
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity of Ir(I)-aminophosphane platforms towards oxidants†‡
Marco
Palmese
 ,
Jesús J.
Pérez-Torrente
and
Vincenzo
Passarelli
*
,
Jesús J.
Pérez-Torrente
and
Vincenzo
Passarelli
*
Departamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), Universidad de Zaragoza-CSIC, C/Pedro Cerbuna 12, ES-50009 Zaragoza, Spain. E-mail: passarel@unizar.es
First published on 5th September 2023
Abstract
The iridium(I)-aminophosphane complex [Ir{κ3C,P,P′-(SiNP-H)}(cod)] has been prepared by reaction of [IrCl(cod)(SiNP)] with KCH3COO. DFT calculations show that this reaction takes place through an unexpected outer sphere mechanism (SiNP = SiMe2{N(4-C6H4Me)PPh2}2; SiNP-H = CH2SiMe{N(4-C6H4Me)PPh2}2). The reaction of [IrCl(cod)(SiNP)] or [Ir{κ3C,P,P′-(SiNP-H)}(cod)] with diverse oxidants has been explored, yielding a range of iridium(III) derivatives. On one hand, [IrCl(cod)(SiNP)] reacts with allyl chloride rendering the octahedral iridium(III) derivative [IrCl2(η3-C3H5)(SiNP)], which, in turn, reacts with tert-butyl isocyanide yielding the substitution product [IrCl(η3-C3H5)(CNtBu)(SiNP)]Cl via the observed intermediate [IrCl2(η1-C3H5)(CNtBu)(SiNP)]. On the other hand, the reaction of [Ir{κ3C,P,P′-(SiNP-H)}(cod)] with [FeCp2]X (X = PF6, CF3SO3), I2 or CF3SO3CH3 results in the metal-centered two-electron oxidation rendering a varied assortment of iridium(III) compounds. [Ir{κ3C,P,P′-(SiNP-H)}(cod)] reacts with [FeCp2]+ (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) in acetonitrile affording [Ir{κ3C,P,P′-(SiNP-H)}(CH3CN)3]2+ isolated as both the triflato and the hexafluorophosphato derivatives. Also, the reaction of [Ir{κ3C,P,P′-(SiNP-H)}(cod)] with I2 (1:1) yields a mixture of iridium(III) derivatives, namely the mononuclear compound [IrI(κ2P,P′-SiNP)(η2,η3-C8H11)]I, containing the η2,η3-cycloocta-2,6-dien-1-yl ligand, and two isomers of the dinuclear derivative [Ir2{κ3C,P,P′-(SiNP-H)}2(μ-I)3]I, the first species being isolated in low yield. DFT calculations indicate that [IrI(κ2P,P′-SiNP)(η2,η3-C8H11)]I forms as the result of a bielectronic oxidation of iridium(I) followed by the deprotonation of the cod ligand by iodide and the protonation of the methylene moiety of the [Ir{κ3C,P,P′-(SiNP-H)}] platform by the newly formed HI. Finally, the oxidation of [Ir{κ3C,P,P′-(SiNP-H)}(cod)] by methyl triflate proceeds via a hydride abstraction from the cod ligand, with the elimination of methane and the formation of the η2,η3-cycloocta-2,6-dien-1-yl ligand with the concomitant two-electron oxidation of the iridium centre. The crystal structures of selected compounds have been determined.
:2) in acetonitrile affording [Ir{κ3C,P,P′-(SiNP-H)}(CH3CN)3]2+ isolated as both the triflato and the hexafluorophosphato derivatives. Also, the reaction of [Ir{κ3C,P,P′-(SiNP-H)}(cod)] with I2 (1:1) yields a mixture of iridium(III) derivatives, namely the mononuclear compound [IrI(κ2P,P′-SiNP)(η2,η3-C8H11)]I, containing the η2,η3-cycloocta-2,6-dien-1-yl ligand, and two isomers of the dinuclear derivative [Ir2{κ3C,P,P′-(SiNP-H)}2(μ-I)3]I, the first species being isolated in low yield. DFT calculations indicate that [IrI(κ2P,P′-SiNP)(η2,η3-C8H11)]I forms as the result of a bielectronic oxidation of iridium(I) followed by the deprotonation of the cod ligand by iodide and the protonation of the methylene moiety of the [Ir{κ3C,P,P′-(SiNP-H)}] platform by the newly formed HI. Finally, the oxidation of [Ir{κ3C,P,P′-(SiNP-H)}(cod)] by methyl triflate proceeds via a hydride abstraction from the cod ligand, with the elimination of methane and the formation of the η2,η3-cycloocta-2,6-dien-1-yl ligand with the concomitant two-electron oxidation of the iridium centre. The crystal structures of selected compounds have been determined.
Introduction
In the last decade, aminophosphanes have attracted increasing attention due to their modular synthesis1 and their consequent suitability to tailor the steric and electronic properties of the resulting metal complex. Actually, a variety of mono and bidentate aminophosphano ligands have been reported, giving rise to a range of metal–ligand architectures2–5 with applications including catalysis,2 bond activation,2a,3 redox-active multimetallic systems4 and metaloenzyme mimics.5 On this background, our group has focussed on the preparation of rhodium6 and iridium7 derivatives with the aminophosphano ligands RNP (R = H, SiMe3) and SiNP (SiMe2{N(4-tolyl)PPh2}2, Fig. 1) showing that besides the foreseeable coordination through phosphorus atom(s) of either monodentate RNP (A, Fig. 1) or bidentante SiNP ligands (B), intramolecular C–H oxidative addition processes give rise to unexpected κ2C,P (C) and κ3C,P,P′ platforms (D).
Relevant to this paper, it is worth a mention that, so far, the κ3C,P,P′ coordination of SiNP has been found to be promoted by π-acceptor ligands like carbon monoxide,7atert-butyl isocyanide7d and trimethyl phosphite,7b with the concomitant formation of an iridium(III)-hydride moiety (Scheme 1). Also, in our hands, the reactivity of the resulting iridium(III) complexes with substrates like alkynes or alkenes was observed to be absent plausibly due to the substitutional inertness of the ancillary ligands.7a,b,d Thus, we decided to explore alternative routes leading to the Ir{κ3C,P,P′-(SiNP-H)} platform. More specifically, on one hand, we envisioned that the introduction of an allyl group at the Ir(κ2P,P′-SiNP) moiety might trigger the deprotonation of one SiCH3 group along with the elimination of propene, rendering the desired Ir{κ3C,P,P′-(SiNP-H)} platform (SiNP-H = CH2SiMe{N(4-C6H4Me)PPh2}2). On the other hand, we decided to explore the use of acetate as a Brønsted base in order to access the Ir{κ3C,P,P′-(SiNP-H)} platform. Thus, we report herein the reaction of [IrCl(cod)(SiNP)] with either allyl chloride or potassium acetate and the study of the reactivity of the resulting metal complexes.
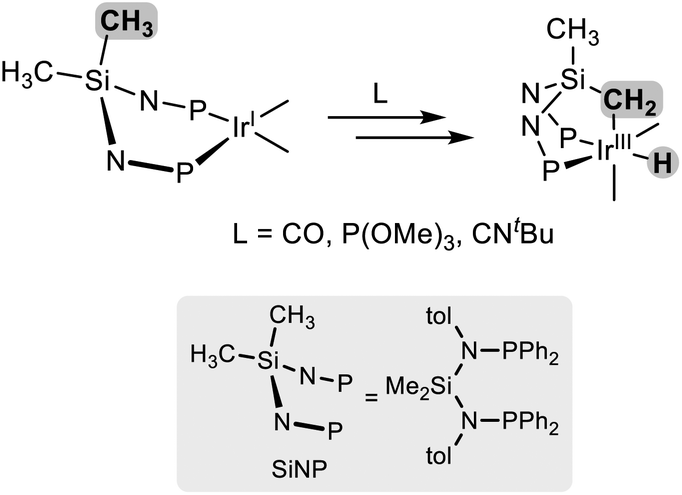 | ||
| Scheme 1 Formation of the [Ir{κ3C,P,P′-(SiNP-H)}] platform upon oxidative addition of the SiCH2-H bond to iridium(I).7a,b,d | ||
Results and discussion
Reaction of [IrCl(cod)(SiNP)] with allyl chloride
The iridium(I) complex [IrCl(cod)(SiNP)] reacts with allyl chloride affording almost quantitatively the iridium(III) derivative [IrCl2(η3-C3H5)(SiNP)] (1) as a result of the formal oxidative addition of the carbon–chlorine bond to iridium(I) and the release of the cod ligand (Scheme 2). | ||
| Scheme 2 Preparation of [IrCl2(η3-C3H5)(SiNP)] (1). | ||
As shown in Fig. 2, the crystal structure of 1 reveals a distorted octahedral coordination polyhedron at the metal centre, similar to that already reported for [RhCl2(η3-C3H5)(SiNP)].6 Indeed, the SiNP exhibits a κ2P,P′ coordination [P1–Ir–P2 92.08(3)°] and the chlorido ligands adopt a mutually cis disposition [Cl1–Ir–Cl2 88.10(3)°] rendering a see–saw IrP2Cl2 fragments, with the η3-allyl completing the coordination sphere of the metal. The resulting complex is chiral (Δ configuration is shown in Fig. 2), however it is worth noting that both enantiomers Δ and Λ are present in the crystal as a consequence of the centrosymmetric space group, namely, P21/c. Reasonably, as a consequence of the different trans influence of phosphorus (P2) and chlorine (Cl1), a non-symmetric coordination of the η3-allyl ligand is observed. As a matter of fact, different metal–carbon bond lengths [Ir–C41 2.138(4), Ir–C42 2.188(4), Ir–C43 2.277(4) Å] are observed along with different carbon–carbon bonds [C41–C42 1.440(5), C42–C43 1.388(6) Å], similar to that observed in iridium-η3-allyl systems reported in the literature.8 Finally, as previously observed in related Ir-SiNP complexes,7d,e N1 and N2 exhibits an almost planar geometry ( ,
,  ) with nitrogen–phosphorus (1.689 Å, av.), nitrogen–silicon (1.762 Å, av.) and nitrogen–carbon (1.438 Å, av.) bond lengths which suggest that nitrogen–phosphorus backdonation is operative to some extent and is responsible for the planar geometry of both N1 and N2.
) with nitrogen–phosphorus (1.689 Å, av.), nitrogen–silicon (1.762 Å, av.) and nitrogen–carbon (1.438 Å, av.) bond lengths which suggest that nitrogen–phosphorus backdonation is operative to some extent and is responsible for the planar geometry of both N1 and N2.
 | ||
| Fig. 2 ORTEP plot of 1. For clarity, most hydrogen atoms are omitted, and tolyl and phenyl groups are shown in a wireframe style. Thermal ellipsoids are at 50% probability. Selected bond lengths (Å) and angles are (°): C41–Ir 2.138(4), C42–Ir 2.188(4), C43–Ir 2.277(4), P1–Ir 2.2676(9), P2–Ir 2.2959(9), Cl1–Ir 2.4469(9), Cl2–Ir 2.4566(9), C42–C41 1.440(5), C43–C42 1.388(6), N1–P1 1.681(3), N1–Si 1.758(3), N2–P2 1.697(3), N2–Si 1.765(3), C15–N1 1.455(4), C34–N2 1.461(4), P1–Ir–P2 92.08(3), Cl1–Ir–Cl2 88.10(3), P1–Ir–Cl2 176.63(3), C41–Ir–Cl1 160.35(10), C43–Ir–P2 160.13(11), N1–Si–N2 107.12(14), C1–Si–C2 108.27(18), C34–N2–P2 118.1(2), C15–N1–P1 120.6(2), C15–N1–Si 112.5(2), P1–N1–Si 126.94(18), C34–N2–Si 110.7(2), P2–N2–Si 125.34(17). | ||
As for the solution structure of 1, at 298 K sharp well-shaped 31P{1H} doublets at 30.6 and 24.3 ppm (2JPP = 25.2 Hz) are observed suggesting the presence of two non-equivalent phosphorus atoms occupying mutually cis coordination sites. On the other hand, broad 1H signals between 6 and 8 ppm suggests that at room temperature the rotation of the tolyl and the phenyl groups around the N–C and P–C bonds, respectively, may be hindered. On this ground, the spectroscopic characterization of 1 was carried out at 233 K. At that temperature, two 1H and two 13C{1H} resonances for the SiCH3 groups are observed (δH, δC: 0.83, 2.4; −0.35, 2.7 ppm) confirming the presence of two different ligands at the axial positions (taking the IrP2 plane as the equatorial plane). As for the η3-allyl moiety, three 13C resonances are observed at 102.9 (C2), 72.1 (C1) and 43.2 ppm (C3) along with five 1H signals (δH 4.84, C2H; 4.03 and 3.94, C1H2; 2.06 and 1.43 ppm, C3H2).
Contrary to our expectation, compound 1 is thermally stable in toluene, neither elimination of propene nor decomposition being observed (1H NMR) even after 18 h heating at reflux. In view of this thermal stability, the reaction of 1 with tert-butyl isocyanide was carried out in order to explore its reactivity. As a matter of fact, 1 reacts with tert-butyl isocyanide smoothly yielding [IrCl(η3-C3H5)(CNtBu)(κ2P,P′-SiNP)]Cl (2Cl) as isocyanide ex/isocyanide exchange (Scheme 3).
 | ||
| Scheme 3 Reaction of 1 with tert-butyl isocyanide. | ||
The 31P{1H} NMR spectrum of 2Cl contains two doublet at 28.3 and 25.9 ppm (2JPP = 20.9 Hz) confirming the κ2P,P′ coordination of SiNP. The 1H and 13C{1H} NMR spectra are also indicative of the presence of the η3-allyl moiety (δH 4.89, C2H; 3.86, 3.72, C1H; 2.31, 2.00 ppm, C3H; δC 108.3, C2; 63.6, C1; 60.1 ppm, C3) and of the tert-butyl isocyanide ligand (δH = 2.04, δC = 31.0 ppm). Interestingly, when the formation of 2Cl was monitored by NMR spectroscopy, [IrCl2(η1-C3H5)(CNtBu)(κ2P,P′-SiNP)] (3) was observed as an intermediate (Scheme 3). In our hands, 3 could eventually be isolated along with around 20% of 1 (Fig. 3). Nonetheless, it was fully characterised in solution at 233 K, even in the presence of 1. The 31P{1H} singlet at 31.9 ppm is indicative of two equivalent phosphorus atoms and, as a consequence, of a symmetric environment at the metal centre. Accordingly, two equivalent tolyl moieties are observed (δH 2.09, δC 20.9 ppm, CH3tol). Also, two 1H signals at 0.47 (δC 4.5 ppm) and −0.05 ppm (δC 3.4 ppm) point at two non-equivalent SiCH3 groups, confirming the presence of two different axial ligands (taking the IrP2 plane as the equatorial plane). As for the allyl group, the 1H and 13C{1H} NMR spectra show a pattern different from those observed for 1 and 2Cl, which indicates an η1 coordination. Indeed, two 13C signals at 148.2 and 108.3 ppm are indicative of an uncoordinated olefinic CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 group, whereas the signals at 3.34 (1H) and 5.3 ppm (13C) have been assigned to the IrCH2 moiety. The tert-butyl isocyanide ligand (δH 1.06; δC 29.5 ppm, CH3) completes the coordination sphere of the metal centre.
CH2 group, whereas the signals at 3.34 (1H) and 5.3 ppm (13C) have been assigned to the IrCH2 moiety. The tert-butyl isocyanide ligand (δH 1.06; δC 29.5 ppm, CH3) completes the coordination sphere of the metal centre.
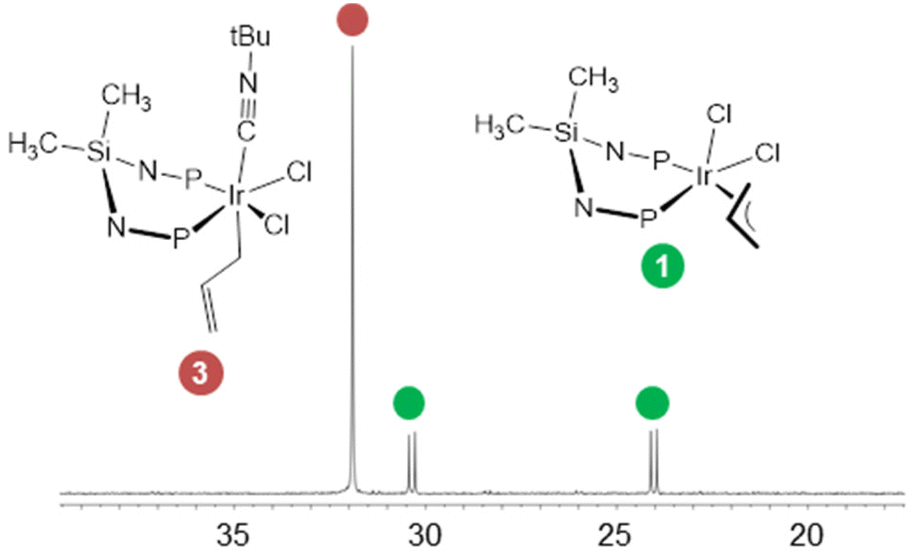 | ||
| Fig. 3 31P{1H} NMR spectrum of 3 in the presence 20 mol% of 1 (CD2Cl2, 233 K). | ||
On this ground, the Cl−/CNtBu exchange reaction between 1 and tert-butyl isocyanide should follow an associative pathway taking advantage of the η3 → η1 → η3 haptotropic shift of the allyl ligand (Scheme 4). In this regard, DFT calculations were performed in order to shed light on the reaction sequence leading to 2Cl. Firstly, the η3 → η1 shift of the allyl ligand in 1 renders the coordinatively unsaturated intermediate I. In turn, I isomerises to II, which ultimately reacts with tert-butyl isocyanide yielding the observed intermediate 3. Afterwards, 3 should release one chlorido ligand (3 → IIICl) and finally the η1 → η3 shift should give rise to 2Cl. As for the calculated relative Gibbs free energy of the proposed intermediates, the formation of both 3 and 2Cl from 1 is calculated to be exergonic in agreement with the observed course of the reaction of 1 with tert-butyl isocyanide. As far as the calculated relative stability of 2Cl vs. 3 is concerned, unaccounted solvation effects and/or underestimated ion pair stabilization for 2Cl may be responsible for the calculated higher stability of 3 with respect to 2Cl.
 | ||
| Scheme 4 Reaction sequence for the reaction of 1 with tert-butyl isocyanide along with the calculated values of relative Gibbs free energy (kcal mol−1, B97D3/def2svp). | ||
Depending on the topology of the η3 → η1 shift in 1, IV may also form (Scheme 4). Nonetheless, its formation is calculated to be significantly more endergonic than the formation of I and II, thus ruling out IV as a possible intermediate.
Synthesis of [Ir{κ3C,P,P′-(SiNP-H)}(cod)]
The reaction of [IrCl(cod)(SiNP)] with potassium acetate in dichloromethane smoothly affords the metallated iridium(I) derivative [Ir{κ3C,P,P′-(SiNP-H)}(cod)] (4) as a result of the deprotonation of one SiCH3 moiety (Scheme 5). Remarkably, as mentioned before, previously reported Ir{κ3C,P,P′-(SiNP-H)} platforms7a,b,d were obtained via SiCH2-H oxidative addition to iridium(I) and, as a result, contained the iridium(III)-hydride moiety (Scheme 1), whereas in this case, for the first time, the reaction implies the base-assisted metal–carbon bond formation (vide infra), with no change of the metal oxidation state. The crystal structure of 4 reveals a distorted bipyramidal geometry at the metal centre (TBPY-5-23) with the ligand SiNP-H occupying three mutually cis positions [C1–Ir–P1 85.89(11)°, C1–Ir–P2 81.96(11)°, P1–Ir–P2 98.82(4)°] and the cod ligand completing the coordination sphere [CT1–Ir–CT2 84.910(15)°] (Fig. 4). Similar to what previously observed in the related iridium(I) complex [Ir{κ3C,P,P′-(SiNP-H)}(CO)2],7a the distance Ir–CT2 is longer than Ir–CT1 [Ir–CT1 2.0153(3), Ir–CT2 2.1007(3) Å] due to the high trans influence of the methylene group. Accordingly, the bond C41–C42 [1.447(6) Å] is longer than C45–C46 [1.409(6) Å] indicating a higher degree of backdonation to the coordinated olefinic bond lying in the equatorial plane. As for the κ3C,P,P′-(SiNP-H) backbone, when compared with the uncomplexed SiNP ligand,7d the wider angle C1–Si–C2 [123.3(2)°] and the smaller angles P1–N1–Si [111.41(18)°] and P2–N2–Si [111.47(19)°] are reasonably the consequence of the metallation and the subsequent formation of two fused five-member cycles [SiNP: C1–Si–C2 111.48(10)°, P1–N1–Si 121.40(9)°, P2–N2–Si 120.94(9)°].7d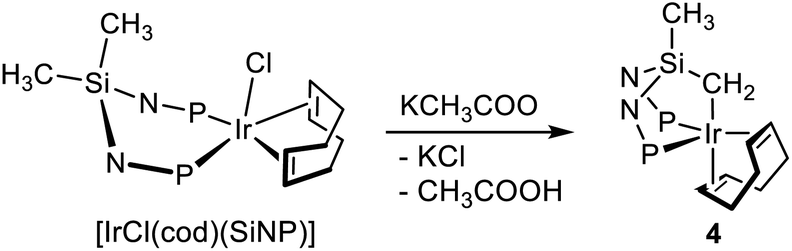 | ||
| Scheme 5 Synthesis of [Ir{κ3C,P,P′-(SiNP-H)}(cod)] (4). | ||
 | ||
| Fig. 4 ORTEP plot of 4. For clarity, most hydrogen atoms are omitted, and tolyl and phenyl groups are shown in a wireframe style. Thermal ellipsoids are at 50% probability. Selected bond lengths (Å) and angles are (°): C1–Ir 2.167(4), P1–Ir 2.2976(10), P2–Ir 2.3451(10), Ir–CT1 2.0153(3), Ir–CT2 2.1007(3), C41–C42 1.447(6), C45–C46 1.409(6), N1–P1 1.701(3), N1–Si 1.759(3), N2–P2 1.704(3), N2–Si 1.770(4), C1–Ir–P1 85.89(11), C1–Ir–P2 81.96(11), P1–Ir–P2 98.82(4), CT1–Ir–CT2 84.910(15), CT1–Ir–C1 86.76(11), CT2–Ir–C1 171.59(11), CT1–Ir–P1 125.65(3), CT1–Ir–P2 133.11(3), C1–Si–C2 123.3(2), N1–Si–N2 113.45(16), C15–N1–P1 123.5(3), C15–N1–Si 124.4(3), P1–N1–Si 111.41(18), C34–N2–P2 124.5(3), C34–N2–Si 119.0(3), P2–N2–Si 111.47(19). CT1 and CT2 are the centroids of C41 and C42, and of C45 and C46, respectively. | ||
The solution structure of 4 is similar to that observed in the solid state. The 31P{1H} singlet at 47.1 ppm along with the 1H/1H{31P} triplet/singlet at 0.14 ppm and the 1H singlet at −0.83 ppm, assigned to the SiCH2Ir and SiCH3 moieties, respectively, confirm that the κ3C,P,P′ coordination is preserved in a symmetrical metal environment. Accordingly, two 1H signals are observed at 3.60 and 2.60 ppm for the cod ligand. Notably, no cross peaks are observed between them in either the 1H–1H COSY or the 1H–1H NOESY spectra, thus ruling out the square pyramidal geometry SPY-5-32 for 4 in solution. Accordingly, the SPY-5-32 isomer was calculated to be less stable than 4 (TBPY-5-23) by 11.2 kcal mol−1 (vide infra).
The course of the formation of 4 was explored by means of DFT calculations (Fig. 5). Two mechanisms were considered,9 one based on the well-established acetate-assisted C–H activation, and the other on the outer sphere deprotonation of the SiCH3 moiety of [IrCl(cod)(SiNP)] by acetate. Fig. 5 shows the Gibbs free energy profiles for the two explored routes along with the structures of calculated transition states.
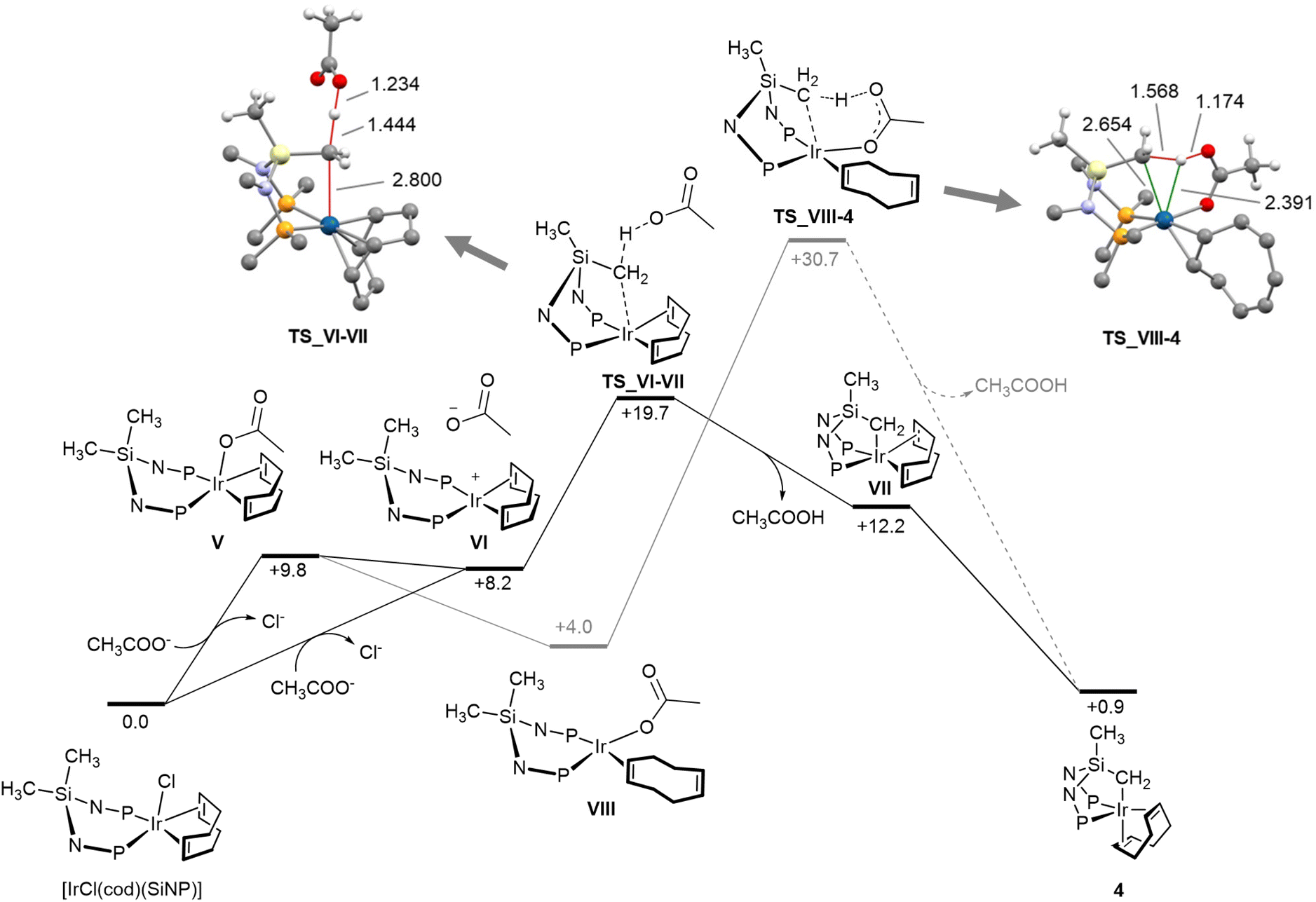 | ||
| Fig. 5 Gibbs free energy profiles for the formation of [Ir{κ3C,P,P′-(SiNP-H)}(cod)] (4) (black, outer sphere deprotonation; gray, acetate-assisted CH activation; kcal mol−1, B97D3/def2svp), along with the calculated structures of TS_VI-VII and TS_VIII-4 and selected interatomic distances (Å). Gray, carbon; white, hydrogen; violet, nitrogen; red, oxygen; yellow, silicon; orange, phosphorus, blue, iridium. | ||
Considering the outer sphere mechanism, the first step should be the chloride abstraction/substitution to yield the pentacoordinate acetato derivative V or the square planar intermediate VI, or an equilibrium mixture of the two. In turn, VI should undergo metallation via an outer sphere interaction of the SiCH3 moiety with free acetate ion. Indeed, acetate deprotonates the SiCH3 group as long as the metal–carbon bond forms (TS_VI-VII). As a result, the square pyramidal derivative VII (SPY-5-32) forms which ultimately isomerises to the observed more stable trigonal bypiramidal product 4 (TBPY-5-23). The overall transformation [IrCl(cod)(SiNP)] + CH3COO− → 4 + CH3COOH + Cl− is calculated to be slightly endergonic (ΔGr = +0.9 kcal mol−1), thus the formation of solid potassium chloride should be decisive for the outcome of the reaction. Regarding the base-assisted C–H bond cleavage mechanism, starting from pentacoordinate acetato derivative V, the preliminary η2, η2 → η2 haptotropic shift of the cod ligand should take place (V → VIII) in order to allow the required arrangement of the C–H bond, the metal centre and the acetato ligand. Once the square planar intermediate VIII forms, the acetate assisted deprotonation/metalation of the SiCH3 moiety takes place via the transition state TS_VIII-4. A thorough examination of interatomic distances in TS_VIII-4 (Fig. 5) indicates that no metal–hydrogen interaction should exist thus ruling out any oxidative character of the SiCH2-H bond cleavage.9 From an energy standpoint, the acetate-assisted C–H activation mechanism is not a competitive route and should be ruled out, since the TS_VIII-4 (+30.7 kcal mol−1) is not accessible under the experimental conditions. On the other hand, the calculated activation barrier (TS_VI-VII, +19.7 kcal mol−1) for the outer sphere mechanism nicely fits in with the observed outcome under the experimental conditions.
The endergonic character of the formation of 4 prompted us to carry out the reaction of 4 with triflic acid, as a strong Brønsted acid, in order to establish if the metalation of SiNP could be reverted, i.e. the Ir-CH2Si could be protonated. As a matter of fact, when CF3SO3H was added to a CD2Cl2 solution of 4 (1:1 molar ratio) in an NMR tube, the instantaneous complete conversion of 4 to [Ir(cod)(SiNP)]+ was observed, as established by comparison with the 31P{1H} and 1H NMR data previously reported for [Ir(cod)(SiNP)]+.7a Accordingly, on top of that, the reaction 4 + CF3SO3H → [Ir(cod)(SiNP)]+ + CF3SO3− was calculated to be highly exergonic (−22.7 kcal mol−1) likely as a consequence of the strong acidic character of CF3SO3H. In this respect, it is worth mentioning that in a previous study7d the reactivity of the iridium(III) derivative [IrH{κ3C,P,P′-(SiNP-H)}(CNtBu)2][PF6] towards Brønsted acids, such as HBF4, HPF6 and CF3COOH, was explored, showing that the Ir-CH2Si bond is not reactive, rather the Si–N bonds break as a result of the formal addition of HF (formed in situ by reaction of PF6− with either CF3COOH or HBF4). On these grounds, the stability of the Ir{κ3C,P,P′-(SiNP-H)} platform towards Brønsted acids seems to be subtly determined by the oxidation state of the metal centre, the metal–carbon bond being reactive towards H+ only in the case of iridium(I).
Reaction of [Ir{κ3C,P,P′-(SiNP-H)}(cod)] with oxidants
In order to expand the family of iridium complexes with the κ3C,P,P′-(SiNP-H) ligand, the reactivity of 4 towards oxidants, namely [FeCp2]X (X = PF6, CF3SO3), I2 or CF3SO3Me, was explored.
4 undergoes a two-electron oxidation when treated with [FeCp2]+ (Ir:Fe 1:2 molar ratio) in acetonitrile affording the octahedral cation [Ir{κ3C,P,P′-(SiNP-H)}(CH3CN)3]2+ (52+) isolated as either the trifluromethanesulfonate or the hexafluorophosphate salt (Scheme 6).
 | ||
| Scheme 6 Synthesis of [Ir{κ3C,P,P′-(SiNP-H)}(CH3CN)3][X]2 (5[X]2). | ||
Crystal structure of 5[CF3SO3]2 shows an octahedral coordination polyhedron at the metal centre with a facial coordination of the κ3C,P,P′-(SiNP-H) ligand [C1–Ir–P1 85.81(9)°, C1–Ir–P2 83.12(8)°, P1–Ir–P2 96.27(3) Å] (Fig. 6). The three remaining coordination sites are occupied by acetonitrile ligands. Noticeably, as a consequence of the higher trans influence of the metallated SiCH2 moiety, the iridium–nitrogen N42–Ir [2.117(3) Å] is longer than N45–Ir [2.071(3) Å] and N48–Ir [2.091(3) Å]. When comparing the κ3C,P,P′-(SiNP-H) ligand in 52+ and 4, regardless of the oxidation state of the metal centre, the IrCH2SiCH3(NP)2 backbones are virtually superimposable, minor differences being observed exclusively in the orientation of the phenyl and tolyl groups, probably as a consequence of the steric hindrance of ancillary ligands and/or the coordination polyhedron. The NMR spectra of 52+ confirm that the solid state structure is preserved in solution. Actually, two sets of signals are observed in both the 1H and the 13C{1H} NMR spectra for the acetonitrile ligand, namely at δH 2.24 ppm and δC 3.2 ppm for the acetonitrile ligands trans to phosphorus and at δH 2.44 and δC 3.9 ppm for the acetonitrile ligand trans to carbon. As for the SiNP-H ligand, in agreement with the proposed structure, one 31P{1H} NMR signal at 18.2 ppm indicates the equivalence of the two phosphorus atoms. Also, the 1H and 13C{1H} signals at 1.46 and at −19.6 ppm, respectively, are assigned to the metallated methylene group, whereas the 1H and 13C{1H} signals at 0.36 and −1.3 ppm, respectively, are assigned to the SiCH3 group.
 | ||
| Fig. 6 ORTEP plot of 52+ in 5[CF3SO3]2. For clarity, most hydrogen atoms are omitted, and tolyl and phenyl groups are shown in a wireframe style. Thermal ellipsoids are at 50% probability. Selected bond lengths (Å) and angles are (°): N42–Ir 2.117(3), N45–Ir 2.071(3), N48–Ir 2.091(3), P1–Ir 2.2994(8), P2–Ir 2.3012(8), C1–Ir 2.105(3), N1–P1 1.687(2), N1–Si 1.757(3), N2–P2 1.678(2), N2–Si 1.780(2), C1–Ir–P1 85.81(9), C1–Ir–P2 83.12(8), P1–Ir–P2 96.27(3), C1–Ir–N42 175.88(10), N45–Ir–P1 171.92(7), N48–Ir–P2 168.94(7). | ||
The treatment of 4 with I2 (1:1) affords a mixture of three products, namely the iridium(III) derivatives [IrI(κ2P,P′-SiNP)(η2,η3-C8H11)]I (6I) and two isomers of the dinuclear species [Ir2{κ3C,P,P′-(SiNP-H)}2(μ-I)3]I (7aI and 7bI), in a molar ratio 6+:7a+:7b+ 54:33:13 (Scheme 7, top). The mixture of 7aPF6 and 7bPF6 was independently prepared either by reaction of 4 with [FeCp2][PF6] in acetone and subsequent addition of NaI, or by direct reaction of 5[PF6]2 with NaI (Ir:I = 2:3) (Scheme 7 and Fig. 7). On the other hand, 6I was isolated in low yields from the mixture 6I + 7aI + 7bI upon recrystallization from CH3CN/Et2O (Fig. 7).
 | ||
| Scheme 7 (Top) Synthesis of 6I, 7aX and 7bX (X = I, PF6). (Bottom) Selected NMR data for 6+ [δC, along with JCP, italics, and δH (CD2Cl2)]. | ||
 | ||
| Fig. 7 31P{1H} NMR spectra (CD2Cl2, 298 K) of (A) the solid mixture resulting from the reaction of 4 with I2; (B) 7a+ + 7b+ obtained from the reaction of 5[PF6]2 with NaI; (C) 6I after recrystallization of a mixture of 6I + 7aI + 7bI from CH3CN/Et2O. | ||
The crystal structure of 6I shows a virtually octahedral geometry at the metal centre with a bidentate κ2P,P′ coordination of SiNP [P1–Ir 2.3215(13) Å, P2–Ir 2.3442(13) Å, P1–Ir–P2 91.53(4)°] (Fig. 8). The iodido ligand lies cis to both phosphorus atoms [I1–Ir 2.7247(4) Å, P1–Ir–I1 90.94(3)°, P2–Ir–I1 93.04(3)°] with the cycloocta-2,6-dien-1-yl ligand formally occupying the three remaining coordination sites. Notably, the olefinic moiety lies trans to one phosphorus atom [Ir–CT 2.2494(2) Å, CT–Ir–P1 171.79(3)°], and the allyl moiety spans the remaining coordination sites trans to P2 and I1 [C43–Ir 2.294(5) Å, C42–Ir 2.200(5) Å, C41–Ir 2.278(6) Å, C41–Ir–I1 155.72(14)°, C43–Ir–P2 169.58(15)°]. To the best of our knowledge, only few examples of cod activation leading to the formation of the cycloocta-2,6-dien-1-yl ligand have been reported for iridium complexes.10 NMR spectra indicate that the solution structure of 6+ is similar to that observed in the solid state. As a matter of fact, two 31P{1H} NMR doublets (δP 32.1, 23.2 ppm, 2JPP = 25.3 Hz) are observed for the two non-equivalent phosphorus atoms. In addition, the 1H and 13C{1H} NMR signals assigned to the C8H11 ligand (Scheme 7, bottom) confirm the presence of the cycloocta-2,6-dien-1-yl ligand.
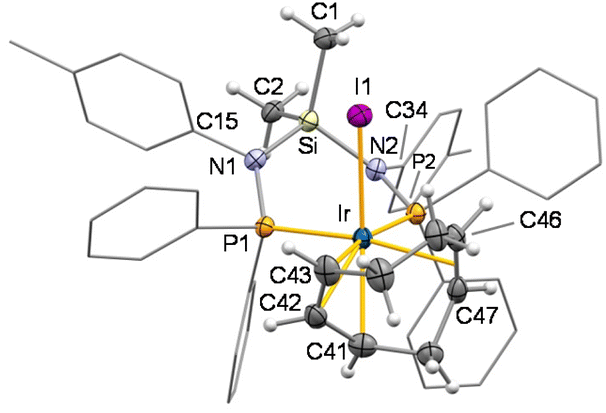 | ||
| Fig. 8 ORTEP plot of 6+ in 6I. For clarity, most hydrogen atoms are omitted, and tolyl and phenyl groups are shown in a wireframe style. Thermal ellipsoids are at 50% probability. Selected bond lengths (Å) and angles are (°): P1–Ir 2.3215(13), P2–Ir 2.3442(13), I1–Ir 2.7247(4), C41–Ir 2.278(6), C42–Ir 2.200(5), C43–Ir 2.294(5), Ir–CT 2.2494(2), C41–C42 1.422(8), C42–C43 1.404(8), C46–C47 1.377(8), C41–Ir–I1 155.72(14), C42–Ir–I1 123.76(16), C43–Ir–I1 90.23(16), P1–Ir–P2 91.53(4), P1–Ir–I1 90.94(3), P2–Ir–I1 93.04(3), CT–Ir–I1 97.262(10), CT–Ir–P1 171.79(3), CT–Ir–P2 88.27(3), C41–Ir–P2 108.65(14), C42–Ir–P2 142.90(17), C43–Ir–P2 169.58(15), C15–N1–P1 118.2(3), C15–N1–Si 112.2(3), P1–N1–Si 129.3(3), C34–N2–P2 116.5(3), C34–N2–Si 111.6(3), P2–N2–Si 131.6(3). CT, centroid of C46 and C47. | ||
Regardless of the synthetic route, a mixture of 7a+ and 7b+ (70:30, aprox.) was always obtained and, in our hands, it could not be separated. In addition, heating a solution containing a mixture of 7a+ and 7b+ resulted in the coalescence of their 31P signals, suggesting that an equilibrium between the two isomers could be operative in solution. Finally, the MALDI-TOF mass spectra uniquely revealed one molecular peak at m/z 2040.8 (calcd. 2041.1, [M]+) which suggests that 7a+ and 7b+ might be isomers. The crystal structure of 7a+ (in 7aPF6) shows a dinuclear [Ir(μ-I)3Ir] core [see Fig. 9 for Ir–I bond lengths and Ia–Ir–Ib angles] featuring a non-interaction intermetallic distance [Ir1⋯Ir2 3.7116(16) Å], with one κ3C,P,P′-(SiNP-H) ligand completing the coordination sphere of each metal centre. The methylene moieties lie trans to the same iodido ligand (I2) rendering a virtual C2v symmetry with two equivalent phosphorus atoms. As a consequence of the higher trans influence of the SiCH2 group when compared to the PPh2N moiety, the Ir1–I2 [2.7909(9) Å] and Ir2–I2 [2.8105(9) Å] bond lengths are longer than the remaining iridium–iodine bond lengths [Ir1–I1 2.7308(8), Ir1–I3 2.7386(9), Ir2–I1 2.7342(8), Ir2–I3 2.7316(8) Å]. The NMR spectra of 7a+ suggest that its structure in solution is similar to that in the solid state. Actually, one 31P{1H} singlet was observed at 20.7 ppm along with one 1H and one 13C{1H} signal for the methyl group of the tolyl fragment [δH 2.05; δC 21.5 ppm] confirming the equivalence of the two SiNP arms. On the other hand, the resonances at δH 2.05 and 0.00 ppm as well as at δC −11.7 and −0.6 ppm are assigned to the IrCH2Si and SiCH3 moieties, respectively.
 | ||
| Fig. 9 ORTEP plot of 7a+ in 7aPF6. For clarity, most hydrogen atoms are omitted, and tolyl and phenyl groups are shown in a wireframe style. Thermal ellipsoids are at 50% probability. Selected bond lengths (Å) and angles are (°): I1–Ir1 2.7308(8), I1–Ir2 2.7342(8), I2–Ir1 2.7909(9), I2–Ir2 2.8105(9), I3–Ir2 2.7316(8), I3–Ir1 2.7386(9), P1–Ir1 2.3037(15), P2–Ir1 2.2902(15), P3–Ir2 2.2886(15), P4–Ir2 2.3096(15), C1–Ir1 2.132(5), C41–Ir2 2.149(5), C1–Ir1–P2 83.36(13), C1–Ir1–P1 84.84(14), P2–Ir1–P1 97.36(6), C41–Ir2–P3 84.65(13), C41–Ir2–P4 84.44(14), P3–Ir2–P4 98.25(6), I1–Ir1–I3 77.36(3), I1–Ir1–I2 80.95(3), I3–Ir1–I2 80.80(3), I3–Ir2–I1 77.42(3), I3–Ir2–I2 80.57(3), I1–Ir2–I2 80.54(3). | ||
As for 7b+, full characterization could not be achieved. Nonetheless, in view of the two resonances at δP 22.0 and 21.0 ppm (Δν1/2 = 50 Hz), the SiCH2 moieties are proposed to lie trans to different bridging iodine atoms at the [Ir(μ-I)3Ir] core, thus rendering a virtual C2 symmetry with two non-equivalent phosphorus atoms slowly exchanging on the NMR timescale at 298 K. As a confirmation, the relative Gibbs free energy for 7a+ (0.0) and 7b+ (+1.3 kcal mol−1) nicely confirms that 7b+ should be observed in solution along with 7a+ (vide infra).
The course of the reaction of 4 with I2 has been investigated by DFT calculations (Scheme 8). As a result of the two-electron oxidation of 4, the iridium(III) species IX2+ is assumed to be the starting point of two independent pathways, leading to either 6+ or 7a/b+, respectively. As for the formation of 7a/b+, the cod ligand of IX+ should be displaced by incoming iodide rendering the dinuclear species 7a+ or 7b+. On the other hand, when dealing with the formation of 6+, the iodido ligand should dissociate (IX+ → X2+I−) and eventually act as a base, abstracting H+ from the cod ligand of X2+ (X2+I− → XI+ + HI). Thereafter, the intermediate XI+, that contains the η2,η3-cycloocta-2,6-dien-1-yl ligand, should react with the newly formed HI undergoing the protonation of the methylene group IrCH2Si, finally yielding 6+ (the structures of the transition state TS_X2+I−-XI+ and TS_XI+-6+ are shown in Scheme 8). For the sake of comparison, the isomer XII+ of [IrI(κ2P,P′-SiNP)(η2,η3-C8H11)]+ was calculated to be 9.1 kcal mol−1 less stable than 6+, which rules out its presence in solution in agreement with the observed NMR spectra.
 | ||
| Scheme 8 (Top) Reaction sequences for the formation of 7a + 7b+ (left) and 6+ (right) with relative Gibbs free energy of intermediates (kcal mol−1, B97D3/def2svp). (Bottom) Calculated structures of the transition states TS_X2+I−-XI+ and TS_XI+-6+ along with selected interatomic distances (Å). Gray, carbon; white, hydrogen; violet, nitrogen; yellow, silicon; orange, phosphorus; blue, iridium; purple, iodine. | ||
The reaction of 4 with methyl triflate also resulted in the formal two-electron oxidation of iridium. As a matter of fact, upon reaction of 4 with methyl triflate (1:1) for 4 days at room temperature, [Ir{κ3C,P,P′-(SiNP-H)}(η2,η3-C8H11)][CF3SO3] (8CF3SO3) forms along with methane (observed via both GC and 1H NMR spectroscopy) as a consequence of the unusual hydride abstraction from the cod ligand (Scheme 9). 8+ contains the iridium(III)-κ3C,P,P′-(SiNP-H) platform and the cycloocta-2,6-dien-1-yl ligand (C8H11) exhibiting a η2,η3 coordination. The 1H and 31P{1H} NMR spectra indicate that the κ3C,P,P′ coordination of the ligand SiNP-H is preserved and experiments a non-symmetric environment. Actually, two 31P{1H} signals (35.4, 31.5 ppm; 2JPP = 9.2 Hz) are observed along with two 1H multiplets for the non-equivalent hydrogen atoms of the methylene group IrCH2Si (0.55, 0.46 ppm, δC −13.8 ppm). Fig. 10 (top) shows a selection of 1H and 13C{1H} NMR data for the ligand C8H11 of 8+. It is worth a mention that the putative isomer XIII+ exhibiting the olefinic group at one of the coordination sites trans to phosphorus and the allyl moiety formally spanning the remaining sites trans to phosphorus and trans to CH2 was not observed (Scheme 9). Actually, confirming the proposed structure for 8+, XIII+ was calculated to be 5 kcal mol−1 less stable than 8+ (Scheme 9).
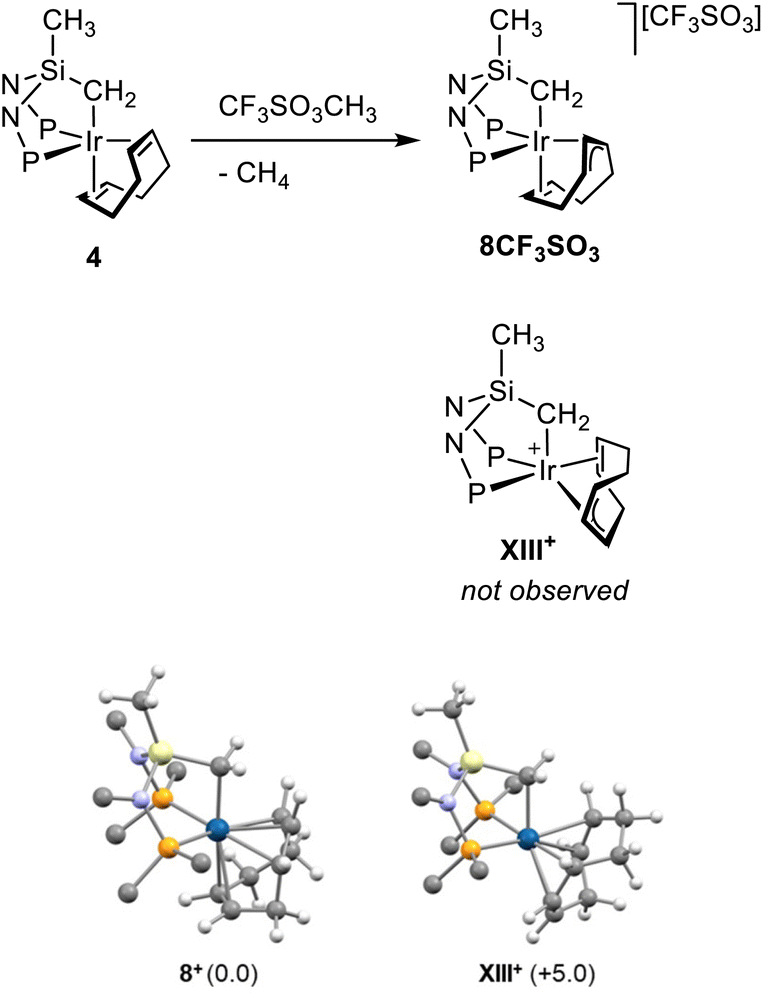 | ||
| Scheme 9 (Top) Synthesis of [Ir{κ3C,P,P′-(SiNP-H)}(η2,η3-C8H11)][CF3SO3] (8CF3SO3). (Bottom) Calculated structures for 8+ and XIII+ along with relative Gibbs free energy (kcal mol−1, B97D3/def2svp). For clarity, only ipso atoms of phenyl and tolyl groups are shown. Gray, carbon; white, hydrogen; violet, nitrogen; yellow, silicon; orange, phosphorus, blue, iridium. | ||
 | ||
| Fig. 10 (Top) Selected NMR data for 8+ [δC, along with JCP, italics, and δH]. (Bottom) Gibbs free energy profile for the reaction of 4 with methyl triflate (kcal mol−1, B97D3/def2svp) along with the calculated structure of TS_4-8+ and selected interatomic distances (Å). Gray, carbon; white, hydrogen; violet, nitrogen; red, oxygen; pale green, fluorine; yellow, silicon; orange, phosphorus; dark yellow, sulphur; blue, iridium. | ||
The mechanism of the formation of 6+ was explored by means of DFT calculations showing that it consists of a straightforward hydride abstraction from the coordinated cod promoted by methyl triflate, ultimately acting as an electrophile (Fig. 10), and goes through the transition state TS_4-8+ (+28.9 kcal mol−1).
Conclusions
The iridium(I)-aminophoshane derivative [IrCl(cod)(SiNP)] is a versatile precursor for the preparation of a range of iridium(III) complexes with the SiNP ligand coordinated either κ2P,P′ or κ3C,P,P′. The oxidative addition of allyl chloride to [IrCl(cod)(SiNP)] affords the thermally stable [IrCl2(η3-C3H5)(SiNP)] (1) which, in turn, reacts with tert-butyl isocyanide rendering the substitution product [IrCl(η3-C3H5)(CNtBu)(SiNP)]Cl (2Cl). The observed intermediate [IrCl2(η1-C3H5)(CNtBu)(SiNP)] (3) as well as DFT calculations point out that the mechanism is associative. In this connection, the η3 ⇄ η1 haptotropic shift of the allyl ligand is key for the outcome of the reaction.The iridium(I) pentacoordinate [Ir{κ3C,P,P′-(SiNP-H)}(cod)] (4) is smoothly obtained treating [IrCl(cod)(SiNP)] with potassium acetate via an outer sphere mechanism in which the acetate ion deprotonate the SiCH3 group while the carbon–iridium bond forms. The two-electron oxidation of [Ir{κ3C,P,P′-(SiNP-H)}(cod)] (4) gives rise to a variety of iridium(III) derivatives. The reaction of 4 with ferrocenium in acetonitrile leads to the oxidation of iridium(I) to iridium(III) rendering the octahedral cation [Ir{κ3C,P,P′-(SiNP-H)}(CH3CN)3]2+ (52+) as a result of the substitution of the cod ligand by three acetonitrile ligands. Alternatively, I2 readily oxidises 4 yielding a mixture of iridium(III) complexes as a consequence of either cod activation or cod substitution, namely [IrI(κ2P,P′-SiNP)(η2,η3-C8H11)]+ (6+) and [Ir2{κ3C,P,P′-(SiNP-H)}2(μ-I)3]+ (7a+/7b+), respectively. As a matter of fact, once [Ir{κ3C,P,P′-(SiNP-H)}(cod)] is oxidised, I− is able to either displace cod affording dinuclear species 7a+/7b+ or shuttle one H+ ion from cod to the κ3C,P,P′-(SiNP-H) ligand in a step-wise fashion, eventually affording the iridium(III) complex 6+. Finally, methyl triflate is also able to oxidise 4 affording 8+ as a result of the hydride abstraction from the cod ligand.
Experimental
General section
All the operations were carried out using standard Schlenk tube techniques under an atmosphere of pre-purified argon or in a Braun glove-box under argon. Solvents were dried and purified according to standard procedures and distilled under argon, or obtained oxygen-and water-free from a solvent purification system (Innovative Technologies). The compounds SiMe2{N(4-C6H4CH3)(PPh2)}2 (SiNP)6 and [IrCl(cod)(SiNP)]7a were prepared according to the literature. NMR spectra were recorded with Bruker spectrometers (AV300, AV400, or AV500) and are referred to SiMe4 (1H, 13C), and H3PO4 (31P). The proposed 1H, 13C, and 31P assignment relies on the combined analysis of 1D [1H, 1H{31P}, 13C{1H}-apt, 31P{1H}] and 2D NMR spectra (1H–1H COSY, 1H–1H NOESY, 1H–13C HSQC, 1H–13C HMBC, 1H–31P HMBC). Fig. 11 shows the numbering scheme used for the assignment of NMR signals in 1, 2Cl, 6I, and 8CF3SO3. Whenever labels P1 and P2, are used for non-equivalent phosphorus atoms, superscript labels “tol-P1/2” and “PhP1/2” are used for hydrogen and carbon atoms belonging to the tolyl and phenyl groups attached/linked to the phosphorus atom P1/P2, respectively. | ||
| Fig. 11 Numbering scheme for the assignment of NMR signals for 1, 2+, 6+, and 8+. | ||
MALDI-TOF mass spectra were recorded using a Bruker AutoFLEX III-TOF/TOF using trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) as a matrix.
:1 = 4:1, 60.1 mg). NMR data for 3: 1H NMR (CD2Cl2, 233 K): δH 7.62 (m, 4H, o-PPh), 7.49–7.33 (10H tot: 4H, o-PPh; 4H, m-PPh, 2H, p-PPh), 7.25 (t, 2H, 3JHH = 7.2 Hz, p-PPh), 7.10 (t, 4H, 3JHH = 7.2 Hz, m-PPh), 6.74–6.68 (5H tot: 1H, C2Hall, 2H, C2Htol; 2H, C3Htol), 6.59 (d, 2H, 3JHH = 8.2 Hz, C3Htol), 6.34 (d, 2H, 3JHH = 8.2 Hz, C2Htol), 5.18 (d, 1H, 3JHH trans = 17.1 Hz, C3Hall trans), 5.03 (d, 1H, 3JHH cis = 9.7 Hz, C3Hall cis), 3.34 (br, 2H, C1Hall), 2.09 (s, 6H, CH3tol), 1.06 (s, 9H, CNtBu), 0.47 (s, 3H, CH3Si), −0.05 (s, 3H, CH3Si); 13C{1H} NMR (CD2Cl2, 233 K): δC 148.2 (C2, all), 138.6 (t, 3JCP = 4.3 Hz, C1, tol), 136.7 (t, 2JCP = 4.4 Hz, C2, PhP), 136.5 (C4, tol), 133.4 (C2, tol), 133.1 (t, 2JCP = 4.1 Hz, C2, PhP), 132.3 (C2, tol), 130.7 (C2, PhP), 130.5 (C2, PhP), 129.1 (C3, tol), 127.4 (t, 3JCP = 5.4 Hz, C3, PhP), 126.0 (t, 3JCP = 5.4 Hz, C3, PhP), 108.3 (C3, all), 56.9 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) NtBu), 29.5 (CH3CNtBu), 20.9 (CH3tol), 5.28 (C1, all), 4.5 (CH3Si), 3.4 (CH3Si); 31P{1H} NMR (CD2Cl2, 233 K): δP 31.9 (2P, SiNP).
Nax), 121.7 (t, 3JCP = 9.7, CH3Neq), 21.2 (CH3tol), 3.9 (H3CNax), 3.2 (H3CNeq), −1.32 (t, 3JCP = 6.8 Hz, CH3Si), −19.6 (t, 2JCP = 4.0 Hz, CH2Si). 31P{1H} NMR (CD2Cl2, 298 K): δP 18.2 (2P, SiNP), −145.5 (hept, 2P, 1JPF = 710.8 Hz, PF6−).
:7aI:7bI = 54:33:13, 31P). A small amount (22 mg) of analytically and spectroscopically pure 6I was recovered by crystallization in acetonitrile/Et2O. Found: C 48.89, H 4.32, N 2.29. Calcd for C48H51I2IrN2P2Si (1191.99 g mol−1): C 48.37, H 4.31, N 2.35. 1H NMR (CD2Cl2, 298 K): δH 8.08–7.83 (br, 4H tot; 2H, o-P1Ph; 2H, o-P2Ph), 7.69–7.60 (2H tot, 1H, p-P1Ph; 1H, p-P2Ph), 7.47 (td, 2H, 3JHH = 7.9 Hz, 4JHP = 2.8 Hz, m-P2Ph), 7.43–7.31 (8H tot; 4H, m-P1Ph; 2H, m-P2Ph; 1H, p-P1Ph; 1H, p-P2Ph), 7.27 (m, 2H, o-P1Ph), 7.19 (dd, 2H, 3JHP = 11.1 Hz, 3JHH = 7.9 Hz, o-P2Ph), 7.03 (dt, 1H, 3JHH = 8.0 Hz, 4JHP = 2.0 Hz, C2Htol-P1), 6.96 (dt, 1H, 3JHH = 8.2 Hz, 4JHP = 1.7 Hz, C2Htol-P2), 6.88 (dd, 1H, 3JHH = 8.0 Hz, 5JHP = 1.0 Hz, C3Htol-P1), 6.74 (dd, 1H, 3JHH = 8.2 Hz, 5JHP = 0.9 Hz, C3Htol-P2), 6.64 (dd, 1H, 3JHH = 8.2 Hz, 5JHP = 1.2 Hz, C3Htol-P1), 6.52 (dd, 1H, 3JHH = 8.2 Hz, 5JHP = 1.4 Hz, C3Htol-P2), 6.09 (dt, 1H, 3JHH = 8.2 Hz, 4JHP = 1.9 Hz, C2Htol-P1), 6.06 (dt, 1H, 3JHH = 8.2 Hz, 4JHP = 1.7 Hz, C2Htol-P2), 5.08 (m, 1H, C1Hcod), 4.17 (dd, 1H, 3JHP = 14.1 Hz, 3JHH = 6.0 Hz, C2Hcod), 3.89 (m, 1H, C3Hcod), 3.67–3.59 (2H tot; 1H, C5Hcod; 1H, C6Hcod), 3.39 (br, 1H, C4Hcod), 3.35–3.19 (2H tot; 1H, C4Hcod; 1H, C7Hcod), 3.03 (m, 1H, C7Hcod), 2.48 (m, 1H, C8Hcod), 2.16 (s, 3H, CH3tol-P1), 2.06 (s, 3H, CH3tol-P2), 1.69 (m, 1H, C8Hcod), 1.28 (s, 3H, CH3Si), −0.35 (s, 3H, CH3Si). 13C NMR (CD2Cl2, 298 K): δC 138.9 (d, 2JCP = 9.1 Hz, C1, tol-P2), 138.3 (d, 2JCP = 8.3 Hz, C1, tol-P1), 138.0 (d, 5JCP = 1.5 Hz, C4, tol-P1), 137.8 (d, 5JCP = 1.7 Hz, C4, tol-P2), 135.2 (br, C2, PhP1; C2, PhP2), 134.9 (d, 2JCP = 12.1 Hz, C2, PhP2), 133.10 (C4, PhP1), 133.03 (C4, PhP2), 132.83 (d, 3JCP = 2.4 Hz, C2, tol-P1), 132.75 (d, 2JCP = 9.2 Hz, C2, PhP2), 132.5 (C2, tol-P2), 132.4 (d, 3JCP = 3.1 Hz, C2, tol-P2), 131.9 (d, 3JCP = 3.0 Hz, C2, tol-P1), 130.18 (C3, tol-P1), 130.10 (C3, tol-P1), 130.03 (C3, tol-P2), 129.96 (d, 3JCP = 11.6 Hz, C3, PhP2), 129.88 (C3, tol-P2), 129.4 (d, 3JCP = 11.1 Hz, C3, PhP1), 128.4 (d, 3JCP = 11.3 Hz, C3, PhP2), 128.2 (d, 3JCP = 10.3 Hz, C3, PhP2), 111.7 (d, 2JCP = 14.3 Hz, C6, cod), 92.2 (C2, cod), 83.6 (d, 2JCP = 28.1 Hz, C1, cod), 64.0 (dd, 2JCP = 5.4; 2.9 Hz, C5, cod), 34.5 (d, 2JCP = 3.4 Hz, C3, cod), 33.3 (d, 3JCP = 4.5 Hz, C7, cod), 28.2 (C8, cod), 21.32 (CH3tol-P1), 21.23 (CH3tol-P2), 19.8 (C4, cod), 5.6 (CH3Si), 5.0 (CH3Si). 31P NMR (CD2Cl2, 298 K): δP 32.1 (d, 2JPP = 25.3 Hz, P1), 23.2 (d, 2JPP = 25.3 Hz, P2).
:20 mL). The solution was dried in vacuo affording a colorless solid finally identified as [Ir{κ3C,P,P′-(SiNP-H)}(η2,η3-C8H11)][OTf] (8OTf) (59.4 mg, 45% yield). Found: C 54.27, H 4.53, N 2.56. Calcd for C49H50F3IrN2O3P2SSi (1086.24 g mol−1): C 54.18, H 4.64, N 2.58. 1H NMR (CD2Cl2, 298 K): δH 7.90 (ddd, 2H. 3JHP = 10.4 Hz, 3JHH = 7.6 Hz, 5JHP = 1.2 Hz, o-P2Ph), 7.82 (ddd, 2H. 3JHP = 11.2 Hz, 3JHH = 7.6 Hz, 5JHP = 1.9 Hz, o-P2Ph), 7.65–7.56 (6H tot; 2H, o-P1Ph; 2H, m-P2Ph; 2H, p-P2Ph), 7.55–7.45 (6H tot; 2H, m-P1Ph; 2H, m-P2Ph; 2H, p-P1Ph), 7.07 (td, 2H, 3JHH = 7.6 Hz, 4JHP = 2.5 Hz, o-P1Ph), 7.02 (d, 2H, 3JHH = 8.2 Hz, C3Htol-P2), 6.70 (d, 2H, 3JHH = 8.2 Hz, C3Htol-P1), 6.69 (d, 2H, 3JHH = 8.2 Hz, C2Htol-P1), 6.49 (ddd, 2H, 3JHP = 10.9 Hz, 3JHH = 7.6 Hz, 5JHP = 1.1 Hz, o-P1Ph), 6.28 (d, 2H, 3JHH = 8.2 Hz, C2Htol-P2), 4.37–4.32 (2H tot; 1H, C2Hcod; 1H, C6Hcod), 4.08 (m, 1H, C1Hcod), 3.76 (m, 1H, C5Hcod), 3.38 (m, 1H, C4Hcod), 2.94–2.83 (2H tot; 1H, C7Hcod; 1H, C4Hcod), 2.52 (m, 1H, C7Hcod), 2.30 (s, 3H, CH3tol-P2), 2.26 (m, 1H, C8Hcod), 2.15 (m, 1H, C3Hcod), 2.10 (s, 3H, CH3tol-P1), 1.68 (m, 1H, C8Hcod), 0.55 (ddd, 1H, 2JHH = 12.4 Hz, 3JHP = 8.3 Hz, 3JHP = 3.0 Hz, SiC
NtBu), 29.5 (CH3CNtBu), 20.9 (CH3tol), 5.28 (C1, all), 4.5 (CH3Si), 3.4 (CH3Si); 31P{1H} NMR (CD2Cl2, 233 K): δP 31.9 (2P, SiNP).
Nax), 121.7 (t, 3JCP = 9.7, CH3Neq), 21.2 (CH3tol), 3.9 (H3CNax), 3.2 (H3CNeq), −1.32 (t, 3JCP = 6.8 Hz, CH3Si), −19.6 (t, 2JCP = 4.0 Hz, CH2Si). 31P{1H} NMR (CD2Cl2, 298 K): δP 18.2 (2P, SiNP), −145.5 (hept, 2P, 1JPF = 710.8 Hz, PF6−).
:7aI:7bI = 54:33:13, 31P). A small amount (22 mg) of analytically and spectroscopically pure 6I was recovered by crystallization in acetonitrile/Et2O. Found: C 48.89, H 4.32, N 2.29. Calcd for C48H51I2IrN2P2Si (1191.99 g mol−1): C 48.37, H 4.31, N 2.35. 1H NMR (CD2Cl2, 298 K): δH 8.08–7.83 (br, 4H tot; 2H, o-P1Ph; 2H, o-P2Ph), 7.69–7.60 (2H tot, 1H, p-P1Ph; 1H, p-P2Ph), 7.47 (td, 2H, 3JHH = 7.9 Hz, 4JHP = 2.8 Hz, m-P2Ph), 7.43–7.31 (8H tot; 4H, m-P1Ph; 2H, m-P2Ph; 1H, p-P1Ph; 1H, p-P2Ph), 7.27 (m, 2H, o-P1Ph), 7.19 (dd, 2H, 3JHP = 11.1 Hz, 3JHH = 7.9 Hz, o-P2Ph), 7.03 (dt, 1H, 3JHH = 8.0 Hz, 4JHP = 2.0 Hz, C2Htol-P1), 6.96 (dt, 1H, 3JHH = 8.2 Hz, 4JHP = 1.7 Hz, C2Htol-P2), 6.88 (dd, 1H, 3JHH = 8.0 Hz, 5JHP = 1.0 Hz, C3Htol-P1), 6.74 (dd, 1H, 3JHH = 8.2 Hz, 5JHP = 0.9 Hz, C3Htol-P2), 6.64 (dd, 1H, 3JHH = 8.2 Hz, 5JHP = 1.2 Hz, C3Htol-P1), 6.52 (dd, 1H, 3JHH = 8.2 Hz, 5JHP = 1.4 Hz, C3Htol-P2), 6.09 (dt, 1H, 3JHH = 8.2 Hz, 4JHP = 1.9 Hz, C2Htol-P1), 6.06 (dt, 1H, 3JHH = 8.2 Hz, 4JHP = 1.7 Hz, C2Htol-P2), 5.08 (m, 1H, C1Hcod), 4.17 (dd, 1H, 3JHP = 14.1 Hz, 3JHH = 6.0 Hz, C2Hcod), 3.89 (m, 1H, C3Hcod), 3.67–3.59 (2H tot; 1H, C5Hcod; 1H, C6Hcod), 3.39 (br, 1H, C4Hcod), 3.35–3.19 (2H tot; 1H, C4Hcod; 1H, C7Hcod), 3.03 (m, 1H, C7Hcod), 2.48 (m, 1H, C8Hcod), 2.16 (s, 3H, CH3tol-P1), 2.06 (s, 3H, CH3tol-P2), 1.69 (m, 1H, C8Hcod), 1.28 (s, 3H, CH3Si), −0.35 (s, 3H, CH3Si). 13C NMR (CD2Cl2, 298 K): δC 138.9 (d, 2JCP = 9.1 Hz, C1, tol-P2), 138.3 (d, 2JCP = 8.3 Hz, C1, tol-P1), 138.0 (d, 5JCP = 1.5 Hz, C4, tol-P1), 137.8 (d, 5JCP = 1.7 Hz, C4, tol-P2), 135.2 (br, C2, PhP1; C2, PhP2), 134.9 (d, 2JCP = 12.1 Hz, C2, PhP2), 133.10 (C4, PhP1), 133.03 (C4, PhP2), 132.83 (d, 3JCP = 2.4 Hz, C2, tol-P1), 132.75 (d, 2JCP = 9.2 Hz, C2, PhP2), 132.5 (C2, tol-P2), 132.4 (d, 3JCP = 3.1 Hz, C2, tol-P2), 131.9 (d, 3JCP = 3.0 Hz, C2, tol-P1), 130.18 (C3, tol-P1), 130.10 (C3, tol-P1), 130.03 (C3, tol-P2), 129.96 (d, 3JCP = 11.6 Hz, C3, PhP2), 129.88 (C3, tol-P2), 129.4 (d, 3JCP = 11.1 Hz, C3, PhP1), 128.4 (d, 3JCP = 11.3 Hz, C3, PhP2), 128.2 (d, 3JCP = 10.3 Hz, C3, PhP2), 111.7 (d, 2JCP = 14.3 Hz, C6, cod), 92.2 (C2, cod), 83.6 (d, 2JCP = 28.1 Hz, C1, cod), 64.0 (dd, 2JCP = 5.4; 2.9 Hz, C5, cod), 34.5 (d, 2JCP = 3.4 Hz, C3, cod), 33.3 (d, 3JCP = 4.5 Hz, C7, cod), 28.2 (C8, cod), 21.32 (CH3tol-P1), 21.23 (CH3tol-P2), 19.8 (C4, cod), 5.6 (CH3Si), 5.0 (CH3Si). 31P NMR (CD2Cl2, 298 K): δP 32.1 (d, 2JPP = 25.3 Hz, P1), 23.2 (d, 2JPP = 25.3 Hz, P2).
:20 mL). The solution was dried in vacuo affording a colorless solid finally identified as [Ir{κ3C,P,P′-(SiNP-H)}(η2,η3-C8H11)][OTf] (8OTf) (59.4 mg, 45% yield). Found: C 54.27, H 4.53, N 2.56. Calcd for C49H50F3IrN2O3P2SSi (1086.24 g mol−1): C 54.18, H 4.64, N 2.58. 1H NMR (CD2Cl2, 298 K): δH 7.90 (ddd, 2H. 3JHP = 10.4 Hz, 3JHH = 7.6 Hz, 5JHP = 1.2 Hz, o-P2Ph), 7.82 (ddd, 2H. 3JHP = 11.2 Hz, 3JHH = 7.6 Hz, 5JHP = 1.9 Hz, o-P2Ph), 7.65–7.56 (6H tot; 2H, o-P1Ph; 2H, m-P2Ph; 2H, p-P2Ph), 7.55–7.45 (6H tot; 2H, m-P1Ph; 2H, m-P2Ph; 2H, p-P1Ph), 7.07 (td, 2H, 3JHH = 7.6 Hz, 4JHP = 2.5 Hz, o-P1Ph), 7.02 (d, 2H, 3JHH = 8.2 Hz, C3Htol-P2), 6.70 (d, 2H, 3JHH = 8.2 Hz, C3Htol-P1), 6.69 (d, 2H, 3JHH = 8.2 Hz, C2Htol-P1), 6.49 (ddd, 2H, 3JHP = 10.9 Hz, 3JHH = 7.6 Hz, 5JHP = 1.1 Hz, o-P1Ph), 6.28 (d, 2H, 3JHH = 8.2 Hz, C2Htol-P2), 4.37–4.32 (2H tot; 1H, C2Hcod; 1H, C6Hcod), 4.08 (m, 1H, C1Hcod), 3.76 (m, 1H, C5Hcod), 3.38 (m, 1H, C4Hcod), 2.94–2.83 (2H tot; 1H, C7Hcod; 1H, C4Hcod), 2.52 (m, 1H, C7Hcod), 2.30 (s, 3H, CH3tol-P2), 2.26 (m, 1H, C8Hcod), 2.15 (m, 1H, C3Hcod), 2.10 (s, 3H, CH3tol-P1), 1.68 (m, 1H, C8Hcod), 0.55 (ddd, 1H, 2JHH = 12.4 Hz, 3JHP = 8.3 Hz, 3JHP = 3.0 Hz, SiC![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) aHbIr), 0.46 (ddd, 1H, 2JHH = 12.4 Hz, 3JHP = 7.0 Hz, 3JHP = 2.4 Hz, SiCHabIr), 0.12 (s, 3H, SiCH3). 13C{1H} NMR (CD2Cl2, 298 K): δC 143.1 (dd, 1JCP = 51.5 Hz, 3JCP = 2.6 Hz, C1, PhP1), 139.2 (d, 2JCP = 8.0 Hz, C1, tol-P1), 138.3 (d, 2JCP = 10.0 Hz, C1, tol-P2), 137.4 (d, 5JCP = 1.6 Hz, C4, tol-P2), 136.0 (d, 2JCP = 11.9 Hz, C2, PhP1), 135.5 (C4, tol-P1), 133.7 (d, 2JCP = 9.4 Hz, C2, PhP2), 133.3 (d, 2JCP = 10.7 Hz, C2, PhP2), 133.1 (d, 4JCP = 2.1 Hz, C4, PhP1), 132.7 (d, 4JCP = 2.5 Hz, C4, PhP2), 132.6 (d, 4JCP = 2.4 Hz, C4, PhP1), 131.8 (d, 4JCP = 1.8 Hz, C4, PhP1), 130.9 (d, 4JCP = 2.2 Hz, C3, tol-P1), 130.7 (d, 4JCP = 1.2 Hz, C3, tol-P2), 130.2 (d, 2JCP = 10.2 Hz, C2, PhP1), 129.9 (C2, tol-P1), 129.7 (d, 3JCP = 9.9 Hz, C3, PhP1), 129.6 (d, 3JCP = 8.7 Hz, C3, PhP2), 129.4 (d, 3JCP = 10.7 Hz, C3, PhP2), 128.9 (d, 3JCP = 10.6 Hz, C3, PhP1), 128.1 (d, 3JCP = 4.1 Hz, C2, tol2), 106.1 (d, 2JCP = 2.0 Hz, C2 cod), 98.8 (C6 cod), 71.0 (d, 2JCP = 35.7 Hz, C1 cod), 53.9 (d, 2JCP = 3.6 Hz, C5 cod), 49.2 (d, 2JCP = 17.2 Hz, C3 cod), 34.2 (d, 3JCP = 5.3 Hz, C7 cod), 29.4 (d, 3JCP = 3.4 Hz, C8 cod), 21.4 (CH3tol-P2), 21.0 (CH3tol-P1), 20.5 (d, 3JCP = 4.5 Hz, C4 cod), −1.6 (t, 3JCP = 6.9 Hz, CH3Si), −13.8 (t, 2JCP = 4.1 Hz, CH2Si). 31P{1H} NMR (CD2Cl2, 298 K): δP 35.4 (d, 1P, 2JPP = 9.1 Hz, SiNP1), 31.5 (d, 1P, 2JPP = 9.2 Hz, SiNP2).
19 and refined by full matrix least-squares on F2 with SHELXL-2014,20 under WinGX.21 In the case of 6 and 7 the program SQUEEZE22 was used to treat the residual electron density.
360/9414 [R(int) = 0.0405], Tmax/Tmin 0.4045/0.3179, data/restraints/parameters 9414/35/536, GooF(F2) = 1.035, R1 = 0.0304 [I > 2σ(I)], wR2 = 0.0762 (all data), largest diff. peak/hole 2.050/−1.461 e Å−3. CCDC deposit number 2282657.‡
aHbIr), 0.46 (ddd, 1H, 2JHH = 12.4 Hz, 3JHP = 7.0 Hz, 3JHP = 2.4 Hz, SiCHabIr), 0.12 (s, 3H, SiCH3). 13C{1H} NMR (CD2Cl2, 298 K): δC 143.1 (dd, 1JCP = 51.5 Hz, 3JCP = 2.6 Hz, C1, PhP1), 139.2 (d, 2JCP = 8.0 Hz, C1, tol-P1), 138.3 (d, 2JCP = 10.0 Hz, C1, tol-P2), 137.4 (d, 5JCP = 1.6 Hz, C4, tol-P2), 136.0 (d, 2JCP = 11.9 Hz, C2, PhP1), 135.5 (C4, tol-P1), 133.7 (d, 2JCP = 9.4 Hz, C2, PhP2), 133.3 (d, 2JCP = 10.7 Hz, C2, PhP2), 133.1 (d, 4JCP = 2.1 Hz, C4, PhP1), 132.7 (d, 4JCP = 2.5 Hz, C4, PhP2), 132.6 (d, 4JCP = 2.4 Hz, C4, PhP1), 131.8 (d, 4JCP = 1.8 Hz, C4, PhP1), 130.9 (d, 4JCP = 2.2 Hz, C3, tol-P1), 130.7 (d, 4JCP = 1.2 Hz, C3, tol-P2), 130.2 (d, 2JCP = 10.2 Hz, C2, PhP1), 129.9 (C2, tol-P1), 129.7 (d, 3JCP = 9.9 Hz, C3, PhP1), 129.6 (d, 3JCP = 8.7 Hz, C3, PhP2), 129.4 (d, 3JCP = 10.7 Hz, C3, PhP2), 128.9 (d, 3JCP = 10.6 Hz, C3, PhP1), 128.1 (d, 3JCP = 4.1 Hz, C2, tol2), 106.1 (d, 2JCP = 2.0 Hz, C2 cod), 98.8 (C6 cod), 71.0 (d, 2JCP = 35.7 Hz, C1 cod), 53.9 (d, 2JCP = 3.6 Hz, C5 cod), 49.2 (d, 2JCP = 17.2 Hz, C3 cod), 34.2 (d, 3JCP = 5.3 Hz, C7 cod), 29.4 (d, 3JCP = 3.4 Hz, C8 cod), 21.4 (CH3tol-P2), 21.0 (CH3tol-P1), 20.5 (d, 3JCP = 4.5 Hz, C4 cod), −1.6 (t, 3JCP = 6.9 Hz, CH3Si), −13.8 (t, 2JCP = 4.1 Hz, CH2Si). 31P{1H} NMR (CD2Cl2, 298 K): δP 35.4 (d, 1P, 2JPP = 9.1 Hz, SiNP1), 31.5 (d, 1P, 2JPP = 9.2 Hz, SiNP2).
19 and refined by full matrix least-squares on F2 with SHELXL-2014,20 under WinGX.21 In the case of 6 and 7 the program SQUEEZE22 was used to treat the residual electron density.
360/9414 [R(int) = 0.0405], Tmax/Tmin 0.4045/0.3179, data/restraints/parameters 9414/35/536, GooF(F2) = 1.035, R1 = 0.0304 [I > 2σ(I)], wR2 = 0.0762 (all data), largest diff. peak/hole 2.050/−1.461 e Å−3. CCDC deposit number 2282657.‡
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 13.1665(17) Å, b = 13.8662(18) Å, c = 15.261(3) Å, α = 108.940(2)°, β = 90.289(2)°, γ = 118.034(2)°, V = 2284.3(6) Å3, Z = 1, Dcalc = 1.549 g cm−3, μ = 3.231 mm−1, F(000) = 1074, colourless prism 0.320 × 0.200 × 0.070 mm3, θmin/θmax 1.437/26.371°, index ranges: −16 ≤ h ≤ 16, −16 ≤ k ≤ 17, −19 ≤ l ≤ 18, reflections collected/independent 20795/9338 [R(int) = 0.0354], Tmax/Tmin 0.5490/0.4262, data/restraints/parameters 9338/1/529, GooF(F2) = 1.038, R1 = 0.0326 [I > 2σ(I)], wR2 = 0.0847 (all data), largest diff. peak/hole 2.621/−1.325 e Å−3. CCDC deposit number 2282655.‡
030/12697 [R(int) = 0.0512], Tmax/Tmin 0.6205/0.4784, data/restraints/parameters 12697/0/646, GooF(F2) = 1.014, R1 = 0.0298 [I > 2σ(I)], wR2 = 0.0598 (all data), largest diff. peak/hole 1.039/−0.685 e Å−3. CCDC deposit number 2282656.‡
, a = 10.3680(8) Å, b = 14.1270(11) Å, c = 19.6166(16) Å, α = 100.9810(10)°, β = 98.2830(10)°, γ = 103.0900(10)°, V = 2694.2(4) Å3, Z = 2, Dcalc = 1.783 g cm−3, μ = 4.040 mm−1, F(000) = 1412, orange prism, 0.250 × 0.140 × 0.090 mm3, θmin/θmax 2.056/26.372°, index ranges −12 ≤ h ≤ 12, −17 ≤ k ≤ 17, −24 ≤ l ≤ 24, reflections collected/independent 39544/10963 [R(int) = 0.0353], Tmax/Tmin 0.5264/0.3558, data/restraints/parameters 10963/0/540, GooF(F2) = 1.052, R1 = 0.0372 [I > 2σ(I)], wR2 = 0.0949 (all data), largest diff. peak/hole 2.043/−1.903 e Å−3. CCDC deposit number 2282659.‡
741/15721 [R(int) = 0.0406], Tmax/Tmin 0.3444/0.2500, data/restraints/parameters 15721/4/913, GooF(F2) = 1.016, R1 = 0.0303 [I > 2σ(I)], wR2 = 0.0753 (all data), largest diff. peak/hole 2.080/−1.144 e Å−3. CCDC deposit number 2282658.‡
, a = 13.1665(17) Å, b = 13.8662(18) Å, c = 15.261(3) Å, α = 108.940(2)°, β = 90.289(2)°, γ = 118.034(2)°, V = 2284.3(6) Å3, Z = 1, Dcalc = 1.549 g cm−3, μ = 3.231 mm−1, F(000) = 1074, colourless prism 0.320 × 0.200 × 0.070 mm3, θmin/θmax 1.437/26.371°, index ranges: −16 ≤ h ≤ 16, −16 ≤ k ≤ 17, −19 ≤ l ≤ 18, reflections collected/independent 20795/9338 [R(int) = 0.0354], Tmax/Tmin 0.5490/0.4262, data/restraints/parameters 9338/1/529, GooF(F2) = 1.038, R1 = 0.0326 [I > 2σ(I)], wR2 = 0.0847 (all data), largest diff. peak/hole 2.621/−1.325 e Å−3. CCDC deposit number 2282655.‡
030/12697 [R(int) = 0.0512], Tmax/Tmin 0.6205/0.4784, data/restraints/parameters 12697/0/646, GooF(F2) = 1.014, R1 = 0.0298 [I > 2σ(I)], wR2 = 0.0598 (all data), largest diff. peak/hole 1.039/−0.685 e Å−3. CCDC deposit number 2282656.‡
, a = 10.3680(8) Å, b = 14.1270(11) Å, c = 19.6166(16) Å, α = 100.9810(10)°, β = 98.2830(10)°, γ = 103.0900(10)°, V = 2694.2(4) Å3, Z = 2, Dcalc = 1.783 g cm−3, μ = 4.040 mm−1, F(000) = 1412, orange prism, 0.250 × 0.140 × 0.090 mm3, θmin/θmax 2.056/26.372°, index ranges −12 ≤ h ≤ 12, −17 ≤ k ≤ 17, −24 ≤ l ≤ 24, reflections collected/independent 39544/10963 [R(int) = 0.0353], Tmax/Tmin 0.5264/0.3558, data/restraints/parameters 10963/0/540, GooF(F2) = 1.052, R1 = 0.0372 [I > 2σ(I)], wR2 = 0.0949 (all data), largest diff. peak/hole 2.043/−1.903 e Å−3. CCDC deposit number 2282659.‡
741/15721 [R(int) = 0.0406], Tmax/Tmin 0.3444/0.2500, data/restraints/parameters 15721/4/913, GooF(F2) = 1.016, R1 = 0.0303 [I > 2σ(I)], wR2 = 0.0753 (all data), largest diff. peak/hole 2.080/−1.144 e Å−3. CCDC deposit number 2282658.‡
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministerio de Ciencia e Innovación MCIN/AEI/10.13039/501100011033, under the project PID2019-103965GB-I00, and the Departamento de Ciencia, Universidad y Sociedad del Conocimiento del Gobierno de Aragón (group E42_23R) is gratefully acknowledged.References
- (a) G. Ewart, D. S. Payne, A. L. Porte and A. P. Lane, J. Chem. Soc., 1962, 3984 RSC; (b) H. H. Sisler and N. L. Smith, J. Org. Chem., 1961, 26, 611 CrossRef CAS; (c) W. A. Hart and H. H. Sisler, Inorg. Chem., 1964, 3, 617–622 CrossRef CAS.
- Selected references: (a) K. M. Gramigna, D. A. Dickie, B. M. Foxman and C. M. Thomas, ACS Catal., 2019, 9, 3153–3164 CrossRef CAS; (b) A. M. Lifschitz, N. A. Hirscher, H. B. Lee, J. A. Buss and T. Agapie, Organometallics, 2017, 36, 1640–1648 CrossRef CAS; (c) A. Prades, S. Núñez-Pertíñez, A. Riera and X. Verdaguer, Chem. Commun., 2017, 53, 4605–4608 RSC; (d) S. A. Bartlett, J. Moulin, M. Tromp, G. Reid, A. J. Dent, G. Cibin, D. S. McGuinness and J. Evans, Catal. Sci. Technol., 2016, 6, 6237–6246 RSC; (e) S. Orgué, T. León, A. Riera and X. Verdaguer, Org. Lett., 2015, 17, 250–253 CrossRef PubMed; (f) W. K. Walker, B. M. Kay, S. A. Michaelis, D. L. Anderson, S. J. Smith, D. H. Ess and D. J. Michaelis, J. Am. Chem. Soc., 2015, 137, 7371–7378 CrossRef CAS PubMed; (g) W. K. Walker, D. L. Anderson, R. W. Stokes, S. J. Smith and D. J. Michaelis, Org. Lett., 2015, 17, 752–755 CrossRef CAS PubMed; (h) F. Trentin, A. M. Chapman, A. Scarso, P. Sgarbossa, R. A. Michelin, G. Strukul and D. F. Wass, Adv. Synth. Catal., 2012, 354, 1095–1104 CrossRef CAS; (i) L. E. Bowen, M. Charernsuk, T. W. Hey, C. L. McMullin, A. G. Orpen and D. F. Wass, Dalton Trans., 2010, 39, 560–567 RSC; (j) B. R. Aluri, N. Peulecke, B. H. Müller, S. Peitz, A. Spannenberg, M. Hapke and U. Rosenthal, Organometallics, 2010, 29, 226–231 CrossRef CAS.
- Selected references: (a) A. Aloisi, É. Crochet, E. Nicolas, J.-C. Berthet, C. Lescot, P. Thuéry and T. Cantat, Organometallics, 2021, 40, 2064–2069 CrossRef CAS; (b) H. Zhang, G. P. Hatzis, C. E. Moore, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2019, 141, 9516–9520 CrossRef CAS PubMed; (c) H. Zhang, B. Wu, S. L. Marquard, E. D. Litle, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Organometallics, 2017, 36, 3498–3507 CrossRef CAS; (d) J. P. Krogman, B. M. Foxman and C. M. Thomas, Organometallics, 2015, 34, 3159–3166 CrossRef CAS.
- Selected references: (a) C. Mu, J. He, S. Lü, J. Yang, Y. Xie, K. Hu, P. Yan and Y.-L. Li, Polyhedron, 2021, 200, 115087 CrossRef CAS; (b) L.-C. Song, L.-D. Zhang, W.-W. Zhang and B.-B. Liu, Organometallics, 2018, 37, 1948–1957 CrossRef CAS; (c) L.-C. Song, X.-F. Han, W. Chen, J.-P. Li and X.-Y. Wang, Dalton Trans., 2017, 46, 10003–10013 RSC.
- Selected references: (a) B. S. Mitchell, W. Kaminsky and A. Velian, Inorg. Chem., 2021, 60, 6135–6139 CrossRef CAS PubMed; (b) J. A. Kephart, A. C. Boggiano, W. Kaminsky and A. Velian, Dalton Trans., 2020, 49, 16464–16473 RSC; (c) B. A. Barden, G. Culcu, J. P. Krogman, M. W. Bezpalko, G. P. Hatzis, D. A. Dickie, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2019, 58, 821–833 CrossRef CAS PubMed; (d) G. Culcu, D. A. Iovan, J. P. Krogman, M. J. T. Wilding, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2017, 139, 9627–9636 CrossRef CAS PubMed; (e) S. Todisco, V. Gallo, P. Mastrorilli, M. Latronico, N. Re, F. Creati and P. Braunstein, Inorg. Chem., 2012, 51, 11549–11561 CrossRef CAS PubMed.
- V. Passarelli and F. Benetollo, Inorg. Chem., 2011, 50, 9958–9967 CrossRef CAS PubMed.
- (a) V. Passarelli, J. J. Pérez-Torrente and L. A. Oro, Inorg. Chem., 2014, 53, 972–980 CrossRef CAS PubMed; (b) V. Passarelli, J. J. Pérez-Torrente and L. A. Oro, Dalton Trans., 2015, 44, 18596–18606 RSC; (c) V. Passarelli, J. J. Pérez-Torrente and L. A. Oro, Dalton Trans., 2016, 45, 951–962 RSC; (d) M. Palmese, J. J. Pérez-Torrente and V. Passarelli, Dalton Trans., 2022, 51, 7142–7153 RSC; (e) M. Palmese, J. J. Pérez-Torrente and V. Passarelli, Dalton Trans., 2022, 51, 12334–12351 RSC.
- Selected references: (a) S. W. Kim, C. C. Meyer, B. K. Mai, P. Liu and M. J. Krische, ACS Catal., 2019, 9, 9158–9163 CrossRef CAS PubMed; (b) D. C. Schmitt, A.-M. R. Dechert-Schmitt and M. J. Krische, Org. Lett., 2012, 14, 6302–6305 CrossRef CAS PubMed; (c) I. S. Kim, M.-Y. Ngai and M. J. Krische, J. Am. Chem. Soc., 2008, 130, 14891–14899 CrossRef CAS PubMed.
- K. M. Altus and J. A. Love, Commun. Chem., 2021, 4, 173 CrossRef PubMed.
- (a) C. Tejel, M. A. Ciriano, V. Passarelli, J. A. López and B. De Bruin, Chem. – Eur. J., 2008, 14, 10985–10998 CrossRef CAS PubMed; (b) M. Martin, W. Sola, O. Torres, P. Plou and L. A. Oro, Organometallics, 2003, 22, 5406–5417 CrossRef CAS; (c) R. Dorta and A. Togni, Organometallics, 1998, 17, 5441–5444 CrossRef CAS; (d) G. W. Bushnell, D. O. Kim Fjeldsted, S. R. Stobart and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1983, 580–581 RSC; (e) D. R. Russell and P. A. Tucker, J. Organomet. Chem., 1977, 125, 303–312 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, C. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1997, 107, 8554–8560 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- SAINT+: Area-Detector Integration Software, version 6.01, Bruker AXS, Madison, WI, 2001 Search PubMed.
- G. M. Sheldrick, SADABS program, University of Göttingen, Göttingen, Germany, 1999 Search PubMed.
- G. M. Sheldrick, SHELXS 97, Program for the Solution of Crystal Structure, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Guido Pampaloni, mentor and friend, on the occasion of his retirement, in recognition of his remarkable contributions to coordination and organometallic chemistry, and his extraordinary commitment to the education of generations of chemists. |
| ‡ Electronic supplementary information (ESI) available: 1H, 13C{1H}-apt and 31P{1H} NMR spectra. Coordinates of calculated structures (xyz). CCDC 2282655–2282659. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02361c |
| This journal is © The Royal Society of Chemistry 2023 |