
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBeryllium compounds for the carbon–halogen bond activation of phenyl halides: the role of non-innocent ligands†
Daniel E.
Trujillo-González
 ab,
Gerardo
González-García
a,
J. Oscar C.
Jiménez-Halla
*a and
Miquel
Solà
*b
ab,
Gerardo
González-García
a,
J. Oscar C.
Jiménez-Halla
*a and
Miquel
Solà
*b
aDepartamento de Química, Campus Guanajuato, Universidad de Guanajuato, Noria Alta S/N, CP 36050, Guanajuato, Gto, Mexico. E-mail: jjimenez@ugto.mx
bInstitut de Química Computacional i Catàlisi and Departament de Química, Universitat de Girona, C/ Maria Aurèlia Capmany, 69, 17003 Girona, Catalonia, Spain. E-mail: miquel.sola@udg.edu
First published on 22nd August 2023
Abstract
Beryllium is a metallomimetic main-group element, i.e., it behaves similarly to transition metals (TMs) in some bond activation processes. To investigate the ability of Be compounds to activate C–X bonds (X = F–I), we have computationally investigated, using DFT methods, the reaction of (CAAC)2Be (CAAC = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethylpyrrolidin-2-ylidene) and a series of five-membered heterocyclic beryllium bidentate ligands with phenyl halides. We have analysed all plausible reaction mechanisms and our results show that, after the initial C–X oxidative addition, migration of the phenyl group occurs towards the less electronegative heteroatom. Our theoretical study highlights the important role of bidentate non-innocent ligands in providing the required electrons for the initial Ph–X oxidative addition. In contrast, the monodentate ligand, CAAC, does not favour this oxidative addition.
Introduction
In the last decade, advancement of s- and p-block chemistry has taken place as a consequence of the potential of some of these elements to replicate the behaviour of transition metals (TMs) in catalysis, the so-called metallomimetic chemistry.1 In the last few years, we have witnessed several studies reporting on the ability of the main-group elements to activate C–H and H–H bonds2–4 among others. Alkaline-earth metals (usually Mg, Ca, Sr, and Ba compounds) have been shown to carry out hydroamination, heterofunctionalization, and cross-metathesis reactions,5 which were only attributed to TMs in the past. Among these elements, beryllium is less preferred to be explored experimentally since it is toxic to human health (long-term exposure causes berylliosis).6–9 However, nowadays it is possible to develop beryllium chemistry with appropriate safety conditions.8Despite its toxicity, beryllium and its compounds have caught the attention of researchers in the last decades due to their applications in materials science and nuclear physics.6,7,10 However, until 2015, less than 180 beryllium compounds, which contain Be–N, Be–C and Be–P bonds, were reported according to the Cambridge structural database.11
Beryllium dihalides form stable complexes with Lewis bases.12–14 Thus, different ligands are used to stabilize the empty orbitals of the beryllium atom. For instance, Frenking and coworkers analyzed a tetracoordinate beryllium compound that was synthesized by the reaction between BeCl2 and two molecules of bis(diphenylphosphino)methane in dichloromethane (Chart 1a).15 Using toluene as a solvent, Petz and coworkers reported a tricoordinate beryllium compound obtained from the reaction between BeCl2 and C(PPh3)2 (Chart 1b).16 Also, Buchner and coworkers carried out two reactions: beryllium dichloride in the presence of PMe3 in benzene led to (PMe3)2BeCl2 and beryllium dichloride reacted with bis(diphenylphosphino)propane in benzene to give the heterocyclic beryllium compound (CH2)3(Ph2P)2BeCl2 (a six membered ring, Chart 1c).17 Paparo and Jones made a ligand substitution in Et2OBeX2 (where X = Br, I) with diamines or diazabutadienes (DBAs) to prepare five-membered heterocyclic beryllium rings (Chart 1d).18 The possibility of obtaining beryllium compounds without Be–halogen bonds was shown by Arrowsmith and Braunschweig in the synthesis of L2Be (L = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethylpyrrolidine-2-ylidene (CAAC), Chart 1e). First, they coordinated CAAC with the BeCl2 in benzene, and then the second CAAC molecule was coordinated using KC8 and EtO2.19 Paparo, Smith, and Jones carried out a reaction between BeBr2(TMEDA) (TMEDA = N,N,N′,N′-tetramethylethylenediamine) and :Al(DIPNacNac) (DIPNacNac = (((2,6-diisopropylphenyl)NCMe)2CH)−) in toluene, to get the first compound with a Be–Al bond (Chart 1f).20 Puchta, Buchner, and co-workers synthesized mono- and dinuclear beryllium halides [(PMe3)2BeX2] and [(PMe3)BeX2]2, through the coordination of PMe3 with the BeCl2 in benzene (Chart 1g).21 Finally, Paparo, Jones, and co-workers synthesized a beryllium naphthalenediyl complex through the reaction between K2(C10H8)2(THF), that allowed the removal of the bromide ligands, and the stabilized NHC-BeBr2 (Chart 1h).22
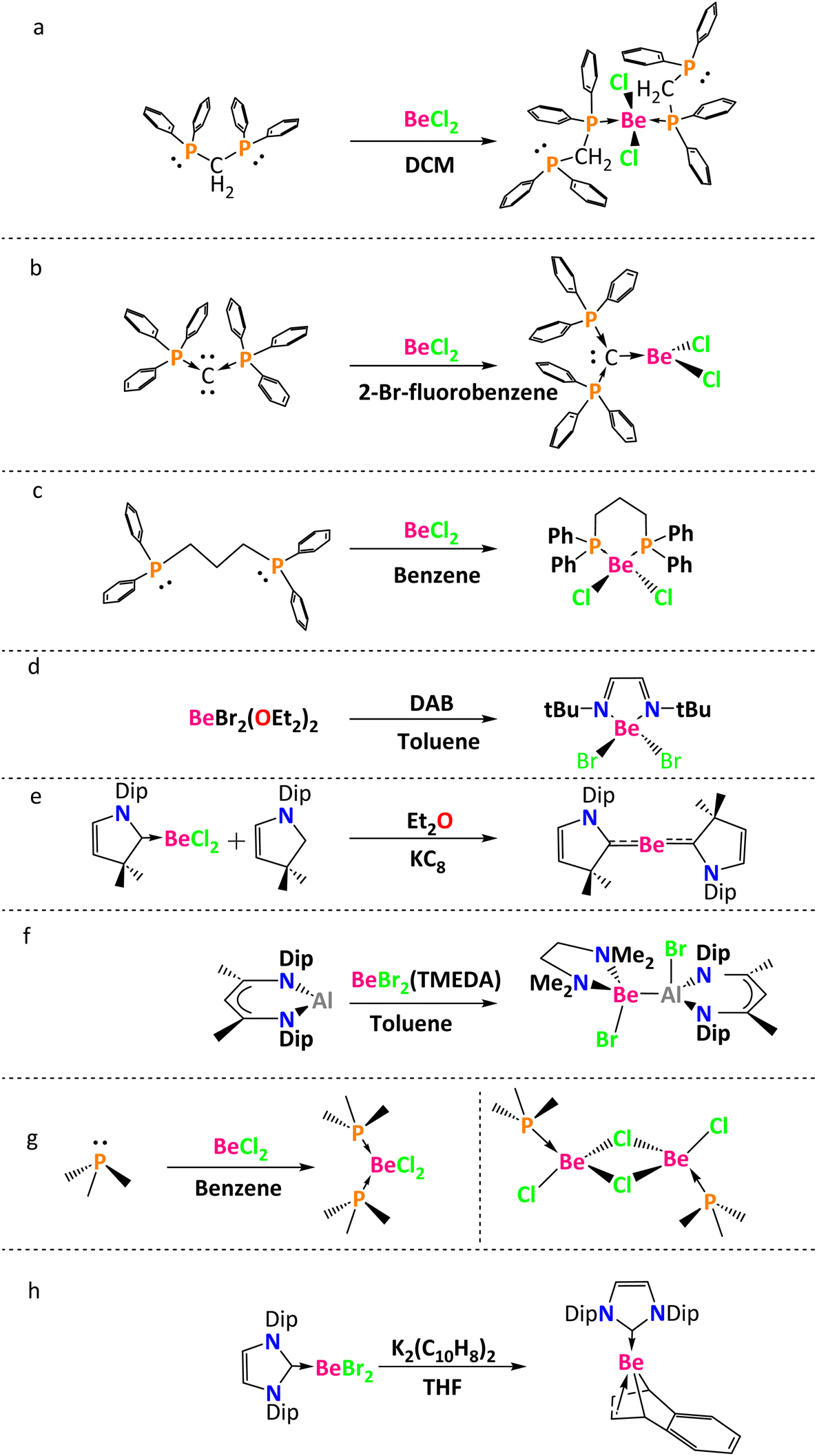 | ||
| Chart 1 Beryllium compounds reported with different ligands. | ||
The importance of choosing an appropriate ligand is not only for better stabilization but also because beryllium compounds can activate bonds23–25 and fixate small molecules.26 Therefore, taking advantage of the preference of Be to form bonds with halogens, we also aimed to tackle an environmental problem due to the use of phenyl halides (which are toxic, carcinogenic, and mutagenic).27,28 On the other hand, aromatic halides are important in organic synthesis chemistry, but a limitation is found due to the inertness of the halogen–carbon bond.29 Regarding the activation of C–X bonds, we can mention a series of examples provided by Matsubara and co-workers who carried out the amination of haloarenes using a nickel catalyst.30 Also, Wu's group made fluorenes and polyarenes using 1-bromonaphthalene, diphenylethyne, and a palladium catalyst.31 Even, an alloy of palladium and gold carried out the C–Cl and C–Br bond activations (more reactive for Ar–Cl than Ar–Br).32 Bickelhaupt, Hamlin, and co-workers studied the carbon(spn)–halogen bond activation using a Pd catalyst and found a decrement in the activation energies following the order C(spn)–F > C(spn)–Cl > C(spn)–Br > C(spn)–I.33 Also, Harder and co-workers reported the hydrohalogenation of aromatic halides (with F, Cl, Br, and I) with alkaline-earth compounds (AeH, Ae = Ca, Sr, and Ba). They found that the conversion rate increases with the metal size (Ca < Sr < Ba) and halogen size (F < Cl < Br < I).34
Herein, using (CAAC)2Be, NtBuNtBuBe (NtBuNtBu = (1E,2E)-N1,N2-di-tert-butylethane-1,2-diimine), NNBe (NN = (1E,2E)-N1,N2-dimethylethane-1,2-diimine), NPBe (NP = (1E,2E)-N-methyl-2-(methylphosphaneylidene)ethan-1-imine), NOBe (NO = (E)-2-(methylimino)acetaldehyde) and NSBe (NS = (E)-2-(methylimino)-ethanethial), we report on some plausible mechanistic proposals for the Ph–X (where X = F, Cl, Br, I) bond activation (see Scheme 1) and the effect of using mono- or bidentate ligands as Lewis bases to stabilize the empty orbitals of Be, adding an example of the ability of beryllium to act as other metals like Sr, Ca, Ba, Pd, and Ni. It is worth noting that among the different beryllium rings used, NtBuNtBuBeBr2 was prepared experimentally.18 The NtBuNtBuBe species can be obtained by reduction of NtBuNtBuBeBr2 using solid potassium (vide infra).
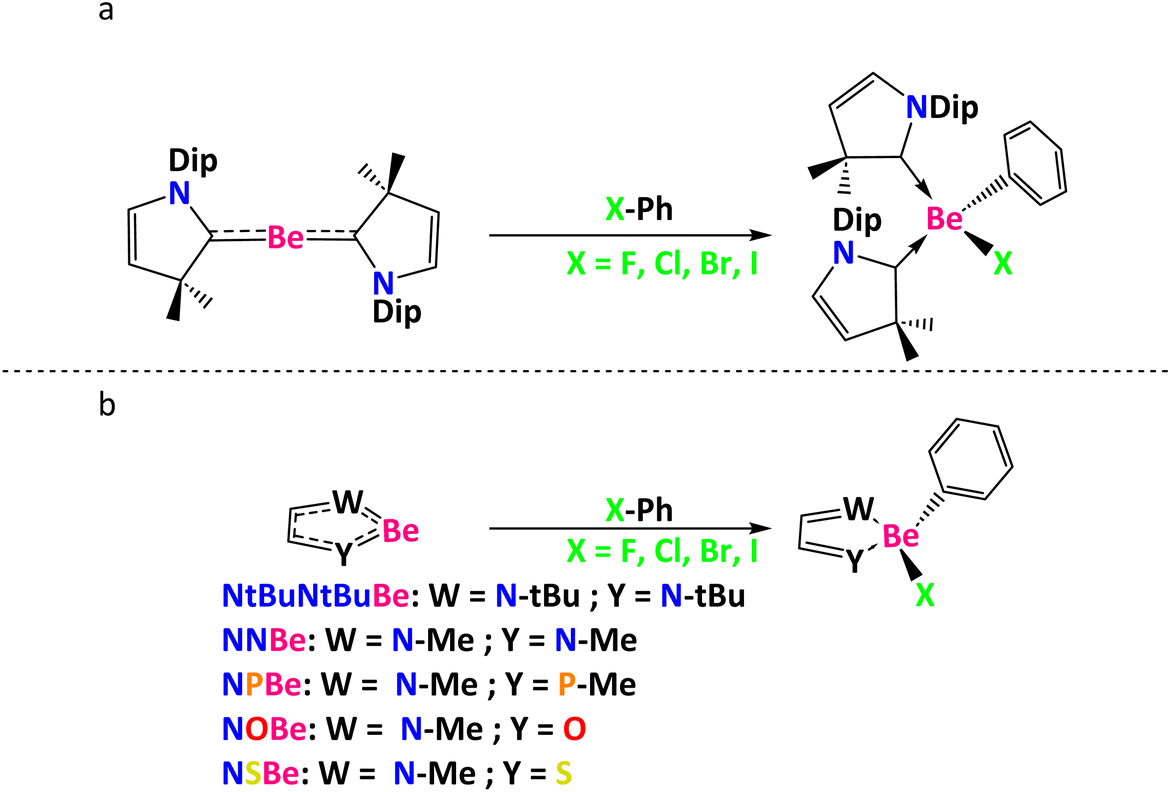 | ||
| Scheme 1 C–X bond activations reported in this work (a) using (CAAC)2Be and (b) using five-membered heterocyclic rings. Dip = 2,6-diisopropylphenyl. | ||
Computational details
All DFT calculations were performed using the Gaussian09 program.35 Geometries were optimized using Head-Gordon's hybrid functional that contains range-separated dispersion corrections (ωB97X-D).36 The electronic configuration of the molecules was represented using the basis sets (def2-SVPP) developed by Ahlrichs and co-workers.37,38 The unrestricted formalism was used for the calculation of open-shell species. All the stationary points were classified according to their harmonic frequencies: only one imaginary frequency for transition states and positive values for all the frequencies for reactants, intermediates, and products. Gibbs energies were computed from the corrected electronic energies at the ωB97X-D/def2-TZVP(SMD:toluene)//ωB97X-D/def2-SVPP level obtained with the Gaussian09 program and including solvent effects with the solvation model based on solute electron density (SMD)39 considering toluene as a solvent. Added corrections of the zero-point energy, thermal contributions to the internal energy, and the entropy term were computed at 298.15 K with the ωB97X-D/def2-SVPP method considering an ideal gas under standard conditions. For species NNBe and Ph–F, we performed additional calculations at the ωB97X-D/def2-TZVP(SMD:toluene)//ωB97X-D/def2-TZVP level of theory (see Fig. S7†). Differences in the energy profiles obtained by optimizing the geometries at the ωB97X-D/def2-SVPP or ωB97X-D/def2-TZVP level of theory are less than 1 kcal mol−1, thus justifying the use of the ωB97X-D/def2-SVPP method for geometry optimizations. For a few elementary reaction steps, despite many attempts, we have been unable to locate the transition state (TS). In these cases, we have obtained an approximate relative electronic energy (ΔE) for the TS based on a linear transit that connects reactants and products. For these approximate TSs (marked with an asterisk in the reaction profiles), we provide the relative electronic energy obtained at the ωB97X-D/def2-TZVP(SMD:toluene)//ωB97X-D/def2-SVPP level. As they are not strict stationary points, Gibbs energies cannot be obtained with standard procedures.Nucleus independent chemical shifts (NICS) were calculated at the B3PW91/6-311+G**//ωB97X-D/def2-SVPP level. Using the atomic partitioning obtained with the AIMALL program,40 the multicenter indices (MCIs)41–43 were computed with the ESI-3D44–46 program at the ωB97X-D/def2-SVPP level. The wavefunction and the electronic structure were analyzed at the ωB97X-D/def2-TZVPP//ωB97X-D/def2-SVPP level of theory with the ORCA47 and the IBOview48 programs.
Using the ωB97X-D/def2-SVPP optimized geometries, energy decomposition analysis (EDA) was performed with the BLYP49,50 functional and the TZ2P basis set with the Amsterdam density functional (ADF2019) software package.51,52 The all-electron basis set used, denoted TZ2P, is of triple-ζ quality with two sets of polarization functions for all atoms. Standard convergence criteria and a fine grid were used. Dispersion forces were included via Grimme's dispersion correction scheme (DFT-D3(BJ)),53 which contains the damping function proposed by Becke and Johnson.54 In EDA, the interaction energy, ΔEint, corresponds to the actual energy change when the geometrically deformed fragments are combined to form the overall complex. ΔEint can further be decomposed within the framework of the canonical Kohn–Sham molecular orbital (MO) model.55–59 The first term, ΔVelstat, corresponds to the classical electrostatic interaction between the unperturbed charge distributions of the fragments in the geometry they possess in the complex. This term is usually attractive. The Pauli repulsion, ΔEPauli, between these fragments comprises the destabilizing interactions, associated with the Pauli principle for fermions, between occupied orbitals and is responsible for the steric repulsion. The orbital interaction, ΔEoi, between these fragments in any MO model, and therefore also in Kohn–Sham theory, accounts for bond pair formation, charge transfer (empty/occupied orbital mixing between different fragments) and polarization (empty/occupied orbital mixing on one fragment due to the presence of another fragment). Finally, the ΔEdisp term accounts for attractive dispersion interactions.
Results and discussion
First, we carried out geometry optimizations for reactants (Fig. S1†). We tested different electronic configurations to find the ground state. NtBuNtBuBe, NNBe, NPBe, NOBe, and NSBe are closed-shell singlets, whereas (CAAC)2Be is an open-shell broken symmetry singlet (the singlet closed-shell is higher in energy by 8.7 kcal mol−1, see Table S1†).60 For the (CAAC)2Be complex, the Be–C distance is 1.646 Å (exp. 1.664 Å;19 theor. 1.644 Å).60 The Be–N bond lengths in all the rings vary from 1.516 to 1.536 Å (theor. 1.535 Å for NNBe61), while the P–Be, O–Be and S–Be bond lengths are 2.046, 1.464, and 2.046 Å, respectively. The C–C bond lengths oscillate from 1.354 to 1.360 Å and shorter C–C bond lengths are found when W and Y shown in Scheme 1 are different (see Fig. S1†).The electronic structure for each ring was investigated with the IBOview program. The N, O, and S atoms tend to delocalize their electron pair inside the ring (to carbon and beryllium atoms) while P does not share its lone pair (see Fig. 1). Other works reported five-membered rings with a non-planar phosphine moiety.62,63 Using effective oxidation states (EOSs) defined by Salvador et al.,64 we determined that the EOS of the Be atom is 2+ at all stationary points that were located (also in (CAAC)2Be). We also carried out EDA for the NNBe taking Be as one of the fragments and the ligand as the other one. We considered the Be fragment in different oxidation and spin states: Be0 (singlet and triplet), Be1+ (doublet) as reported by Parameswaran's group for a similar compound, BeN2(CH3)2C2H2,61 and Be2+ (singlet). Taking the oxidation state from the lowest stabilizing ΔEoi value,65,66 we also found that the oxidation state of Be is 2+ (see Fig. S2†). This agrees with the intrinsic bond orbitals (IBOs) displayed in Fig. 1 that indicate that Be does not have a lone pair.
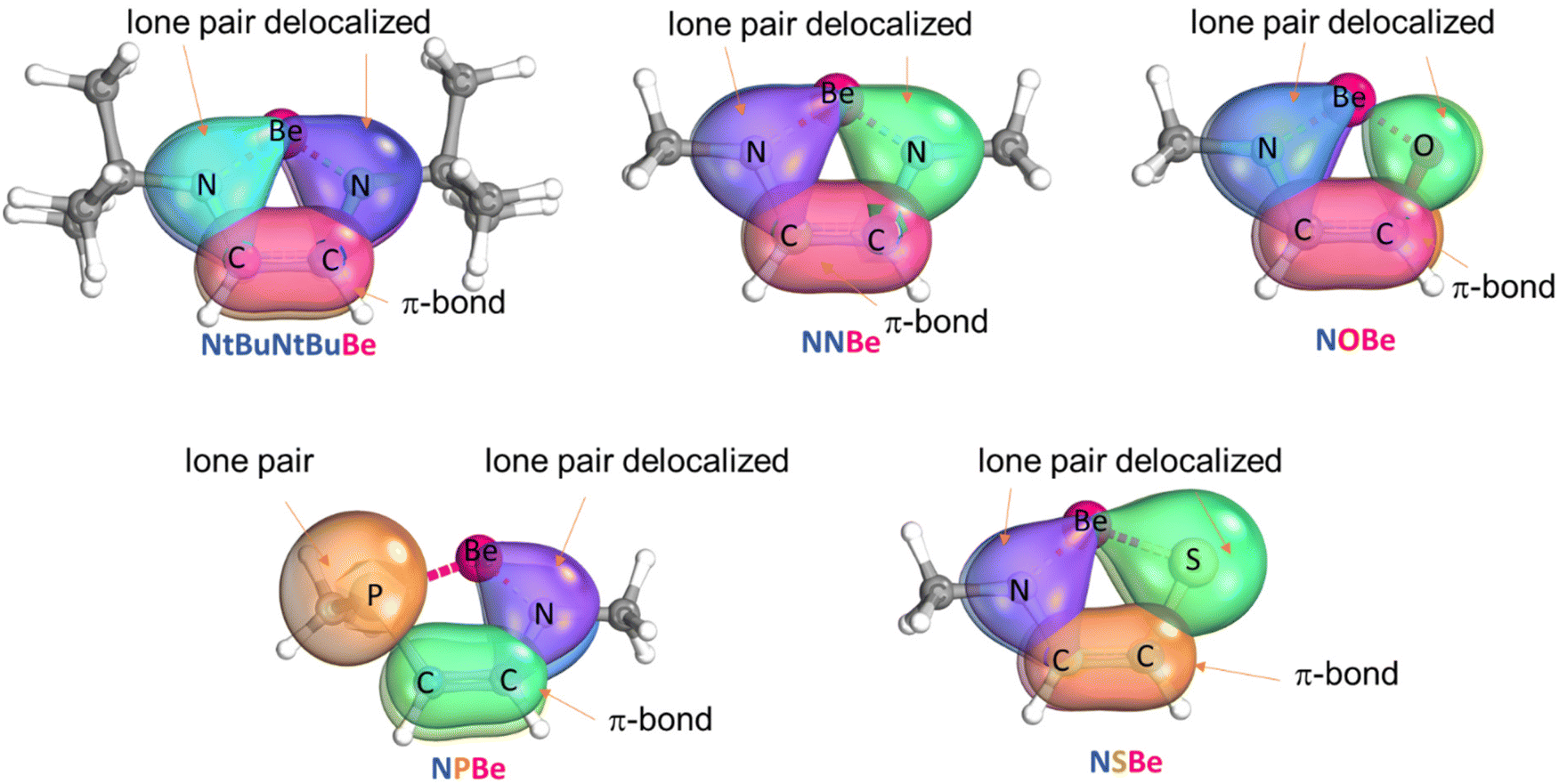 | ||
| Fig. 1 IBOs for heterocyclic beryllium compounds. | ||
Due to the electron delocalization, we considered the possibility of these compounds to have an aromatic character (rings with formally 6π-electrons); we calculated the NICS index at the B3PW91/6-311+G**//ωB97X-D/def2-SVPP level of theory. In accordance with our results, the NICS(0) values are −8.3 (benzene), −9.2 (NtBuNtBuBe), −8.7 (NNBe), −8.0 (NSBe), −6.3 (NOBe) and −2.9 (NPBe) ppm. All of the rings except NPBe show relatively large negative values that indicate the aromatic character of these rings with six π-electrons61,67 (for NICS(1)zz values, see Table S2†). In the NPBe system, the phosphorus electron pair is not involved in the π-system, which quenches the aromaticity of this ring. Our NICS result of NNBe agrees with that reported by Parameswaran's group (NICSzz(1) = −9.6).61 The aromatic character of the rings, indicated by NICS results, is further substantiated by the MCI values (see Table S2†). From the computed normalized MCI values (MCI1/n)68 we obtained the following order of aromaticity: benzene (0.59) > NNBe (0.51) ≈ NtBuNtBuBe (0.50) ≈ NOBe (0.50) ≈ NSBe (0.50) > NPBe (0.47), which agrees with the NICS ordering. On the other hand, the isomerization stabilization energies (ISEs)69 provide a different order of aromaticity, that is, NSBe > NPBe > NOBe > NNBe, and prove that the aromatic stabilization energy of these systems is relatively low (Table S4†).70
Before starting with the activation of the Ph–X bonds, we calculated the reduction of NtBuNtBuBeBr2 using a K10 cluster of Cs symmetry71 to get NtBuNtBuBe, two molecules of KBr, and a K8 cluster of C2v symmetry (Scheme 2).71 The Gibbs reaction energy is clearly favorable according to our calculations (ΔG(SMD = toluene) = −43.4 kcal mol−1). Thus, we now discuss the reaction mechanism for the halogen–carbon bond activation using the NtBuNtBuBe ring (Fig. 2).
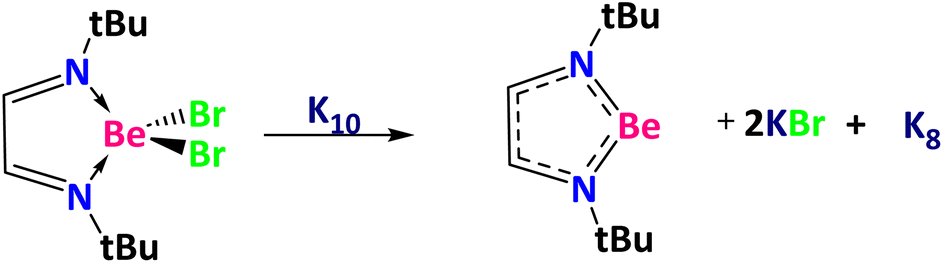 | ||
| Scheme 2 NtBuNtBuBeBr2 reduction to NtBuNtBuBe. | ||
 | ||
| Fig. 2 Reaction mechanism determined at the ωB97X-D/def2-TZVP(SMD:toluene)//ωB97X-D/def2-SVPP level. X = F (green), Cl (pink), Br (brown), and I (purple). | ||
The reaction starts with the donor–acceptor interaction between one of the lone pairs of the halogen and the LUMO of NtBuNtBuBe (1.1a, Fig. 2) that is mainly located on the Be atom.61 This step is exergonic for F but not for Cl, Br, and I. Through the transition state (TS) 1.2a, that describes the Ph–X bond cleavage with ΔG‡ = 27.2 (F), 27.0 (Cl), 22.9 (Br), and 21.6 (I) kcal mol−1, we got 1.3a (the process is kinetically more favorable in the following order: I > Br > F ∼ Cl). Intermediate 1.3a is characterized by a tetracoordinate beryllium compound with a 2+ oxidation state of Be. Next, the phenyl group migrates from Be to N (TS 1.4a with ΔG‡ = 21.3 (F), 24.1 (Cl), 25.2 (Br), and 27.8 (I) kcal mol−1) to get the product 1.5a. According to EOS, Be does not change the oxidation state of 2+ along the reaction. The electron density needed for the bond activation comes from the ligand that plays a non-innocent role (vide infra).
The Gibbs reaction energy is exergonic for all halogens, with the exergonicity decreasing as the halogen becomes heavier. Except for I, the Gibbs energy barriers to generate product 1.5a from 1.3a are lower in energy than those of the 1.1a → 1.3a process. The TOF-determining intermediate (TDI)72 is 1.1a for F or reactants for Cl and Br and the TOF-determining transition state (TDTS) is 1.2a for F, Cl, and Br, whereas for I the TDI is 1.3a and the TDTS is 1.4a. The energetic span (TDTS–TDI energy difference) values are 27.2 (F), 27.0 (Cl), 22.9 (Br), and 27.8 (I) kcal mol−1. According to our results, the most favorable activation takes place for Ph–Br. Fig. S5† shows other reaction paths that were analyzed for this NtBuNtBuBe system but were ruled out because they were found to be higher in energy than those presented in Fig. 2.
The second reaction mechanism that we have studied corresponds to the NNBe ring (Fig. 3). As before, the reaction starts with the halogen coordination with the Be moiety in an exergonic process. This interaction is more favorable in NNBe than in NtBuNtBuBe because in the latter the LUMO is located higher in energy by about 0.11 eV.
 | ||
| Fig. 3 Reaction mechanism determined at the ωB97X-D/def2-TZVP(SMD:toluene)//ωB97X-D/def2-SVPP level. X = F (green), Cl (pink), Br (brown), and I (purple). | ||
To get 2.3a (favorable for all halogens in the order F > Br > Cl > I), it is necessary to surmount the TS 2.2a corresponding to the bond activation with ΔG‡ = 23.6 (F), 20.6 (Cl), 17.2 (Br), and 15.6 (I) kcal mol−1. The lower barriers for NNBe than those for NtBuNtBuBe are attributed to the higher steric hindrance of the TS 1.2a in the NtBuNtBuBe species. For the NNBe compound, we carried out an IBO analysis along the reaction coordinate for the Ph–F bond activation (Fig. S4†). In NNBe-FPh (2.1a), the ring redistributes electron density from the C–C π bond to the antibonding Ph–F and results in 2.3a (Fig. S4a†). In this way, the Be atom does not change its oxidation state. It is the ligand that promotes the oxidative addition of Ph–F by becoming oxidized. The ligand participates in the redox process as a redox non-innocent ligand that behaves as an electron reservoir.73,74 During the process, the ligand loses its aromaticity (see Table S2†).
Through the TS 2.4a, which corresponds to the phenyl group migration from Be to N (with ΔG‡ = 18.5 (F), 22.1 (Cl), 23.0 (Br), and 24.6 (I) kcal mol−1), we got the final product 2.5a. The reaction energies are more exergonic for F than for Cl, Br, and I. To pass from 2.3a to 2.5a, the N–C π bond donates electron density to the Be–Ph σ* antibonding orbital that, as a result, is weakened, favoring the formation of the N–Ph bond (Fig. S4b†).
The energetic spans, which are determined by the intermediate 2.1a and the TS 2.2a for F and the intermediate 2.3a and the TS 2.4a for Cl, Br, and I, are 23.6 (F), 22.1 (Cl), 23.0 (Br), and 24.6 (I). The change of the substituent tBu by Me favors the reaction by decreasing the Gibbs energy barriers and increasing the exergonicity of the reaction.
The next mechanism is for the NPBe reaction (Fig. 4). In an exergonic step the reaction starts with the formation of the intermediate 3.1a (favorable in the following order: I > F > Cl > Br). Through the TS 3.2a, that corresponds to the oxidative addition of the Ph–X bond (with ΔG‡ = 23.4 (F), 23.1 (Cl), 21.4 (Br) and 20.4 (I) kcal mol−1), we got the product 3.4a (except for F, for which after the TS we got 3.3a and then 3.4a in a barrierless process). An alternative pathway involves crossing the TS 3.2b that describes the phenyl group migration from the halogen to nitrogen (with ΔG‡ = 28.6 (F), 32.7 (Cl), and 31.4 (Br) and ΔE‡ of 25.3 (F), 31.1 (Cl), 43.0 (Br) and 43.3 (I) kcal mol−1), through which we got the product 3.4b. The difference between 3.4a and 3.4b is the position of the phenyl group, on P or N, respectively. The Gibbs reaction energies are favorable in both cases but it is more for 3.4a than for 3.4.b (in the two reactions the exergonicity follows the order: F > Cl > Br > I). The reason for the higher stability of 3.4a than that of 3.4b is attributed to the greater capability of the phosphine group to share electron density in comparison to that of the amine (phosphorus is softer and less electronegative than nitrogen).75 Also, the delocalization of the N electron pair over the ring stabilizes the product while the electron pair of P cannot be delocalized.
 | ||
| Fig. 4 Reaction mechanism determined at the ωB97X-D/def2-TZVP(SMD:toluene)//ωB97X-D/def2-SVPP level. X = F (green), Cl (pink), Br (brown), and I (purple). | ||
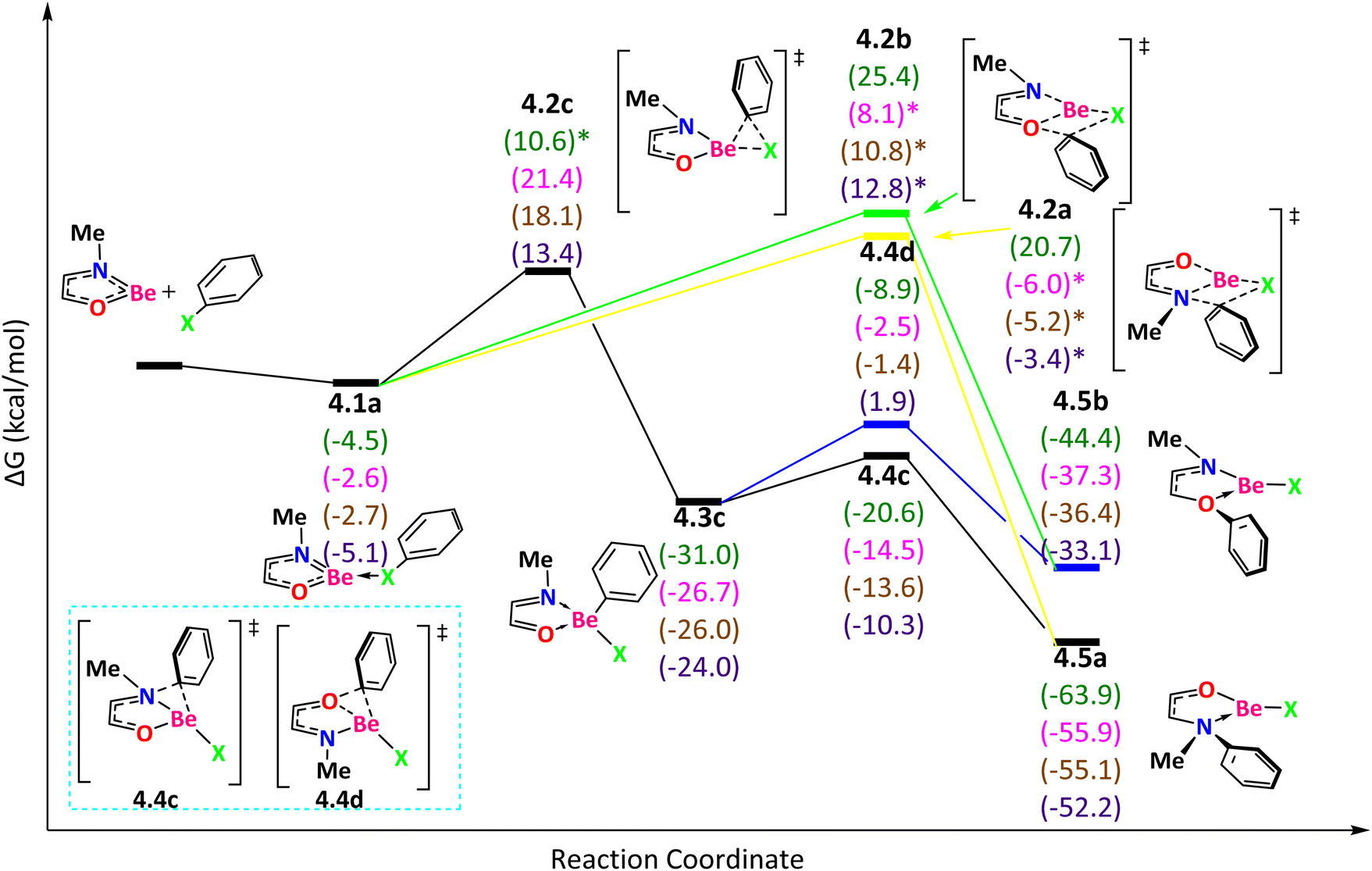
When Y contains an element of group 16 (O or S), we found additional paths that are not present in the reaction mechanisms discussed above. For NOBe (Fig. 5), the reaction starts with an exergonic step to form 4.1a, favorable in the following order: I > F > Br ∼ Cl. If the reaction proceeds through the TS 4.2a, with ΔG‡ = 25.2 (F) (ΔE‡ = 24.0 (F), 9.1 (Cl), 9.5 (Br), and 14.0 (I) kcal mol−1), the phenyl group migrates from the halogen to N and the product 4.5a is obtained. We have explored alternative pathways but all of them were found to be higher in energy. For instance, if the reaction goes through the TS 4.2b, the phenyl group migrates from the halogen to O to generate 4.5b with ΔG‡ = 29.9 (F) (ΔE‡ = 29.8 (F), 23.2 (Cl), 25.6 (Br), and 30.2 (I) kcal mol−1). Now, if the reaction passes through the TS 4.2c, with ΔG‡ = 24.0 (Cl), 20.8 (Br), and 18.5 (I) kcal mol−1 (ΔE‡ = 27.6 (F), 24.0 (Cl), 20.5 (Br), 18.9 (I) kcal mol−1), the phenyl group migrates from the halogen to Be producing the tetracoordinate beryllium compound 4.3c with the following exergonicity order: F > Cl > Br > I. From that intermediate, if the phenyl group migrates from Be to N (the TS 4.4c with ΔG‡ = 10.4 (F), 12.2 (Cl), 12.4 (Br), and 13.7 (I) kcal mol−1), the product is 4.5a. The other TS 4.4d (with ΔG‡ = 22.1 (F), 24.2 (Cl), 24.6 (Br) and 25.9 (I) kcal mol−1), that describes the phenyl group migration from Be to O, leads to the product 4.5b. The product 4.5a is thermodynamically more stable than 4.5b. In conclusion, the expected main product for all halogens is 4.5a obtained by surmounting the TS 4.2a.
 | ||
| Fig. 5 Reaction mechanism determined at the ωB97X-D/def2-TZVP(SMD:toluene)//ωB97X-D/def2-SVPP level. X = F (green), Cl (pink), Br (brown), and I (purple). | ||
For the last bidentate ligand, NSBe (Fig. 6), the reaction starts with an exergonic step to form 5.1a, favorable in the following order: I > F > Cl > Br. If the reaction passes through the TS 5.2a, with ΔG‡ = 29.2 (F) and 32.8 (Cl) (ΔE‡ = 27.6 (F), 30.7 (Cl), 14.8 (Br), and 17.4 (I) kcal mol−1), the phenyl group migrates from the halogen to N and the product 5.5a is obtained. If the reaction goes through the TS 5.2b, with ΔG‡ = 29.7 (F) (ΔE‡ = 29.4 (F), 24.2 (Cl), 21.5 (Br) and 21.8 (I) kcal mol−1), the phenyl group migrates from the halogen to S and the product 5.5b is generated. Now, if the reaction passes through TS 5.2c, with ΔG‡ = 30.6 (Cl), 26.6 (Br) and 25.1 (I) kcal mol−1, the phenyl group migrates from the halogen to Be and the tetracoordinate beryllium compound 5.3c is produced with the following exergonicity order: Cl > Br > I. In the case of F, all the attempts to locate 5.2c ended in 5.2a. Therefore, we conclude that the route through 5.2c is not possible for Ph–F activation. From the intermediate 5.3c, if the phenyl group migrates from Be to N (TS 5.4d with ΔG‡ = 10.7 (Cl), 11.1 (Br) and 13.0 (I) kcal mol−1), the product is 5.5a. The other TS 5.4c (with ΔG‡ = 0.8 (Cl), 1.4 (Br) and 3.1 (I) kcal mol−1), that describes the phenyl group migration from Be to S, leads to the product 5.5b. The product 5.5a is more favorable than 5.5b.
 | ||
| Fig. 6 Reaction mechanism determined at the ωB97X-D/def2-TZVP(SMD:toluene)//ωB97X-D/def2-SVPP level. F (green), Cl (pink), Br (brown), and I (purple). The dashed lines indicate that no intermediate or TS exists for that halogen. | ||
In all cases, during the first step corresponding to the oxidative addition, the aromaticity of the five-membered heterocyclic ring is lost (Table S2†). As said before, the loss of aromatic stabilization energy when going from reactants to x.2a is higher in NSBe (11.9 kcal mol−1) than in NOBe (5.7 kcal mol−1) or NNBe (3.8 kcal mol−1). Interestingly, for systems with similar steric hindrance such as NSBE, NOBe and NNBe, the five-membered heterocyclic rings with higher aromatic stabilization energy in the reactant are those having the larger Gibbs energy barriers for the oxidative addition (Table S4†), as expected from the fact that during the oxidative addition the five-membered heterocyclic ring loses its aromaticity.
In general, when the TS describes the Ph–X bond activation (where Ph is bonded to Be, N, P, O, or S), the activation energy is highest for X = F and it is due to the higher strength of the Ph–F bond (see Table S3†). The higher the Ph–X bonding energy, the higher the energy barrier is. For the same reason (stronger Be–F bonds), usually the case X = F provides the most exergonic processes (see Table S3†). Other reaction paths involving bicyclic intermediates were tested but they were found to be highly endergonic and they were not further analyzed (Fig. S5†).
Finally, the (CAAC)2Be system was studied (Fig. 7). The reaction starts with a concerted transition state that describes the Ph–X bond activation with ΔG‡ = 49.2 (F), 53.4 (Cl), 53.2 (Br) and 53.0 (I) kcal mol−1. We found high Gibbs energy barriers in this process despite lack of aromaticity in (CAAC)2Be. These high energy barries are due to the fact that the (CAAC)2Be system must deform significantly by decreasing the ∠CBeC angle to provide space for the interaction of the LUMO of (CAAC)2Be with the HOMO of the Ph–X molecule (Fig. S6†). The deformation energy values are 14.1 (F), 15.1 (Cl), 15.7 (Br), and 16.7 (I) kcal mol−1. Once 6.1a is surmounted, formation of 6.2a is highly exergonic with ΔG‡ = 14.9 (F), 16.3 (Cl), 16.9 (Br) and 17.1 (I) kcal mol−1 (6.3a), and the phenyl group in 6.2a migrates from the Be to the carbenic carbon to generate 6.4a. The Gibbs reaction energies are exergonic in the following order: F > Cl > Br > I.
 | ||
| Fig. 7 Reaction mechanism determined at the ωB97X-D/def2-TZVP(SMD:toluene)//ωB97X-D/def2-SVPP level. X = F (green), Cl (pink), Br (brown), and I (purple). | ||
Conclusions
The reaction mechanisms of the phenyl–halogen bond activation using beryllium compounds were studied. The analysed beryllium five-membered rings have a closed-shell singlet ground state, whereas the (CAAC)2Be species has an open-shell singlet as the ground state. The beryllium five-membered ring exhibits aromaticity due to the delocalization of electrons in the cycle. According to the energy profiles, the activation of Ph–X bonds involves exergonic reactions, favorable in the following order: NPBe > NNBe > NOBe > NtBuNtBuBe > NSBe > CAAC2Be, as the halogen becomes heavier. In many cases, the rate determining step is the oxidative addition that occurs in the first step of the reaction. In general, the higher the aromatic stabilization energy of the five-membered heterocyclic ring, the greater the energy barrier for the oxidative addition. This process also benefits from the reduction of the steric hindrance of the compounds. Interestingly, Be(II) retains the oxidation state along the whole reaction path and the electrons needed for the oxidative addition are given by the ligands that have a key role in the reaction. In this sense, the metallomimetic character of the beryllium complexes is more provided by the ligands than by the element itself. For the (CAAC)2Be species, the Gibbs energy barriers are high because of the need to distort the reactants to allow the interaction with the Ph–X species.Data availability
The data that support the findings of this study are available in the ESI† of this article.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
M. S. thanks the Spanish Ministerio de Ciencia e Innovación (project PID2020-13711GB-I00) and the Generalitat de Catalunya (project 2021SGR623). D. E. T. G., G. G.-G., and J. O. C. J.-H. acknowledge the facilities of the DCNyE, the Chemistry Department and the National Laboratory UG-CONACyHT (LACAPFEM) at the University of Guanajuato. D. E. T. G. thanks CONACyHT for a PhD fellowship (822937), DAIP and IQCC for the computational time in beta cluster and the fellowship provided by the GRCT090 group.References
- M. R. Buchner and L. R. Thomas-Hargreaves, Dalton Trans., 2021, 50, 16916–16922 RSC.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- C. Weetman, Chem. – Eur. J., 2021, 27, 1941–1954 CrossRef CAS PubMed.
- M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231–8261 CrossRef PubMed.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- K. A. Walsh and E. E. Vidal, Beryllium Chemistry and Processing, ASM International, Materials Park, Ohio, 2009 Search PubMed.
- R. Puchta, Nat. Chem., 2011, 3, 416–416 CrossRef CAS PubMed.
- M. R. Buchner, Chem. – Eur. J., 2019, 25, 12018–12036 CrossRef CAS PubMed.
- Beryllium: Mineralogy, Petrology, and Geochemistry, ed. E. S. Grew, Mineralogical Society Of America, Washington, DC, 2002 Search PubMed.
- D. Naglav, M. R. Buchner, G. Bendt, F. Kraus and S. Schulz, Angew. Chem., Int. Ed., 2016, 55, 10562–10576 CrossRef CAS PubMed.
- K. J. Iversen, S. A. Couchman, D. J. D. Wilson and J. L. Dutton, Coord. Chem. Rev., 2015, 297, 40–48 CrossRef.
- J. L. Dutton and G. Frenking, Angew. Chem., Int. Ed., 2016, 55, 13380–13382 CrossRef CAS PubMed.
- P. Parameswaran and G. Frenking, J. Phys. Chem. A, 2010, 114, 8529–8535 CrossRef CAS PubMed.
- D. E. Trujillo-González, G. González–García, T. A. Hamlin, F. M. Bickelhaupt, H. Braunschweig, J. O. C. Jiménez–Halla and M. Solà, Eur. J. Inorg. Chem., 2023, 26, e202200767 CrossRef.
- G. Frenking, N. Holzmann, B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2010, 636, 1772–1775 CrossRef CAS.
- W. Petz, K. Dehnicke, N. Holzmann, G. Frenking and B. Neumüller, Z. Anorg. Allg. Chem., 2011, 637, 1702–1710 CrossRef CAS.
- M. R. Buchner, M. Müller and S. S. Rudel, Angew. Chem., Int. Ed., 2017, 56, 1130–1134 CrossRef CAS PubMed.
- A. Paparo and C. Jones, Chem. – Asian J., 2019, 14, 486–490 CrossRef CAS PubMed.
- M. Arrowsmith, H. Braunschweig, M. A. Celik, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, T. Kramer, I. Krummenacher, J. Mies, K. Radacki and J. K. Schuster, Nat. Chem., 2016, 8, 890–894 CrossRef CAS PubMed.
- A. Paparo, C. D. Smith and C. Jones, Angew. Chem., Int. Ed., 2019, 58, 11459–11463 CrossRef CAS PubMed.
- M. R. Buchner, D. Ćoćić, S. I. Ivlev, N. Spang, M. Müller and R. Puchta, Dalton Trans., 2023, 52, 5287–5296 RSC.
- A. Paparo, A. J. R. Matthews, C. D. Smith, A. J. Edwards, K. Yuvaraj and C. Jones, Dalton Trans., 2021, 50, 7604–7609 RSC.
- M. Arrowsmith, M. S. Hill, G. Kociok-Köhn, D. J. MacDougall and M. F. Mahon, Angew. Chem., Int. Ed., 2012, 51, 2098–2100 CrossRef CAS PubMed.
- M. R. Buchner, N. Spang, M. Müller and S. S. Rudel, Inorg. Chem., 2018, 57, 11314–11317 CrossRef CAS PubMed.
- K. G. Pearce, M. S. Hill and M. F. Mahon, Chem. Commun., 2023, 59, 1453–1456 RSC.
- T. J. Hadlington and T. Szilvási, Nat. Commun., 2022, 13, 461 CrossRef CAS PubMed.
- M. A. Keane, Appl. Catal., A, 2004, 271, 109–118 CrossRef CAS.
- K. V. Murthy, P. M. Patterson and M. A. Keane, J. Mol. Catal. A: Chem., 2005, 225, 149–160 CrossRef CAS.
- V. V. Grushin and H. Alper, Chem. Rev., 1994, 94, 1047–1062 CrossRef CAS.
- K. Matsubara, K. Ueno, Y. Koga and K. Hara, J. Org. Chem., 2007, 72, 5069–5076 CrossRef CAS PubMed.
- C.-W. Lee, E.-C. Liu and Y.-T. Wu, J. Org. Chem., 2015, 80, 10446–10456 CrossRef CAS PubMed.
- R. N. Dhital, K. Bobuatong, M. Ehara and H. Sakurai, Chem. – Asian J., 2015, 10, 2669–2676 CrossRef CAS PubMed.
- B. P. Moloto, P. Vermeeren, M. Dalla Tiezza, C. Esterhuysen, F. M. Bickelhaupt and T. A. Hamlin, Eur. J. Org. Chem., 2022, e202200722 CrossRef CAS.
- M. Wiesinger, B. Rösch, C. Knüpfer, J. Mai, J. Langer and S. Harder, Eur. J. Inorg. Chem., 2021, 2021, 3731–3741 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J.-D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- T. A. Keith, AIMAll (Version 19.10.12), TK Gristmill Software, Overland Park KS, USA, 2019 (https://aim.tkgristmill.com) Search PubMed.
- M. Giambiagi, M. Segre De Giambiagi, C. D. Dos Santos Silva and A. Paiva De Figueiredo, Phys. Chem. Chem. Phys., 2000, 2, 3381–3392 RSC.
- P. Bultinck, R. Ponec and S. Van Damme, J. Phys. Org. Chem., 2005, 18, 706–718 CrossRef CAS.
- F. Feixas, E. Matito, J. Poater and M. Solà, Chem. Soc. Rev., 2015, 44, 6434–6451 RSC.
- E. Matito, M. Solà, P. Salvador and M. Duran, Faraday Discuss., 2007, 135, 325–345 RSC.
- E. Matito, M. Duran and M. Solà, J. Chem. Phys., 2005, 122, 014109 CrossRef PubMed; Erratum, íbid., 2005, 125, 059901 Search PubMed.
- E. Matito, ESI-3D: Electron Sharing Indices Program for 3D Molecular Space Partitioning, Institute of Computational chemistry and Catalysis (IQCC), University of Girona, Catalonia, Spain, 2006; https://iqcc.udg.es/~eduard/ESI (accessed 12th July 2023) Search PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. Fonseca Guerra, J. G. Snijders, G. te Velde and E. J. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. R. Johnson and A. D. Becke, J. Phys. Chem., 2005, 123, 024101 CrossRef PubMed.
- P. Vermeeren, S. C. C. van der Lubbe, C. Fonseca Guerra, F. M. Bickelhaupt and T. A. Hamlin, Nat. Protoc., 2020, 15, 649–667 CrossRef CAS PubMed.
- M. Bickelhaupt, J. Comput. Chem., 1999, 20, 114–128 CrossRef.
- W.-J. van Zeist and F. M. Bickelhaupt, Org. Biomol. Chem., 2010, 8, 3118 RSC.
- F. M. Bickelhaupt and E. J. Baerends, in Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistry, ed. K. B. Lipkowitz and D. B. Boyd, Wiley-VCH, New York, 1999, pp. 199–212 Search PubMed.
- T. A. Hamlin, P. Vermeeren, C. Fonseca Guerra and F. M. Bickelhaupt, in Complementary Bonding Analysis, ed. S. Grabowsky, De Gruyter, 2021, pp. 199–212 Search PubMed.
- M. Gimferrer, S. Danés, E. Vos, C. B. Yildiz, I. Corral, A. Jana, P. Salvador and D. M. Andrada, Chem. Sci., 2022, 13, 6583–6591 RSC.
- S. Parambath, J. Narayanan S. J. and P. Parameswaran, Dalton Trans., 2023, 52, 3378–3385 RSC.
- H. Jacobsen, J. Organomet. Chem., 2005, 690, 6068–6078 CrossRef CAS.
- W. W. Schoeller and D. Eisner, Inorg. Chem., 2004, 43, 2585–2589 CrossRef CAS PubMed.
- E. Ramos-Cordoba, V. Postils and P. Salvador, J. Chem. Theory Comput., 2015, 11, 1501–1508 CrossRef CAS PubMed.
- C. R. Landis, R. P. Hughes and F. Weinhold, Organometallics, 2015, 34, 3442–3449 CrossRef CAS.
- P. Jerabek, P. Schwerdtfeger and G. Frenking, J. Comput. Chem., 2019, 40, 247–264 CrossRef CAS PubMed.
- M. Solà, A. I. Boldyrev, M. K. Cyrański, T. M. Krygowski and G. Merino, Aromaticity and Antiaromaticity: Concepts and Applications, John Wiley & Sons, Ltd, Chichester, West Sussex, 2023 Search PubMed.
- J. Cioslowski, E. Matito and M. Solà, J. Phys. Chem. A, 2007, 111, 6521–6525 CrossRef CAS PubMed.
- P. V. R. Schleyer and F. Pühlhofer, Org. Lett., 2002, 4, 2873–2876 CrossRef CAS PubMed.
- M. K. Cyrański, Chem. Rev., 2005, 105, 3773–3811 CrossRef PubMed.
- S. Abdalla, M. Springborg and Y. Dong, Surf. Sci., 2013, 608, 255–264 CrossRef CAS.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- V. Lyaskovskyy and B. De Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- L. A. Berben, B. De Bruin and A. F. Heyduk, Chem. Commun., 2015, 51, 1553–1554 RSC.
- H. Anane, S. El Houssame, A. El Guerraze, A. Jarid, A. Boutalib, I. Nebot-Gil and F. Tomás, J. Mol. Struct. (THEOCHEM), 2004, 709, 103–107 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt02251j |
| This journal is © The Royal Society of Chemistry 2023 |