(1) | 179.3(2) |
In all imidoyl complexes, the coordination sphere of the Pd atom was essentially planar, and the interligand angles did not depart significantly from 90°. The Pd-donor distances were unexceptional and varied only slightly over the entire series, except for the Pd–Y bonds. Notably, the influence of the Y atom on the Pd–P bond in the trans position was also small (the Pd–P bond was approximately 0.02 Å longer in 5a/5b than in 4a/4b due to the larger trans influence18 of the S-donor group). In addition, the Pd–PPh3 bonds were somewhat longer than the Pd–PMe3 bonds for steric reasons.
The Pd–C (≈1.97–1.99 Å) and C1–N1 (≈1.27 Å) bond lengths in 4 and 5 were similar to those reported for [Pd{C(R1)NR2}Cl(PR3)2] (R1 = 2-pyridyl, R2 = 4-MeOC6H4, PR3 = PMe2Ph;19 R1 = Me, R2 = 2,6-Me2C6H3, (PR3)2 = dppe;20 and R1 = Me, R2 = 2-vinylphenyl, PR3 = PEt321) and for the chelate compounds obtained from 1′-(diphenylphosphino)-1-isocyanoferrocene,7 while the C1–N1 bond lengths did not differ much from those of ferrocene imines FcCHNAr (Ar = aryl).22 The arrangements at the C1N1 bonds were anti (torsion angles C2–C1–N1–C11 are 176–179°) and the attached ferrocene units were rotated by 21–25° with respect to the {N1,C1,C2} plane (the range of the dihedral angles between the C(11–15) ring and the carbene plane is given), thereby maintaining conjugation.23 The ferrocene moieties adopted their usual geometries with similar Fe–C distances and negligible tilt angles (<3°). Finally, the C6H4 ring attached to the carbene C1 atom was twisted by approximately 70° from the plane defined by the palladium and its four ligating atoms and by 71–89° from the “imidoyl” {N1,C1,C2} plane.
The structures of aminocarbene complexes 6a·CHCl3 and 7a are displayed in Fig. 3 (for additional structure plots, see the ESI†). Inspection of the data outlined in Table 2 and comparison with those of 4 and 5 revealed that N-methylation to give aminocarbene complexes affected the coordination sphere of Pd(II) only marginally. Compared to the respective imidoyl precursors, the carbene complexes showed shorter Pd–Cl bonds (by 0.05–0.06 Å), presumably due to the decreased trans influence,24 and slightly shorter Pd–C distances (0.01–0.03 Å) due to increased Pd–C bond orders. Correspondingly, the N1–C1 bond was longer (≈0.03 Å), which suggested partial loss of the double bond (imine) character; similar elongation was observed for the N1–C11 bond. The presence of a third substituent at the N atom resulted in opening of the N1–C1–C2 angle to 120°, while the C1–N1–C11 angle remained almost unchanged. Individual parameters compared well with the values reported for similar compounds featuring cyclic pyridine-2-ylidene ligands.25
 | ||
| Fig. 3 Molecular structures of 6a·CHCl3 and 7a (for displacement ellipsoid plot and additional structural diagrams, see the ESI†). The counter ions ([BF4]−) and the solvent molecule were omitted for clarity. | ||
| Parameter | 6a·CHCl3 | 6b | 7a | 7b |
|---|---|---|---|---|
| a φ is the torsion angle C11–N1–C1–C2; C2 is the pivotal carbon atom from the C(2-2) phenyl ring bonding to C1. b The Pd-bound SMe group was disordered and was refined over two positions; parameters for the dominant orientation are given. | ||||
| Y | N2 | N2 | S1 | S1 |
| Pd1–C1 | 1.978(2) | 1.954(2) | 1.973(3) | 1.960(3) |
| Pd1–Y | 2.187(2) | 2.211(2) | 2.365(1) | 2.395(1) |
| Pd1–P1 | 2.2725(8) | 2.2606(5) | 2.301(1) | 2.273(1) |
| Pd1–Cl1 | 2.3635(7) | 2.3590(5) | 2.3547(9) | 2.350(1) |
| C1–Pd1–Y | 86.8(1) | 86.92(7) | 87.2(1) | 86.3(1) |
| C1–Pd1–P1 | 93.85(8) | 92.34(6) | 93.8(1) | 93.3(1) |
| Cl1–Pd1–Y | 91.77(6) | 92.40(4) | 87.48(4) | 90.81(4) |
| Cl1–Pd1–P1 | 87.60(3) | 88.32(2) | 91.53(4) | 89.90(4) |
| C1–N1 | 1.298(4) | 1.305(2) | 1.301(4) | 1.304(5) |
| N1–C11 | 1.431(4) | 1.436(2) | 1.431(4) | 1.440(5) |
| N1–C21 | 1.491(3) | 1.487(3) | 1.533(4) | 1.512(6) |
| N1–C1–C2 | 120.1(2) | 120.8(2) | 119.4(3) | 120.1(3) |
| C1–N1–C11 | 123.6(2) | 122.8(2) | 123.1(3) | 123.3(3) |
| φ | 170.4(2) | 177.5(2) | –177.6(3) | 177.1(3) |
Even in 6 and 7, the phenylene rings were twisted with respect to the coordination plane (by 64–73°) and to the carbene moiety {N1,C1,C2} (by 75–84°). The ferrocene units remained virtually unperturbed (with tilt angles less than ≈3.5°), and their C(11–15) rings were rotated by 26.0(4)° (in 7b) to 38.5(4)° (in 7a) from the {N1,C1,C2} planes.
Description of the bonding situation
The similarity of the molecular structures led us to examine the bonding situations in the imidoyl and carbene complexes using theoretical methods and compounds 4a and 6a as the representative pair. An analysis using the intrinsic bond orbital (IBO)26 approach has already revealed different electron sharing between the individual donor atoms and the Pd(II) centers. Specifically, the IBOs corresponding to the coordination bonds between palladium and the hard donors (N and Cl) in complex 4a showed greater charge localisation on the donor atoms [N(1.70)/Pd(0.17), Cl(1.76)/Pd(0.16)], which was consistent with their lower polarizabilities and indicated higher ionicity of the Pd–N and Pd–Cl dative bonds. As such, these bonds are more easily deformed, which was reflected by the relatively larger differences between the calculated and experimental bond distances. Conversely, electron sharing between the soft phosphine and imidoyl ligands and Pd(II) was considerably larger and increased from the phosphine [P(1.49)/Pd(0.42)] to the imidoyl [C(1.15)/Pd(0.81)] moiety, suggesting more covalent bonding in the latter case (Fig. 4). The IBO analysis also indicated negligible π-backbonding from Pd to the phosphorus and the imidoyl carbon and exposed the presence of a lone electron pair on the imidoyl nitrogen (vide infra). | ||
| Fig. 4 Intrinsic bond orbitals relevant to the Pd–C bonds in 4a and 6a (values in parentheses indicate the fraction of σ and π bonding electrons assigned to the individual atoms). For additional diagrams, see ESI.† | ||
Carbene formation by methylation of the imidoyl moiety was accompanied by a change from an X-type Pd–C interaction in 4a (σ-bonded imidoyl carbon) to an L-type interaction27 in 6a [C(1.39)/Pd(0.52); donation of the carbene lone pair]. Notably, the altered nature of the Pd–C bond also affected the other Pd-donor interactions. This influence was particularly evident for the trans-chloride [Cl(1.67)/Pd(0.26)] and cis-PPh3 ligands [P(1.39)/Pd(0.55)], while the NMe2 moiety was less affected [N(1.68)/Pd(0.19)].
Subsequent topological analysis of the calculated electron density using Bader's Quantum theory of atoms in molecules (QTAIM)28 supported the results obtained with the IBO approach. Low electron densities (ρbcp) and positive values for the electron density Laplacians (∇2ρbcp) at the bond critical points (bcp) between palladium and the coordinated atoms in both model complexes (Table 3) pointed to closed-shell interactions (either ionic, dative, or van der Waals), in this case a donor–acceptor bond involving a heavy metal element.29 The potential-to-kinetic energy density ratios (|Vbcp|/Gbcp) fell into the intermediate region (2 > |Vbcp|/Gbcp > 1) in all cases. While covalent interactions are indicated by a |Vbcp|/Gbcp ratio >2, interactions with pronounced ionic character are indicated by |Vbcp|/Gbcp < 1.30 Thus, the ratio |Vbcp|/Gbcp close to 1 found for the Pd–N and Pd–Cl bonds in 4a and 6a suggested increased ionicity. This finding was supported by the increased ratio of kinetic energy density to electron density (Gbcp/ρbcp) at the corresponding bond critical points. In contrast, the parameters determined for the Pd–C and Pd–P bonds, together with the more negative ratios of the total energy density to electron density, Hbcp/ρbcp, pointed to higher covalency.
| Compound | Bond | Bond length [Å] | ρ bcp [e Å−3] | ∇ 2 ρ bcp [e Å−5] | H bcp [a.u.] | |Vbcp|/Gbcp [a.u.] | G bcp/ρbcp [a.u.] | H bcp/ρbcp [a.u.] | DI | |
|---|---|---|---|---|---|---|---|---|---|---|
| Exp. | Calc.a | |||||||||
| a Calculated at the B3LYP(d3bj)/def2-TZVPP:sdd(Pd); a.u. = atomic units. | ||||||||||
| 4a | Pd–C | 1.986 | 2.000 | 0.139 | 0.150 | –0.065 | 1.64 | 0.73 | –0.47 | 0.894 |
| Pd–P | 2.266 | 2.273 | 0.108 | 0.112 | –0.045 | 1.62 | 0.68 | –0.42 | 0.890 | |
| Pd–N | 2.201 | 2.248 | 0.071 | 0.247 | –0.012 | 1.16 | 1.04 | –0.17 | 0.463 | |
| Pd–Cl | 2.416 | 2.445 | 0.065 | 0.195 | –0.013 | 1.21 | 0.94 | –0.2 | 0.613 | |
| 6a | Pd–C | 1.978 | 1.978 | 0.142 | 0.260 | –0.065 | 1.50 | 0.92 | –0.46 | 0.925 |
| Pd–P | 2.273 | 2.291 | 0.107 | 0.081 | –0.044 | 1.68 | 0.61 | –0.41 | 0.871 | |
| Pd–N | 2.187 | 2.240 | 0.073 | 0.244 | –0.013 | 1.18 | 1.01 | –0.18 | 0.482 | |
| Pd–Cl | 2.363 | 2.358 | 0.079 | 0.219 | –0.019 | 1.27 | 0.94 | –0.24 | 0.733 | |
The conversion of 4a into 6a was manifested mainly by greater electron density depletion at the bond critical points (as expressed by the Laplacian of electron density). The bond critical points for the Pd–C bonds in both complexes were close to the nodal surface in the Laplacian distribution (Fig. 5). In such cases, the profile of the Laplacian along the whole bond path could provide useful information.31 Indeed, a comparison of the Laplacian profiles along the Pd–C bond path plotted against the normalised bond distance (Fig. 6) showed that carbene formation was associated not only with electron density depletion at the bond critical point but also with a significant charge accumulation in the region of the carbene lone electron pair.
 | ||
| Fig. 5 (top) Contour plots of the electron density ρ in the plane defined by P, Pd and C atoms and (bottom) Laplacian contours (∇2ρ; positive – full red lines, negative – dashed blue lines) for 4a and 6a (atomic critical points are shown yellow circles, bond critical points as blue circles, and bond paths corresponding to the Pd–C bonds as brown lines). | ||
 | ||
| Fig. 6 Plots of electron density (ρ, left) and its Laplacian (∇2ρ, right) along the normalised bond length of the Pd–C bond in 4a (blue line) and 6a (red line). Weak vertical lines represent the locations of the bond critical points. | ||
Electrochemistry
The redox properties of complexes 4–7 were studied by cyclic voltammetry on a glassy carbon disc electrode in dichloromethane containing Bu4N[PF6] as the supporting electrolyte. The redox behaviour of the imidoyl complexes 4 and 5 was generally similar (Fig. 7 and S13,† data in Table 4). Initially, the compounds underwent reversible redox changes (O1/R1) at approximately 0 V vs. the ferrocene/ferrocenium reference,32 which is significantly less than for FcNC itself (E°′ ≈ 0.30 V in MeCN/Bu4N[BF4]).11a This redox process, which was negligibly affected by the nature of the chelating ligand LCY and the phosphine, was attributed to one-electron oxidation of the ferrocenyl group. Such assignment was supported by DFT calculations, which showed that the HOMO of the model compound 4a was localised predominantly on the ferrocene unit (Fig. 8). An analysis of the frontier molecular orbitals using natural atomic orbitals (NAO) showed that the HOMO involved bonding overlap of the iron 3d orbitals (≈75%) with the π-systems of the cyclopentadienyl rings (≈10%, 2p of C). In contrast, the LUMO exhibited a high degree of delocalisation and was spanned across palladium (≈21%, 4d, 5s and 5p) and the donor atoms, including the carbene carbon (≈12%, 2s and 2p of C), phosphorus (≈8%, 3s and 3p of P), amine nitrogen (≈5%, 2s and 2p of N) and chloride (≈3%, 3s and 3p).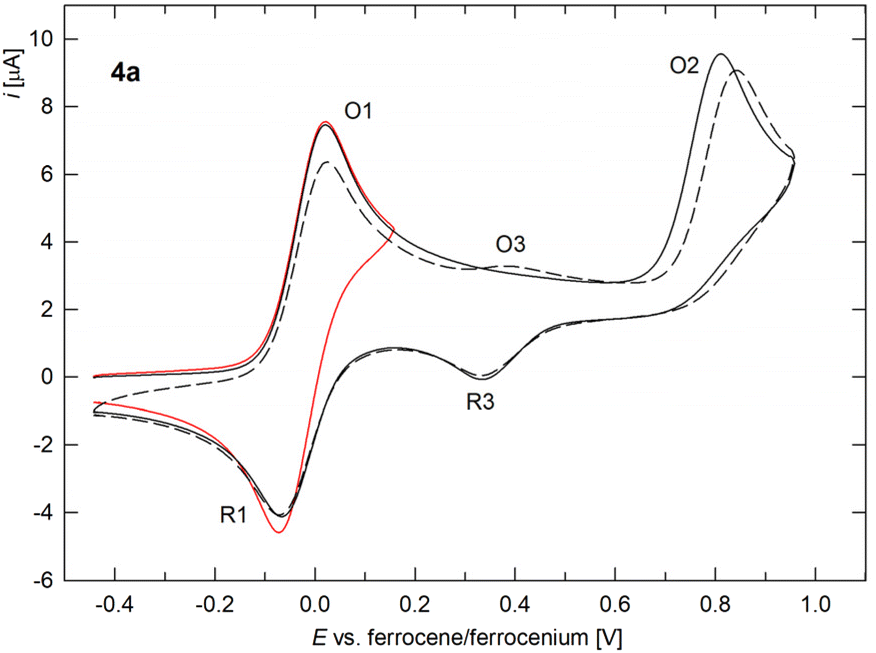 | ||
| Fig. 7 Cyclic voltammograms of 4a recorded at a glassy carbon electrode in dichloromethane containing Bu4N[PF6] as the supporting electrolyte and a 100 mV s−1 scan rate. The second scan is shown by a dashed line. | ||
 | ||
| Fig. 8 Frontier orbitals of 4a and the cation in 6a (isosurfaces at ±0.05 a.u. are shown). | ||
| Compound | Ferrocene oxidation E° (O1/R1) | Additional processes |
|---|---|---|
| a The potentials are given in volts relative to the ferrocene/ferrocenium reference (for details, see Experimental). The potentials for the reversible redox processes were determined as the average of the anodic (Epa) and cathodic (Epc) peak potentials, E°′ = ½(Epa + Epc). The peaks in the cyclic voltammograms were separated by 90–100 mV due to the large resistance. The decamethylferrocene standard showed similar values. | ||
| 4a | −0.03 | E pa(O2) ≈ 0.9, Epc(R3) ≈ 0.35 |
| 4b | 0.00 | E pa(O2) ≈ 0.8, Epc(R3) ≈ 0.39 |
| 5a | 0.00 | E pa(O2) ≈ 0.9, Epc(R3) ≈ 0.34 |
| 5b | 0.01 | E pa(O2) ≈ 0.9, Epc(R3) ≈ 0.37 |
| 6a | 0.43 | E pc(R2) = −1.68 |
| 6b | 0.46 | E pc(R2) = −1.89 |
| 7a | 0.44 | E pc(R2) = −1.59 |
| 7b | 0.46 | E pc(R2) = −1.79 |
At higher potentials, compounds 4 and 5 displayed an additional, ill-defined irreversible oxidation (O2) peak at approximately 0.8–0.9 V. The oxidative wave was associated with a weak cathodic counterwave (R3) during the reverse scan, and upon repeated scanning, a weak oxidative wave (O3) was detected (hardly discernible for some compounds), most likely associated with the cathodic event R3. The waves R3 and O3 were attributed to electrochemically generated species. Overall, this suggested that the second oxidation was followed by chemical steps that produced another redox-active species (ECE mechanism). For PMe3 complexes 4b and 5b, the second wave was composite, followed by a more or less separated postwave, and for compound 5b, two weak reduction waves were observed and attributed to the generated species. Notably, no defined waves were detected in the cathodic region for 4 and 5. Typically, several weak reduction waves were observed, attributable to electrochemically generated and/or decomposition products (see ESI†).
In the accessible potential range, the carbene complexes 6 and 7 displayed one reversible redox change (waves O1 and R1) and one irreversible, probably two-electron reduction (R2; Fig. 9 and S14,†Table 4). The chemical reactions associated with reduction R1 (e.g., decomposition of the electrogenerated species) produced additional redox-active species, which were oxidised during the following scans, giving rise to weak, broad waves at potentials lower than those for O1. Notably, the voltammogram shown in Fig. 9 did not change when the scan direction was reversed (i.e., when the cathodic region was scanned first). However, the weak peak(s) due to the additional species were only observed after traversing the reduction wave R2.
 | ||
| Fig. 9 Cyclic voltammograms of 6a, recorded at a glassy carbon electrode in dichloromethane containing Bu4N[PF6] as the supporting electrolyte and a 100 mV s−1 scan rate. The first and second scan are as a red solid line and a black dashed line, respectively; the initial scan direction is indicated with an arrow. | ||
For the PMe3 complexes 6b and 7b, an additional irreversible oxidation wave was observed just before O1 (see ESI†). In addition, 6b and 7b showed further irreversible oxidations (R2 and R3) when the scan range was extended towards more negative potentials. No similar waves were observed for the analogous PPh3 complexes.
Based on an inspection of the frontier orbitals (Fig. 8), oxidation O1 was also attributed to the ferrocene/ferrocenium couple. Even in this case, the HOMO of 6a was localised almost exclusively on the ferrocene moiety (≈87%, 3d of Fe; ≈10%, 2p of C). The shift of the first oxidation towards more positive potentials (by approximately 0.4 V compared to the imidoyl complexes 4 and 5) corresponded with the cationic nature of the carbene complexes, which made electron removal more difficult. Conversely, reduction R1 was strongly affected by the LCY and phosphine ligands and was probably a Pd/carbene-centred redox process (likely a Pd(II)-to-Pd(0) reduction33). Indeed, the LUMO of 6a was an anti-bonding orbital with major contributions from the carbon (≈43%, 2p) and nitrogen (≈10%, 2p) atoms of the carbene moiety, along with smaller admixtures of palladium (≈8%, 4d) and iron (≈5%, 3d) atoms.
Reactions of the imidoyl complexes with Brønsted and Lewis acids
The results of the IBO analysis suggesting the presence of a lone electron pair at the imidoyl nitrogen and previous reports describing the preparation of multinuclear complexes34 and protic carbenes from imidoyl complexes35 led us to further explore the reactivity of the representative complex 4a.Initially, we treated complex 4a with in situ-generated (Ph3P)Au+ in dichloromethane-methanol (Scheme 4). To our surprise, the reaction did not afford any Pd–Au complex but rather produced a burgundy red compound (see the UV-vis spectrum in Fig. 1), which was isolated in 52% yield by chromatography (23% after crystallisation) and structurally authenticated as (Z)-2,2-dimethyl-1-(ferrocenylimino)isoindolin-2-ium tetrafluoroborate (8) using spectroscopic methods and X-ray crystallography (Fig. 10). The addition of Ag[BF4] (1 equiv.) to 4a in CDCl3 led to a nearly 90% NMR yield of 8 (70% isolated yield at the 25 μmol scale). Attempts to similarly react complex 5a (Ag[BF4]/CDCl3) were unsuccessful, most likely due to the different basicity of the sulfur atom in the SMe group replacing the NMe2 moiety.
 | ||
| Scheme 4 Reaction of 4a with Lewis and Brønsted acids to give 8. | ||
 | ||
| Fig. 10 Molecular structure of 8. Selected distances and angles (in Å and deg): N1–C1 1.256(2), N1–C11 1.404(2), C1–N1–C11 123.2(1), N2–C1 1.530(2), N2–C8 1.518(2), N2–C9 1.496(2), N2–C10 1.506(2), C1–N2–C8 105.2(1) and C9–N2–C10 109.8(1); the carbon atoms forming the indolinium moiety, C(1–8), were coplanar to within 0.01 Å, and the N2 atom was displaced by 0.364(1) Å from this plane; the Fe1–C(11–15) bond lengths range 2.038(2)–2.055(2) Å, and the dihedral angle of the cyclopentadienyl planes was 2.5(1)°. | ||
Subsequently, we treated 4a with selected protic acids. While the addition of HCl (dioxane solution) to 4a resulted in rapid decomposition, the reaction with NH4[BF4] in dichloromethane-methanol again produced 8 in approximately 26% yield (based on NMR analysis; protic carbene analogous to 6a was not detected). No reaction was observed with NH4Cl and K[BF4], very likely due to the very limited solubilities of these salts and the absence of H+ in the latter case.
The formation of 8 can be explained by reductive elimination36 of the N,C-chelating imidoyl ligand from 4a, presumably initiated by halide removal with an inorganic salt or Ag/Au reagents. The structure of 8 combining a quaternary nitrogen atom and an imine moiety is unique. While structurally related compounds, e.g., aryl-Cr(CO)3 complexes with oxo-pyrrolium ylide substituents, were previously obtained from thermally induced reactions of chromium aminocarbene complexes with alkynes,37 the only similar compounds appear to be 2-oxoindolinium salts obtained by intramolecular cyclisation of 1-diazo-2-[2-(dialkylamino)phenyl]-2-oxoethylphosphonates proceeding via carbene intermediates38,39 and 1-iminoisoindolines prepared in a conventional manner from 2-(chloromethyl)benzonitrile and amines.40
Conclusions
In summary, we demonstrated that Pd(II) complexes formed by orthopalladation of benzylic amines and thioethers and subsequent bridge cleavage with phosphines underwent facile insertion of isocyanoferrocene into their Pd–C bonds to provide structurally rigid, C,Y-chelating imidoyl complexes (Y = N, S) with pendant, redox-active ferrocenyl substituents. These imidoyl complexes were smoothly alkylated at the imine nitrogen with the Meerwein salt to provide the corresponding cationic aminocarbene complexes. Cyclic voltammetry supported by DFT calculations suggested that the primary oxidation of these compounds occurred at the peripheral ferrocene moiety. Furthermore, DFT calculations were used to rationalise the collected structural information. In particular, they showed that the imidoyl-to-carbene conversion represented the transformation of an X-type (σ-organyl) into an L-type (π-coordinated carbene) ligand. When attempting to synthesise protic carbene (analogous to 6a) and heterometallic complexes from imidoyl complex 4a, we noted that the compound rather underwent reductive elimination of the chelating imidoyl ligand, leading to 1-(ferrocenylimino)isoindolinium salt 8.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Charles University Research Centre Program (project no. UNCE/SCI/014) and by the Grant Agency of Charles University (project no. 222120). Computational resources were provided by the e-INFRA CZ project (ID 90254), supported by the Ministry of Education, Youth, and Sports of the Czech Republic.References
- R. Ramozzi, N. Chéron, B. Braïda, P. C. Hiberty and P. Fleurat-Lessard, New J. Chem., 2012, 36, 1137 RSC.
- V. P. Boyarskiy, N. A. Bokach, K. V. Luzyanin and V. Y. Kukushkin, Chem. Rev., 2015, 115, 2698 CrossRef CAS PubMed and references cited therein.
- L. Malatesta, in Progr. Inorg. Chem, ed. F. A. Cotton, Interscience Publishers, London, UK, 1959, vol. 1, pp. 283–379 Search PubMed.
- R. A. Michelin, A. J. L. Pombeiro and M. F. C. G. da Silva, Coord. Chem. Rev., 2001, 218, 75 CrossRef CAS.
- For reviews focused on carbene complexes, see: (a) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS PubMed; (b) P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862 CrossRef; (c) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS PubMed; (d) P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5, 711 CrossRef CAS PubMed.
- For examples, see: (a) K. Hiraki, Y. Fuchita and T. Masumoto, Bull. Chem. Soc. Jpn., 1980, 53, 1171 CrossRef CAS; (b) D. L. Reger and J. E. Collins, J. Organomet. Chem., 1995, 491, 159 CrossRef CAS.
- K. Škoch, I. Císařová, J. Schulz, U. Siemeling and P. Štěpnička, Dalton Trans., 2017, 46, 10339 RSC.
- (a) K. Škoch, I. Císařová, F. Uhlík and P. Štěpnička, Dalton Trans., 2018, 47, 16082 RSC; (b) K. Škoch, J. Schulz, I. Císařová and P. Štěpnička, Organometallics, 2019, 38, 3060 CrossRef.
- Complexes with C,N-ligands: (a) Y. Yamamoto and H. Yamazaki, Inorg. Chim. Acta, 1980, 41, 229 CrossRef CAS; (b) J. Dupont and M. Pfeffer, J. Chem. Soc., Dalton Trans., 1990, 3193 RSC; (c) A. Zografidis, K. Polborn, W. Beck, B. A. Markies and G. van Koten, Z. Naturforsch., 1994, 49, 1494 CrossRef CAS; (d) A. Böhm, K. Polborn, K. Sünkel and W. Beck, Z. Naturforsch., B: J. Chem. Sci., 1998, 53, 448 CrossRef; (e) J.-F. Ma, Y. Kojima and Y. Yamamoto, J. Organomet. Chem., 2000, 616, 149 CrossRef CAS; (f) K.-E. Lee, H.-T. Jeon, S.-Y. Han, J. Ham, Y.-J. Kim and S. W. Lee, Dalton Trans., 2009, 6578 RSC. Complexes with C,S-ligands: (g) J. Dupont, M. Pfeffer, J. C. Daran and Y. Jeannin, Organometallics, 1987, 6, 899 CrossRef CAS. For related systems, see: (h) L. Canovese, F. Visentin, C. Santo, C. Levi and A. Dolmella, Organometallics, 2007, 26, 5590 CrossRef CAS.
- J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527 CrossRef CAS PubMed.
- (a) T. El-Shihi, F. Siglmüller, R. Herrmann, M. F. N. N. Carvalho and A. J. L. Pombeiro, J. Organomet. Chem., 1987, 335, 239 CrossRef CAS; (b) G. R. Knox, P. L. Pauson, D. Willison, E. Solčániová and Š. Toma, Organometallics, 1990, 9, 301 CrossRef CAS.
- For examples of studies focused on the coordination behaviour and reactivity of FcNC, see: (a) Y.-C. Huang, W.-Y. Lan, W.-M. Ching and S. C. N. Hsu, New J. Chem., 2020, 44, 18242 RSC; (b) M. V. Barybin, T. C. Holovics, S. F. Deplazes, G. H. Lushington, D. R. Powell and M. Toriyama, J. Am. Chem. Soc., 2002, 124, 13668 CrossRef CAS PubMed; (c) V. N. Nemykin, A. A. Purchel, A. D. Spaeth and M. V. Barybin, Inorg. Chem., 2013, 52, 11004 CrossRef CAS PubMed; (d) J. Mahrholdt, J. Noll, M. Korb, T. Rüffer and H. Lang, Inorg. Chim. Acta, 2022, 534, 120829 CrossRef CAS; (e) S. D. Waniek, C. Förster and K. Heinze, Eur. J. Inorg. Chem., 2022, e202100905 CrossRef CAS; (f) P. Vosáhlo, M. Franc, P. Harmach, J. Schulz and P. Štěpnička, J. Organomet. Chem., 2023, 1000, 122874 CrossRef.
- For an overview of the chemistry of ferrocene-based carbenes, see: U. Siemeling, Eur. J. Inorg. Chem., 2012, 3523 CrossRef CAS.
- For similar reactions involving [(LCN)PdCl]2, see: (a) A. E. Kelly, S. A. Macgregor, A. C. Willis, J. H. Nelson and E. Wenger, Inorg. Chim. Acta, 2003, 352, 79 CrossRef CAS; (b) A. Mentes, R. D. W. Kemmitt, J. Fawcett and D. R. Russell, J. Mol. Struct., 2004, 693, 241 CrossRef CAS.
- D. C. D. Butler and C. J. Richards, Organometallics, 2002, 21, 5433 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part A: Theory and Applications in Inorganic Chemistry, Wiley, Hoboken, NJ, 6th edn, 2009, ch. 2.6.1, pp. 192–204 Search PubMed.
- D. R. Scott and R. S. Becker, J. Chem. Phys., 1961, 35, 516 CrossRef CAS.
- (a) T. G. Appleton, H. C. Clark and L. E. Manzer, Coord. Chem. Rev., 1973, 10, 335 CrossRef CAS; (b) F. R. Hartley, Chem. Soc. Rev., 1973, 2, 163 RSC.
- B. Crociani, M. Sala, A. Polo and G. Bombieri, Organometallics, 1986, 5, 1369 CrossRef CAS.
- G. R. Owen, R. Vilar, A. J. P. White and D. J. Williams, Organometallics, 2002, 21, 4799 CrossRef CAS.
- K. Onitsuka, M. Yamamoto, S. Suzuki and S. Takahashi, Organometallics, 2002, 21, 581 CrossRef CAS.
- For examples, see: (a) J. Silver, J. R. Miller, A. Houlton and M. T. Ahmet, J. Chem. Soc., Dalton Trans., 1994, 3355 RSC; (b) V. Kovač, A. Višnjevac, V. Rapić and B. Kojić-Prodić, J. Mol. Struct., 2004, 687, 107 CrossRef; (c) W. Imhof, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m461 CrossRef CAS PubMed.
- (a) L. Lin, A. Berces and H.-B. Kraatz, J. Organomet. Chem., 1998, 556, 11 CrossRef CAS; (b) L. Dufková, H. Matsumura, D. Nečas, P. Štěpnička, F. Uhlík and M. Kotora, Collect. Czech. Chem. Commun., 2004, 69, 351 CrossRef; (c) T. A. Fernandes, H. Solařová, I. Císařová, F. Uhlík, M. Štícha and P. Štěpnička, Dalton Trans., 2015, 44, 3092 RSC.
- S. Fuertes, A. J. Chueca and V. Sicilia, Inorg. Chem., 2015, 54, 9885 CrossRef CAS PubMed.
- S. K. Schneider, P. Roembke, G. R. Julius, H. G. Raubenheimer and W. A. Herrmann, Adv. Synth. Catal., 2006, 348, 1862 CrossRef CAS.
- (a) G. Knizia, J. Chem. Theor. Comput., 2013, 9, 4834 CrossRef CAS PubMed; (b) G. Knizia and J. E. M. N. Klein, Angew. Chem., Int. Ed., 2015, 54, 5518 CrossRef CAS PubMed.
- M. L. H. Green, J. Organomet. Chem., 1995, 500, 127 CrossRef CAS.
- (a) R. F. W. Bader, Chem. Rev., 1991, 91, 893 CrossRef CAS; (b) R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9 CrossRef CAS.
- (a) D. Cremer and E. Kraka, Angew. Chem., Int. Ed. Engl., 1984, 23, 627 CrossRef; (b) P. Macchi, D. M. Proserpio and A. Sironi, J. Am. Chem. Soc., 1998, 120, 13429 CrossRef CAS; (c) P. Macchi and A. Sironi, Coord. Chem. Rev., 2003, 238–239, 383 CrossRef CAS; (d) D. Stalke, Chem. – Eur. J., 2011, 17, 9264 CrossRef CAS PubMed.
- (a) R. Bianchi, G. Gervasio and D. Marabello, Inorg. Chem., 2000, 39, 2360 CrossRef CAS PubMed; (b) E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529 CrossRef CAS.
- For examples, see: (a) M. W. Stanford, J. I. Schweizer, M. Menche, G. S. Nichol, M. C. Holthausen and M. J. Cowley, Angew. Chem., Int. Ed., 2019, 58, 1329 CrossRef CAS PubMed; (b) A. Münch, L. Knauer, H. Ott, C. Sindlinger, R. Herbst-Irmer, C. Strohmann and D. Stalke, J. Am. Chem. Soc., 2020, 142, 15897 CrossRef PubMed; (c) B. Lindquist-Kleissler, J. S. Wenger and T. C. Johnstone, Inorg. Chem., 2021, 60, 1846 CrossRef CAS PubMed.
- G. Gritzner and J. Kůta, Pure Appl. Chem., 1984, 56, 461 CrossRef.
- C. Amatore, M. Azzabi and A. Jutand, J. Organomet. Chem., 1989, 363, C41 CrossRef CAS.
- (a) T. Nakajima, M. Tsuji, N. Hamada, Y. Fukushima, B. Kure and T. Tanase, J. Organomet. Chem., 2014, 768, 61 CrossRef CAS; (b) B. Crociani, R. Bertani, T. Boschi and G. Bandoli, J. Chem. Soc., Dalton Trans., 1982, 1715 RSC; (c) R. Uson, J. Fornies, P. Espinet, E. Lalinde, P. G. Jones and G. M. Sheldrick, J. Organomet. Chem., 1983, 253, C47 CrossRef CAS; (d) M. Tanabiki, K. Tsuchiya, Y. Motoyama and H. Nagashima, Chem. Commun., 2005, 3409 RSC.
- D. L. Reger and J. E. Collins, J. Organomet. Chem., 1995, 491, 159 CrossRef CAS.
- (a) W. J. Marshall and V. V. Grushin, Organometallics, 2003, 22, 1591 CrossRef CAS; (b) A. C. Albéniz, P. Espinet, R. Manrique and A. Pérez-Mateo, Chem. – Eur. J., 2005, 11, 1565 CrossRef PubMed; (c) P. Veit, C. Förster and K. Heinze, Beilstein J. Org. Chem., 2016, 12, 1322 CrossRef CAS PubMed.
- (a) E. Chelain, R. Goumont, L. Hamon, A. Parlier, M. Rudler, H. Rudler, J.-C. Daran and J. Vaissermann, J. Am. Chem. Soc., 1992, 114, 8088 CrossRef CAS; (b) E. Chelain, A. Parlier, M. Audouin, H. Rudler, J.-C. Daran and J. Vaissermann, J. Am. Chem. Soc., 1993, 115, 10568 CrossRef CAS.
- F. Léost, B. Chantegrel and C. Deshayes, Tetrahedron Lett., 1997, 53, 7557 CrossRef.
- For the formation of 2-oxoindole derivatives by reductive elimination from Pd(II) complexes, see: R. Frutos-Pedreño, P. González-Herrero and J. Vicente, Organometallics, 2013, 32, 4664 CrossRef.
- F. S. Babichev and A. K. Tyltin, Ukr. Khim. Zh., 1976, 36, 62 Search PubMed and references therein.
Footnote |
| † Electronic supplementary information (ESI) available: Complete experimental procedures and characterisation data, summary of crystallographic parameters, additional structure diagrams and cyclic voltammograms, results from IBO analysis, and copies of the NMR spectra. CCDC 2288861–2288870. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02717a |
| This journal is © The Royal Society of Chemistry 2023 |