
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Water-assisted synthesis of stable and multicolored CsPbX3@SiO2 core–shell nanoparticles as fluorescent probes for biosensing†
Cynthia
Collantes
 a,
William
Teixeira
a,
Victoria
González-Pedro
*ab,
María-José
Bañuls
ab,
Pedro
Quintero-Campos
a,
Sergi
Morais
abc and
Ángel
Maquieira
abc
a,
William
Teixeira
a,
Victoria
González-Pedro
*ab,
María-José
Bañuls
ab,
Pedro
Quintero-Campos
a,
Sergi
Morais
abc and
Ángel
Maquieira
abc
aInstituto Interuniversitario de Investigación de Reconocimiento Molecular y Desarrollo Tecnológico (IDM), Universitat Politècnica de València-Universitat de València, Camino de Vera s/n, E46022 València, Spain. E-mail: vigonpe@upvnet.upv.es
bDepartamento de Química, Universitat Politècnica de València, Camino de Vera s/n, E46022 València, Spain
cUnidad Mixta UPV-La Fe, Nanomedicine and Sensors, IIS La Fe, Valencia, Spain
First published on 15th November 2023
Abstract
Colloidal lead halide perovskite nanocrystals are highly luminescent materials with great promise as fluorescent probes in biosensing as long as their intrinsic instability in aqueous media is effectively addressed. In this study, we successfully prepared stable and multicolored CsPbX3@SiO2 (X = Cl/Br, Br and I) core–shell nanoparticles through a simple method based on the water-induced transformation of Cs4PbX6 into CsPbX3, combined with sol–gel procedures. We observed that the concentration of the Cs4PbX6 precursor plays a crucial role in the formation of isolated nanospheres with uniform silica coating and in controlling the number of core-free particles. Furthermore, our research expands this approach to other halide compositions, resulting in multicolored core–shell nanoparticles with emission wavelengths ranging from 490 to 700 nm, average sizes below 30 nm, and photoluminescence quantum yields close to 60%. Unlike in previous reports, the silica coating boosts the photoluminescence quantum yields compared to uncoated counterparts and provides increased structural stability for more than four days. Moreover, a controlled thermal treatment confers water stability to the as-synthesized nanoparticles. To establish the feasibility of the developed materials as fluorescent probes, we successfully demonstrated their specific recognition of a humanized antibody (omalizumab) used in treating patients with severe allergic asthma. This work paves the way to develop in vitro tests using CsPbX3@SiO2 core–shell nanoparticles as fluorogenic probes.
1. Introduction
Colloidal nanocrystals (NCs) of CsPbX3 metal halide perovskites (MHPs) have garnered significant attention in multiphoton-imaging applications and lighting materials. These NCs possess remarkable properties, including an ultrahigh photoluminescence (PL) quantum yield (∼99%),1 tunable emission properties with a wide color gamut,2 and unprecedented multi-photon absorption cross-sections.3 Moreover, their ease of synthesis and cost-effectiveness further contribute to their appeal.4 As a result, these NCs have emerged as promising luminescent tags for imaging, biosensing, and clinical diagnosis. However, two main challenges need to be addressed to establish their viability as luminescent probes. First, the synthesis of water-stable NPs is essential to ensure their stability and functionality in aqueous media. Second, it is crucial to fabricate nano-sized monodisperse particles that remain structurally intact under various chemical environments and processing conditions, enabling precise biomolecule conjugation at a single particle level.5,6To overcome this matter, the surface of the NCs needs to be protected with materials that chemically prevent water from reaching the NC surface. A wide range of materials including metal oxides, polymers, MOFs, metal chalcogenides, and perovskite derivatives have been used as shells to protect the surface of perovskite NCs.7 Among various encapsulants, SiO2 has received significant interest because of its transparency, biocompatibility, and chemical stability. However, coating SiO2 shells on MHP NCs is challenging because the hydrolysis reaction requires some amount of water that degrades the NCs. Several attempts have been made to coat SiO2 on MHP NCs to reduce or eliminate the amount of water, yielding multiple NC-embedded SiO2 matrices.8–16 Another approach that is often used is the in situ crystallization of perovskite NCs inside mesoporous silica nanoparticles by intercalating salt precursors and thermal treatment. The studies showed that mesoporous encapsulation enhances the stability of MHPs and prevents halide ion exchange when NCs of two different halides are mixed.17,18
In an alternative promising strategy for encapsulating metal halide perovskites, several research groups have exploited the transformative properties of metal halides, specifically the 0D Cs4PbBr6 into 3D CsPbBr3 transformation features to synthesize core–shell nanoparticles and nanocomposites. This transformation can be achieved by creating a Pb-enriched environment19 or a Cs-deficient situation where the CsPbX3 phase is more stable. For example, in the nonpolar-water interphase, CsX ions are stripped from the Cs4PbX6 due to their ionic nature and high solubility in water,20 resulting in luminescent CsPbBr3 NCs. Other approaches involve the use of Cs+-adsorbent materials such as Prussian blue21 and thermal annealing as a physical strategy to induce phase conversion.22 In addition to these approaches, Udayabhaskararao et al. described spontaneous transformation from CsPbX3 into Cs4PbX6 NCs (and vice versa) by adjusting the oleylamine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :oleic acid ratio.23 Recently, Baranov et al. reported that poly(maleic anhydride-1-alt-octadecene) (PMAO) triggered phase transformation24 where the polymer destabilizes the Cs4PbX6 NC surface by the removal of olefin ligands, inducing the formation of CsPbBr3 NCs.
:oleic acid ratio.23 Recently, Baranov et al. reported that poly(maleic anhydride-1-alt-octadecene) (PMAO) triggered phase transformation24 where the polymer destabilizes the Cs4PbX6 NC surface by the removal of olefin ligands, inducing the formation of CsPbBr3 NCs.
The route of transformation from 0D into 3D results in the production of CsPbBr3 NCs that exhibit improved stability in polar media.20 This issue presents an opportunity to apply the systematic sol–gel process to grow a silica (SiO2) shell on the NC surface, thereby enhancing their stability towards moisture. Some studies have successfully combined transformation-triggering conditions with a sol–gel process. For instance, Hu et al. encapsulated CsPbX3 (X = Cl/Br, Br, Br/I and I) into a Janus-type SiO2 heterostructure of 18 nm with a PLQY of 80%. To obtain entirely coated NPs, Li et al. incorporated a pre-silanization step of Cs4PbBr6 NCs with partially hydrolyzed TMOS (PH-TMOS).25 This method introduces silanization seeds on the MHP NC surface before phase conversion and encapsulation, giving CsPbBr3@SiO2 core–shell NPs of 60 nm with a PLQY of 65%. In an alternative approach, Park et al. used nitric acid to accelerate the hydrolysis of tetraethyl orthosilicate (TEOS).26 However, instead of obtaining isolated colloidal NPs, they obtained composites. In this research line, Rossi et al. exploited a transformation route triggered by maleic anhydride, synthesizing CsPbBr3@SiO2 NPs of 22–33 nm with a PLQY of 1–4%.27
In this study, we have successfully synthesized multi-colored CsPbX3@SiO2 (X = Cl/Br, Br and I) core–shell NPs with remarkable and tunable emission wavelengths, which offer excellent potential as a multimodal fluorescent tag for imaging and biosensing, in health or biotechnological applications. Our work is based on the water-triggered phase transformation of 0D Cs4PbBr6 NCs into CsPbBr3 NCs in the presence of TMOS. Moreover, long-term stable and water-dispersible perovskite NPs were also obtained under a controlled thermal treatment. Finally, to demonstrate the suitability of these materials as fluorescent labels, as-synthesized NPs were conjugated to a humanized antibody, and the specific recognition of its paratope was proved in a direct immunoassay as a proof of concept.
2. Experimental section
2.1. Materials
Cesium carbonate (Cs2CO3, 99.9%, Sigma Aldrich), lead bromide (PbBr2, ≥98%, Sigma Aldrich), lead iodide (PbI2, 98.5%, Alfa Aesar), lead chloride (PbCl2, 99%, Alfa Aesar), 1-octadecene (ODE, 90% tech, Sigma Aldrich), oleic acid (OA, tech. 90%, Alfa Aesar), oleylamine (OAm, approximate C18-content 80–90%, Sigma Aldrich), tetramethyl orthosilicate (TMOS, 99%, ACROS Organics), toluene and hexane.2.2. Synthesis of Cs4PbX6 NCs (X = Br, I and mixed composition of Cl/Br)
Cs4PbX6 NCs were prepared via the hot-injection method described by Akkerman with some modifications.19 Briefly, a 100 mL 3-neck flask containing 0.2 mmol PbX2 (0.1 mmol of each lead halide for the mixed composition), 5 mL of ODE and 0.2 mL of OA was heated under mild stirring and vacuum at 120 °C for 30 min. Then, 1.5 mL of OAm was injected under an inert atmosphere until the solution became clear, indicating that PbX2 was entirely dissolved, and the flask was cooled down to room temperature.The flask was heated again under vacuum and mild stirring to reach the optimal temperature (60 °C for PbI2, 80 °C for PbBr2, and 100 °C for the mixture PbCl2 + PbBr2), and it was left for 15 more min. After an injection of 0.5 mL of Cs–OA (400 mg of Cs2CO3 previously dissolved in 8 mL of OA under vacuum at 100 °C), the solution turned turbid within 15–30 s, and finally, the flask was cooled down to room temperature. The total volume was distributed into four microtubes and centrifuged at 4000 rpm for 5 min. The sediment was dispersed in ∼5 mL of anhydrous hexane. The concentration of the stock solution was determined by weighing mass residues after solvent removal during the purification step: 25.74 ± 3.38 mg mL−1 for Cs4PbBr6, 35.19 ± 1.07 mg mL−1 for Cs4PbI6 and 28.75 ± 1.54 mg mL−1 for Cs4Pb(ClxBr6−x).
2.3. Synthesis of CsPbX3@SiO2 core–shells (X = Br, I and mixed composition of Cl/Br)
To prepare core–shell NPs, 2 μL of partially hydrolyzed TMOS (prepared by adding 2 mL of toluene, 12 μL of Milli-Q H2O filtered with a 0.45 μm filter and 198 μL of TMOS into a glass vial and stirring at 500 rpm for 18 h) was added to 2 mL of a solution of Cs4PbX6 NCs. After stirring at 500 rpm for 5 h, 10 μL of TMOS was added under magnetic stirring at 900 rpm for 1 min. Afterward, 300 μL of Milli-Q H2O was added under magnetic stirring at 1200 rpm for 2 min. The vial remained undisturbed overnight. Core–shell NPs were purified by centrifugation at 9000 rpm for 5 min.To obtain water-resistant core–shell NPs, 5 mL of the NCs@SiO2 NPs was placed in a porcelain plate and calcined at 565 °C for 10 min with a mean heating rate of 1 °C min−1 in a muffle furnace under an air atmosphere. After cooling to room temperature, the sample was dispersed in water and ultrasonicated for 30 min before TEM preparation.
2.4. Immunoassay
(i) Preparation of core–shell NPs derivatized with amine groups (MHP@SiO 2 –NH2). 10.5 mg of CsPbX3@SiO2 NPs and 6 μL of (3-aminopropyl)triethoxysilane were added to 1 mL of anhydrous toluene and stirred for 5 h. Functionalized NPs were purified by centrifugation at 9000 rpm and resuspended first in ethanol and then in 3.75 mL of phosphate buffered saline. (ii) Preparation of NP-αIgE perovskite–omalizumab conjugates. 25 μL of omalizumab (humanized antibody used in asthma treatment) at 1.5 mg mL−1 was added to MHP@SiO2–NH2 NPs and incubated overnight at 4 °C. Conjugated NPs were centrifuged, and the resultant sediment was dispersed in 250 μL of 10 mM sodium phosphate buffer, 150 mM NaCl, and 0.05% Tween 20, pH 7 (PBS-T). (iii) Direct immunoassay. A black polystyrene 96-well plate was coated overnight at 4 °C with 5 mg L−1 of a specific paratope receptor (100 μL per well) in 50 mM sodium carbonate/bicarbonate buffer, pH 9.6. Ovalbumin was used (5 mg L−1) as the negative control. The plate was washed four times with PBS-T. Then, 100 μL per well of the NP-αIgE conjugates were added and incubated for 1 h at room temperature. After washing the plate six times with PBS-T, PL spectra were measured using an EnSpire multimode plate reader (PerkinElmer).2.5. Characterization methods
UV–vis absorption spectra were recorded using a UV–visible spectrophotometer (Agilent 8453, Agilent Technologies). The photoluminescence spectra were obtained using a spectrofluorometer (PTI QMA4, Horiba) at an excitation wavelength of 355 nm for all spectra. The PLQY was determined by using quinine sulfate in 0.5 M H2SO4 (Φ = 0.45) as a standard for the CsPb(ClxBr3−x) and CsPbBr3 NPs at an excitation wavelength of 310 nm. TEM images were taken using a transmission electron microscope at 120 kV (JEM-1400, Flash) and a field emission transmission electron microscope at 200 kV equipped with an X-ray detector (JEM 2100F, JEOL).3. Results and discussion
For the preparation of core–shell NPs, Cs4PbX6 NCs (X = Cl/Br, Br, and I) were synthesized according to the procedure described by Akkerman et al.19 The synthesis yielded NPs that presented characteristic absorption peaks at 305, 314, and 367 nm (Fig. S1†) and exhibited hexagonal or rhombohedral morphology with mean particle sizes of 18.97 ± 2.84 nm, 14.83 ± 1.62 nm, and 17.59 ± 2.99 nm, respectively (Fig. S2†).The Cs4PbX6 NCs were first silanized using partially hydrolyzed TMOS (PH TMOS). At this step, the silanol groups of TMOS, attached to the Cs4PbX6 NC surface, replaced the original hydrophobic olefin precursor and introduced growth sites for the condensation of the SiO2 network. In the second step, silane-derivatized NCs were treated with a TMOS aqueous solution, which promoted the 0D to 3D phase transition via CsX stripping and shell formation around the transformed NCs via condensation of TMOS.25 A schematic representation of the formation mechanism of core–shell NPs from Cs4PbX6 NCs is illustrated in Fig. 1.
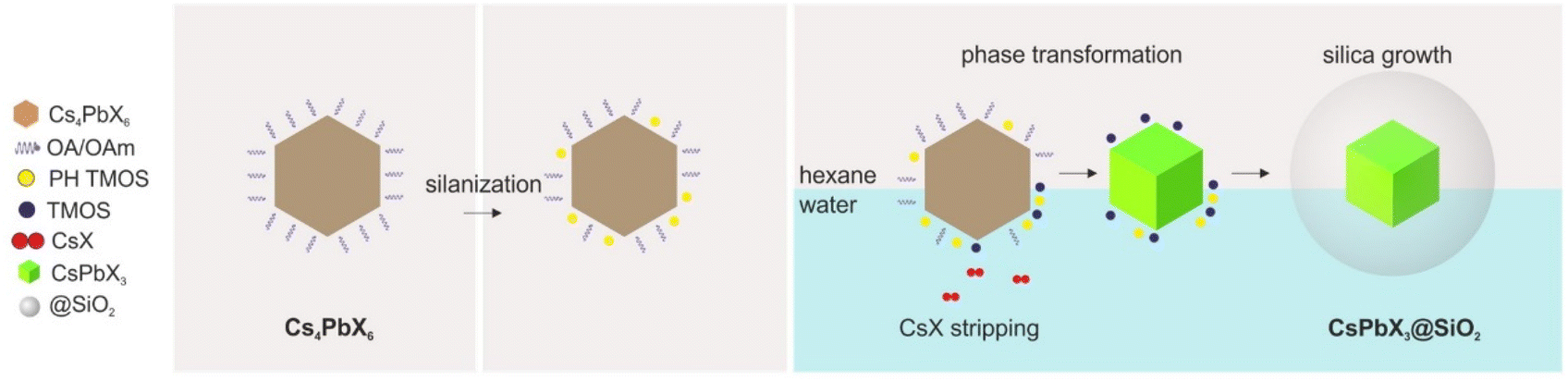 | ||
| Fig. 1 Scheme of the core–shell NP formation mechanism. | ||
To optimize the Cs4PbX6 NC concentration for the synthesis of CsPbX3@SiO2 NPs, serial dilutions were tested, starting from the following concentrations of the purified NCs: 14.4, 25.7 and 17.6 mg mL−1 for Cs4Pb (ClxBr6−x), Cs4PbBr6 and Cs4PbI6, respectively. Fig. S3† summarizes the transmission electron microscopy (TEM) results of three representative concentrations per composition, and Table S1† provides structural features of the resulting core–shell NPs.
It should be observed that, independent of the halide composition, high starting material concentrations produce a heterogeneous population of NPs, containing uncoated crystals of different sizes and shapes and silica spheres with or without a nanocrystal core. It may be associated with the more extensive formation of CsPbBr3 NCs, which reduces the coating efficiency of the silane, leading to the evolution of the uncoated NPs into other perovskite structures.28 At an intermediate concentration, uniform-sized monodisperse NPs are prevalent, while at low concentrations, the presence of core-free silica nanoparticles increases. It could be explained that decreasing CsPbBr3 NCs would facilitate homogeneous nucleation, resulting in the prevalence of core-free NPs.
Fig. S3† shows how remarkable the reduction of the core size is when comparing the intermediate and the lowest concentrations (from 11 to 3 nm in CsPb(ClxBr3−x)@SiO2, from 4 to 2 nm in CsPbBr3@SiO2 and from 2 nm to almost undetectable or coreless nanospheres in CsPbI3@SiO2). This behavior is due to the increase in the water:Cs4PbX6 ratio, which shifts the equilibrium of the CsX stripping reaction (Cs4PbBr6 → CsPbBr3 + 3CsX), producing smaller core particles.
Regarding the emission properties, the right panel of Fig. S3† shows the PL intensity and maximum wavelength for different tested serial dilutions. It must be noticed that the emission peak presents similar features for the intermediate and lower concentrations, while a slight blue shift is observed for the highest one. This shift could be attributed to the heterogeneous NP population obtained under the synthesis conditions.
With regard to the particle uniformity and PL emission intensity, we chose 7.2 mg mL−1, 3.2 mg mL−1 and 2.2 mg mL−1 Cs4PbX6 (X = Cl/Br, Br and I) as the optimal concentrations for preparing CsPbX3@SiO2 NPs. Fig. 2 shows the absorption/emission spectra and TEM images of the resulting nanoparticles. Table 1 presents information about the emission peak, full width at half maximum (fwhm), PLQY, mean particle diameter, core size, and number of cores per particle, including NP features of the most representative works in the literature. From these data, it is noteworthy that all samples showed relatively sharp peaks with the full width at half-maximum in the 22 to 33 nm range. Our developed CsPbX3@SiO2 core–shell NPs presented the tiniest particle sizes reported in the literature by water-assisted synthesis methodologies while preserving the emission quantum yield. In addition, the methods described are extended to other halide compositions of NPs, obtaining multi-colored NPs with a color gamut from blue to red. Concerning the PLQY, coated NPs exhibited an enhancement of emission efficiency of 44% and 32% regarding the uncoated CsPb(ClxBr3−x) and CsPbBr3 counterparts. These results highlight the beneficial role of silica in shallow defect passivation of CsPbX3 NCs formed via the water-induced transformation of Cs4PbX6 NCs. It should be considered that, according to the literature, NPs of comparable sizes to ours are poor emitters.27 Paying attention to our data, it is worth mentioning that we synthesized core–shell NPs that cover all the practical needs to be exploited as a fluorescent label for biosensing, such as tunable emission properties through the visible region, sharp emission peaks, and reduced particle size (20–30 nm). Although studies exist that describe core–shell NPs with significant PLQYs and within those sizes, they are devoted to Br compositions, do not report evidence on the formation of core–shell NPs of other halide compositions, and do not provide size dispersion analysis.29–31
 | ||
| Fig. 2 Optical and structural characterization of the CsPbX3@SiO2 NPs. (a) UV–vis absorption and PL spectra recorded at an excitation wavelength of 355 nm, (b) low and (c) high magnification TEM images and (d) histogram showing the particle size distribution. | ||
| Composition | Emission peak (nm) | Fwhm (nm) | PLQY | Particle size (nm) | Core size (nm) | Cores/particle (nm) | Ref. |
|---|---|---|---|---|---|---|---|
| a Not reported. The PLQY for CsPbI3 was not determined due to the low sample stability. | |||||||
| Cs 4 PbBr 6 to CsPbBr 3 phase transition | |||||||
| CsPb(ClxBr3−x)@SiO2 | 492 | 26 | 18% | 30.06 ± 2.98 | 11.62 ± 2.1 | 1 | This work |
| CsPbBr3@SiO2 | 506 | 22 | 57% | 22.15 ± 1.94 | 4.26 ± 0.9 | 1 | |
| CsPbI3@SiO2 | 689 | 33 | N.Ra | 33.89 ± 4.82 | 2.05 ± 0.46 | 5 | |
| CsPbBr3@SiO2 | 521 | 16.4 | 65% | 60 | 12.25 | 1 | 25 |
| CsPbBr3@SiO2 | 507 | 20 | 8% | 17 | 10 | 1 | 27 |
| Ligand assisted reprecipitation | |||||||
| CsPbBr3@SiO2 | 501 | 22 | 88 | 26 | 10.5 | 1 | 29 |
| Hot injection | |||||||
| CsPbBr3@SiO2 | 519 | 16 | 87 | NR | NR | 1 | 30 |
The core dimensions’ size-shrinkage of CsPbX3 compared to their precursor NCs is explained by the CsX-stripping mechanism.20,24,25,32 Cs4PbX6 NCs are CsX-rich perovskite structures; therefore, upon water treatment, stripping of CsX occurs because of the ionic nature of Cs4PbX6 and the very high solubility of CsX in water, which lead to the decomposition of Cs4PbX6 and the formation of CsPbX3 NCs. During the process, the rhombohedral Cs4PbX6 NCs are converted into simple cubic structured CsPbX3 NCs, and this could be confirmed by the shrinkage of the particle size.20 In our work, the reported size reduction (32%, 75%, and 88% for X = Cl/Br, Br, and I) is more significant than the one described in the literature. This fact might be caused by the harsher conditions in our method, which ended up etching, to a greater extent, the 3D nanocrystal surface. On the other hand, there also appeared to exist a correlation between the halide nature and the size and number of cores per particle. We attributed this behavior to the relative extrinsic stability of ternary cesium halide perovskites towards water and the oxidant environment, which decreases in the order CsPbCl3 > CsPbBr3 > CsPbI3. This behavior may be associated with substituting larger I, by Br or Cl, leading to the reduction of lattice constants and transition to the cubic phase, which is a more compact and stable structure than the tetragonal pseudo-cubic phase.33,34 Hence, the degradation of the as-formed NCs is accelerated in iodine species, leading to crystal decomposition, core size reduction before silica shell formation, and simultaneous encapsulation of several cores per particle.
Table S2 and Fig. S4† show the coexistence and ratio of core–shell to empty SiO2 nanoparticles for each composition. The results are 29.1% in CsPbClxBr3−x@SiO2, 78.2% in CsPbBr3@SiO2, and 6.8% in CsPbI3@SiO2. The presence of core-free silica nanoparticles in the samples could be explained according to the LaMer theory,35,36 which claims that to ensure coated perovskite NCs without core-free SiO2 particles, the monomer concentration must fulfill the conditions of C < Chomo throughout the reaction process, where Chomo is the homogeneous nucleation TMOS concentration threshold. In our method to prevent perovskite degradation, the concentration of TMOS is large, thus core–shell and core-free nanoparticles co-exist in the final product. Future research is mandatory to achieve effective coating without core-free silica NPs as long as they lead to a loss of the effective fluorescence signal. This matter could be addressed by the following: (i) the fractionated drop method, which can always meet the above-mentioned conditions, in which fresh TMOS is added after the previous TMOS is consumed,37 and (ii) replacing TMOS with TEOS, which presents slower condensation rates, thus reducing the homogeneous nucleation threshold.
A set of coated and uncoated NP samples with comparable concentrations (i.e., equal absorbance values at 310 nm) were prepared, and their PL was recorded for fresh samples and after four days (storage RH 60%) to evaluate structural stability. The PL spectra and pictures of the samples under UV-light are presented in Fig. 3. Remarkably, PL intensity diminished faster over time in the uncoated NPs. CsPb(ClxBr3−x) NCs suffered a decay of 75%, and the emission peak is blue-shifted from 488 to 458 nm after four days. It could be associated with a reduction in the amount of Br in CsPbX3 that blue-shifts the emission peak. In the case of CsPb(ClxBr3−x)@SiO2 NPs, it barely moved 7 nm and boosted its PL intensity up to 60%; this increase in emission yields over time could be attributed to halide diffusion and rearrangement on nanoparticles confined in the perovskite NCs, reducing nonradiative losses. The photoluminescence of CsPbBr3 NCs was quenched after one day, while CsPbBr3@SiO2 maintained strong green emission after four days. These results indicate that the SiO2 coating conferred structural stability at a certain degree against moisture, except for the iodine samples, which both quickly suffered from degradation. The latter could be attributed to the iodine-based perovskites’ poor phase and chemical stability, making it difficult to obtain stable NCs in the red region, also called the “perovskite red wall”.38
 | ||
| Fig. 3 Emission spectra recorded at an excitation wavelength of 355 nm for fresh (orange) and samples stored at RH 60% (blue) of non-encapsulated CsPbX3 NCs (a, c and e) and CsPbX3@SiO2 NPs (b, d, and f) prepared at equal concentrations. | ||
Finally, despite the structural stability conferred by the SiO2 shell coating, it could not withstand the diffusion of water molecules. Consequently, the developed core–shell NPs suffered fast degradation in water. Intending to obtain water-resistant NPs, we performed a straightforward approach based on strategic silica pore collapse under thermal treatment.39 For this purpose, the core–shell NPs were thermally treated at 565 °C. Under these conditions, the pores of the silica shell transformed into a compact network. In parallel, the sublimated perovskite NCs were retained inside the silica structure. In the cooling down process, CsPbX3 NCs were formed again, conferring bright photoluminescence and complete stability against water media to the resulting nanoparticles.
The thermally treated particles maintained their stability in water for four days (Fig. 4). Elemental EDS analysis of calcined samples is depicted in Fig. S4.† The EDS patterns showed an atomic ratio of O to Si of 2. After extracting the silica contribution from elemental composition analysis we observed that mixed halide CsPb(Br0.7Cl0.3)3 contained 70% bromine and 30% chloride. Besides that, elemental atomic ratios of small nanocores (Cs:Pb:X) trapped inside are (1:0.5:0.5) and (1:1.1:0.3) for CsPbBr3 and CsPb(Br0.7Cl0.3)3, respectively. The deviation from stoichiometry can be attributed to the poor reliability on EDS quantification when the particles are too small and the thickness of the shell is remarkable. According to the literature, our described NPs exhibit more extended stability in pure water solvent (see Table S1 in the ESI†). In this context, it should be stated that conventionally fluorescent probes are presented as lyophilized nanoconjugates in buffer-free media, and immunoassay takes shorter than 1 hour, thus making those NPs suitable for their use as fluorescent probes.
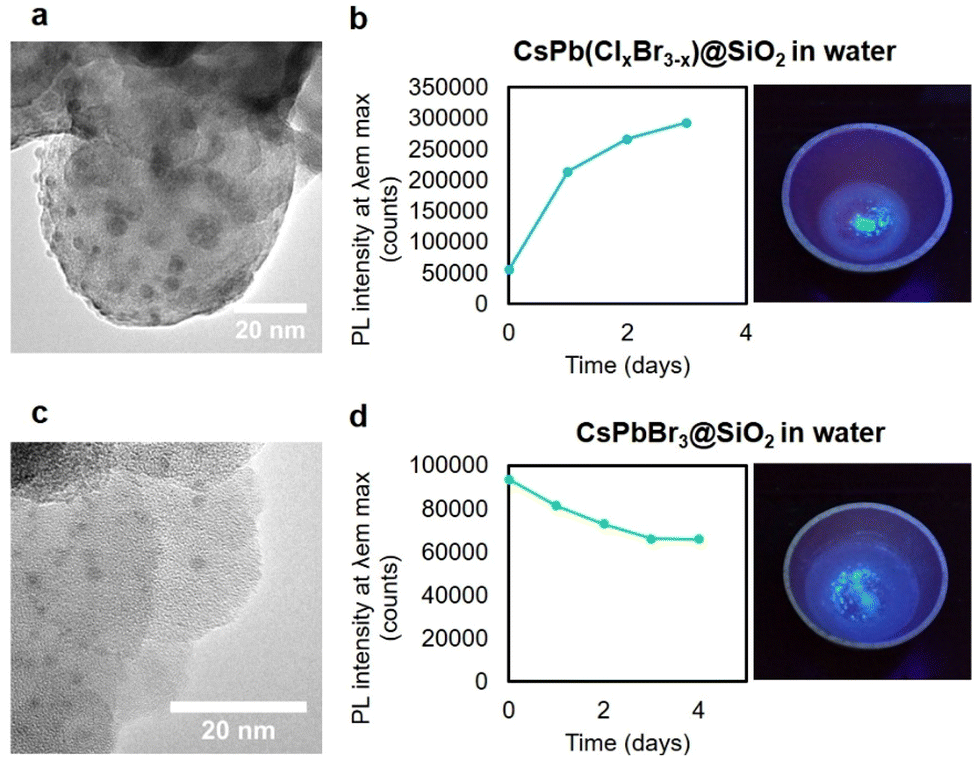 | ||
| Fig. 4 TEM images of the calcined (a) CsPb(Clx/Br3−x)@SiO2 and (c) CsPbBr3@SiO2 NPs and PL intensity at maximum emission versus time in water (b and d). | ||
Furthermore, as proof of concept, we tested the as-synthesized NPs as fluorogenic labels for the in vitro detection of a humanized antibody (Fig. 5a). For this purpose, the core–shell NPs were conjugated using direct adsorption with omalizumab (NP-αIgE), a therapeutic antibody used for treating severe asthma. A multi-well black polystyrene plate was coated with a specific receptor for the paratope of the humanized antibody (5 mg L−1). Then, the coated wells were incubated for 1 h at 25 °C with the fluorogenic conjugate, which selectively recognized the receptor. Finally, the photoluminescence spectra were recorded using the EnSpire multimode plate reader (PerkinElmer). As a negative control, wells were coated with ovalbumin protein at 5 mg L−1 (for further experimental details, see the Experimental section).
 | ||
| Fig. 5 (a) Schematic illustration of the fluorogenic recognition event. (b) Signal to noise ratio, and (c) photoluminescence spectra of the test (colored line) and control assay (black line). | ||
Fig. 5b and c shows the signal-to-noise ratio and photoluminescence spectra for polystyrene wells incubated with NP-αIgE conjugates. Our results confirmed the successful chemical biorecognition of allergen-specific humanized antibodies, achieving a signal-to-noise ratio of ∼10000 and 30000 for CsPb(ClxBr3−x) and CsPbBr3, respectively. Besides, the control experiment showed a negligible photoluminescence signal, revealing excellent selectivity of the assay. Remarkably, CsPb(ClxBr3−x) presented a similar emission wavelength to that of CsPbBr3 (∼505 nm). This fact can be attributed to the poor amount of Cl in the hybrid structure after thermal treatment, as shown in Fig. S4.† We are expanding the methodology to other halide compositions to develop multiplexed immunoassays. Hence, the findings presented herein provide initial evidence of the potential of perovskite nanoparticles as a new fluorogenic probe in immunochemistry. These results pave the way for developing immunoassay tests. As for concerns regarding the toxicity associated with the presence of lead in perovskite compositions, it is essential to note that there is a wide range of in vitro applications in biosensing (luminescent microarrays, flow cytometry, paper-based immunoassays, etc.) and bioimaging (immunostaining, cell histology, in vitro cell tracking, etc.) where lead toxicity is not a limiting factor. Additionally, it is worth mentioning that while this work serves as a proof of concept for the suitability of these materials as fluorescent labels, ongoing research is actively exploring lead-free perovskite NCs with reduced toxicity. Thus, we anticipate that this methodology can soon be extended to lead-free perovskite NPs.
4. Conclusions
In summary, we have developed a simple method for synthesizing CsPbX3 core–shell nanoparticles. This novel approach involves the transformation of water-triggered 0D Cs4PbBr6 into CsPbX3, followed by encapsulation inside a silica shell using alkoxysilane. Our findings indicate that the initial concentration of Cs4PbX6 plays a crucial role in achieving a uniform distribution of monodisperse perovskite core–shell NPs and controlling the number of core-free NPs. By optimizing our method, we successfully obtained core–shell nanoparticles with emissions ranging from 480 to 690 nm. These nanoparticles exhibited small particle sizes (20–30 nm), narrow size distribution cores (2–11 nm), and sharp emission peaks (fwhm of 22–33 nm). Moreover, the core–shell nanoparticles demonstrated structural stability and maintained their luminescence properties under RH of 60% for up to four days. Although previous reports described perovskite core–shell NPs obtained using similar methodologies,25,27 it is important to highlight that our work goes one step ahead and extends this method to other halide compositions, achieving NPs with a color gamut covering most of the visible region. In addition, according to the literature, the size reduction presents a deleterious effect on the PLQY, and CsPbBr3 NPs described in the literature prepared by exploiting transformative features with sizes below 30 nm are poor emitters. Their PLQYs are almost totally quenched upon coating with SiO2. Conversely, our NPs’ present PLQYs of 18% and 57% for CsPb(ClxBr3−x)@SiO2 and CsPbBr3@SiO2versus 10% and 43% of the uncoated counterparts, meaning that the coating yields a recovery factor of 80% and 32%. These data point out the dual functionality of the silica shell, which reduces nonradiative losses via shallow defect passivation of CsPbX3 NCs and prevents their structural degradation.Furthermore, this size reduction has significant implications for practical application in biosensing, as their dimensions are comparable to functional biological units (protein, enzymes, and nucleic acids), allowing for a single tag per biomolecule ratio.6 Additionally, by employing a controlled post-synthetic thermal treatment, we achieved water-resistant NPs through strategic silica pore condensation of the silica shell pores into a compact shell structure. Remarkably, our developed NPs meet the requirements of size, color gamut, and emission yields, along with their mild synthesis conditions, positioning them as promising labels with the potential to outperform organic dyes, fluorescent proteins, and chalcogenide QDs. The water-stable NPs were successfully conjugated with a humanized antibody to evaluate their viability as fluorescent reporters, demonstrating specific recognition of its paratope.
Although further research is required to synthesize water-stable labels of different halide compositions, we believe that MHP NPs will enable the development of sensitive, multicolor, and multiplexed assays, opening exciting possibilities for their applications in life sciences.
Author contributions
C. C. designed the methodology, performed the experiments, and wrote the original draft. W. T. carried out thermal treatment studies to obtain water-stable nanoparticles. P. Q. C and S. M. designed the immunoassay and provided the immunoreagents. V. G. P. conceptualized the investigation and co-wrote, reviewed, and edited the manuscript. M. J. B. and A. M. supervised this study and acquired financial support for the project leading to this publication. All the authors proofread and approved the final manuscript for submission.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the E.U FEDER project ADBIHOL/AEI/10.13039/501100011033 from MCIN/AEI and PROMETEO/2020/094 from Generalitat Valenciana. C. C. thanks the Spanish Ministry of Economy and Competitiveness for her predoctoral contract (BES-2017-080242). W. T. acknowledges the financial support from Universitat Politécnica de Valéncia for the Ph.D. studies (PAID-01-19-06).References
- B. A. Koscher, J. K. Swabeck, N. D. Bronstein and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 6566–6569 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- A. Swarnkar, R. Chulliyil, V. K. Ravi, M. Irfanullah, A. Chowdhury and A. Nag, Angew. Chem., Int. Ed., 2015, 54, 15424–15428 CrossRef CAS PubMed.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- P. Dobosz, S. Morais, R. Puchades and A. Maquieira, Biosens. Bioelectron., 2015, 69, 294–300 CrossRef CAS PubMed.
- S. K. Nune, P. Gunda, P. K. Thallapally, Y. Y. Lin, M. Laird Forrest and C. J. Berkland, Expert Opin. Drug Delivery, 2009, 6, 1175–1194 CrossRef CAS PubMed.
- C. Collantes, W. Teixeira, V. González Pedro, M. J. Bañuls and Á. Maquieira, Appl. Mater. Today, 2023, 31, 101775 CrossRef.
- C. Meng, D. Yang, Y. Wu, X. Zhang, H. Zeng and X. Li, J. Mater. Chem. C, 2020, 8, 17403–17409 RSC.
- Y. Huang, F. Li, L. Qiu, F. Lin, Z. Lai, S. Wang, L. Lin, Y. Zhu, Y. Wang, Y. Jiang and X. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 26384–26391 CrossRef CAS.
- W. Wang, J. Li, P. Ni, B. Liu, Q. Chen, Y. Lu, H. Wu, B. Cao and Z. Liu, ES Mater. Manuf., 2019, 4, 66–73 Search PubMed.
- S. Li, D. Lei, W. Ren, X. Guo, S. Wu, Y. Zhu, A. L. Rogach, M. Chhowalla and A. K. Y. Jen, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- X. Wang, S. Zhuo, J. Fu, X. Li, X. Zhao, H. Jiang, G. Lv, P. Li, J. Li, W. H. Zhang and W. Ma, ACS Appl. Mater. Interfaces, 2023, 15, 20208–20218 CrossRef CAS PubMed.
- Y. Duan, D. Y. Wang and R. D. Costa, Adv. Funct. Mater., 2021, 31, 2104634 CrossRef CAS.
- C. Sun, Y. Zhang, C. Ruan, C. Yin, X. Wang, Y. Wang and W. W. Yu, Adv. Mater., 2016, 28, 10088–10094 CrossRef CAS.
- H. Zhao, L. Wei, P. Zeng and M. Liu, J. Mater. Chem. C, 2019, 7, 9813–9819 RSC.
- W. Song, Y. Wang, B. Wang, Y. Yao, W. Wang, J. Wu, Q. Shen, W. Luo and Z. Zou, Nano Res., 2020, 13, 795–801 CrossRef CAS.
- V. Malgras, S. Tominaka, J. W. Ryan, J. Henzie, T. Takei, K. Ohara and Y. Yamauchi, J. Am. Chem. Soc., 2016, 138, 13874–13881 CrossRef CAS PubMed.
- D. N. Dirin, L. Protesescu, D. Trummer, I. V. Kochetygov, S. Yakunin, F. Krumeich, N. P. Stadie and M. V. Kovalenko, Nano Lett., 2016, 16, 5866–5874 CrossRef CAS PubMed.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS PubMed.
- L. Wu, H. Hu, Y. Xu, S. Jiang, M. Chen, Q. Zhong, D. Yang, Q. Liu, Y. Zhao, B. Sun, Q. Zhang and Y. Yin, Nano Lett., 2017, 17, 5799–5804 CrossRef CAS PubMed.
- F. Palazon, C. Urso, L. De Trizio, Q. Akkerman, S. Marras, F. Locardi, I. Nelli, M. Ferretti, M. Prato and L. Manna, ACS Energy Lett., 2017, 2, 2445–2448 CrossRef CAS.
- F. Palazon, G. Almeida, Q. A. Akkerman, L. De Trizio, Z. Dang, M. Prato and L. Manna, Chem. Mater., 2017, 29, 4167–4171 CrossRef CAS PubMed.
- T. Udayabhaskararao, L. Houben, H. Cohen, M. Menahem, I. Pinkas, L. Avram, T. Wolf, A. Teitelboim, M. Leskes, O. Yaffe, D. Oron and M. Kazes, Chem. Mater., 2018, 30, 84–93 CrossRef CAS.
- D. Baranov, G. Caputo, L. Goldoni, Z. Dang, R. Scarfiello, L. De Trizio, A. Portone, F. Fabbri, A. Camposeo, D. Pisignano and L. Manna, Chem. Sci., 2020, 11, 3986–3995 RSC.
- M. Li, X. Zhang and P. Yang, Nanoscale, 2021, 13, 3860–3867 RSC.
- S. Park, M. N. An, G. Almeida, F. Palazon, D. Spirito, R. Krahne, Z. Dang, L. De Trizio and L. Manna, Nanoscale, 2019, 11, 18739–18745 RSC.
- C. Rossi, R. Scarfiello, R. Brescia, L. Goldoni, G. Caputo, L. Carbone, D. Colombara, L. De Trizio, L. Manna and D. Baranov, Chem. Mater., 2022, 34, 405–413 CrossRef CAS.
- X. Zhang, X. Bai, H. Wu, X. Zhang, C. Sun, Y. Zhang, W. ei Zhang, W. Zheng, W. W. Yu, L. Andrey Rogach, D. Zhang, X. Bai, H. Wu, C. Sun, Y. Zhang, W. W. Yu, W. Zhang, W. Zheng and A. L. Rogach, Angew. Chem., Int. Ed., 2018, 57, 3337–3342 CrossRef CAS PubMed.
- Q. Zhong, M. Cao, H. Hu, D. Yang, M. Chen, P. Li, L. Wu and Q. Zhang, ACS Nano, 2018, 12, 8579–8587 CrossRef CAS PubMed.
- F. Gao, W. Yang, X. Liu, Y. Li, W. Liu, H. Xu and Y. Liu, Chem. Eng. J., 2021, 407, 128001 CrossRef CAS.
- M. R. Kar, R. Chakraborty, U. Patel, S. Ray, T. K. Acharya, C. Goswami and S. Bhaumik, Mater. Today Chem., 2022, 23, 100753 CrossRef CAS.
- H. Hu, L. Wu, Y. Tan, Q. Zhong, M. Chen, Y. Qiu, D. Yang, B. Sun, Q. Zhang and Y. Yin, J. Am. Chem. Soc., 2018, 140, 406–412 CrossRef CAS PubMed.
- C. Zheng and O. Rubel, J. Phys. Chem. C, 2019, 123, 19385–19394 CrossRef CAS.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. Il Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- V. K. Lamer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- H. L. Ding, Y. X. Zhang, S. Wang, J. M. Xu, S. C. Xu and G. H. Li, Chem. Mater., 2012, 24, 4572–4580 CrossRef CAS.
- C. Collantes, V. González Pedro, M.-J. Bañuls and Á. Maquieira, ACS Appl. Nano Mater., 2021, 4, 2011–2018 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, S. Kumar, J. Bär, F. Bertolotti, N. Masciocchi, A. Guagliardi, M. Grotevent, I. Shorubalko, M. I. Bodnarchuk, C. J. Shih and M. V. Kovalenko, ACS Nano, 2017, 11, 3119–3134 CrossRef CAS.
- Q. Zhang, B. Wang, W. Zheng, L. Kong, Q. Wan, C. Zhang, Z. Li, X. Cao, M. Liu and L. Li, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optical and structural characterization of Cs4PbX6 NCs, TEM microscopy and EDS analysis of core–shell NPs, description of optical and structural features of core–shell NPs obtained under different synthesis conditions. See DOI: https://doi.org/10.1039/d3dt02593d |
| This journal is © The Royal Society of Chemistry 2023 |