
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of Mg on the Li ion mobility in Li4−2xMgxP2S6†
Sven
Neuberger
 ,
Neeshma
Mathew
,
Sheyi Clement
Adediwura
and
Jörn
Schmedt auf der Günne
*
,
Neeshma
Mathew
,
Sheyi Clement
Adediwura
and
Jörn
Schmedt auf der Günne
*
University of Siegen, Faculty IV: School of Science and Technology, Department of Chemistry and Biology, Inorganic materials chemistry and Center of Micro- and Nanochemistry and Engineering (Cμ), Adolf-Reichwein-Straße 2, 57068 Siegen, Germany. E-mail: gunnej@chemie.uni-siegen.de
First published on 23rd October 2023
Abstract
Aliovalent substitution of Li in salts by Mg can generate Li vacancies and thus in principle improve the ionic conductivity. In fact the influence of substitution on ionic conductivity is far more complex. Here the impact of Mg substitution on Li ion mobility is studied in the example of Li4P2S6 by a combination of nuclear magnetic resonance experiments on 31P and 7Li at variable temperatures, impedance spectroscopy and X-ray powder diffraction to elucidate the relationship with structural changes and the effect on mobility on different length scales. It is found that substituting Li ions with Mg ions in Li4−2xMgxP2S6 with 0 ≤ x ≤ 0.2 increases the local Li ion mobility up to a certain concentration at which a phase transition induces a different structural realignment of the P2S64− units. The determined activation energies can be assigned to vacancy hopping processes by comparison with nudged elastic band calculations at the density functional level of theory, which shows not only the possibilities but also limitations of substituting Li with Mg.
Introduction
Lithium-rich non-oxido chalcogenide phosphates have become relevant as electrode materials in Li ion batteries and as separator materials in lithium (ion) batteries1 due to their high lithium ion conductivities,2 for example lithium argyrodites Li6PS5X (X = Cl, Br, I)3 or Li10GeP2S12.4 In order to design a compound with high Li ion conductivity, a compound does not only need to be rich in lithium, but also needs to feature high Li ion mobility in two or more dimensions.5 There are mainly two mechanisms for Li ion mobility in ceramic electrolytes,5 which are a vacancy-based and an interstitial site-based ion mobility mechanism. Both mechanisms mainly rely on point defects, such as Frenkel6 and Schottky defects.7 The overall Li ion conductivity for a vacancy-based mechanism is related to the product of the vacancy number density and the mobility of the defects, i.e. in principle a Li-rich compound with many vacancies should be ideal from this point of view.One way to increase Li vacancy concentration of a Li salt is aliovalent doping,8i.e. replacing an atom with one of different valence so that additional defects are produced to compensate for the valence mismatch. Aliovalent substitution of atoms of an anionic sublattice9,10 has been successful for example for Li argyrodites in which a sulfide S2− is substituted by a halide anion X−. In sum the substitution of Li+ and S2− with a halide anion X− and a vacancy □ produced fast ion conductors Li7−x□xPS6−xXx (X = Cl, Br, I).3,11 For complex anions aliovalent substitution of atoms of a bigger formal oxidation number has been successfully applied to different sulfidophosphates by replacing the center atom of a complex anion as in Li10MP2S12 (M = Sn, Ge, Si),4,12,13i.e. substitution of M(IV) and Li+ with M(V) and a vacancy □. A similar strategy is the substitution of Li+ by a metal atom with higher valence, including Mg2+, Ca2+, Fe2+ or Al3+ as shown for Mg2+ doped Li4Ti5O12,14 Li6−x−2yCayPS5−xCl1+x,15 Li7−2xFexPS6,16 or Li5.4Al0.2PS5Br.17 Besides the increase in mobility it has been observed that phase transitions are influenced as in the case of Li7−2xFexPS616 where by partial substitution of Li+ by Fe2+ the high temperature modification of Li7PS6 was stabilized.18
The test system in this study is lithium hexasulfidohypodiphosphate Li4P2S6, for which several crystal structures19–21 have been published on the basis of X-ray and neutron diffraction data with the motivation to resolve the unsatisfactory disorder of P sites in the initially published structure.22 While the motifs of the different structures – a chain-like arrangement of the P2S64− group and Li atoms on octahedral sites – are found in all proposed structural models, only the model in the space group P321 (Z = 3) explains all experimental data including the presence of minimum three P sites as shown by two-dimensional double-quantum 31P NMR.21 The structural model suggests a fully ordered unit cell, where Li ion hopping should involve interstitial sites or vacancies. While for the different models which are inconsistent with the NMR findings, detailed studies20,23 including quantum-chemical calculations have been done to provide estimates for activation energies, but this is not the case for the model in P321. In the course of finalizing the manuscript we became aware of a single-crystal X-ray study in which the authors succeeded to solve the structure of Li4P2S6 from twinned crystals,24 in the space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 (Z = 3), which is in full agreement with the 31P NMR results, but reduces the number of crystallographic orbitals for Li from 4 to 2. In terms of the arrangement of the atoms it only shows minor deviations from the structure described in the space group P321 (Z = 3) to which it is a supergroup, so that both structures converted to the space group P1 produce the same results in the quantum-chemical calculations shown below. It is an open question if a phase transition between the two structures exists. For this reason we have based this contribution on the structure described in P321, which does not change any of the made conclusions about Li mobility though.
m1 (Z = 3), which is in full agreement with the 31P NMR results, but reduces the number of crystallographic orbitals for Li from 4 to 2. In terms of the arrangement of the atoms it only shows minor deviations from the structure described in the space group P321 (Z = 3) to which it is a supergroup, so that both structures converted to the space group P1 produce the same results in the quantum-chemical calculations shown below. It is an open question if a phase transition between the two structures exists. For this reason we have based this contribution on the structure described in P321, which does not change any of the made conclusions about Li mobility though.
Interestingly several groups have reported electrical conductivity values for Li4P2S6 which differ by orders of magnitude in the conductivity values at room temperature (σRT = 1.6 × 10–10 S cm−1 and 4.7 × 10–5 S cm−1)20,25 and also the reported activation energies differ significantly (EA = 0.29 eV to 0.48 eV).19,20 Aliovalent substitution has been tried with magnesium with the compositions Li4−2xMgxP2S6, with x ≥ 1/2, featuring increased ionic conductivities26 of up to σRT ∼ 10–6 S cm−1. For the compositions Li3.33Mg0.33P2S6 and Li2.66Mg0.66P2S6 trigonal disordered crystal structures (both space group P1m)26 were derived from powder diffraction data which were based on the initial model of Li4P2S6 which has P sites with 50% occupancy. Interestingly the high magnesium content phase shows a layered structure as suggested for Mg2P2S627 in the space group C2/m meaning that small quantities of Li doped into Mg2P2S6 cause a phase transition into a phase in the space group P1m. An open question is whether in the phase diagram of Li4P2S6 Mg2P2S6 phases exist, which in the low-Mg doping regime feature an enhanced Li conductivity because of a suppressed phase transition and whether the expected phase transition has a positive or a negative effect on the Li ion conductivity. Furthermore it makes sense to double-check the structural model of the highly substituted phases Li4−2xMgxP2S6 given that the structure of Li4P2S6 in the space group P321 was not known at the time.
The purpose of this study is to evaluate which effects aliovalent doping/substitution with Mg can have on sulfidophosphates both in terms of structure and ionic conductivity. As a model system serves the ordered and not highly conductive crystalline substance Li4P2S6.
Experimental part
Synthesis
All starting materials were stored inside a glove box (MBraun, Garching, Germany) under an argon atmosphere. All sample preparations were carried out in the same glove box under the same conditions.Magnesium powder (8.868 mmol, 500.0 mg, Alfa Aesar, 99.9%) was filed off a bar with a rasp and placed in a graphite boat in a gas flow quartz glass apparatus. The magnesium was heated in a tube furnace at 590 °C under a H2S atmosphere for 17 h to obtain magnesium sulfide MgS.
Li4−2xMgxP2S6 was synthesized according to eqn (1). Red phosphorus (1.115 mmol, 138.1 mg, ACROS Organics, 99.999%), sulfur (1.120 mmol, 287.3 mg, ChemPur, 99.999%), lithium sulfide Li2S (4.353 mmol, 200.0 mg, Alfa Aesar, 99.9%) and magnesium sulfide MgS were thoroughly ground and mixed in an agate mortar and afterwards placed in a graphitized quartz ampoule (8 mm outer diameter). The ampoule was sealed under vacuum and afterwards heated to 800 °C in a tube furnace for seven days.
 | (1) |
NMR spectroscopy
The chemical shift values of 31P and 7Li are reported on a deshielding scale, relative to 85% H3PO4 and to a solution of LiCl in D2O (9.7 mol kg−1), respectively. The 1H resonance of 1% Si(CH3)4 in CDCl3 served as an external secondary reference using the Ξ values for 31P and 7Li as reported by the IUPAC28,29 for all solid-state NMR measurements. All magic angle spinning (MAS) rotors (4 mm, ZrO2) were packed under an argon atmosphere in a glove box. The 31P and 7Li MAS NMR measurements were carried out on a Bruker Avance II spectrometer at a frequency of 121.50 MHz and 116.64 MHz, respectively (B0 = 7.0 T), with a home-built 4 mm McKay probe at a sample spinning frequency of νrot = 10 kHz. For the 31P and 7Li MAS NMR measurements, a repetition delay of 1 s was used. The static variable temperature (VT) 7Li NMR measurements were carried out on a Bruker Avance II NMR spectrometer at a frequency of 116.66 MHz (B0 = 7.0 T) with a home-built static McKay probe in an evacuated quartz ampoule (d = 3 mm). The analysis and fitting of the spectra were done with Deconv2Dxy (version 0.7).30 The static 7Li spectra were deconvoluted with multiple Gaussian functions per spectrum to get a good description of the full lineshape. By using only Gaussian functions for the deconvolution it is straight-forward to determine the second moment of the lineshape.30X-ray powder diffraction
X-ray powder diffraction (XRD) patterns were recorded using a STOE STADI P powder diffractometer (Stoe, Darmstadt, Germany) with Ge(111)-monochromated Cu Kα1 radiation (λ = 1.54056 Å) in Debye–Scherrer geometry (capillary inner diameter: 0.48 mm) with a linear position sensitive detector. A 2Θ range between 10° and 90° with a step size of 0.01° was measured, using a counting time of 120 s per step.Pawley fits and Rietveld analysis
Rietveld refinements were carried out using TOPAS academic V7.1.31 The Li4P2S6 crystal structure21 with the space group P321 (no. 150) was used as a starting model. In the given order, the following parameters were refined: the scale factor and background coefficients using a Chebyshev function with 10 free parameters, the peak shape using the fundamental parameter approach, the zero-shift error and the lattice constants. For the atom positions of the P atoms, split positions were used to determine site preferences of the respective atoms. The anisotropic displacement parameters were fixed to 1. Mg atoms were not refined due to the lack of scattering contrast. Fit indicators of Rwp, Rexp, and GOF were used to assess the quality of the refined structural models.32Electrochemical impedance spectroscopy
The Li3.74Mg0.13P2S6 powder sample was thoroughly ground in an agate mortar and pressed into a cylindrical pellet (diameter d = 13 mm) by applying a pressure load of 4 tons (equivalent to p = 295 MPa) for 20 minutes. The thickness of the pellet was measured with an electronic micrometer screw gauge. Afterwards, the pellet was brought into contact with silver foil (thickness: 0.1 mm, R. Götze GmbH & Co. KG, silver content 935) by pressing silver sheets (diameter d = 13 mm) onto both sides of the pellet (load of 4 tons, equivalent to p = 295 MPa for 20 minutes). Samples were handled inside a glove box (MBraun, Garching, Germany) under an argon atmosphere.Electrochemical impedance spectroscopy (EIS) measurements were performed using an NEISYS electrochemical impedance analyzer (Novocontrol Technologies, Montabaur, Germany). Short and load calibrations were performed before the impedance measurements with a 100 Ω resistor as the load. The prepared pellet was mounted onto a home-made EIS sample cell for impedance measurements. A quasi-four-wire measurement setup in the potentiostatic mode was used, with a perturbation signal of 10 mV amplitude, swept through the 50 mHz to 1 MHz frequency range. The temperature was controlled using variable temperature and flow controller (NMR Service GmbH, Erfurt, Germany) with a constant nitrogen gas flow and a setup with three thermocouples for the sample, the sample holder and the Dewar containing the sample holder. Each temperature point was held for 20 min before every EIS measurement to ensure temperature stability with an accuracy of ±0.1 K throughout the measurement period. The data analysis was performed using home developed software using SciPy libraries33 which implement non-linear least squares fitting and error analysis.
Computational methods
A plane wave (PW) pseudopotential (PP) method based on density functional theory (DFT) using Quantum Espresso 6.8 (QE)34 was employed to calculate the Li-ion diffusion properties in Li4P2S6.The cif file of Li4P2S6 with the space group P321 (ICSD-434755)21 was converted into an input file using cif2cell 2.0.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35 for the QE calculations. All fractional co-ordinates were allowed to relax using the QE relax calculations under the periodic boundary conditions. Optimised pseudopotentials of GBRV type36 were used for the simulations with Monkhorst–Pack scheme37 based optimisation for the selection of automatic k points. For the final simulations a k-mesh of 4 × 4 × 4 over the Brillouin zone, resulting in 36 k-points including the gamma point, was employed with an error of convergence of 0.001 eV. A plane wave kinetic energy cut-off of 60 eV and kinetic energy cut-off for charge density of 525 eV were used with an error of convergence of 0.027 eV.38
35 for the QE calculations. All fractional co-ordinates were allowed to relax using the QE relax calculations under the periodic boundary conditions. Optimised pseudopotentials of GBRV type36 were used for the simulations with Monkhorst–Pack scheme37 based optimisation for the selection of automatic k points. For the final simulations a k-mesh of 4 × 4 × 4 over the Brillouin zone, resulting in 36 k-points including the gamma point, was employed with an error of convergence of 0.001 eV. A plane wave kinetic energy cut-off of 60 eV and kinetic energy cut-off for charge density of 525 eV were used with an error of convergence of 0.027 eV.38
Climbing Image-Nudged Elastic Band (CI-NEB)39 calculations were performed to calculate the activation energy for the supercell containing a 1 × 1 × 2 unit cell.40 The Li-ion diffusion was calculated by creating vacancies in the nearest sites as the first and last images. Eight intermediate images were used along with the first and last images to identify the diffusion pathway for the Li-ion transport.41
Results and discussion
In order to obtain a better understanding of the effect of Mg doping, in the first part the structural impact is studied by X-ray diffraction and 31P magic-angle-spinning (MAS) NMR and in the second part the impact on Li mobility is studied by 7Li NMR, nudged elastic band calculations and impedance spectroscopy (EIS). Finally, the results are discussed in the context with earlier work on this system and their implications on Mg doping in general.Investigation of structural changes
What is apparent from the previous studies is that the limiting phases in the phase diagram i.e. Mg2P2S6 in the space group C2/m27 and Li4P2S6 in the space group Pm1 or P32121 do not share a structure which allows the formation of a single solid solution over the complete range because the P2S6 groups do not have a compatible arrangement relative to each other. What could be shown convincingly in the previous study26 is that even a small amount of substitution of Mg for Li causes a phase transition away from the monoclinic structure of Mg2P2S6. For this reason, the focus here is on the low-substitution regime. Important for the solution of the structure of Li4P2S6 in the space group P321 were three 31P NMR peaks which could be resolved by 2D 31P single-quantum double-quantum correlation NMR experiments. Besides, the structural models need to explain a reflection in the powder diffraction at 16.6°21,24 which was absent in previously suggested models. These markers can thus help to identify a phase transition to a layered structural arrangement of the P2S6 groups.
The 31P MAS NMR spectra of Li4−2xMgxP2S6 (Fig. 1) contain two sharp peaks at δ = 107.3 and 109.2 ppm with a peak area ratio of exactly 1.0:2.0 at magnesium contents below x = 0.07. With higher magnesium concentrations above x > 0.07, the peak area ratio changes and the signals start to merge and become broader indicating higher disorder due to structural changes and defect formation. The 31P MAS NMR spectra over a wider ppm range are depicted in Fig. S1.† Also, the T1 relaxation times of the 31P NMR signals decrease with increasing magnesium content (Fig. S2†). 31P MAS NMR, scanning electron microscopy coupled with energy dispersive X-ray spectroscopy and X-ray diffraction (Fig. S10–S13†) give evidence of a homogeneous compound and do not give evidence for side-phases.
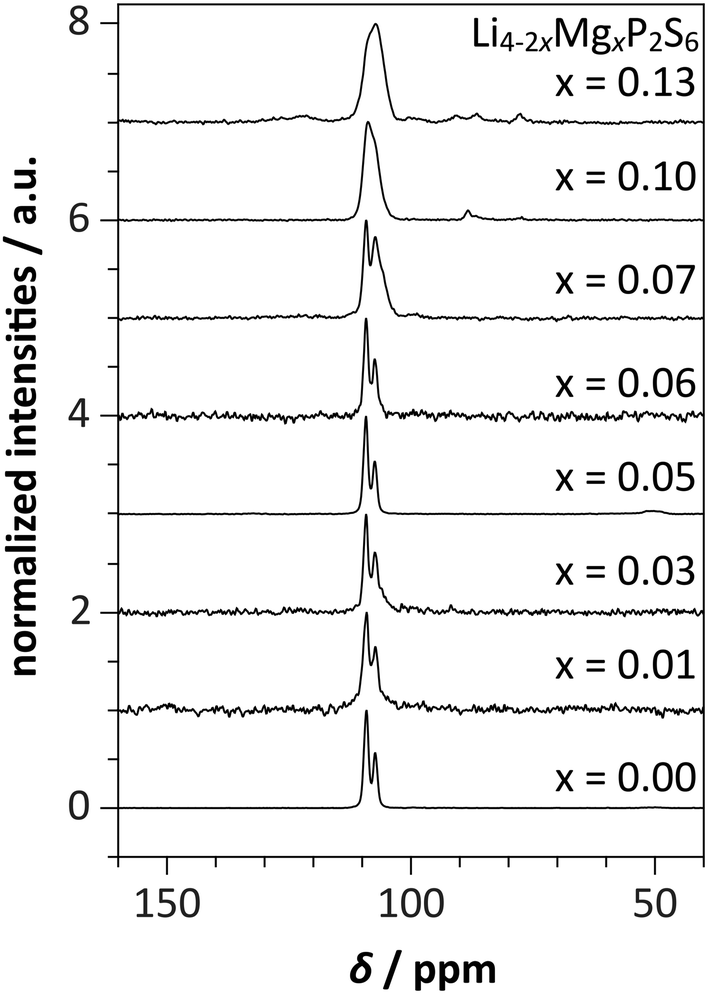 | ||
| Fig. 1 Experimental 31P MAS NMR spectra of Li4−2xMgxP2S6 at a sample spinning frequency νrot of 10 kHz. At low Mg content, the 31P MAS NMR spectra contain the characteristic signals of Li4P2S6 with a peak area ratio of exactly 1.0:2.0, which merge into one broad signal at Mg concentrations of x > 0.06. | ||
The broadening of the 31P NMR resonances is expected for a disordered phase where Li atoms are statistically substituted by Mg atoms even without a change of the crystal structure. However, the change in peak area ratio at values of x bigger than 0.07 is indicative of a structural change, i.e. a change of the space group and or structure type of Li4−2xMgxP2S6. This can be interpreted as a solubility limit of Mg (x ≈ 0.07) in the structure of pure Li4P2S6. The peak broadening is consistent with the formation of a solid solution and not a mixture of ordered phases.
To further analyze the structural changes, Li4−2xMgxP2S6 was analyzed with X-ray powder diffraction. Up to the highest degree of Mg substitution made in this study of x = 0.20, the XRD patterns only show sharp reflections similar to those of Li4P2S621 (Fig. 2a). However, it should be noted that the 101 reflection at a 2Θ angle of 16.6° disappears at high substitution levels (Fig. 2b). These results confirm the structural model proposed for high substitution levels26 in the space group P1m. Helpful in this context are the previous trials for solving the structure of Li4P2S619,20,22 which have suggested layered structures and are consistent with the powder diffractogram with a reflection missing at a 2Θ angle of 16.6°. The results confirm the interpretation of Mg-substitution at high levels by Takada et al. but require a revision in the low-substitution regime, where up to a composition of x = 0.07 a defective structure of Li4P2S6 (Fig. 3 left, a model based on P321) is preferred over the layered arrangement (Fig. 3 right, a model based on P1m (Z = 1)). The full-scale figure is also depicted in Fig. S3.†
 | ||
| Fig. 2 (a) X-ray powder diffraction patterns (Cu Kα1 radiation) of Li4−2xMgxP2S6 with increasing Mg2+ content. The position of the most intense diffraction peak of Li4P2S6 is marked with a red line. All diffraction patterns only show sharp diffraction peaks of Li4P2S6.21 (b) Detailed comparison of the (101) Miller reflection (at 16.6°) and the (110) Miller reflection (at 16.9°) of Li4−2xMgxP2S6 with increasing Mg2+ content. The positions of the two reflections are marked with dashed lines. At higher Mg2+ concentrations the intensity of the (101) Miller reflection decreases until it completely vanishes at x ≥ 0.07. | ||
 | ||
| Fig. 3 Visualization of a 2 × 2 × 2 supercell of Li4−2xMgxP2S6. The structural transformation from the space group P321 (left cells) to a layered structural model occurs due to an incorporation of Mg2+ ions. The doping process leads to a P1m like layered alignment of the P2S64− units similar to that for Mg2P2S6 (space group C2/m) and as expected for the structural model of Li3.33Mg0.33P2S6 (space group P1m).26 The insets show the coordination environments for the Mg2+ ions (orange) for the different structural models. In the space group P321, the coordination polyhedra of Mg2+ are slightly distorted due to a different alignment and thus leads to a different number of P2S64− units surrounding the central atom of the polyhedron. | ||
To understand how the substitution process leads to a phase transition, it is helpful to compare the limiting structures. Following the structural models of Li3.33Mg0.33P2S6 (space group P1m)26 and Mg2P2S6 (space group C2/m),42 the preferred positions for the Mg2+ ions are the sites within the P2S64− layer (Fig. 3 right) while the defects are located in the interlayer. From the relative arrangement of the P2S6 groups it is clear that a solid solution with only gradual changes is impossible.
This interpretation is further corroborated by the results of Rietveld refinements carried out using a structural model with the space group P321 and with extra split positions for the P atoms with variable occupancy in order to trace the structural changes. Due to the lack of scattering contrast only the heavy elements were refined and Mg/Li were not explicitly determined but their atom positions were fixed to the values of the undoped compound with the space group P321. The split position in principle allows for a gradual change of the arrangement from the P321 structure over to the layered arrangement.
The shift of the P1 atoms upon increasing Mg2+ concentration results in an additional crystallographic site P1b shifted by half of the lattice constant in the z-direction in contrast to the original P1a site (Fig. 4). The occupancy factor of the P1a site significantly drops at x > 0.05, which is in agreement with the suggested model. An exemplary structural model is depicted in Fig. 5 for x = 0.13 and Table 1.
 | ||
| Fig. 4 Change of the occupancy factor of the P1a site with increasing Mg2+ concentration; the two dashed lines are a guide to the eye. The occupancy factor is close to 1 at concentrations x ≤ 0.05. At x > 0.05, the occupancy factor decreases, indicating a site preference for the P1b site of the structural model. | ||
 | ||
| Fig. 5 Exemplary structural model of Li3.74Mg0.13P2S6 at room temperature, obtained from Rietveld refinements of X-ray powder data (λ = 1.54060 Å) without refining the Mg2+ ions. The P1a and P1b sites are indicated with different colors to show the shift of the P2S64− ions upon increasing Mg concentration. | ||
| Li3.74Mg0.13P2S6 structure from X-ray powder diffraction (space group P321, no. 150); | ||||||
|---|---|---|---|---|---|---|
|
a = 10. 5211(2) Å; c = 6. 59924 (18) Å |
||||||
| Fit residuals (Rwp, Rexp, GOF): 10.47, 7.80, 1.34 | ||||||
| Atom | Wyckoff site | X | Y | z | Occ. | B eq/Å2 |
| Li1 | 3e | 0.625 | 0 | 0 | 0.935 | 1 |
| Li2 | 3f | 0.631 | 0 | 0.5 | 0.935 | 1 |
| Li3 | 3e | 0.317 | 0 | 0 | 0.935 | 1 |
| Li4 | 3f | 0.329 | 0 | 0.5 | 0.935 | 1 |
| P1a | 2c | 0 | 0 | 0.17 | 0.571(8) | 1 |
| P1b | 2c | 0 | 0 | 0.663 | 0.429(8) | 1 |
| P2 | 2d | 0.333 | 0.666 | 0.663 | 1 | 1 |
| P3 | 2d | 0.333 | 0.666 | 0.335 | 1 | 1 |
| S1 | 6g | 0.104(2) | 0.211(1) | 0.247(1) | 1 | 1 |
| S2 | 6g | 0.114(1) | 0.560(2) | 0.251(1) | 1 | 1 |
| S3 | 6g | 0.452(2) | 0.227(3) | 0.254(1) | 1 | 1 |
Investigation of the Li ion mobility
The questions we wanted to answer are, if in the low Mg substitution regime an increase in Li ion conductivity exists and what can be learned about the Li ion motion in the two different structure types. A critical point in the Mg substitution strategy is that while the vacancy concentration increases with increasing Mg content, Mg could act as a “roadblock” in the pathway of the mobile Li ions because of its higher formal charge. To this end 7Li NMR and nudged elastic-band calculations were utilized to study microscopic Li mobility and electrochemical impedance spectroscopy (EIS) to study macroscopic Li mobility.In such a case, moment analysis provides a handy tool for motion analysis.43–45 The decrease of the second moments M2 of the central transition of the static 7Li signals is indicative of motional narrowing and hence of motional averaging of the anisotropic terms of the chemical shift tensor. For the static 7Li NMR signals, the second moments M2 were calculated to quantify the change in line broadening (Fig. 6a). The static 7Li NMR signals increase and the room temperature 7Li T1 relaxation time shows a change of trend for an x value of about 0.07. This is consistent with the postulated phase change at around the same value as observed by 31P MAS NMR and XRD. If the 7Li quadrupole interaction is assumed to be conserved upon Mg substitution, then the sharper lineshape can be interpreted as a more efficient motional averaging, because of a faster Li mobility. The room temperature 7Li T1 relaxation times can be interpreted in a similar manner assuming that the temperature applies to the low-temperature regime of a plot of relaxation rate versus inverse temperature rate. In that regime according to BPP theory46 an increase in relaxation rate 1/T1 corresponds to a decrease in the correlation time related to Li mobility, i.e. in a crude approximation, a higher relaxation rate is indicative of a higher Li mobility. A consequence of this interpretation is that there is no fast Li conducting phase in the low substitution regime and that the layered structure which is preferred at high substitution levels is beneficial for Li-ion conductivity.
 | ||
| Fig. 6 (a) Second moments M2 of the static 7Li NMR signals of Li4−2xMgxP2S6. M2 first increases with increasing Mg2+ concentration up to x = 0.05. At x ≥ 0.06, the second moments start to decrease. (b) T1 relaxation times of the 7Li NMR signals of Li4−2xMgxP2S6 at 116.66 MHz. The T1 relaxation times decrease with increasing Mg2+ concentration. At x ≥ 0.05, the T1 relaxation time decreases faster until x = 0.07. | ||
Finally, what remains to be clarified are the activation energies for a vacancy based ion conduction mechanism. For this purpose, detailed studies were done on Li3.74Mg0.13P2S6 with the help of static 7Li variable temperature (VT) NMR and line shape analysis as well as electrochemical impedance spectroscopy (EIS). In the static 7Li VT NMR spectra (Fig. 7a), a big change in line shape was noticeable due to motional narrowing.
 | ||
| Fig. 7 (a) Normalized static 7Li VT NMR spectra of Li3.74Mg0.13P2S6 from 173 K (dark blue) to 463 K (red). The static 7Li NMR signals become sharper with increasing temperature. (b) Second moments M2 of the static 7Li VT NMR signals of Li3.74Mg0.13P2S6 (blue points) and Li4P2S6 (black squares) with two fitted lines each. The intersections of two lines for each mark the onset points at the temperature at which the motional narrowing becomes more intense. | ||
The theory devised by Waugh and Fedin43,44 can be used to estimate the activation energy for lithium ion motion with an inaccuracy of 10% using the onset temperature Tonset of this motional process according to eqn (2). Therefore, the onset points for the motional narrowing for Li3.74Mg0.13P2S6 and Li4P2S6 were constructed as an intersection of two linear fits g(T) and h(T) (eqn (S1) and (S2)†).
 | (2) |
Onset temperatures of Tonset = 342.3 K for Li3.74Mg0.13P2S6 and Tonset = 380.6 K for Li4P2S6 were determined, which correspond to activation energies EA of 0.55(5) eV for Li3.74Mg0.13P2S6 and 0.62(6) eV for Li4P2S6, respectively, i.e. a small reduction of the activation energy is observed which describes the local Li ion mobility.
Macroscopic motion was studied by EIS in Li4P2S6 which was required because pure Li4P2S6 did not get characterized together with the Mg substituted compounds.26 The Nyquist plots of both the pure and the Mg substituted compound Li3.74Mg0.13P2S6 (Fig. 8) show only one large resistive contribution to the impedance data, depicting the bulk region based on the effective capacitances obtained from the equivalent circuit analysis (Table 2). To ensure reproducibility of the electrochemical impedance spectroscopy measurements, data from a second heating cycle were also recorded for both compounds, exhibiting the same ionic behavior as the first heating cycle for both compounds. Hence, both compounds did not change during the measurement process (Fig. 9).
 | ||
| Fig. 8 Nyquist plots of the electrochemical impedance spectroscopy measurements of Li4P2S6 and Li3.74Mg0.13P2S6 measured between 304 K and 364 K. The plot shows one half cycle per temperature. The inset shows the circuit configuration used as a fit model for the Nyquist plots consisting of a resistor RT, a constant phase element QT and a constant phase element for the electrode region Qs. | ||
 | ||
| Fig. 9 Arrhenius plot of the logarithmic ion conductivities of Li4P2S6 and Li3.74Mg0.13P2S6 as a function of the reciprocal temperature and the linear fits are indicated with dashed lines. The activation energy for the Li ion motion can be estimated from the slope of a linear fit. | ||
The room temperature Li ion conductivity σ298 K of undoped Li4P2S6 reported here is lower than the previously reported values.19,20,25 Also, the activation energy EEISA of Li4P2S6 is much higher than the ones reported in the literature. Such deviations are not uncommon and a possible explanation for these deviations can be found in the different preparation techniques. The reported 31P NMR spectrum of crystalline Li4P2S6 with an ionic conductivity of 1.6 × 10–10 S cm−1 shows the presence of side phases of Li3PO4 and Li3PS4.20 The reported ionic conductivity of 4.7 × 10–5 S cm−1 resulted from powder samples produced by mechanical milling and they show a low degree of crystallinity in the X-ray diffraction patterns.25 The third reported value of 2.4 × 10–7 S cm−1 results from powder samples, which were post processed by a washing step, ball milling and annealing.19 While it is known that the sample preparation for impedance spectroscopy can lead to significant uncertainty in the conductivity parameter, we ascribe the deviations here mostly to purity, morphology and crystallite sizes of the Li4P2S6 powder samples which have been investigated.
The results from the EIS measurements presented here confirm the assumed increase in ionic conductivity (Table 2) by magnesium substitution.26 Furthermore it is shown that the overall conductivity of pure Li4P2S6 is low while the activation energy agrees reasonably well with those found in previous studies19,20,25 where compounds of good crystallinity and purity were studied i.e. in ref. 20 (Table 2). It can be concluded that the Mg substitution leads to both a phase change and increase of the vacancy concentration which are beneficial for ionic conductivity.
Finally, quantum chemical calculations using the “nudged elastic band” concept were employed to estimate activation energies for individual jump processes which in turn could help to identify the bottlenecks in Li mobility. Simulations in former studies19,20 for pure Li4P2S6 suggested that the lithium ion motion mainly relies on interstitial transport in the x–y-plane of the crystal structure with the space group P1 m, while Li ion diffusion in the z-direction is energetically unfavourable since it involves Li ion jumps between the P2S64− layers. In order to determine the diffusion pathways for Li4P2S6 in the space group P321, Climbing Image-Nudged Elastic Band (CI-NEB) calculations were carried out.
For the structure in the space group P321, potential migration pathways for vacancy mediated diffusion were identified and the activation energies for the local jumps were calculated (Fig. 10).
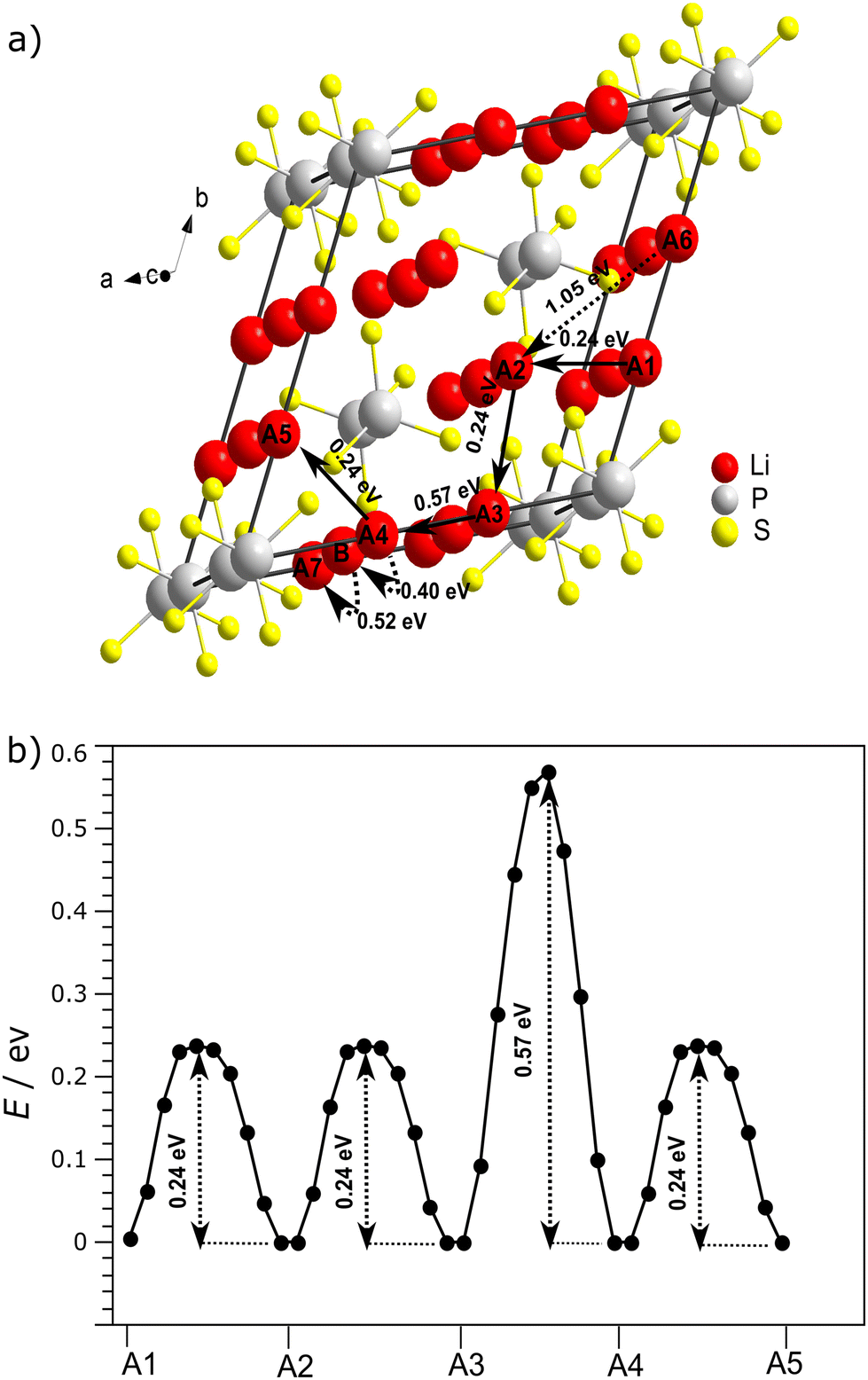 | ||
| Fig. 10 Potential pathways of vacancy-based Li migration and the associated activation energies determined from Nudged Elastic Band calculations. (a) Li-ion transport routes along the a-axis A1 → A2 → A3 → A4 → A5 and along the c-axis A4 → B → A7 within the structure of Li4P2S6 (space group P321). The solid arrows represent the most favorable long-range Li ion diffusion pathway in the sense of featuring the lowest maximum activation energy. (b) The graph shows the energy barriers along the most favorable pathway along the a-axis for transport over a complete unit cell. | ||
Migration routes which can contribute to macroscopic Li transport need to cover a complete unit cell. By the simulations it is found that the maximum activation energy which forms the bottleneck for a migration pathway along the c-axis with 0.52 eV (Fig. S8†) is lower than the corresponding one for a pathway along the a-axis with 0.57 eV (Fig. 10). Both pathways involve more than one elementary hopping process. Certain direct jumps over the center of the void along the P–P axis are penalized by a high activation energy of >1.0 eV (Fig. S9†). It can be concluded that the pathway along the c-axis is preferential and that the Li mobility in Li4P2S6 is slightly anisotropic. The value found by the NEB calculations is in good agreement with the values found by impedance spectroscopy. The NEB simulations indicate only a minor distortion of the P2S6 group in the transition state which indicates that the potential rotation of the PS3-unit around the P–P bond is decoupled from the Li-motion. The mobility of the Mg2+ ion involves a significantly higher activation energy according to NEB calculations than that of the Li+ ion between the same two sites (Fig. S7†), i.e. 1.0 vs. 0.52 eV, respectively. This finding is in agreement with the higher formal charge of the Mg2+ ion and means that the Mg2+ substitution sites need to be circumvented by the mobile Li+ ions for macroscopic transport.
Conclusions
While aliovalent Mg substitution in Li4P2S6 showed a moderate increase in Li ion conductivity, the overall improvement was disappointing. The results indicate that among the factors which explain this observation are the blocking of Li pathways by Mg ions and maybe even more important changes in the crystal structure which can occur even at a low substitution level. For this study it proved beneficial to be able to compare the activation energies for ionic motion from different sources, i.e. impedance spectroscopy, NMR and Nudged Elastic Band calculations.Author contributions
Conceptualization: JSADG; investigation: SN, NM, SCA; writing – original draft; SN, NM, SCA; writing – review and editing: JSADG; visualization: SN, NM, SCA; funding acquisition: JSADG; supervision: JSADG.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Sarah Langenbach and Benjamin W. Lucke for practical support. Financial support by the Deutsche Forschungsgemeinschaft (INST 221/117-1 FUGG) is acknowledged.References
- S. Hama, T. Otomo and K. Kawamoto, JP2014035865(A), 2014.
- P. Knauth, Solid State Ionics, 2009, 180, 911–916 CrossRef CAS.
- H.-J. Deiseroth, S.-T. Kong, H. Eckert, J. Vannahme, C. Reiner, T. Zaiß and M. Schlosser, Angew. Chem., Int. Ed., 2008, 47, 755–758 CrossRef CAS PubMed.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682 CrossRef CAS PubMed.
- H. Yang and N. Wu, Energy Sci. Eng., 2022, 10, 1643–1671 CrossRef CAS.
- J. Frenkel, Z. Phys., 1926, 35, 652–669 CrossRef.
- W. E. Wallace and R. A. Flinn, Nature, 1953, 172, 681–682 CrossRef CAS.
- A. R. West, Chem. Rec., 2006, 6, 206–216 CrossRef CAS PubMed.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- L. Zhou, N. Minafra, W. G. Zeier and L. F. Nazar, Acc. Chem. Res., 2021, 54, 2717–2728 CrossRef CAS PubMed.
- P. Adeli, J. D. Bazak, K. H. Park, I. Kochetkov, A. Huq, G. R. Goward and L. F. Nazar, Angew. Chem., Int. Ed., 2019, 58, 8681–8686 CrossRef CAS PubMed.
- P. Bron, S. Johansson, K. Zick, J. Schmedt auf der Günne, S. Dehnen and B. Roling, J. Am. Chem. Soc., 2013, 135, 15694–15697 CrossRef CAS PubMed.
- J. M. Whiteley, J. H. Woo, E. Hu, K.-W. Nam and S.-H. Lee, J. Electrochem. Soc., 2014, 161, A1812 CrossRef.
- P. Jakes, J. Granwehr, H. Kungl and R.-A. Eichel, Z. Phys. Chem., 2015, 229, 1439–1450 CrossRef CAS.
- P. Adeli, J. D. Bazak, A. Huq, G. R. Goward and L. F. Nazar, Chem. Mater., 2021, 33, 146–157 CrossRef CAS.
- H. Schneider, S. J. Sedlmaier, H. Du, T. Kelley, K. Leitner, J. ter Maat, C. Scordilis-Kelley, A. Mudalige, J. Kulisch and L. Schneider, ChemistrySelect, 2019, 4, 3351–3354 CrossRef CAS.
- Z. Zhang, J. Zhang, H. Jia, L. Peng, T. An and J. Xie, J. Power Sources, 2020, 450, 227601 CrossRef CAS.
- M. S. Whittingham, Y. Song, S. Lutta, P. Y. Zavalij and N. A. Chernova, J. Mater. Chem., 2005, 15, 3362–3379 RSC.
- Z. D. Hood, C. Kates, M. Kirkham, S. Adhikari, C. Liang and N. A. W. Holzwarth, Solid State Ionics, 2016, 284, 61–70 CrossRef CAS.
- C. Dietrich, M. Sadowski, S. Sicolo, D. A. Weber, S. J. Sedlmaier, K. S. Weldert, S. Indris, K. Albe, J. Janek and W. G. Zeier, Chem. Mater., 2016, 28, 8764–8773 CrossRef CAS.
- S. Neuberger, S. P. Culver, H. Eckert, W. G. Zeier and J. Schmedt auf der Günne, Dalton Trans., 2018, 47, 11691–11695 RSC.
- R. Mercier, J. P. Malugani, B. Fahys, J. Douglande and G. Robert, J. Solid State Chem., 1982, 43, 151–162 CrossRef CAS.
- A. R. Stamminger, B. Ziebarth, M. Mrovec, T. Hammerschmidt and R. Drautz, RSC Adv., 2020, 10, 10715–10722 RSC.
- H. Ben Yahia, K. Motohashi, S. Mori, A. Sakuda and A. Hayashi, Z. Kristallogr. - Cryst. Mater., 2023, 238, 209–216 CrossRef CAS.
- H. Nagata and J. Akimoto, ChemistrySelect, 2020, 5, 9926–9931 CrossRef CAS.
- K. Takada, T. Inada, A. Kajiyama, H. Sasaki, S. Kondo, M. Watanabe and M. Kanda, Solid State Ionics, 2002, 147, 23–27 CrossRef CAS.
- W. Klingen, G. Eulenberger and H. Hahn, Naturwissenschaften, 1968, 55, 229–230 CrossRef CAS.
- R. K. Harris, E. D. Becker, de M. S. M. Cabral, P. Granger, R. E. Hoffman and K. W. Zilm, Pure Appl. Chem., 2008, 80, 59–84 CrossRef CAS.
- R. K. Harris and E. D. Becker, J. Magn. Reson., 2002, 156, 323–326 CrossRef CAS.
- D. Jardón-Álvarez and J. Schmedt auf der Günne, Solid State Nucl. Magn. Reson., 2018, 94, 26–30 CrossRef PubMed.
- A. A. Coelho, TOPAS-Academic V7.1 (version V7.18) Coelho Software, Brisbane, 2020 Search PubMed.
- The Rietveld Method, ed. R. A. Young, Oxford University Press, Oxford, New York, 1995 Search PubMed.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S. J. van der Walt, M. Brett, J. Wilson, K. J. Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. J. Carey, İ. Polat, Y. Feng, E. W. Moore, J. VanderPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E. A. Quintero, C. R. Harris, A. M. Archibald, A. H. Ribeiro, F. Pedregosa and P. van Mulbregt, Nat. Methods, 2020, 17, 261–272 CrossRef CAS PubMed.
- P. Giannozzi, O. Baseggio, P. Bonfà, D. Brunato, R. Car, I. Carnimeo, C. Cavazzoni, S. de Gironcoli, P. Delugas, F. Ferrari Ruffino, A. Ferretti, N. Marzari, I. Timrov, A. Urru and S. Baroni, J. Chem. Phys., 2020, 152, 154105 CrossRef CAS PubMed.
- T. Björkman, Comput. Phys. Commun., 2011, 182, 1183–1186 CrossRef.
- K. F. Garrity, J. W. Bennett, K. M. Rabe and D. Vanderbilt, Comput. Mater. Sci., 2014, 81, 446–452 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Xu, K. Zhong, J.-M. Zhang and Z. Huang, Solid State Ionics, 2015, 281, 1–5 CrossRef CAS.
- N. Kuganathan, A. L. Solovjov, R. V. Vovk and A. Chroneos, Heliyon, 2021, 7, e07460 CrossRef CAS PubMed.
- S. Jörgens and A. Mewis, Z. Anorg. Allg. Chem., 2004, 630, 51–57 CrossRef.
- J. S. Waugh and E. I. Fedin, Fiz. Tverd. Tela, 1962, 4, 2233–2237 CAS.
- J. S. Waugh and E. I. Fedin, Phys. Solid State, 1963, 4, 1633–1636 Search PubMed.
- R. Bertermann and W. Müller-Warmuth, Z. Naturforsch., A, 1998, 53, 863–873 CrossRef CAS.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Phys. Rev., 1948, 73, 679–712 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt02624h |
| This journal is © The Royal Society of Chemistry 2023 |