
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnexpected reactivity of cyclometalated iridium(III) dimers. Direct synthesis of a mononuclear luminescent complex†
Jing
Tu
a,
Daniele
Veclani
 b,
Filippo
Monti
*b,
Andrea
Mazzanti
a,
Letizia
Sambri
a,
Nicola
Armaroli
b and
Andrea
Baschieri
*b
b,
Filippo
Monti
*b,
Andrea
Mazzanti
a,
Letizia
Sambri
a,
Nicola
Armaroli
b and
Andrea
Baschieri
*b
aDepartment of Industrial Chemistry “Toso Montanari”, University of Bologna, Via Piero Gobetti 85, 40129 Bologna, Italy
bInstitute for Organic Synthesis and Photoreactivity (ISOF), National Research Council of Italy (CNR), Via Piero Gobetti 101, 40129 Bologna, Italy. E-mail: filippo.monti@isof.cnr.it; andrea.baschieri@isof.cnr.it
First published on 20th September 2023
Abstract
A new synthetic method has been developed for the preparation of unexpected emissive iridium(III) complexes (A and B), directly obtained from the established [Ir(ppy)2(μ-Cl)]2 dimer, under reaction conditions in which such compounds are usually considered stable. Complex A ([Ir(ppy)2(Oppy)], where Hppy = 2-phenylpyridine and HOppy = 2-(o-hydroxyphenyl)pyridine) was obtained from the dimer without the addition of further ancillary ligands in the reaction environment, but in the presence of a basic water environment in 2-ethoxyethanol as solvent at 165 °C. The complex evidences the unexpected insertion of an oxygen atom between the iridium(III) center and the carbon atom of one ppy moiety. Under specific reaction conditions, the mer-[Ir(ppy)3] complex (B) was obtained. The presence of the right amount of water is important to maximize the formation of A relative to B. Both compounds were fully characterized by NMR spectroscopy and mass spectrometry (MS), and the X-ray structure of A was also determined. DFT calculations were used to shed light on the reaction mechanism leading to the unexpected formation of A, suggesting that the Oppy ligand is generated intramolecularly once the [Ir(ppy)2(μ-OH)]2 dimer is formed. The process is probably assisted by a redox reaction involving the second iridium(III) center in the dimer. The electrochemical and photophysical properties of complexes A and B were investigated in comparison with the well-known fac-[Ir(ppy)3] analogue (C). Complex A displays a green emission (λmax = 545 nm) with a photoluminescence quantum yield (PLQY) of nearly 40%, whereas the oxygen-free counterpart B is poorly emissive, exhibiting an orange emission (λmax = 605 nm) with a PLQY below 10%. These findings may pave the way for the direct synthesis of neutral luminescent complexes with the general formula [Ir(C^N)2(OC^N)], even using dimers with non-commercial or highly substituted C^N ligands, without the need for synthesizing the corresponding hydroxyl-substituted ancillary ligand, which may be hardly obtainable.
Introduction
Phosphorescent cyclometalated iridium(III) complexes displaying strong and tunable emission have been the subject of intense research and many interesting applications. They have been extensively used in different fields, such as optoelectronic technology (OLED or LEC),1,2 biocompatible probes for in vitro bioimaging,3–8 photoredox catalysis,9 and sensing10,11 thanks to their peculiar features, such as chemical inertness, good thermal and photostability, and, most importantly, photophysical properties. The latter can be finely tuned with simple chemical modifications of the ligands coordinated with the metal center.Octahedral heteroleptic iridium(III) complexes typically entail two bidentate cyclometalating ligands (HC^N), and an ancillary ligand (X^Y) that saturates the coordination sphere of the iridium center.12 Usually, this type of luminescent compound is obtained in two synthetic steps. First, a μ-dichloro-bridged dimer with the general formula [Ir(C^N)2(μ-Cl)]2 is synthesized by direct cyclometalation of iridium(III) chloride hydrate with a specific HC^N ligand at a relatively high temperature. Then, the obtained precursor is subsequently reacted with an excess of the desired ancillary ligands to give the final complex. This strategy has afforded vast libraries of cationic, neutral and even less explored anionic13,14 luminescent complexes [Ir(C^N)2(X^Y)]n, where n = +1, 0, or −1.12,15
Iridium(III) dimers are generally considered quite stable compounds in the absence of chelating species, also under harsh reaction conditions and high temperatures.
However, during some experiments aimed at the synthesis of new classes of luminescent complexes, we noticed that the earliest example of the iridium(III) dimer, [Ir(ppy)2(μ-Cl)]2, (Hppy = 2-phenylpyridine), exhibited an unexpected reactivity which, to the best of our knowledge, has not been reported yet.
Given the importance of such a class of derivatives, we considered this finding worthy of an in-depth analysis. After an appropriate characterization (see below for all the details), we realized that the degradation of the dimer itself yields a neutral iridium complex with a 2-phenylpyridine derivative as ancillary ligand. The latter contains an unexpected oxygen atom between the iridium(III) center and the carbon in position 2 of the phenyl moiety of the ligand (Scheme 1).
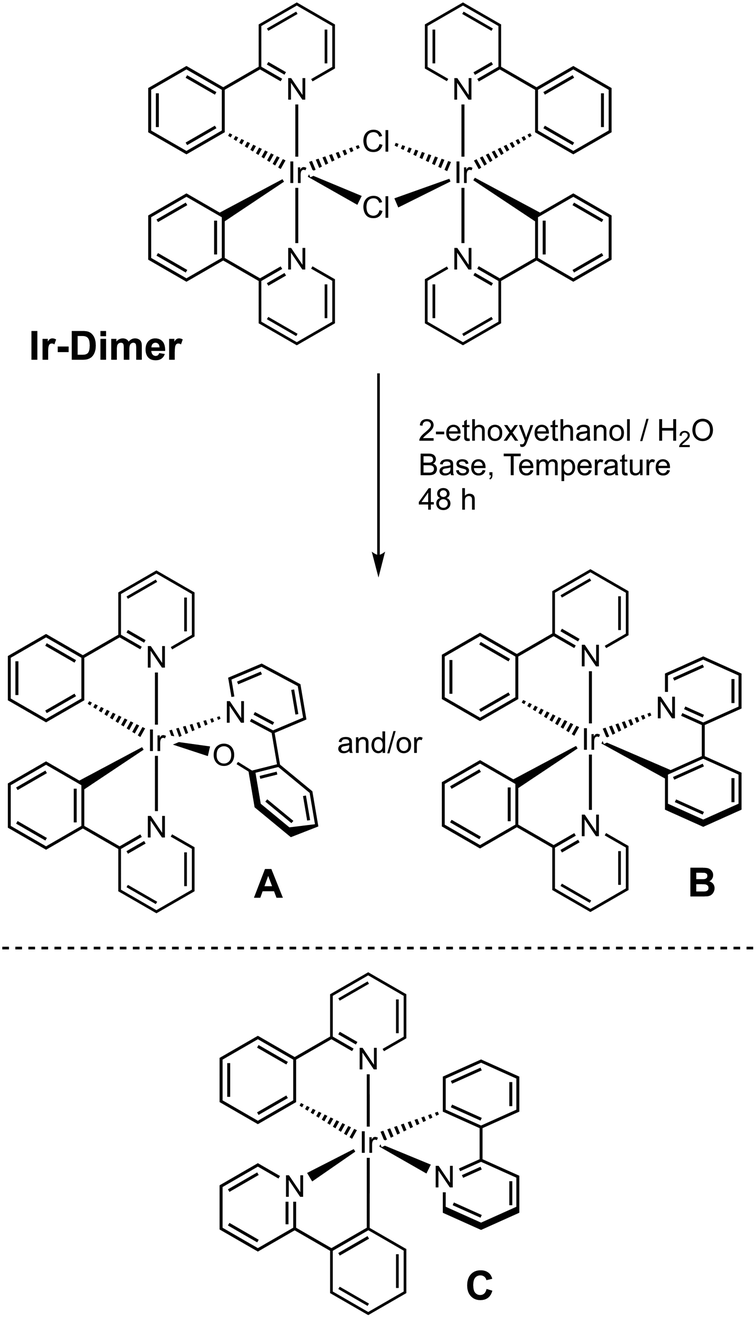 | ||
| Scheme 1 General description for the synthesis of complexes A and B (top); structure of the previously reported archetypal fac-[Ir(ppy)3] complex (C) (bottom). | ||
In other words, starting from a dimer containing only cyclometallating 2-phenylpyridines, we got a luminescent complex carrying a deprotonated 2-(o-hydroxyphenyl)pyridine (HOppy) as an anionic ancillary ligand.
HOppy is a very simple chelating O^N bidentate system already used in several organometallic complexes. In most cases, this compound is used to get metal complexes with the general formula M(O^N)2 or M(O^N)3 (where M = Cu2+,16 Be2+,17 Fe3+, Co3+, Cu2+ and Pd2+,18 Zn2+,19 Cr3+,20 Al3+,21 Mn3+and Ni2+ ions22).
Only recently HOppy has been used as a ligand for the formation of iridium(III) complexes with applications in homogeneous catalysis.23 To the best of our knowledge, the use of HOppy as an ancillary ligand in luminescent iridium(III) complexes has been reported in the literature in only three very recent papers.24–26 On the other hand, with the aim of testing systematic variations in the ancillary ligand donor atoms and bite angles for the metal center chelation, there are numerous examples in the literature of metal complexes obtained using structures similar to HOppy such as 2-(2-hydroxyphenyl)benzoxazole,27 2-(2-hydroxyphenyl)benzothiazole,28–30 and 2-(2-hydroxyphenyl)-1H-benzimidazole.31
In this paper we focus our attention on the unexpected reactivity of the μ-dichloro-bridged iridium(III) dimer with the formula [Ir(ppy)2(μ-Cl)]2 towards the direct synthesis of the emissive neutral complexes [Ir(ppy)2(Oppy)] (A). A series of synthetic conditions were investigated to understand the factors leading to the formation of this unexpected complex and to rationalize the reaction mechanism. During our research, the formation of meridional-[Ir(ppy)3] (B) was also observed. This complex, already known in the literature,32 was used as a reference compound for all subsequent structural, photophysical and electrochemical characterization studies, since it shows a nearly identical structure compared to A, lacking just an oxygen atom (Scheme 1). For the sake of completeness and comparison, also the archetypal facial-[Ir(ppy)3] isomer (C) is included in the characterization (see below).
Experimental section
General information
Analytical grade solvents and commercially available reagents were used as received unless otherwise stated. Chromatographic purifications were performed using 70–230 mesh silica gel. 1H and 13C NMR spectra were recorded on Varian Inova 300 MHz, Varian Mercury 400 MHz, Agilent 500 MHz or Inova 600 MHz spectrometers. Chemical shifts (δ) are reported in ppm relative to residual solvent signals for 1H and 13C NMR (1H NMR: 7.26 ppm for CDCl3, 5.33 ppm for CD2Cl2; 13C NMR: 77.0 ppm for CDCl3, 53.84 ppm for CD2Cl2). 13C NMR spectra were acquired with the 1H broadband decoupled mode. Coupling constants are given in Hz. The abbreviations used to indicate the multiplicity of signals are s, singlet; d, doublet; t, triplet; q, quartet; dd, double doublet; and m, multiplet. The high-resolution mass spectra (HRMS) were obtained with an ESI-QTOF (Agilent Technologies, model G6520A) instrument, and the m/z values are referred to as the monoisotopic mass. ESI-MS analyses were performed by direct injection of acetonitrile solutions of the compounds using a WATERS ZQ 4000 mass spectrometer.The cyclometalated μ-dichloro-bridged iridium(III) precursor [Ir(ppy)2(μ-Cl)]2 (Ir-dimer) was prepared following a reported procedure33,34 by refluxing the IrCl3·xH2O salt and the cyclometalating ligand Hppy = 2-phenylpyridine in a 2-ethoxyethanol/water mixture (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
Synthesis of complexes A and B
Complex A. In a round bottom flask, equipped with a Graham condenser, the Ir-dimer (90 mg, 0.084 mmol, 1.0 equiv.) and K2CO3 (580 mg, 4.2 mmol, 50 equiv.) were dispersed in 2-ethoxyethanol and water (9 + 1.2 mL) and the mixture was heated with an oil bath at 165 °C under stirring for 48 hours. After this period, the solution was cooled to room temperature and H2O (20 mL) was added. The mixture was extracted with DCM (2 × 20 mL). The organic layer was washed with fresh water (2 × 15 mL), dried over Na2SO4 and the solvent evaporated. The crude mixture was purified by column chromatography on silica gel using Et2O/petroleum ether/DCM 5
:4:1 as an eluent, to give A as a yellow solid (14.1 mg, 0.021 mmol, 25% yield). 1H NMR (600 MHz, CD2Cl2) δ 9.11 (dq, J = 5.7, 0.8 Hz, 1H), 8.08 (dq, J = 5.7, 0.8 Hz, 1H), 7.99 (d, J = 8.0 Hz, 1H), 7.80–7.67 (m, 5H), 7.56–7.51 (m, 3H), 7.29 (dd, J = 7.9, 2.0 Hz, 1H), 7.12–7.09 (m, 1H), 6.90–6.81 (m, 3H), 6.74–6.65 (m, 4H), 6.43–6.38 (m, 2H), 6.26 (dd, J = 8.3, 1.1 Hz, 1H), 6.04 (dd, J = 7.6, 1.1 Hz, 1H); 13C NMR (151 MHz, CD2Cl2) δ 170.2 (C), 168.2 (C), 168.1 (C), 157.9 (C), 153.2 (C), 151.0 (CH), 150.9 (C), 150.1 (CH), 149.0 (CH), 145.4 (C), 145.1 (C), 137.6 (CH), 137.4 (CH), 136.7 (CH), 133.6 (CH), 132.5 (CH), 131.6 (CH), 130.6 (CH), 129.5 (CH), 129.1 (CH), 127.6 (C), 125.3 (CH), 124.7 (CH), 124.0 (CH), 122.7 (CH), 122.0 (CH), 121.8 (CH), 121.5 (CH), 121.4 (CH), 120.7 (CH), 119.2 (CH), 118.4 (CH), 114.8 (CH); ESI-MS: 672 [M + H]+; 694 [M + Na]+; HRMS (ESI-QTOF) for C33H24IrN3O: ([M + H]+): m/z calcd: 670.1599; found: 670.1631.
Complex B. In a round bottom flask, equipped with a Graham condenser, the Ir-dimer (90 mg, 0.084 mmol, 1.0 equiv.) and K2CO3 (580 mg, 4.2 mmol, 50 equiv.) were dispersed in 2-ethoxyethanol and water (10 + 0.5 mL) and the mixture was heated with an oil bath at 200 °C under stirring for 48 hours. After this period, the solution was cooled to room temperature and H2O (20 mL) was added. The mixture was extracted with DCM (2 × 20 mL). The organic layer was washed with fresh water (2 × 15 mL), dried over Na2SO4 and the solvent evaporated. The crude mixture was purified by column chromatography on silica gel using Et2O/hexane 3
:2 as an eluent, to give B as an orange solid (15.1 mg, 0.023 mmol, 27% yield). 1H NMR (500 MHz, CDCl3) δ 8.10 (dd, J = 5.9 Hz, 1H), 7.93–7.90 (m, 2H), 7.80–7.72 (m, 3H), 7.69–7.66 (m, 1H), 7.65–7.57 (m, 3H), 7.51 (t, J = 7.5 Hz, 1H), 7.44 (t, J = 7.4 Hz, 1H), 6.99–6.84 (m, 7H), 6.82 (t, J = 7.2 Hz,1H), 6.73–6.67 (m, 2H), 6.61 (d, J = 6.6 Hz, 1H), 6.46 (d, J = 7.2 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 177.8 (C), 175.4 (C), 170.6 (C), 168.5 (C), 167.9 (C), 159.4 (C), 153.3 (CH), 151.2 (CH), 147.8 (CH), 145.3 (C), 144.8 (C), 142.3 (C), 138.0 (CH), 136.4 (CH), 135.5 (CH), 134.0 (CH), 132.8 (CH), 130.6 (CH), 129.9 (CH), 129.5 (CH), 124.2 (CH), 124.1 (CH), 123.9 (CH), 122.2 (CH), 122.0 (CH), 121.2 (CH), 120.8 (CH), 119.0 (CH), 118.8 (CH), 118.5 (CH), 118.3 (CH); ESI-MS: 656 [M + H]+; 678 [M + Na]+; 694 [M + K]+.
Complex A. Ligand 2-(o-hydroxyphenyl) pyridine (HOppy) (14.4 mg, 0.084 mmol, 3 equiv.) was dissolved in DCM/MeOH with a ratio of 1
:1 (4 mL). Then, K2CO3 (116 mg, 0.84 mmol, 30 equiv.) was added and the mixture was stirred for 3 hours at room temperature under a N2 atmosphere. Ir-dimer (30 mg, 0.028 mmol, 1 equiv.) was then added and the mixture was stirred for a further 24 hours. The solvent was evaporated under reduced pressure. H2O (20 mL) was added and the mixture was extracted with DCM (3 × 20 mL). The collected organic phases were washed with brine (20 mL), dried over Na2SO4, and concentrated. The crude mixture was purified by column chromatography on silica gel, with Et2O/hexane at a ratio of 3:2 as an eluent, to give the desired products in 45% yield.
Complex B. In a round bottom flask the Ir-dimer (30 mg, 0.028 mmol, 1 equiv.), 2-phenylpyridine (24 μL, 0.168 mmol, 6 equiv.) and Na2CO3 (35 mg, 0.336 mmol, 12 equiv.) were dispersed in 2-ethoxyethanol (4 mL) and the mixture was stirred for 3 hours at 135 °C under a nitrogen atmosphere in the dark. H2O (20 mL) was added and the mixture was extracted with DCM (3 × 20 mL). The combined organic phase was washed with fresh water (2 × 20 mL) and dried over Na2SO4 and the solvent was evaporated. The crude mixture was purified by column chromatography on silica gel with Et2O/hexane at a ratio of 3
:2 as an eluent to give the desired products in 35% yield.
NMR spectra in CDCl3 of these compounds were in agreement with those of the previously described ones.
Electrochemical characterization
Voltammetric experiments were performed using a Metrohm AutoLab PGSTAT 302N electrochemical workstation in combination with the NOVA 2.1.6 software package. All the measurements were carried out at room temperature in acetonitrile solutions with a sample concentration of approximately 0.5 mM and using 0.1 M tetrabutylammonium hexafluorophosphate (electrochemical grade, TBAPF6) as the supporting electrolyte. Oxygen was removed from the solutions by bubbling nitrogen. All the experiments were carried out using a three-electrode setup (BioLogic VC-4 cell, volume range: 1–3 mL) with a glassy carbon working electrode (having an active surface disk of 1.6 mm diameter), the Ag/AgNO3 redox couple (0.01 M in acetonitrile, with a 0.1 M TBAClO4 supporting electrolyte) as the reference electrode, and a platinum wire as the counter electrode. At the end of each measurement, ferrocene was added as the internal reference. Cyclic voltammograms (CVs) were typically recorded at a scan rate of 100 mV s−1. Osteryoung square-wave voltammograms (OSWVs) were recorded with a scan rate of 25 mV s−1, a SW amplitude of ±20 mV, and a frequency of 25 Hz.Photophysics
Spectroscopic investigations were carried out in spectrofluorimetric grade acetonitrile. The absorption spectra were recorded with a PerkinElmer Lambda 950 spectrophotometer. For the photoluminescence experiments, the sample solutions were placed in fluorimetric Suprasil quartz cuvettes (10.00 mm) and dissolved oxygen was removed by bubbling argon for 30 min. The uncorrected emission spectra were obtained with an Edinburgh Instruments FLS920 spectrometer equipped with a Peltier-cooled Hamamatsu R928 photomultiplier tube (PMT, spectral window: 185–850 nm). An Osram XBO xenon arc lamp (450 W) was used as the excitation light source. The corrected spectra were acquired by means of a calibration curve, obtained using an Ocean Optics deuterium–halogen calibrated lamp (DH-3plus-CAL-EXT). The photoluminescence quantum yields (PLQYs) in solution were obtained from the corrected spectra on a wavelength scale (nm) and measured according to the approach described by Demas and Crosby,36 using an air-equilibrated water solution of tris(2,2′-bipyridyl)ruthenium(II) dichloride as a reference (PLQY = 0.040).37 The emission lifetimes (τs) were measured through the time-correlated single photon counting (TCSPC) technique using a HORIBA Jobin Yvon IBH FluoroHub controlling a spectrometer equipped with a pulsed NanoLED or SpectraLED (λexc = 370 nm) as the excitation source and a red-sensitive Hamamatsu R-3237-01 PMT (185–850 nm) as the detector. The analysis of the luminescence decay profiles was accomplished using the DAS6 Decay Analysis Software provided by the manufacturer, and the quality of the fit was assessed with the χ2 value close to unity and with the residuals regularly distributed along the time axis. To record the 77 K luminescence spectra, samples were put in quartz tubes (2 mm inner diameter) and inserted into a special quartz Dewar flask filled with liquid nitrogen. Experimental uncertainties are estimated to be ±8% for τ determinations, ±10% for PLQYs, ±2 nm and ±5 nm for absorption and emission peaks, respectively.Computational details
Density functional theory (DFT) calculations were performed to investigate both the reaction mechanism and the photophysics of the complexes. All simulations were carried out using the B.01 revision of the Gaussian 16 program package,38 in combination with the M06 global-hybrid meta-GGA exchange–correlation functional.39,40 The fully relativistic Stuttgart/Cologne energy-consistent pseudopotential with the multielectron fit was used to replace the first 60 inner-core electrons of the iridium metal center (i.e., ECP60MDF) and was combined with the associated triple-ζ basis set (i.e., cc-pVTZ-PP basis).41 On the other hand, the Pople 6-31+G (d) basis was used for O atoms while the 6-31G(d) basis was adopted for all the others.42,43 The efficacy of the adopted computational protocol has already been validated in similar studies as reported in the literature.12,44 All the reported complexes were fully optimized without symmetry constraints, using the time-independent DFT approach. Frequency calculations were always used to confirm the nature of every stationary point found by geometry optimizations (i.e., minima or transition states, TSs). Ground-state properties and the reaction mechanism were investigated using spin-restricted DFT; oxidized and reduced species (doublets) and the lowest excited triplet states were optimized using a spin-unrestricted approach. Since solvation plays a key role in influencing the kinetics, thermodynamics and photophysics of metal complexes,45–47 environmental effects have been always considered using the polarizable continuum model (PCM) to simulate 2-methoxyethanol (for the investigation of the reaction mechanism) or acetonitrile (for photophysics) solvation effects.48–50In the reaction-mechanism study, some explicit solvent molecules (water or methanol, as a computationally cheap alternative to 2-ethoxyethanol) were considered; all TSs were modelled in an associative way, as previously adopted in the literature.51,52 Energy profiles were estimated using Gibbs Free energy (G) with the addition of zero-point energy (ZPE) and thermal corrections at 298.15 K in terms of the electronic energy.
To investigate the nature of the emitting states, time-dependent DFT (TD-DFT) calculations,53,54 carried out at the same level of theory used for geometry optimizations, were used to calculate the first 16 triplet excitations and their nature was assessed with the support of Natural Transition Orbital (NTO) analysis.55 The emission energy from the lowest triplet excited states was estimated by subtracting the SCF energy of the emitting state (Tem) in its minimum conformation from that of the singlet ground state having the same geometry and equilibrium solvation of Tem. All the pictures showing molecular geometries, orbitals and spin-density surfaces were created using Visual Molecular Dynamics (VMD) software56 or GaussView 6.57
Results and discussion
Optimization of the reaction conditions for the synthesis of A
As mentioned in the Introduction, some amounts of complex A were found randomly in a reaction mixture made for other purposes. To better understand the conditions that favor its formation, we have optimized the reaction, changing the quantities of previously used reagents, with the aim of increasing the yield of A and rationalizing the reaction mechanism. The main results are listed in Table 1.| Entry | Base (equiv.) | Water (%) | Temp.b (°C) | A (yield %)c | B (yield %)c |
|---|---|---|---|---|---|
| a Ir-dimer 0.028 mmol, VTOT solvent = 3.5 mL, t = 48 h, n.d. = not detected. b Temperature set in the heating system. c Yield of the isolated product after chromatography on silica gel. d t = 24 h. e Reaction performed under a nitrogen atmosphere. f Ir-dimer previously reacted with a silver(I) salt. g Starting from the [Ir(ppy)2(μ-OH)]2 dimer. | |||||
| 1 | K2CO3 (15) | 0 | 110 | n.d. | n.d. |
| 2 | K2CO3 (15) | 10 | 110 | n.d. | n.d. |
| 3 | K2CO3 (50) | 15 | 120 | n.d. | n.d. |
| 4 | K2CO3 (50) | 20 | 150 | n.d. | <5 |
| 5 | K2CO3 (15) | 0 | 150 | n.d. | n.d. |
| 6 | NaOH (50) | 20 | 150 | n.d. | <5 |
| 7 | K2CO3 (50) | 5 | 180 | <5 | 25 |
| 8 | K2CO3 (50) | 5 | 200 | n.d. | 27 |
| 9 | K 2 CO 3 (50) | 12 | 165 | 25 | n.d. |
| 10 | NaOH (50) | 12 | 165 | 24 | 10 |
| 11 | K2CO3 (50) | 0 | 165 | n.d. | <5 |
| 12 | K2CO3 (50) | 12 | 145 | n.d. | n.d. |
| 13 | K2CO3 (50) | 25 | 165 | n.d. | n.d. |
| 14d | K2CO3 (50) | 12 | 165 | 6 | n.d. |
| 15e | K2CO3 (50) | 12 | 165 | 18 | n.d. |
| 16f | K2CO3 (50) | 12 | 165 | n.d. | n.d. |
| 17 | HBF4 (50) | 12 | 165 | n.d. | n.d. |
| 18g | K2CO3 (50) | 0 | 165 | 25 | <5 |
As reported in entry 1, when the Ir-dimer reacts with a base (e.g., K2CO3) at 110 °C in 2-ethoxyethanol as a solvent, nothing happens, even if water is added to the reaction environment (entry 2). Notably, conditions of entry 2 are very similar to those commonly required for the synthesis of iridium(III) dimers.33 Therefore, as expected, in the reaction crude mixture we found unreacted Ir-dimer or species ascribable to the addition of solvent or hydroxyl groups to the dimer, whose instability towards coordinating solvents (e.g. DMSO or DMF) is already known.58,59
Even if the amounts of base and water are increased, and the temperature is raised up to 120 °C, there is no formation of A but only Ir-dimer residues are found (entry 3). Eventually, both the reaction temperature (150 °C) and the quantity of water (20%) were increased. In this case, in addition to Ir-dimer residues, we found traces of an emissive complex, later identified as B (entry 4).
However, when water is removed and the amount of K2CO3 is lowered, complex B is not formed (entry 5). Probably, at least a small amount of water in the reaction environment is needed to solubilize the base. Anyway, if K2CO3 is replaced with a strong base such as NaOH, the results were the same (entry 4 vs. 6).
Upon increasing the temperature to 180 °C (entry 7), we obtained two emissive compounds: complex B with a yield of 25% and traces of complex A. Upon increasing the temperature up to 200 °C only complex B was obtained (entry 8).
These results proved that to promote the formation of A it is necessary to maintain the reaction temperature between 150 °C and 200 °C and to have some water in the reaction environment. The reaction was thus set up with 12% of water at 165 °C. Finally, we have obtained complex A with a relatively good yield (25%) without the presence of complex B (entry 9). As previously observed (entry 4 vs. 6), preserving the same reaction conditions, but replacing the K2CO3 base with NaOH, leads to no substantial changes, but to the formation of a higher amount of by-product B (entry 9 vs. 10).
As a confirmation of the optimized conditions, the same reaction was carried out without water or by decreasing the temperature but no product A was formed (entries 11 and 12). Similarly, even after increasing the quantity of water in solution (entry 13), complex A was not obtained. Probably, too much water in the reaction environment substantially decreases the internal temperature of the reaction. By halving the reaction time (entry 14 vs. 9), complex A is still obtained but in a lower yield (6 vs. 25%, respectively).
When the reaction is carried out under a nitrogen atmosphere and using previously degassed solvents, the obtained results are nearly identical to those obtained under air-equilibrated conditions (compare entries 15 and 9). This proves that the oxygen atom inserted into the Ir–C bond does not come from atmospheric oxygen.
To better understand the reaction mechanism, a test was also performed using an Ir-dimer previously treated with a silver(I) salt. Ag+ ions are able to react with the bridging chlorine of the dimer, resulting in two mononuclear and independent iridium(III) fragments, together with solid AgCl. Under such conditions (entry 16), we did not detect any trace of A, indicating that the formation of such a complex must involve a rearrangement within the ligands of the iridium(III) dimer and does not involve a reaction between ligands coming from two separate complexes.
It should also be mentioned that replacing the base with an acid does not result in the formation of complex A, even using the optimized reaction conditions (entry 17 vs. 9).
Finally, we synthesized the [Ir(ppy)2(μ-OH)]2 dimer following a procedure previously reported in the literature (see Fig. S14†),60 and we used it as starting material instead of the [Ir(ppy)2(μ-Cl)]2 dimer to obtain A (entry 18). In this case the presence of water is not essential, indicating that the oxygen atom in the Oppy ligand comes directly from one of the μ2-bridging OH− ligands in the [Ir(ppy)2(μ-OH)]2 dimer.
Synthesis
In detail, using the optimized reaction conditions, (entry 8 for complex B and entry 9 for complex A), complexes A and B were obtained as air-stable yellow and orange solids, respectively, by careful purification via column chromatography on silica gel (see the Experimental section, method 1). Their structures were elucidated by one-dimensional (1D) NMR experiments (Fig. S1–S13†), ESI-MS spectrometry (Fig. S15 and S16†) and X-ray analyses (see below and the ESI†). Notably, the identification of B was quite easy since it is a well-known compound. In contrast, the determination of the structure of A was more challenging.1H-NMR analysis confirmed that both complexes are asymmetric compounds with 24 different hydrogens. However, the chemical shifts of the protons are different for A and B in CDCl3, allowing us to exclude that they are the same product (compare Fig. S1 and S7†). In addition, for complex A, some protons were superimposed on the residual peak of deuterated chloroform, and therefore the characterization was also carried out in CD2Cl2. Furthermore, we could also exclude the obtainment of the fac-[Ir(ppy)3] complex C, since it has only 8 signals in the 1H-NMR spectrum due to its C3 symmetry.
By means of ESI-MS analysis we identified their molecular weight (Fig. S15 and S16†). In the positive ion mode, both compounds showed peaks at m/z = 501 [(ppy)2Ir]+, m/z = 533 [(ppy)2Ir + MeOH]+ or m/z = 542 [(ppy)2Ir + MeCN]+, ascribed to a structure similar to that of the starting Ir-dimer and derived from sample fragmentations. Although these signals give us useful information on a moiety of the structure of these two complexes, the signals with higher molecular weights are more meaningful. For complex B we identify signals at m/z = 656 [M + H]+, 678 [M + Na]+and 694 [M + K]+, while for complex A we identify signals at m/z = 672 [M + H]+and 694 [M + Na]+. From these results we can state that the complexes are neutral and that their molecular ion (M) is different by 16 g mol−1.
Complex B was identified as mer-[Ir(ppy)3], while for complex A we hypothesized a very similar structure but with an additional oxygen atom. Finally, X-ray analysis (see below for a complete description) confirmed the structure of complex A, obtained with this uncommon synthesis.
In any case, to further demonstrate unequivocally the structure of the complexes, we synthesized them following classical procedures previously reported in the literature24,32,35 (Scheme 2), with only slight modifications (see the Experimental section, method 2).
 | ||
| Scheme 2 Alternative synthesis of the complexes used as a comparison for A and B. | ||
In particular, the Ir-dimer [Ir(ppy)2(μ-Cl)]2 was reacted with an excess of ligand HOppy, previously deprotonated with K2CO3 used as a base. The reaction mixture was kept under stirring for 24 hours under an inert atmosphere. After the usual workup and purification, complex A was obtained in a good yield (45%).
On the other hand, the procedure to get tris-homoleptic complex B reported in the literature was similar to that illustrated in the present paper, but carried out by adding more cyclometalating ligands at lower temperatures and without the addition of water. The Ir-dimer, 2-phenylpyridine and Na2CO3 were dissolved in 2-ethoxyethanol, and the mixture was stirred and heated for 3 hours at 135 °C. Compound B could be isolated in 35% yield. All compounds were fully characterized by 1H and 13C NMR spectroscopy (Fig. S2–S11†).
The NMR spectra of the products obtained using the last procedures, i.e. with the addition of the ancillary ligands to the Ir-dimer, perfectly match those of the products previously obtained by exploiting the synthesis reported in Scheme 1 (compare Fig. S1 and S12† for A, and Fig. S7 and S13† for B).
X-ray characterization
Suitable crystals for X-ray diffraction analysis could be obtained by slow diffusion of diethyl ether vapours in dichloromethane solutions of A. The structure of this complex is reported in Fig. 1 and the related crystallographic data are summarized in Tables S1–S8.† | ||
| Fig. 1 Experimental X-ray structure of complex A. Thermal ellipsoids are at the 50% probability level. Solvent molecules and hydrogens are omitted for clarity. | ||
In the crystal structure (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group), the two nitrogen atoms of the phenylpyridines are arranged in a trans relationship, with Ir–N bond lengths of 2.01 and 2.05 Å. Within the ancillary ligand, the Ir–N bond length is 2.17 Å and the Ir–O one is 2.16 Å. To better accommodate the larger six-membered ring, the two aromatic rings are not coplanar, with a skew angle of 33.5°. Additionally, the 2-hydroxyphenyl ring is strongly distorted from planarity (full structural details are reported in the ESI†).
space group), the two nitrogen atoms of the phenylpyridines are arranged in a trans relationship, with Ir–N bond lengths of 2.01 and 2.05 Å. Within the ancillary ligand, the Ir–N bond length is 2.17 Å and the Ir–O one is 2.16 Å. To better accommodate the larger six-membered ring, the two aromatic rings are not coplanar, with a skew angle of 33.5°. Additionally, the 2-hydroxyphenyl ring is strongly distorted from planarity (full structural details are reported in the ESI†).
DFT calculations: reaction mechanism
DFT methods were employed to investigate the reaction mechanism leading to the formation of [Ir(ppy)2(Oppy)] (A), starting from the iridium(III) dimer [Ir(ppy)2(μ-Cl)]2.Since water seems to play a significant role in the synthesis of A, as experimentally observed in Table 1, the hydrolysis of the Ir-dimer has been considered as the first step of the reaction mechanism. Indeed, hydrolysis reactions often represent a key step of the activation process for metal-containing compounds, with the aqua-complexes being more reactive than their non-aqua counterparts.45–47,51,52 In the present case, the hydrolysis of the Ir-dimer into the chloride-free [Ir(ppy)2(μ-OH)]2 dimer (P2) occurs in two consecutive steps (combined in Step I), implying the substitutions of the two chloride ligands with incoming OH− groups. Both steps are thermodynamically favored, as shown in Fig. S17 and 18 and Table S9† reporting the optimized structures and the free-energy reaction profile.
The first hydrolysis reaction involves the reagent adduct (RA1) where OH− interacts via a hydrogen bond (H-bond) with a μ2-Cl ligand, requiring 10.1 kcal mol−1 (Table S9†). The nature of the associated TS1a was observed by the single-imaginary vibrational mode, which corresponds to the bond breaking/formation process. The calculated activation free energy barrier is 8.9 kcal mol−1, which is in line with the literature.51,52TS1a evolves in an intermediate species (I1, Fig. S17†) where terminal OH and Cl coordinate with the two different Ir(III) atoms, with the breaking of the bridging structures. Intermediate I1 is strongly stabilized in energy, as shown in Fig. S18.† The mixed (μ2-Cl and μ2-OH) dimer (P1) is finally obtained via the product adduct (PA1), which is formed from a second TS (TS1b) showing a ΔG‡ = 11.9 kcal mol−1 (Table S9†).
The second hydrolysis step, leading to the formation of the [Ir(ppy)2(μ-OH)]2 dimer (P2), shows a similar reaction mechanism and free-energy profile (Fig. S18†). In particular, the ΔG‡ values of TS2a and TS2b are 9.4 and 12.0 kcal mol−1, to be compared to 8.9 and 11.9 kcal mol−1 for TS1a and TS1b, respectively (Table S9†).
The energy profiles of the reaction mechanisms for steps II to IV are shown in Fig. 2 (see also Fig. S19 and Table S9†). In the first part of step II, one of the μ2-bridging OH− ligands of P2 migrates as a terminal water molecule onto one of the iridium centers, leading to the formation of I3. Such a process involves an intramolecular proton transfer occurring without any activation energy barrier via the TS3a transition state (Table S9†). The resulting I3 intermediate is composed of a trigonal-bipyramidal iridium(III) unit and an octahedral one (with the attached water ligand), linked together through a μ2-oxo bridge.
 | ||
| Fig. 2 Free-energy profile calculated in 2-methoxyethanol (using PCM) for the reaction steps II to IV, leading to the formation of A, starting from the [Ir(ppy)2(μ-OH)]2 dimer (P2). Gibbs free energies are reported in kcal mol−1, relative to the Ir-dimer. | ||
The second part of step II is the most counterintuitive from a chemical point of view, since it can be thought of as the attack of a ppy carbanion to the μ2-oxo bridge. Indeed, when the Ir–C bond breaks, it does not result in a Ir(III) center and a carbanionic ppy− moiety, but a redox reaction occurs. The trigonal-bipyramidal iridium(III) center, during Ir–C bond breaking (TS3b) loses its highly positive charge and adopts the square planar structure typical of iridium(I) complexes (Fig. S19†).61,62 This reduction induces the oxidation of the previously negatively charged cyclometalated carbon, as highlighted in Fig. S20† reporting the Hirshfeld charges of such atoms. The so-obtained carbocation is then able to evolve into P3, with the formation of the C–O bond of the novel Oppy ligand. The difference in energy between I3 and P3 is only 3.2 kcal mol−1, but the transition state linking such minima exhibits a high energy barrier (ΔG‡ for TS3b = 35.1 kcal mol−1, Table S9†). Such an intramolecular reductive elimination step is able to explain why, if directly starting from the [Ir(ppy)2(μ-OH)]2 dimer, the presence of water is not required in the reaction environment to obtain complex A (Table 1, entry 18).
In step III, P3 interacts (via H-bond) with a solvent molecule (methanol or water) forming RA4 or RA′4 adducts, respectively (Fig. S19†). Notably, such a solvent molecule is able to replace the pyridine moiety of the ppy ligand in the coordination sphere of the Ir(I) center, leading to the formation of I4 and I′4 complexes. This process is the rate determining step (RDS) of the whole reaction mechanism with the highest ΔG‡ value of 36.9 kcal mol−1, calculated for TS4a in methanol. The barrier for the water-based TS′4a is even higher and therefore not further considered (Table S9†). Eventually, the uncoordinated pyridine moiety in I4 is able to substitute the water molecule bound to the Ir(III) center, leading to the formation of PA4, with an energy stabilization of 11.4 kcal mol−1 (Table S9†). Such displacement occurs throughout the transition state TS4b (Fig. S19†) with a ΔG‡ value of 9.0 kcal mol−1, which is comparable to the previously observed hydrolysis reactions (Table S9†). It is worth stressing that, at the end of step III, a ppy ligand has finally migrated from one iridium center to the other.
In the last step IV, the final product A is obtained along with the formation of the Ir(I)-complex by-product (X). The process starts with the migration of the water molecule from the close vicinity of the ppy ligand attached to the octahedral Ir(III) center (as in PA4) to the methanol molecule coordinated to the square-planar Ir(I) ion (as in RA5). Such reorganization of H-bonds induces a stabilization of 5.3 kcal mol−1 (Fig. 2 and Table S9†). The subsequent coordination of the water molecule to the Ir(I) ion leads to the formation of PA5, through the transition state TS5 (ΔG‡ = 11.2 kcal mol−1, Table S9†) with a negligible increase in energy (i.e., 1.8 kcal mol−1, Table S9†). In this compound, the final complexes A and X are already formed and interact via two H-bonds (Fig. S19†). Indeed, when the two products are completely separated in the final step of the reaction mechanism, a destabilization in energy of 9.2 kcal mol−1 is calculated.
In summary, the hydrolysis reactions of Step I are necessary for the activation of the [Ir(ppy)2(μ-Cl)]2 dimer and the formation of the thermodynamically-favored [Ir(ppy)2(μ-OH)]2 dimer (P2). Such reactions occur in the presence of water in a basic environment, since μ2-Cl− ions are replaced by μ2-OH− ones, as already observed in the literature.60,63 As experimentally proved in Table 1 (entry 16), the integrity of the P2 dimer is essential for the reaction progress. The presence of the right amount of water is important to maximize the formation of A at the expense of B (compare entries 9–11 in Table 1). Indeed, only P2 can undergo the proton-transfer reaction leading to the formation of the μ2-oxo bridge I3 (Fig. 2). Temperatures above 165 °C are necessary to overcome the high-energy barriers calculated for TS3b and TS4a in Step II and III. The whole reaction (i.e., Ir-dimer → A + X) is slightly disfavored from the thermodynamic point of view (ΔGr = 3.4 kcal mol−1). However, it must be emphasized that it is entropically favored and solvation can further stabilize the final products.
DFT calculations: ground-state properties
The structural and electronic properties of [Ir(ppy)2(Oppy)] (A) were further investigated by DFT methods and compared to those of the mer-[Ir(ppy)3] complex (B). For comprehensiveness, the archetypal fac-[Ir(ppy)3] isomer (C) was also included in this comparison.The overlap between the experimental X-ray structure of A and the DFT-optimized one is good (Fig. S21†), with only minor inconsistencies between the two geometries. Indeed, the Oppy ligand appears to be slightly more planar in the single-crystal structure, if compared to the DFT-optimized compound (i.e., 35.5° vs. 37.9°). Such a phenomenon can be explained by considering that important intermolecular π–π interactions involving the aromatic ligands are found in the crystal and they may lead to planarization due to staking.
The energy diagrams and the frontier molecular orbitals of A–C are depicted in Fig. 3. For all complexes, as commonly found in cyclometalated iridium(III) systems, the HOMO is predominantly localized on the iridium d orbitals and on the phenyl moieties of the ppy cyclometalating ligands;2,12 notably, in the case of A, such probability density is also spread on the phenoxy ring of the Oppy ligand. The largest HOMO stabilization is observed in fac-[Ir(ppy)3] (C), which displays three nearly degenerate occupied orbitals due to its C3 symmetry. On the other hand, complex A and mer-[Ir(ppy)3] (B) display a higher-lying HOMO located, for both compounds, at +0.10 eV above that of C.
 | ||
| Fig. 3 Energy diagram reporting the frontier Kohn–Sham molecular orbitals of A–C in acetonitrile. Orbitals with a similar topology are plotted with the same colour for an easier comparison. For some relevant orbitals, the corresponding isosurface is also displayed (isovalue = 0.04 e1/2 bohr−3/2). | ||
The effect of the insertion of the bridging oxygen atom in A has more impact on the unoccupied frontier molecular orbitals. Actually, in all of the investigated complexes, the LUMO, LUMO+1 and LUMO+2 remain always localized on the π* orbitals of the pyridine moiety of the three cyclometalated ligands, as commonly found in similar systems.64 Anyway, comparing A and B, a stabilization of about 0.1 eV is observed in the LUMO and LUMO+1 (centered on the two ppy ligands in both A and B), while the LUMO+2 (i.e., localized on the Oppy ligand of A) appears to be slightly destabilized compared to the corresponding orbital in mer-[Ir(ppy)3] (B). In contrast, no considerable effects are found due to mer-to-fac isomerization and the LUMO of fac-[Ir(ppy)3] (C) is destabilized by just 0.04 eV if compared to that of the mer-counterpart (B).
Electrochemistry
To explore the effect of the presence of the extra oxygen atom on the electronic properties of complex A, square-wave and cyclic voltammetry experiments were carried out in room-temperature acetonitrile solutions on all complexes (Fig. 4 and S22,† respectively) and the recorded redox potentials are collected in Table 2, relative to the Fc/Fc+ couple. | ||
| Fig. 4 Square-wave voltammograms of complexes A–C (0.5 mM) in acetonitrile solution at 298 K. | ||
| E ox (ΔEp)a [V (mV)] | E red (ΔEp)a [V (mV)] | ΔEredoxb [V] |
|
|---|---|---|---|
| a The reported potential values are obtained by square-wave and cyclic voltammetry and reported vs. the ferrocene/ferrocenium couple is used as an internal reference. The values in parentheses are the peak-to-peak separation from cyclic voltammetry (ΔEp); redox processes are reversible, unless otherwise stated (irr.). b ΔEredox = Eox − Ered. | |||
| A | +0.30 (irr.) | −2.576 (118), −2.802 (129) | 2.88 |
| B | +0.222 (67) | −2.634 (80), −2.878 (120) | 2.86 |
| C | +0.328 (69) | −2.669 (85), −2.902 (88) | 3.00 |
For all complexes, the oxidation process can be formally attributed to the Ir(III)/Ir(IV) redox couple, as already known from the literature2,12 and corroborated by the spin-density distributions calculated on the DFT-optimized oxidized radicals (Fig. S23,† top). The highest oxidation potential is found for fac-[Ir(ppy)3] (C), as expected by its more stabilized HOMO (Fig. 3). In contrast, the mere analysis of the HOMO levels is not able to explain the lower oxidation potential of mer-[Ir(ppy)3] (B) with respect to A (i.e., +0.224 vs. +0.304 V, respectively, Table 2), since the two complexes have the same HOMO energy. Only by taking into account the whole oxidation process vs. the ferrocene/ferrocenium couple (see the Experimental section for further details), DFT calculations provide a very accurate estimation of the oxidation potentials, resulting in a predicted Eox = +0.190 V for B and +0.324 V for A. Finally, it should be emphasized that the presence of the oxidized Oppy ligand in A not only increases the oxidation potential of the complex, but leads to irreversibility (Fig. S24†), while for both isomers of [Ir(ppy)3] (B and C) the oxidation process is fully reversible (Fig. S22†).65
As far as the cathodic region is concerned, the reduction processes in all complexes are reversible and can be ascribed to the reduction of the ppy ligands, as indicated by unrestricted DFT calculations (Fig. S23,† bottom). In this case, there is a direct correlation between the LUMO energy of the complexes and their reduction potential, with C having the most destabilized LUMO and the most negative Ered, and A displaying the lowest LUMO and the less negative reduction potential (compare Fig. 3 and Table 2). Indeed, considering the whole reduction process, the DFT estimates of Ered are remarkable: −2.529 V, −2.620 V and −2.672 V for A, B and C, to be compared to the experimental values of −2.576 V, −2.634 V and −2.669 V (Table 2).
Photophysical properties and excited-state calculations
The UV-Vis absorption spectra of complexes A–C were recorded in acetonitrile solution at room temperature (Fig. 5). The absorption spectra of the mer-[Ir(ppy)3] complex (B) and of the corresponding fac-isomer (C) are in line with literature data and were extensively commented elsewhere,32 so that band attributions and further remarks are not discussed here. On the other hand, it is worth mentioning that the absorption profile of [Ir(ppy)2(Oppy)] (A) strongly resembles that of parent complex B, but its molar absorption coefficients are generally lower, probably due to the lack of the conjugation of the Oppy ligand which can lead to a decrease in the intensity of the corresponding π–π* transitions. | ||
| Fig. 5 Absorption spectra of complexes A–C in acetonitrile solution at 298 K. The lowest-energy transitions are magnified in the inset. | ||
In order to get a deeper insight into the excited state properties of A–C, the lowest-lying triplet states of all the complexes were explored using TD-DFT methods. Tables S10–S12† show the lowest triplet excitations, reported in terms of Natural Transition Orbital (NTO) couples.55 A concise representation of the lowest triplet vertical excitations from the ground-state minimum is reported in Fig. 6 for all the investigated complexes.
 | ||
| Fig. 6 Energy diagram of the lowest-lying triplet states for complexes A–C, computed in acetonitrile as vertical excitations from the respective ground-state minimum-energy geometries. | ||
TD-DFT calculations indicate that fac-[Ir(ppy)3] (C) displays the highest-lying triplets of the series, which are found in groups of three nearly degenerate states due to C3 symmetry (Fig. 6). By removing such symmetry, as in the mer-[Ir(ppy)3] isomer (B), the corresponding triplet levels are more separated, with T1 and T2 lowering their energy by 0.08 and 0.03 eV, respectively, if compared with T1 in C. An even more pronounced stabilization of T1 and T2 is observed by passing from B to [Ir(ppy)2(Oppy)] (A). Indeed, in such an oxo-complex, T1 and T2 are further stabilized by approx. 0.05 eV, if compared to their analogues in B, despite preserving the same predominant 3LC nature on the trans-ppy ligands. On the other hand, T3 in A is centered on the Oppy ligand and experiences only a stabilization of 0.03 eV if compared to T3 in B, which is centered on the equatorial ppy ligand (Tables S10 and S11†).
Normalized emission spectra of complexes A–C are reported in Fig. 7, both in acetonitrile solution at 298 K and in butyronitrile glass at 77 K. The corresponding photophysical parameters are summarized in Table 3.
 | ||
| Fig. 7 Normalized emission spectra of complexes A–C in acetonitrile at 298 K (solid) and in butyronitrile glass at 77 K (dashed). Sample concentration: ≈15 μM. | ||
| CH3CN oxygen-free solution 298 K | BuCN glass 77 K | ||||||
|---|---|---|---|---|---|---|---|
| λ em [nm] | PLQYa [%] | τ [μs] | k r [105 s−1] | k nr [105 s−1] | λ em [nm] | τ [μs] | |
| a λ exc = 365 nm. b λ exc = 370 nm. c Radiative constant: kr = PLQY/τ. d Non-radiative constant: knr = 1/τ − kr. | |||||||
| A | 545 | 37.7 | 1.41 | 2.67 | 4.42 | 520 | 4.21 |
| B | 605 | 9.6 | 0.42 | 2.29 | 21.5 | 583 | 1.23 |
| C | 530 | 70.3 | 1.46 | 4.82 | 2.03 | 512 | 6.15 |
The novel [Ir(ppy)2(Oppy)] complex (A) displays emission properties that are somehow in between that of the already-known fac- and mer-[Ir(ppy)3] complexes (C and B).66,67 Indeed, A displays an emission profile that is very similar to the one of C, with just a minor redshift of 0.06 eV. Compared to the B analogue, the presence of the oxygen atom in [Ir(ppy)2(Oppy)] is able to impart a strong blue-shift of the emission and induce a remarkable increase in the photoluminescence quantum yield (i.e., λem = 545 vs. 605 nm and PLQY = 37.7 vs. 9.6% in Avs.B, Table 3).
Such experimental evidence seems to be in contrast to the scenario described in Fig. 6 by TD-DFT calculations, where A is estimated to show the lowest-lying T1 of the series at the ground-state minimum-energy geometry. However, if the triplet excited states are fully relaxed, DFT calculations are able to predict the correct trend of the experimental emission spectra. Indeed, the adiabatic energy difference between S0 and T1 was calculated to be 2.42, 2.35 and 2.55 eV for A, B and C, respectively (Fig. S25†). The gap is obviously reduced if considering the T1 → S0 vertical transition with non-equilibrium solvation (see the Experimental section for further details), leading to estimated phosphorescence at 2.14, 1.74 and 2.27 eV for A, B and C. If compared to the experimental mean-photon energy (Fig. S26†), the calculated phosphorescence is virtually perfect in the case of the fac-[Ir(ppy)3] complex (C) and underestimated by just 0.07 eV for [Ir(ppy)2(Oppy)] (A), while a larger underestimation is computed for the emission of mer-[Ir(ppy)3] (B).
As already stated, DFT calculations indicate that the nature of all the emitting states is 3LC (mainly localized on the ppy ligands) and this is also corroborated by the minor rigidochromic shift observed in the emission spectra recorded at 77 vs. 298 K (Fig. 7). The progressive loss of vibronic resolution by passing from fac-[Ir(ppy)3] (C) to [Ir(ppy)2(Oppy)] (A) and, then, to the mer-[Ir(ppy)3] complex (B), can be qualitatively explained by considering that the ppy ligands are all equivalent in C (due to its C3 symmetry in S0), leading to perfectly isoenergetic ppy-centered emitting triplets (all displaying C1 symmetry after relaxation). In contrast, such triplets are no longer equivalent in A and B, but sufficiently close in energy to be thermally equilibrated (Fig. S25†). Accordingly, they can be all emissive, leading to a progressively broader and less vibronically resolved spectrum (Fig. 7).
Conclusions
A serendipitous procedure for the synthesis of [Ir(ppy)2(Oppy)] (A) has been discovered and subsequently optimized. Starting from the Ir-dimer [Ir(ppy)2(μ-Cl)]2, it is possible to obtain neutral luminescent complexes without adding any further ancillary organic ligands to the reaction environment. An accurate modulation of the reaction conditions allows maximizing the formation of A at the expense of the mer-[Ir(ppy)3] complex (B). Surprisingly, in the structure of A, there is an oxygen atom bridging the iridium(III) center and the usually cyclometalating carbon of the 2-phenylpyridine ligand. A DFT investigation on the reaction mechanism was able to rationalize the experimental findings, explaining the feasibility of an apparently counterintuitive oxygen insertion. The so-obtained [Ir(ppy)2(Oppy)] complex displays a more blue-shifted emission with respect to the oxygen-free analogue mer-[Ir(ppy)3] complex (B), with superior PLQYs. In conclusion, this work introduces a non-standard methodology for the direct synthesis of new luminescent iridium(III) complexes based on the use of common iridium dimers as sole starting materials.This synthetic protocol has been tested and optimized here using just pristine 2-phenylpyridine as the prototypical cyclometalating ligand used in neutral iridium(III) complexes. Anyway, such an approach could be further extended to iridium(III) dimers with non-commercial or highly substituted ligands for obtaining complexes with the general formula [Ir(C^N)2(OC^N)]. Indeed, if using the standard approach, it would be necessary to react the [Ir(C^N)2(μ-Cl)]2 dimer with the corresponding hydroxyl-substituted ligand (here a relatively trivial 2-(o-hydroxyphenyl) pyridine), but such an ancillary ligand may not be easily obtainable; this work indicates that an alternative synthetic pathway could be explored.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the CNR (PHEEL and Progetti@CNR – RIPRESA) and funded by MUR-PNRR (NEST – Network 4 Energy Sustainable Transition, Extended Partnership – PE0000021) and PNRR Mission 04, Component 2, Investment 1.5 (project ECOSISTER – Ecosystem for Sustainable Transition in Emilia-Romagna, grant number ECS00000033 within Spoke 1 – Materials for sustainability and ecological transition and Spoke 2 – Clean energy production, storage and saving). Funding from the University of Bologna is also gratefully acknowledged.References
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed.
- R. D. Costa, E. Orti, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178–8211 CrossRef CAS PubMed.
- Y. You, J. Chin. Chem. Soc., 2018, 65, 352–367 CrossRef CAS.
- K. K. Lo, Acc. Chem. Res., 2015, 48, 2985–2995 CrossRef CAS PubMed.
- D. L. Ma, W. L. Wong, W. H. Chung, F. Y. Chan, P. K. So, T. S. Lai, Z. Y. Zhou, Y. C. Leung and K. Y. Wong, Angew. Chem., Int. Ed., 2008, 47, 3735–3739 CrossRef CAS PubMed.
- A. Maity, J. S. Choi, T. S. Teets, N. Deligonul, A. J. Berdis and T. G. Gray, Chem. – Eur. J., 2013, 19, 15924–15932 CrossRef CAS PubMed.
- K. Lo, K. Tsang, K. Sze, C. Chung, T. Lee, K. Zhang, W. Hui, C. Li, J. Lau and D. Ng, Coord. Chem. Rev., 2007, 251, 2292–2310 CrossRef CAS.
- A. Baschieri, S. Muzzioli, V. Fiorini, E. Matteucci, M. Massi, L. Sambri and S. Stagni, Organometallics, 2014, 33, 6154–6164 CrossRef CAS.
- M. H. Shaw, J. Twilton and D. W. MacMillan, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed.
- Q. Zhao, F. Li, S. Liu, M. Yu, Z. Liu, T. Yi and C. Huang, Inorg. Chem., 2008, 47, 9256–9264 CrossRef CAS PubMed.
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- F. Monti, A. Baschieri, L. Sambri and N. Armaroli, Acc. Chem. Res., 2021, 54, 1492–1505 CrossRef CAS PubMed.
- S. Y. Takizawa, R. Kano, N. Ikuta and S. Murata, Dalton Trans., 2018, 47, 11041–11046 RSC.
- E. Matteucci, A. Baschieri, A. Mazzanti, L. Sambri, J. Avila, A. Pertegas, H. J. Bolink, F. Monti, E. Leoni and N. Armaroli, Inorg. Chem., 2017, 56, 10584–10595 CrossRef CAS PubMed.
- Iridium(III) in Optoelectronic and Photonics Applications, ed. E. Zysman-Colman, John Wiley & Sons, Hoboken, New Jersey, 2017, vol. 1 Search PubMed.
- L. Jenšovský, Collect. Czech. Chem. Commun., 1957, 22, 1564–1568 CrossRef.
- L. Kábrt and Z. Holzbecher, Collect. Czech. Chem. Commun., 1976, 41, 540–547 CrossRef.
- P. Ganis, A. Saporito, A. Vitagliano and G. Valle, Inorg. Chim. Acta, 1988, 142, 75–79 CrossRef CAS.
- M. E. Burin, V. A. Ilichev, A. P. Pushkarev, D. L. Vorozhtsov, S. Y. Ketkov, G. K. Fukin, M. A. Lopatin, A. A. Nekrasov and M. N. Bochkarev, Org. Electron., 2012, 13, 3203–3210 CrossRef CAS.
- D. A. Bardwell, D. Black, J. C. Jeffery, E. Schatz and M. D. Ward, J. Chem. Soc., Dalton Trans., 1993, 2321–2327 RSC.
- N. Grossmann, A. Magri, M. Laux, B. Stadtmuller, P. Thielen, B. Schafer, O. Fuhr, M. Ruben, M. Cinchetti and M. Aeschlimann, Dalton Trans., 2016, 45, 18365–18376 RSC.
- D. A. Bardwell, J. C. Jeffery and M. D. Ward, Inorg. Chim. Acta, 1995, 236, 125–130 CrossRef CAS.
- Y. Hwang, Y. Park, Y. B. Kim, D. Kim and S. Chang, Angew. Chem., Int. Ed., 2018, 57, 13565–13569 CrossRef CAS PubMed.
- P. N. Lai and T. S. Teets, Chem. – Eur. J., 2019, 25, 6026–6037 CrossRef CAS PubMed.
- P.-B. Si, H.-F. Zhe, A.-H. Zhou, X.-Q. Liu, M.-Y. Teng, M.-Z. Rong, Y.-F. Wang, Q. Wang, Z.-L. Wang and J. Zhang, New J. Chem., 2021, 45, 18796–18804 RSC.
- A.-H. Zhou, T. Han, P.-B. Si, X.-Q. Liu, M.-Y. Teng, G.-L. Huang, B. Liu, Q. Wang and J. Zhang, J. Organomet. Chem., 2023, 985, 122596 CrossRef CAS.
- J. Massue, G. Ulrich, F. Monti and A. Barbieri, Eur. J. Inorg. Chem., 2020, 2020, 1775–1782 CrossRef CAS.
- J. Cerezo, A. Requena, J. Zuniga, M. J. Piernas, M. D. Santana, J. Perez and L. Garcia, Inorg. Chem., 2017, 56, 3663–3673 CrossRef CAS PubMed.
- Y. Hao, X. Guo, L. Lei, J. Yu, H. Xu and B. Xu, Synth. Met., 2010, 160, 1210–1215 CrossRef CAS.
- H. R. Park and Y. Ha, Mol. Cryst. Liq. Cryst., 2012, 567, 149–155 CrossRef CAS.
- M. A. Katkova, T. V. Balashova, V. A. Ilichev, A. N. Konev, N. A. Isachenkov, G. K. Fukin, S. Y. Ketkov and M. N. Bochkarev, Inorg. Chem., 2010, 49, 5094–5100 CrossRef CAS PubMed.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377–7387 CrossRef CAS PubMed.
- M. Nonoyama, Bull. Chem. Soc. Jpn., 1974, 47, 767–768 CrossRef CAS.
- S. Sprouse, K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 2002, 106, 6647–6653 CrossRef.
- J.-Y. Tsai, M. S. Barone, A. Tamayo and M. E. Thompson, US Pat, US7598388B2, 2009 Search PubMed.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Rev. B.01, Gaussian Inc., Wallingford CT, USA, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- D. Figgen, K. A. Peterson, M. Dolg and H. Stoll, J. Chem. Phys., 2009, 130, 164108 CrossRef PubMed.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS.
- A. Di Girolamo, F. Monti, A. Mazzanti, E. Matteucci, N. Armaroli, L. Sambri and A. Baschieri, Inorg. Chem., 2022, 61, 8509–8520 CrossRef CAS PubMed.
- D. Lovison, D. Alessi, L. Allegri, F. Baldan, M. Ballico, G. Damante, M. Galasso, D. Guardavaccaro, S. Ruggieri, A. Melchior, D. Veclani, C. Nardon and W. Baratta, Chem. – Eur. J., 2022, 28, e202200200 CrossRef CAS PubMed.
- D. Veclani, A. Melchior, M. Tolazzi and J. P. Ceron-Carrasco, J. Am. Chem. Soc., 2018, 140, 14024–14027 CrossRef CAS PubMed.
- D. Veclani, M. Tolazzi, J. P. Ceron-Carrasco and A. Melchior, J. Chem. Inf. Model., 2021, 61, 4391–4399 CrossRef CAS PubMed.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- C. J. Cramer and D. G. Truhlar, Continuum Solvation Models, in Solvent Effects and Chemical Reactivity, ed. O. Tapia and J. Bertrán, Springer Netherlands, 2002, vol. 17, pp. 1–80 Search PubMed.
- I. Ritacco, N. Russo and E. Sicilia, Inorg. Chem., 2015, 54, 10801–10810 CrossRef CAS PubMed.
- F. Ponte, I. Ritacco, G. Mazzone, N. Russo and E. Sicilia, Inorg. Chim. Acta, 2018, 470, 325–330 CrossRef CAS.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- A. D. Laurent, C. Adamo and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16, 14334–14356 RSC.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6, Semichem Inc., Shawnee Mission KS, USA, 2016 Search PubMed.
- M. Lepeltier, T. K. M. Lee, K. K. W. Lo, L. Toupet, H. Le Bozec and V. Guerchais, Eur. J. Inorg. Chem., 2007, 2007, 2734–2747 CrossRef.
- B. Schmid, F. O. Garces and R. J. Watts, Inorg. Chem., 2002, 33, 9–14 CrossRef.
- B. Beyer, C. Ulbricht, A. Winter, M. D. Hager, R. Hoogenboom, N. Herzer, S. O. Baumann, G. Kickelbick, H. Görls and U. S. Schubert, New J. Chem., 2010, 34, 2622–2633 RSC.
- L. Rubio-Perez, M. Iglesias, J. Munarriz, V. Polo, V. Passarelli, J. J. Perez-Torrente and L. A. Oro, Chem. Sci., 2017, 8, 4811–4822 RSC.
- S. Nückel and P. Burger, Organometallics, 2001, 20, 4345–4359 CrossRef.
- F. Scarpelli, A. Ionescu, L. Ricciardi, P. Plastina, I. Aiello, M. La Deda, A. Crispini, M. Ghedini and N. Godbert, Dalton Trans., 2016, 45, 17264–17273 RSC.
- J. Frey, B. F. Curchod, R. Scopelliti, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and E. Baranoff, Dalton Trans., 2014, 43, 5667–5679 RSC.
- J. C. Deaton, R. H. Young, J. R. Lenhard, M. Rajeswaran and S. Huo, Inorg. Chem., 2010, 49, 9151–9161 CrossRef CAS PubMed.
- T. Hofbeck and H. Yersin, Inorg. Chem., 2010, 49, 9290–9299 CrossRef CAS PubMed.
- N. Ide, N. Matsusue, T. Kobayashi and H. Naito, Thin Solid Films, 2006, 509, 164–167 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2249817. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02689b |
| This journal is © The Royal Society of Chemistry 2023 |