
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe(II) complexes of pyridine-substituted thiosemicarbazone ligands as catalysts for oxidations with hydrogen peroxide†
Carmen E.
Castillo‡
 a,
Miguel A.
Gonzálvez‡
b,
Andrés G.
Algarra
a,
M. Jesús
Fernández-Trujillo
a,
Montserrat
Ferrer
bc,
Manuel
Martínez
*bc and
Manuel G.
Basallote
*a
a,
Miguel A.
Gonzálvez‡
b,
Andrés G.
Algarra
a,
M. Jesús
Fernández-Trujillo
a,
Montserrat
Ferrer
bc,
Manuel
Martínez
*bc and
Manuel G.
Basallote
*a
aDepartamento de Ciencia de los Materiales e Ingeniería Metalúrgica y Química Inorgánica, Instituto de Biomoléculas (INBIO), Universidad de Cádiz, Apartado 40, E-11510 Puerto Real, Cádiz, Spain. E-mail: manuel.basallote@uca.es
bDepartament de Química Inorgànica i Orgànica, Secció de Química Inorgànica, Universitat de Barcelona, Martí i Franquès 1-11, E-08028 Barcelona, Spain. E-mail: manel.martinez@qi.ub.edu
cInstitute of Nanoscience and Nanotechnology (IN2UB), Universitat de Barcelona, 08028 Barcelona, Spain
First published on 26th September 2023
Abstract
The reaction of three [FeII(TSC)2] complexes, where TSC is a pyridine-substituted thiosemicarbazone of the HDpT or HBpT families, with H2O2 in acetonitrile solution does not result in the accumulation of the corresponding [FeIII(TSC)2]+ complexes. Instead, a mixture of diamagnetic low-spin FeII species is generated. According to the MS spectra, those species result from the sequential addition of up to five oxygen atoms to the complex. This capability for the addition of oxygen atoms suggested that oxygen atom transfer to external substrates may be possible, and these TSC complexes were tested in the oxidation of thioanisole and styrene with H2O2. As hypothesized, the complexes are active in both the oxidation of thioanisole to its sulfoxide and styrene to benzaldehyde, with time scales indicating the participation of the species containing added oxygen atoms. Interestingly, the free thiosemicarbazone ligands and the [Zn(Dp44mT)2] complex also catalyse the selective sulfoxidation of thioanisole, but they are ineffective in catalysing styrene oxidation to benzaldehyde. These findings open up new directions for the development of thiosemicarbazone-based metal catalysts for oxidation processes.
Introduction
Thiosemicarbazones (TSCs) and their metal complexes have been a topic of interest for many years because of their biological relevance.1 These organic molecules show anti-tumor activity,2 with some TSCs having entered into clinical trials.3 Interestingly, some studies have suggested that this fact can be related to chelation of intracellular iron,4,5 the FeIII/II redox activity of these compounds playing a pivotal role in the generation of reactive oxygen species (ROS) involved in redox signaling.6,7 The fact that the metals coordinated to this family of compounds can be involved in the oxidation of thioamines and organothiols is also relevant, especially when the coordinated metal shows easy redox cycling properties.8,9 In this respect, the fact that [FeII(TSC)2] complexes of the 2-pyridylketone family have a reversible FeIII/FeII redox cycle within an accessible window, is relevant for any of the situations stated above.10,11 The oxidation of the ferrous complexes of this family of thiosemicarbazones by oxygen in aqueous and methanol solutions has already been studied to ascertain the formation of ROS in the reaction medium.12,13 In these studies, after FeII to FeIII oxidation with dioxygen, the ROS found was hydrogen peroxide, and attempts to clarify the reaction of H2O2 with the original FeII or oxidised FeIII complexes have not been successful because of the observation of complex spectral changes, often with little reproducibility.In contrast with the abundant studies on their biological properties, the catalytic behaviour of thiosemicarbazone complexes has been much less explored, although some examples of homogeneous catalytic processes have been reported.14 Most of this reactivity refers to C–C cross-coupling reactions, with catalytic oxidation processes being limited to a few examples of alcohol oxidations to their corresponding aldehydes and ketones,15,16and olefin epoxidations.17–19 Thus, the [RuH(CO)(PPh3)2(TSC)] complexes, where TSC is a bidentate anthracenealdehyde thiosemicarbazone, catalyse the selective oxidative cleavage of a variety of alkenes including styrene using NaIO4 as the oxidant in acetonitrile/water solution.20
In this report, we present the results obtained for the oxidation with H2O2 in acetonitrile solution of the [FeII(TSC)2] complexes shown in Scheme 1. Although intuitively direct oxidation of the FeII complexes to FeIII species is expected to occur upon reaction with H2O2, the NMR and ESI-MS monitoring of their reaction with H2O2 indicate that the process involves the addition of several oxygen atoms to the complex with no neat oxidation of the metal centre. Furthermore, the complexes have been found to be catalytically active for sulfoxidation of thioanisole and the cleavage of styrene into benzaldehyde, which open new possibilities for the use of thiosemicarbazone complexes in oxidation processes.
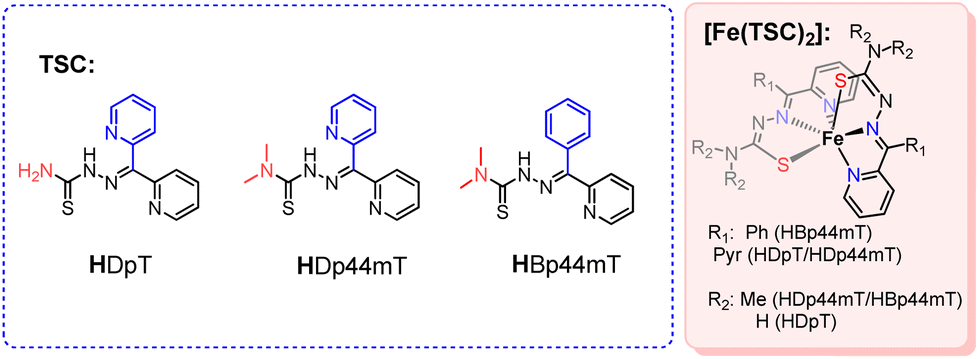 | ||
| Scheme 1 Employed thiosemicarbazone ligands and their FeII complexes. | ||
Results and discussion
Reaction of [FeII(TSC)2] complexes with H2O2 in acetonitrile solution
UV-Vis spectroscopy monitoring of the reaction of the FeII bis-thiosemicarbazone complexes derived from the deprotonation of the HDpT, HDp44mT, and HBp44mT ligands (Scheme 1) with an excess of H2O2 in acetonitrile solution revealed that formation of the known [FeIII(TSC)2]+ species2,11–13,21 only occurs after several days. At shorter times the reaction mixture shows complex spectral changes (Fig. 1) that could not be satisfactorily fitted to a feasible kinetic model. This fact prompted us to find information by studying the process using other feasible techniques.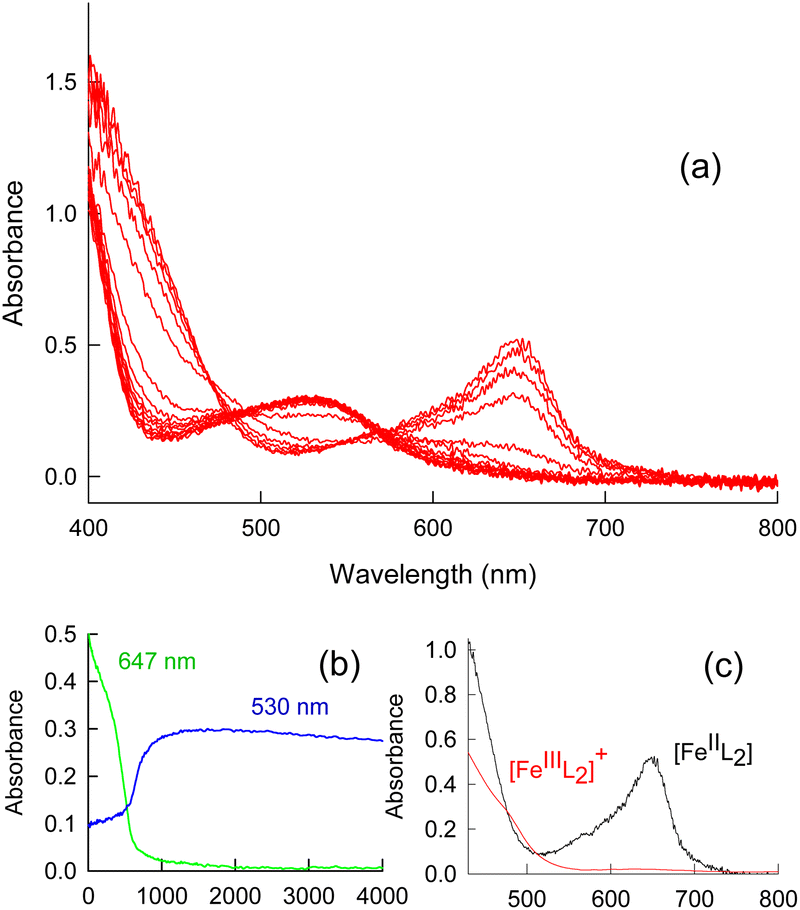 | ||
| Fig. 1 (a) Typical time-resolved UV-Vis spectral changes observed during the first three days of the reaction of the [Fe(Dp44mT)2] complex (5 × 10−5 M) with H2O2 (0.04 M) in acetonitrile solution at 15 °C. (b) Absorbance/time traces at 647 and 530 nm for the spectral changes in (a). (c) Comparison of the spectra for the FeII and FeIII complexes under the same conditions. | ||
Given the fact that accumulation of the [FeIII(TSC)2]+ species does not seem to occur at the early stages of the reaction, the evolution of the NMR spectra of reaction mixtures of the iron complexes with hydrogen peroxide were monitored over time.
The results indicate that, although the spectra of the solutions aged for 2–3 weeks do not show any signal (in agreement with formation of the final FeIII complexes), at shorter time scales all the signals in the NMR spectra appear within the spectral window typical of diamagnetic species, and with chemical shifts close to those observed for the starting complexes (Fig. 2 and S1, S2†). The spectra thus suggest the formation of distinct low spin FeII compounds during the reaction. On the other hand, the ESI-MS monitoring (Fig. 3) shows that after 30 minutes of reaction of [Fe(Dp44mT)2] with H2O2, the signal of the initial complex at m/z = 624 is the major component. Although this signal corresponds to the FeIII complex, the mass spectrum of all the [FeII(TSC)2] complexes having fully substituted amine groups have been found to show only the signal of the oxidised [FeIII(TSC)2]+, even in absence of any oxidant added.10 Furthermore, some new weak signals corresponding to the addition of oxygen atoms are also observed at this time. After 24 hours, the signal for the starting complex has disappeared and signals due to species resulting from the addition of up to five oxygen atoms dominate the spectrum (see Table S1† for detailed m/z values of the peaks observed and their assignment). As the NMR spectral data indicate that they do not correspond to paramagnetic FeIII or FeIV intermediates, they must be low spin FeII complexes with different forms of the oxidised ligand/s having extensive incorporation of oxygen atoms. Identical behaviour is observed for the [Fe(Bp44mT)2] complex (Fig. S3†). Interestingly, for the equivalent reactions on the [Fe(DpT)2] complex the MS signals (obtained in the negative ESI-MS spectra, as they result from deprotonation of the unsubstituted –NH2 units, Fig. S4†) show that the process is much faster than for the other complexes, as after 2 hours of reaction with H2O2 0.05 M the intensity of the signal for the species with four oxygens added (631 m/z−) is already dominant. In any case, the addition of up to five oxygen atoms is also confirmed.
 | ||
| Fig. 2 1H NMR spectra recorded during the reaction of complex [Fe(Dp44mT)2] (2 × 10−3 M) with an excess of H2O2 (0.25 M) in CD3CN solution at 25 °C. The bottom spectrum corresponds to the starting complex in the absence of added H2O2. The intense signal at ca. 9 ppm corresponds to the added hydrogen peroxide. | ||
 | ||
| Fig. 3 ESI-MS spectra recorded during the reaction of complex [Fe(Dp44mT)2] (2 × 10−3 M) with an excess of H2O2 (0.25 M) in CH3CN solution at 25 °C. The bottom spectrum (A) was obtained 30 min after mixing, and the top spectrum (B) after 24 hours. | ||
As a whole, the results arising from the monitoring of the reaction of the three ferrous complexes with H2O2 in acetonitrile solution indicate that the reaction only leads to accumulation of FeIII complexes after long periods of time (up to 2–4 weeks in some cases, see Fig. S5†), which suggests that they can even be formed by O2 oxidation when most of the hydrogen peroxide has been decomposed.12,13 At shorter reaction times, a mixture of low spin FeII complexes with different forms of the oxidised thiosemicarbazone ligands can be observed, differing in the number of added oxygen atoms.
Addition of two oxygen atoms to metal-TSC complexes has already been observed with a coordinated sulfone being formed.22 Furthermore, in other instances, four oxygen atoms produce a SO42− anion after desulfurisation of the ligand, and the molecular formula showing the loss of the sulphur atom has been observed.9,23,24 However, the present results indicate a quite different behaviour, species with a one-by-one increase of the number of oxygen atoms are detected, with some even having more than the four expected from oxidation to sulfone of the two TSC ligands within the complex. That is, a complex mixture of species that differ in the number of oxygen atoms is formed without any evidence of ligand fragmentation or FeII oxidation, a behaviour that to our knowledge has not been previously reported.
Given the complex mixture of intermediates formed and the impossibility to crystallize any of them, we decided to look for additional information by carrying out DFT optimizations of the geometries resulting from the addition of up to five oxygen atoms at the different possible reacting sites of the TSC ligands within the [FeII(Dp44mT)2] complex. The results are included in the ESI† and they show that a variety of attacks at different sites are thermodynamically favored. Notably, the formation of one and two sulfone moieties by addition of two and four oxygen atoms (Fig. 4), respectively, leads to especially stable intermediates compared with all the remaining isomers (see Fig. S22†), which is in agreement with experimental observations for related complexes.22
 | ||
| Fig. 4 Most stable computed isomers of [Fe(Dp44mT)2O2] and [Fe(Dp44mT)2O4] featuring one and two SO2 groups, respectively. For simplicity, H atoms were not drawn. | ||
[FeII(TSC)2] catalytic oxidation of organic substrates
Given the extensive and gradual incorporation of oxygen atoms in the [FeII(TSC)2] systems indicated above, the possibility that these could be shuttled to substrates can be reasonably expected. Consequently, we decided to check the possible activity of the complexes in catalytic oxygen atom transfer (OAT) processes. Using hydrogen peroxide as the oxidant of thioanisole, we found that all the FeII complexes selectively catalyse the formation of methyl phenyl sulfoxide, with no sulfone being observed (Fig. S6†). The results in Table 1 show that the yields are, in all cases, significantly higher than for a blank experiment, and that no meaningful relationship can be found between the structure of the catalyst and the activity observed. Nevertheless, comparison of the yields after 1 and 5 days does reveal significant differences, presumably caused by the different kinetics of formation of the active intermediates (fastest for [Fe(DpT)2], and slowest for [Fe(Bp44mT)2], see above). This fact is confirmed by comparison of entries 2 and 3 in Table 1, where higher yields are obtained when the [Fe(Dp44mT)2] complex has been incubated with H2O2 until a clear change in the colour of the solution is observed. The data thus indicates that the species, or mixture of species, produced by oxygen addition to the ligand play an important role in the catalytic process.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a
a
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Sulfoxide yield (%) | TONb (5 days) | |
| 1 day | 5 days | |||
|
a The percentage of oxidation product formed was calculated by integrating the 1H NMR peaks corresponding to starting material and oxidation product, and taking the ratio of the normalized peak areas.
b mmoles of thioanisole converted in 5 days by mmol of catalyst.
c The complex was incubated with H2O2 until appearance of the maximum intensity of the band at 530 nm (see Fig. 1).
|
||||
| 1 | No catalyst | 4 | 18 | — |
| 2 | [Fe(Dp44mT)2] | 20 | 55 | 84 |
| 3 | [Fe(Dp44mT)2]c | 32 | 79 | 121 |
| 4 | [Fe(Bp44mT)2] | 37 | 33 | 50 |
| 5 | [Fe(DpT)2] | 44 | 66 | 101 |
In view of the results obtained for the oxidation of thioanisole, the use of the same complexes in the catalytic oxidation of styrene was explored. When we tested the [Fe(TSC)2] complexes in this reaction, we observed that they successfully catalyse (Table 2) the formation of benzaldehyde with high selectivity (Fig. S7 and S8†). Catalytic oxidation of styrene into benzaldehyde is an industrially attractive reaction pathway,25 the use of green oxidants as O2 or H2O2 being much desired.26 Nevertheless, the development of catalysts for the selective oxidation to benzaldehyde is challenging, since a large variety of oxidised species can form (Fig. S9†). Even so, some catalysts with a higher selectivity towards benzaldehyde using different oxidants have been reported.25,27–31 The results indicate that the [Fe(TSC)2] complexes show a lower activity than that found for some of the reported catalysts, but the capability of these complexes to carry out selective oxidative cleavage of styrene into benzaldehyde is remarkable. As for the thioanisole data, no meaningful differences are observed between the three FeII complexes studied. Interestingly though, comparison of entries 2 and 3 indicates that aging the solution containing [Fe(Dp44mT)2] and H2O2 causes a decrease of the catalytic activity towards benzaldehyde formation, which is the opposite of that observed for the sulfoxidation of thioanisol. These results suggest that the active species for the two reactions is not the same, and that an earlier (less oxygenated) intermediate is the responsible of oxidative cleavage of styrene. It is also important to note that formation of the epoxide was not observed in any of the experiments, and the use of styrene oxide as substrate produced negative results (entry 4, Table 2), indicating that the formation of benzaldehyde does not involve an initial epoxidation. On the other hand, although optimization of the catalytic performance was out of the scope of the present work, some experiments using the [Fe(Bp44mT)2] complex showed that changing the temperature, the concentrations of the oxidant or the substrate, or increasing the reaction time do not lead to significant changes (Table S2†), the complex concentration being the only factor that causes a significant improvement in the catalytic performance (Table S2,† and entries 5 and 6 in Table 2). To our knowledge, there are only a few cases in which TSC complexes have been found capable to oxidize styrene to benzaldehyde. Some [MoIVO2(bis-TSC)] complexes catalyse oxidative cleavage of styrene to benzaldehyde using tBuOOH, although the process is proposed to go via the formation of styrene oxide,19 and [MoIVO2(TSC)L] complexes with tridentate thiosemicarbazones catalyse the conversion of styrene to styrene oxide and benzaldehyde using H2O2 as oxidant. In all these cases, the catalytic process is proposed to go through oxidation of the complexes to form MoVI intermediates capable of carrying out oxygen atom transfer (OAT) to the alkene.32
a
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Conversion (%) | Selectivity (%) | TONb (5 days) | ||
| 1 day | 5 days | 1 day | 5 days | |||
|
a The percentage of oxidation products formed was calculated by integrating the 1H NMR peaks corresponding to starting material and oxidation products, and taking the ratio of the normalized peak areas.
b mmols of styrene converted by mmol of catalyst.
c The complex was incubated with H2O2 until appearance of the maximum intensity of the band at 530 nm (see Fig. 1).
d Experiment using styrene oxide instead of styrene; a complex mixture of products is formed not including benzaldehyde.
e Experiment using [catalyst] = 4.2 × 10−3 M, [styrene] = [H2O2] = 0.32 M.
|
||||||
| 1 | Blank | 0 | 0 | 0 | 0 | 0 |
| 2 | [Fe(Dp44mT)2] | 8 | 29 | 49 | 66 | 44 |
| 3 | [Fe(Dp44mT)2]c | 11 | 25 | 43 | 54 | 38 |
| 4 | [Fe(Dp44mT)2]d | 13 | 25 | 0 | 0 | 38 |
| 5 | [Fe(Bp44mT)2] | 5 | 11 | 72 | 62 | 17 |
| 6 | [Fe(Bp44mT)2]e | 16 | 35 | 79 | 55 | 27 |
| 7 | [Fe(DpT)2] | 9 | 14 | 62 | 68 | 21 |

As a whole, the time scales of the catalytic oxidations and the formation of FeII complexes with several added oxygen atoms suggest that these species play some role in the catalytic processes. Nevertheless, although the NMR data indicate that there is no accumulation of products resulting from the oxidation of the FeII centre, the possibility of the active species being transients resulting from metal oxidation, with the opening of a chelate ring, cannot be ruled out. Actually, the formation of transient FeIII(OOH) and FeIV(O) species upon reaction of FeII complexes with H2O2 is well illustrated.33–37 Nevertheless, the absence of styrene oxide in the reaction products as well as the inability of the complexes to catalyse the conversion of the epoxide into benzaldehyde suggests the operation of a mechanism in which the added oxygen atoms also play some role.
Catalytic activity of the free TSCs and the [Zn(Dp44mT)2] complex on substrate oxidation
All the results collected so far, especially those related to the ESI-MS experiments, suggest that the structural unit of the TSC ligands is preserved during the reaction of their FeII complexes with H2O2, and that the oxidation products result from extensive oxygen addition to the ligands without oxidation of the metal centre. Obvious questions are whether coordination to the metal centre is indeed required for the formation of these oxygenated species and if the free ligands are also active as catalysts. With these possibilities in mind, we decided to evaluate the catalytic activity of both the thiosemicarbazone ligands and the redox-inactive known [Zn(Dp44mT)2] complex under the same conditions used for the FeII complexes.38 The results indicated that although the ligands and the ZnII complex show similar (in some cases even higher) catalytic activity in the sulfoxidation of thioanisole (Table 3), they are completely inactive for the formation of benzaldehyde from styrene. Coordination to the metal centre clearly regulates the nature and reactivity of the intermediates formed, leading to dramatic changes in the catalytic performance.a
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Sulfoxide yieldb (%) | TONc (5 days) | |
| 1 day | 5 days | |||
|
a The percentage of oxidation product formed was calculated by integrating the 1H NMR peaks corresponding to starting material and oxidation product, and taking the ratio of the normalized peak areas.
b Amount of thioanisole converted to methyl phenyl sulfoxide.
c mmoles of substrate converted by mmol of catalyst in 5 days.
|
||||
| 1 | Blank | 4 | 18 | — |
| 2 | HDp44mT | 66 | 70 | 107 |
| 3 | HBp44mT | 24 | 98 | 149 |
| 4 | HDpT | 44 | 74 | 113 |
| 5 | [Zn(Dp44mT)2] | 31 | 48 | 73 |
We consequently checked the reactivity of the free TSC ligands and [Zn(Dp44mT)2] with H2O2 using both 1H NMR and ESI-MS techniques. Even though the NMR spectra obtained during the reaction of the free thiosemicarbazones with hydrogen peroxide (Fig. S10–S12†) show the formation of a mixture of species that maintain the general structure and N⋯H hydrogen bonding (signals in the 12–15 ppm region), the MS data indicate a behaviour very different from that of the iron complexes. For all the three free ligands, signals of products resulting from thiosemicarbazone desulfurization and ligand fragmentation are observed (Fig. S13–S15†). The desulfurization process observed can be considered the result of H2O2 attack to the sulphur resulting in the sulphate elimination previously reported in the literature.9,23,24 Similarly, for the [Zn(Dp44mT)2] complex, the MS spectra (Fig. S16†) shows that addition of H2O2 leads to rapid desulfurization and TSC dissociation processes; after 24 hours the solution contains almost exclusively free ligand. Thus, the observed catalytic activity of the [Zn(Dp44mT)2] complex in the oxidation of thioanisole can be attributed to free HDp44mT. Importantly, in contrast to the behaviour observed for the iron complexes, no evidence of the formation of species resulting from extensive addition of oxygen atoms is obtained neither for the ligands nor for [Zn(Dp44mT)2], clearly indicating that formation of the oxygen-rich species is necessary for styrene oxidation.
Conclusions
The reactivity of pyridine-substituted thiosemicarbazones and their iron complexes with H2O2 has been found to behave very differently from that reported with O2.12,13 Reaction of the [FeII(TSC)2] complexes with H2O2 does not lead to the oxidation of the metal centre, but produces instead a mixture of low spin FeII species resulting from the sequential addition of oxygen atoms without fragmentation of the coordinated ligands. In contrast, the reaction of free TSCs ligands with H2O2 results in an attack to the S atom, producing desulfurization and oxidative fragmentation of the molecules. In the case of [Zn(Dp44mT)2] the reaction leads to desulfurization and TSC dissociation.Both the TSC ligands and their iron and zinc complexes catalyse the selective formation of methyl phenyl sulfoxide without further oxidation to sulfone, which indicates that sulfoxidation does not require the participation of the metal centre and occurs via ligand-based intermediates. In sharp contrast, the activity towards styrene oxidation to benzaldehyde only takes place when the FeII complexes are utilised. The free TSC ligands and [Zn(Dp44mT)2] are completely inactive, indicating a definitive involvement of iron coordination in the reactivity. The formation of stable FeII intermediates with several added oxygen atoms to the TSC ligands appears to be responsible of the catalytic oxidative cleavage of styrene into benzaldehyde. Contrarily, addition of more than one oxygen atom to the free ligands leads to desulfurization and the activity is lost. In this respect, the lack of activity of the zinc complex towards styrene oxidation can also be the result of its inherent lability, which causes dissociation of the coordinated TSC. In any case, it appears that the presence of the redox active iron centre is required for the formation of the oxygen-rich intermediates, as those species are not formed with the zinc complex.
These results represent a new perspective on thiosemicarbazone-based metal catalysts for oxidation processes. The large number of TSC ligands and complexes available, and the significant effect of metal coordination in the formation of oxidized intermediates, opens the possibility of developing more selective processes using other TSC ligands and metal ions. The striking change observed in the oxidative capability of the complexes with respect to the free ligands indicates a possible novel pathway for the interaction between TSCs, metal ions and ROS in biological systems.
Experimental section
Materials
The thiosemicarbazones of the di-2-pyridylketone (HDpT, HDp44mT) and 2-benzoylpyridine families (HBpT), along with their FeII and ZnII complexes, were prepared and characterised as described in the literature.10,21,38 Hydrogen peroxide solutions were prepared by using commercial 30% (w/w) stabilised aqueous H2O2 solutions. All other reagents were commercially available and used as received.Monitoring of the reaction of the complexes with H2O2
For NMR spectral monitoring of the reactions with H2O2, the required amount of oxidant was added to a solution of the corresponding free TSC or metal complex in CD3CN. The 1H NMR spectra at different reaction times were recorded using a Bruker 400 MHz or 500 MHz instruments available at the Unitat d'RMN – Centres Científics i Tecnològics (CCiTUB) of the Universitat de Barcelona and the Servicios Centralizados de Ciencia y Tecnología of the Universidad de Cádiz.Catalytic activity
The experiments on catalytic oxidation of substrates were monitored using NMR. For that purpose, solutions of the TSC or of the corresponding metal complexes were prepared in the same way than previously described for monitoring the oxidation with H2O2, the only difference being that the substrate (thioanisole or styrene) was added before oxidant addition. The samples were thermostated at 50 °C and the NMR spectra were recorded immediately upon mixing, and after one and five days later. Selected experiments in which the reaction conditions were modified were also performed using the [Fe(Bp44mT)2] complex. The amounts of products formed were determined by integration of the corresponding NMR signals using the solvent signal as internal reference. Whereas for the case of thioanisole the signals for the substrate and its oxidation product are well separated and allow for easy integration, some overlapping signals are observed in the case of styrene, so in this case results are subject to larger errors. This was especially true in the case of spectra recorded after one day, where the amount of products formed is very small. In any case, the formation of benzaldehyde was confirmed by also recording the 13C NMR spectra.Author contributions
Conceptualization, project administration, and supervision: M. M., and M. G. B.; formal analysis, investigation and data curation: C. E. C., M. A. G., A. G. A., M. J. F. R., and M. F.; funding acquisition: A. G. A., C. E. C., M. M. and M. G. B.; visualization, and writing – review & editing: C. E. C., M. A. G., A. G. A., M. J. F. R., M. F., M. M. and M. G. B.; writing – original draft: A. G. A., M. M., and M. G. B. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministerio de Ciencia, Innovación y Universidades for projects PID2019-107006GB-C21 and PID2019-107006GB-C22 is gratefully acknowledged. Carmen E. Castillo also knowledge the 2014–2020 ERDF operational program and the Department of Economy, Knowledge, Business and University of the regional government of Andalusia, project reference: FEDER-UCA18-106753. The authors also thank the Universidad de Cádiz for providing the computational resources and the NMR and mass spectrometry facilities.References
- F. A. French, E. J. Blanz, S. C. Shaddix and R. W. Brockman, J. Med. Chem., 1974, 17, 172–181 CrossRef PubMed.
- Y. Yu, D. S. Kalinowski, Z. Kovacevic, A. R. Siafakas, P. J. Jansson, C. Stefani, D. B. Lovejoy, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2009, 52, 5271–5294 CrossRef.
- V. Pósa, A. Stefanelli, J. H. B. Nunes, S. Hager, M. Mathuber, N. V. May, W. Berger, B. K. Keppler, C. R. Kowol, É. A. Enyedy and P. Heffeter, Cancers, 2022, 14, 4455 CrossRef.
- C. E. Cooper, G. R. Lynagh, K. P. Hoyes, R. C. Hider, R. Cammack and J. B. Porter, J. Biol. Chem., 1996, 271, 20291–20299 CrossRef PubMed.
- P. V. Bernhardt, M. Martínez, C. Rodríguez and M. Vazquez, Dalton Trans., 2012, 41, 2122–2130 RSC.
- Y. Yu, E. Gutierrez, Z. Kovacevic, F. Saletta, P. Obeidy, Y. Suryo Rahmanto and D. R. Richardson, Curr. Med. Chem., 2012, 19, 2689–2702 CrossRef CAS.
- D. Galaris, V. Skiada and A. Barbouti, Cancer Lett., 2008, 266, 21–29 CrossRef CAS.
- E. I. Stiefel and K. Matsumoto, Transition Metal-Sulfur Chemistry, Washington, DC, 1996 Search PubMed.
- P. Gómez-Saiz, J. García-Tojal, V. Diez-Gómez, R. Gil-García, J. L. Pizarro, M. I. Arriortua and T. Rojo, Inorg. Chem. Commun., 2005, 8, 259–262 CrossRef.
- D. R. Richardson, P. C. Sharpe, D. B. Lovejoy, D. Senaratne, D. S. Kalinowski, M. Islam and P. V. Bernhardt, J. Med. Chem., 2006, 49, 6510–6521 CrossRef CAS PubMed.
- D. R. Richardson, D. S. Kalinowski, V. Richardson, P. C. Sharpe, D. B. Lovejoy, M. Islam and P. V. Bernhardt, J. Med. Chem., 2009, 52, 1459–1470 CrossRef CAS.
- M. A. Gonzálvez, A. G. Algarra, M. G. Basallote, P. V. Bernhardt, M. J. Fernández-Trujillo and M. Martínez, Dalton Trans., 2019, 48, 16578–16587 RSC.
- P. V. Bernhardt, M. A. Gonzálvez and M. Martínez, Inorg. Chem., 2017, 56, 14284–14290 CrossRef CAS PubMed.
- S. Priyarega, J. Haribabu and R. Karvembu, Inorg. Chim. Acta, 2022, 532, 120742 CrossRef CAS.
- M. Mohamed Subarkhan and R. Ramesh, Spectrochim. Acta, Part A, 2015, 138, 264–270 CrossRef CAS PubMed.
- M. Ulaganatha Raja, N. Gowri and R. Ramesh, Polyhedron, 2010, 29, 1175–1181 CrossRef CAS.
- Z. Moradi-Shoeili, D. M. Boghaei, M. Amini, M. Bagherzadeh and B. Notash, Inorg. Chem. Commun., 2013, 27, 26–30 CrossRef CAS.
- J. Pisk, J. C. Daran, R. Poli and D. Agustin, J. Mol. Catal. A: Chem., 2015, 403, 52–63 CrossRef CAS.
- Z. Moradi-Shoeili and M. Zare, Kinet. Catal., 2018, 59, 203–210 CrossRef CAS.
- S. Muthumari and R. Ramesh, ChemistrySelect, 2018, 3, 3036–3041 CrossRef CAS.
- D. S. Kalinowski, Y. Yu, P. C. Sharpe, M. Islam, Y.-T. Liao, D. B. Lovejoy, N. Kumar, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2007, 50, 3716–3729 CrossRef CAS PubMed.
- R. Acharyya, S. Dutta, F. Basuli, S. M. Peng, G. H. Lee, L. R. Falvello and S. Bhattacharya, Inorg. Chem., 2006, 45, 1252–1259 CrossRef CAS.
- P. Gómez-Saiz, J. García-Tojal, M. A. Maestro, F. J. Arnaiz and T. Rojo, Inorg. Chem., 2002, 41, 1345–1347 CrossRef.
- R. Pedrido, M. J. Romero, M. R. Bermejo, M. Martínez-Calvo, A. M. González-Noya and G. Zaragoza, Dalton Trans., 2009, 8329–8340 RSC.
- T. A. G. Duarte, A. P. Carvalho and L. M. D. R. S. Martins, Catal. Sci. Technol., 2018, 8, 2285–2288 RSC.
- F. Nami, A. G. Mojarrad and S. Zakavi, Polyhedron, 2021, 207, 115377 CrossRef CAS.
- S. T. Liu, K. V. Reddy and R. Y. Lai, Tetrahedron, 2007, 63, 1821–1825 CrossRef CAS.
- B. Feng, Z. Hou, X. Wang, Y. Hu, H. Li and Y. Qiao, Green Chem., 2009, 11, 1446–1452 RSC.
- E. A. Nyawade, H. B. Friedrich, B. Omondi and P. Mpungose, Organometallics, 2015, 34, 4922–4931 CrossRef CAS.
- N. S. Lawal, H. Ibrahim and M. D. Bala, Molecules, 2022, 27, 4941 CrossRef CAS PubMed.
- V. Kogan, M. M. Quintal and R. Neumann, Org. Lett., 2005, 7, 5039–5042 CrossRef CAS.
- T. M. Asha, M. Sithambaresan and M. R. Prathapachandra Kurup, Polyhedron, 2019, 171, 530–541 CrossRef CAS.
- J. Chen, A. Draksharapu, D. Angelone, D. Unjaroen, S. K. Padamati, R. Hage, M. Swart, C. Duboc and W. R. Browne, ACS Catal., 2018, 8, 9665–9674 CrossRef CAS PubMed.
- K. Cheaib, M. Q. E. Mubarak, K. Sénéchal-David, C. Herrero, R. Guillot, M. Clémancey, J. M. Latour, S. P. de Visser, J. P. Mahy, F. Banse and F. Avenier, Angew. Chem., Int. Ed., 2019, 58, 854–858 CrossRef CAS.
- J. Serrano-Plana, F. Acuña-Parés, V. Dantignana, W. N. Oloo, E. Castillo, A. Draksharapu, C. J. Whiteoak, V. Martin-Diaconescu, M. G. Basallote, J. M. Luis, L. Que, M. Costas and A. Company, Chem. – Eur. J., 2018, 24, 5331–5340 CrossRef CAS.
- A. Bohn, C. Chinaux-Chaix, K. Cheaib, R. Guillot, C. Herrero, K. Sénéchal-David, J. N. Rebilly and F. Banse, Dalton Trans., 2019, 48, 17045–17051 RSC.
- K. Chen, M. Costas and L. Que, Dalton Trans., 2002, 672–679 RSC.
- A. E. Stacy, D. Palanimuthu, P. V. Bernhardt, D. S. Kalinowski, P. J. Jansson and D. R. Richardson, J. Med. Chem., 2016, 59, 4965–4984 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional ESI-MS and 1H NMR spectra. See DOI: https://doi.org/10.1039/d3dt02442c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |