
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe inhibitory effects of platinum(II) complexes on amyloid aggregation: a theoretical and experimental approach†
Sara
La Manna
a,
Valentina
Roviello
b,
Vittoria
Monaco
cd,
James A.
Platts
e,
Maria
Monti
cd,
Elisabetta
Gabano
 f,
Mauro
Ravera
g and
Daniela
Marasco
*a
f,
Mauro
Ravera
g and
Daniela
Marasco
*a
aDepartment of Pharmacy, University of Naples Federico II, 80131, Naples, Italy. E-mail: daniela.marasco@unina.it; Tel: +39-081-2532043
bDepartment of Chemical, Materials, and Industrial Production Engineering (DICMaPI), University of Naples Federico II, 80125 Naples, Italy
cDepartment of Chemical Sciences, University of Naples Federico II, 80126, Naples, Italy
dCEINGE Biotecnologie Avanzate “Franco Salvatore” S.c.a r.l., 80131, Naples, Italy
eSchool of Chemistry, Cardiff University, Park Place, Cardiff, CF10 3AT, UK
fDipartimento per lo Sviluppo Sostenibile e la Transizione Ecologica, University of Piemonte Orientale, Piazza S. Eusebio 5, 13100, Vercelli, Italy
gDepartment of Sciences and Technological Innovation, University of Piemonte Orientale, Viale Michel 11, 15121 Alessandria, Italy
First published on 25th August 2023
Abstract
Platinum (Pt)(II) square planar complexes are well-known anticancer drugs whose Mechanism of Action (MOA) are finely tuned by the polar, hydrophobic and aromatic features of the ligands. In the attempt to translate this tunability to the identification of potential neurodrugs, herein, four Pt(II) complexes were investigated in their ability to modulate the self-aggregation processes of two amyloidogenic models: Sup35p7–13 and NPM1264–277 peptides. In particular, phenanthriplatin revealed the most efficient agent in the modulation of amyloid aggregation: through several biophysical assays, as Thioflavin T (ThT), electrospray ionization mass spectrometry (ESI-MS) and ultraviolet-visible (UV-vis) absorption spectroscopy, this complex revealed able to markedly suppress aggregation and to disassemble small soluble aggregates. This effect was due to a direct coordination of phenanthriplatin to the amyloid, with the loss of several ligands and different stoichiometries, by the formation of π–π and π–cation interactions as indicated from molecular dynamic simulations. Presented data support a growing and recent approach concerning the repurposing of metallodrugs as potential novel neurotherapeutics.
Introduction
Transition metal coordination compounds have unique and distinctive features including variable oxidation states, diverse range of geometries, coordination numbers and ligands.1 A vast library of metal complexes was designed and explored over many years for diverse therapeutic applications in the field of cancer, microbial and viral infections, diabetes and neurodegenerative diseases (NDDs)2 and many studies outlined that coordination chemistry plays a crucial role in the definition of the mechanisms of action (MOAs) of metallodrugs. MOAs generally depend on both the binding preferences of transition metals for biomolecules and the regulation of ligands exchanges with functional groups coming from proteins and oligonucleotides.3 In spite of deep clinical diversity, NDDs share several events as the cellular accumulation of intrinsically disordered proteins and the dysregulation of the homeostasis of several metal ions in the brain, and, till now, no definite cures are available for these disorders, even if considerable advances in understanding the molecular mechanisms at the basis of these diseases and in the development of therapeutics have been carried out.4The self- or hetero-assembly of amyloidogenic systems, as the Aβ-amyloid (Amyloid beta (Aβ or Abeta)), α-synuclein, huntingtin, tau and islet amyloid polypeptides, to form oligomers and fibrils is directly linked to NDDs as Alzheimer's, Parkinson's, and Huntington's diseases, frontotemporal dementia and type II diabetes.5 Often the structural details of the oligomers/fibers formed by full-length proteins are elusive and very difficult to study, while, the investigations of the self-assemblies of protein fragments, as model, allow to deepen structural, biophysical and biological properties of in vivo amyloids.5
In the context of drug discovery processes in early steps of amyloid aggregation, metallodrugs can be employed. After the pioneering study of Barnham et al.6 many Pt complexes were investigated for their inhibitory properties of amyloid aggregation:7 examples can be traced back to complexes containing heteroaromatic ligands such as pyridine or its derivatives,8 imidazole,9 thiazole, pyrazoles, quinoline and isoquinoline, tetrazoles and triazoles.10 In detail, phenanthroline(phen)-Pt(II) complexes with two monodentate ligands exhibited inhibitory effects toward the aggregation of Aβ1–4011 and prion protein (PrP) fragments.12 The inhibition mechanism depends on multiple factors as the coordination of the metal center, the 1st and 2nd electrostatic spheres around the ion, hydrogen bond networks and van der Waals interactions. Examples of multifactorial amyloid inhibition are several Co-13 and Pt-compounds14 bearing polyaromatic ligands: they demonstrated reduction of Aβ aggregation through a coordinative mechanisms aided by the formation of π–π stacking interactions with aromatic side chains.15 Two glycoconjugate pentacoordinate Pt-complexes were analyzed in their capacity to affect the self-aggregation processes of two fragments of the C-terminal region of Aβ-peptide, Aβ21–40 and Aβ25–35. The water-soluble complex, named 1Ptdep, inhibited the aggregates through the direct binding to Aβ-peptides, drastically reducing the morphological amyloid features of fibers.16
On the basis of these promising results, herein we investigated the ability of four Pt(II) complexes to modulate the aggregation of two amyloid models, NPM1264–277 and Sup35p7–13, whose sequences are reported in Table 1. In the screening of agents able to modulate amyloid aggregation, often protein/peptide, even if not directly involved in neurodegeneration, are employed as models of amyloids. This is the case of Nucleophosmin 1 (NPM1), which is not an amyloid protein strictly speaking, but presents an amyloid-prone fragment, including 264–277 residues.17 In the recent past, we tested the ability of several metal-based CORM (carbon monoxide releasing molecules) to modulate the amyloid aggregation NPM1264–277.18,19 Conversely, the heptapetide GNNQQNY, spanning residues 7–13 of the Yeast Prion Protein Sup35p (Sup35p7–13), is directly involved in the aggregation of Sup35p since it is located in the prion-determining N-terminal domain (PrD),20 which demonstrated able to form amyloid fibrils.21,22
| Peptide | Sequence |
|---|---|
| NPM1264–277 | Ac-VEAKFINYVKNCFR-NH2 |
| Sup35p7–13 | Ac-GNNQQNY-NH2 |
In a recent study, we have investigated the ability of a series of square planar Pt(II)-complexes to inhibit the aggregation of amyloid peptides. The pyridine-based cationic, [PtCl(tpy)]+, called Pt-terpy, exhibited good inhibitory effects and the ability to reduce the cytotoxicity of amyloid in human SH-SY5Y neuroblastoma cells.23 To deepen the MOAs of similar compounds, in the present study we investigated the effects of square planar Pt(II) complexes differing in their charge: positive for two complexes and neutral for other two compounds. In detail, we analyzed the (SP-4-3)-diamminechlorido (quinoline)platinum(II) nitrate (quinoplatin, 1) and (SP-4-3)-diamminechlorido(phenanthridine)platinum(II) nitrate (phenanthriplatin, 2)24 and the enantiomeric (SP-4-2)-dichlorido(1,1′-binaphthalene-2,2′-diamine)platinum(II), 3R and 3S, respectively (Fig. 1). These complexes contain a chiral ligand (i.e., 1,1′-binaphthalene-2,2′-diamine, DABN) and the R isomer demonstrated to interact with the G-quadruplex structure AG3(TTAGGG)3 less efficiently than its S counterpart.25–27
 | ||
| Fig. 1 Structure of the Pt(II) complexes investigated in the present work; DABN = 1,1′-binaphthalene-2,2′-diamine. | ||
The ability of Pt(II) complexes to interfere with the aggregation of amyloid models was investigated via a range of spectroscopic and biophysical techniques as wells as by molecular dynamic (MD) studies.
Experimental section
Peptide synthesis
Peptides were synthesized as already reported.19 both in the acetylated and amidated form. The sequences are reported in Table 1, after purification they were lyophilized and treated with HFIP to ensure a monomeric state and then stored at −20 °C until use.Synthesis of complexes
Complexes 1–3 were synthesized according to literature procedures. Briefly, (SP-4-2)-diamminedichloridoplatinum(II), cisplatin, was reacted with AgNO3 in DMF to remove one chloride ion, then the N-heterocyclic ligand was added in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio and the reaction was stirred for 16 h at 55 °C. The following purification steps gave 1 and 2 as white solids (yield: 70% and 59% for 1 and 2, respectively).26 Complexes 3R and 3S were synthesized directly by reacting equimolar quantities of K2PtCl4 and the R- or S-isomers of 1,1′-binaphthalene-2,2′-diamine (DABN) in DMF at 40 °C in the dark overnight. The following purification steps gave 3R and 3S as yellow solids (yield: 78% and 72% for the R and S-isomers counterparts, respectively).26 The stabilities of Pt complexes were already reported in ref. 25–27
:1 molar ratio and the reaction was stirred for 16 h at 55 °C. The following purification steps gave 1 and 2 as white solids (yield: 70% and 59% for 1 and 2, respectively).26 Complexes 3R and 3S were synthesized directly by reacting equimolar quantities of K2PtCl4 and the R- or S-isomers of 1,1′-binaphthalene-2,2′-diamine (DABN) in DMF at 40 °C in the dark overnight. The following purification steps gave 3R and 3S as yellow solids (yield: 78% and 72% for the R and S-isomers counterparts, respectively).26 The stabilities of Pt complexes were already reported in ref. 25–27
Fluorescence assays
ThT assay was carried out on fluorescence reader Envision 2105 (PerkinElmer) in black plates (96 well) under stirring. Measurements were collected every 2 min (λex 440 nm and λem 483 nm). Assays were performed at 25 °C employing a peptide concentration of 100 μM for NPM1264–277 and 400 μM for Sup357–13 in 50 mM borate (pH 8.5) and 50 mM phosphate buffer (pH 7.4) respectively, for both a ThT final concentration of 50 μM. Pt-complexes stock solutions (50 mM in water for 1, 10 mM in water for 2 and 50 mM in DMSO for 3R and 3S) were opportunely diluted to obtain employed ratios with respect to amyloid peptides. Disaggregation assays were performed on a Jasco FP 8300 spectrofluorometer (JASCO, Tokyo, Japan) in a 1 cm cuvette under magnetic stirring (λex = 440 nm, λem = 483 nm). Spectra were recorded every 15 min and assays were performed in duplicates, and intensities were reported as averaged values and transformed as percentages of aggregated fraction. Indicating Fmax as the maximum reached fluorescence signal and n is a cooperativity parameter t1/2 value is the time at which F is equal to one-half of Fmax through the fitting of the data of F versus time through the empirical Hill equation as follows:
UV-vis absorption spectroscopy
UV/vis titrations of Pt-complexes with NPM1264–277 were carried out employing a BioDrop Duo UV Visible Spectrophotometers (Cambridge, United Kingdom). To a fixed concentration of the Pt(II) complexes (50 μM). Repeated additions of 1.25 μL of NPM1264–277 stock solution (2 mM) in water, kept at 0 °C, were performed till reaching a ratio of 1:2.5 complex:NPM1264–277. Spectra were registered in the 250–500 nm range, upon each addition. For 2, EC50 value was derived from non-linear regression of the data employing log [inhibitor] vs. response and “dose–response stimulation equation” of GraphPad program.
Circular dichroism
CD spectra of NPM1264–277 (100 μM) in 10 mM borate buffer pH 8.5, alone or at 1:1 molar ratio with metal complexes, were registered on a Jasco J-815 spectropolarimeter (JASCO, Tokyo, Japan), at 25 °C using a 0.1 cm path-length quartz cuvette. Deconvolutions of CD spectra were obtained by BESTSEL software (https://bestsel.elte.hu/).
ESI-MS analysis
NPM1264–277 at 5 μM in 15 mM AMAC (Ammonium acetate) buffer pH = 7.0, was incubated at two different times (0 and 4 h) with 1:5 ratios with 2 compound and the obtained mixtures were analyzed by native ESI-MS on a Q-ToF Premier (Waters, Milliford, MA, USA) mass spectrometer. The analyses were carried out by direct injection at 10 μL min−1, setting the source parameters at 3 kV for capillary voltage and 42 kV for cone voltage. The acquisition range was scanned from 100 to 2000 m/z in 1 s and the raw data were processed with MassLynx 4.1 software (Waters, Milliford, MA, USA). Fosforic acid solution at 50% (v/v) in acetonitrile was used for the instrument calibration.
Scanning electron microscopy
Samples (100 μL) containing NPM1264–277 (100 μM) alone or mixed with Pt-complexes at a 1:5 ratio, at two different times of aggregation (t 2 h for 3S and 4 h for 1 and 2), were dropped on stubs and introduced into chamber of field emission scanning (Nova NanoSem 450 FEI/ThermoFisher Scientific), to obtain SEM micrographs at 3.00 and 5.00 kV in high vacuum mode, with an Everhart Thornley Detector (ETD) and the Through the Lens Detector (TLD).
Computational methods
The structure of NPM1264–277 was built in extended (φ = ψ = 180°) form and capped with amide groups. Its geometry was optimised at GFN2-xTB level in implicit model of water, in the xtb package. Potential Pt binding sites were explored by manually attaching 2 to different polar atoms in backbone and side chains, followed by an optimisation at the same level. Optimized geometries were imported into Amber1628 using the tleap utility, assigning parameters for the ff14SB29 forcefield in GBSA implicit model of aqueous solvation. For Pt-bound peptide, parameters for metal, ligands and bound Cys were obtained using the MCPB.py30 approach, in which bonded parameters were extracted from DFT calculations using the Seminario method,31 while non-bonded parameters were taken from RESP calculations. These were combined with ff14SB for proper comparison with metal-free simulations, and key data are reported in the Table S2.† All systems were minimised with 1000 steps of steepest descent. Three independent MD trajectories were then generated, first by heating to 300 K over 1 ns with a further 1 ns equilibration, both with positional restraints on all atoms, then with 100 ns of unrestrained MD. RMSD data (ESI Fig. S1†) indicate that stable trajectories were established within a few ns. Analysis of all 300 ns of trajectory data was performed using cpptraj,25 concentrating on secondary structure and clustering: the latter using a k-means approach with 10 clusters.
Results and discussion
Pt(II) complexes interfere with amyloid aggregation
To analyze the effects of Pt-compounds on the self-recognition process of amyloid systems, Thioflavin T (ThT) binding assay was employed;32 and both inhibitory (Fig. 2) and disaggregating (Fig. 3) experiments were carried out. The time courses of the ThT fluorescence of NPM1264–277 and Sup35p7–13 peptides alone and incubated with complexes are reported in Fig. 2; two peptide:Pt-complex molar ratios were analyzed, 1:1 and 1:5. For NPM1264–277, in the absence of complexes, the starting value of fluorescence was different from 0 for an immediate partial oligomerization during sample preparation17 (Fig. 2A and B) and the time intervals required to reach saturated ThT signal appeared fast, ∼10 min, while a slight delay is observed, ∼1.5 h, for the presence of DMSO (Fig. 2C and D) that is the control condition required for compounds 3R and 3S. From the comparison of time-course profiles, all complexes were able to inhibit the aggregation of NPM1264–277 even if to different extents: between the charged complexes, the most effective inhibitor was found to be complex 2, since it inhibited NPM1264–277 aggregation of ∼50% at 1:1 and ∼70% at 1:5 ratio (Fig. 2B), whereas the inhibitory effect of 1 was significantly less at both ratios (Fig. 2A). Unlike 1 and 2, no difference in the inhibition effects of 3R and 3S was detected that was of ∼50%; for both compounds in both ratios (Fig. 2C and D).
 | ||
| Fig. 2 Inhibitory effects of Pt-complexes on amyloid aggregation. Time course of ThT fluorescence emission intensity, reported as % of aggregated fraction, of NPM1264–277 (upper panel) and Sup35p7–13 (lower panel) upon incubation with: (A and E) 1; (B and F) 2; (C) 3S; (D) 3R at 1:1 and 1:5 peptide to Pt compound ratios. | ||
 | ||
| Fig. 3 Disaggregation effects of Pt-complexes 1–3. Percentage of aggregated fraction, for NPM1264–277 (A) and Sup35p7–13 (B) before and after the addition of Pt complexes 1–3 at 1:5 ratio. The addition of Pt-complexes is indicated by an arrow. | ||
By comparing aggregation times of NPM1264–277 and Sup35p7–13 peptides alone, great differences are observable: indeed in the case of Sup35p7–13, the time of the half reduction of the signal in the amyloid growth phase, named t½, was evaluated of ∼2.5 h (Fig. 2E and F). Noticeably the sudden decrease of folded fraction after ∼2.5 h is likely due formation of less soluble species. Thus, differently from NPM1264–277, these features hamper performance of inhibitory assays with 3R and 3S complexes which are stable for only two hours.26 Consequently, the effects of only complexes 1 and 2 preincubated with Sup35p7–13 were evaluated. Compound 1 appeared almost ineffective while 2 showed an inhibition of ∼75% (Fig. 2E and F), at both molar ratios. No interference signals with ThT were observed for all complexes for the entire durations of the analyses.
Having assessed the different abilities of metal complexes to suppress amyloid aggregation, we evaluated if they are also able to disaggregate soluble pre-formed oligomers,33 monitoring the ThT signals after the addition of metal compounds to NPM1264–277 and Sup35p7–13 aggregates at 1:5 ratio (Fig. 3). On the basis of different aggregation kinetics (see above), Sup35p7–13 was pre-aggregated in the absence of the complexes for 3 hours, while NPM1264–277, which is partially aggregated at t = 0, was allowed to further aggregate for 10 min before starting experiments. Upon the addition of Pt-complexes to both amyloids, a decrease in ThT fluorescence intensity was observed with a greater effect of 2 with respect to 1. Indeed, while the addition of 1 caused a disaggregating effect on NPM1264–277 and Sup35p7–13 of 30% and 60%, respectively, 2 showed a stronger effect by inducing a reduction of 40% and 70%, respectively. Also the neutral complexes 3R and 3S showed a strong disaggregating capacity that led to a reduction of ThT signal of ∼60% for NPM1264–277 and 45% for Sup35p7–13.
On the basis of similar effects of Pt-complexes observed on NPM1264–277 and Sup35p7–13, we focused only on NPM1264–277, since its longer sequence with respect to the heptapeptide Sup35p7–13, makes it more sensitive in subsequent conformational studies. ThT results indicated similar inhibitory effects provided similar inhibitory effects, hence we continued with compounds 1 and 2 which demonstrated more stable during time.26
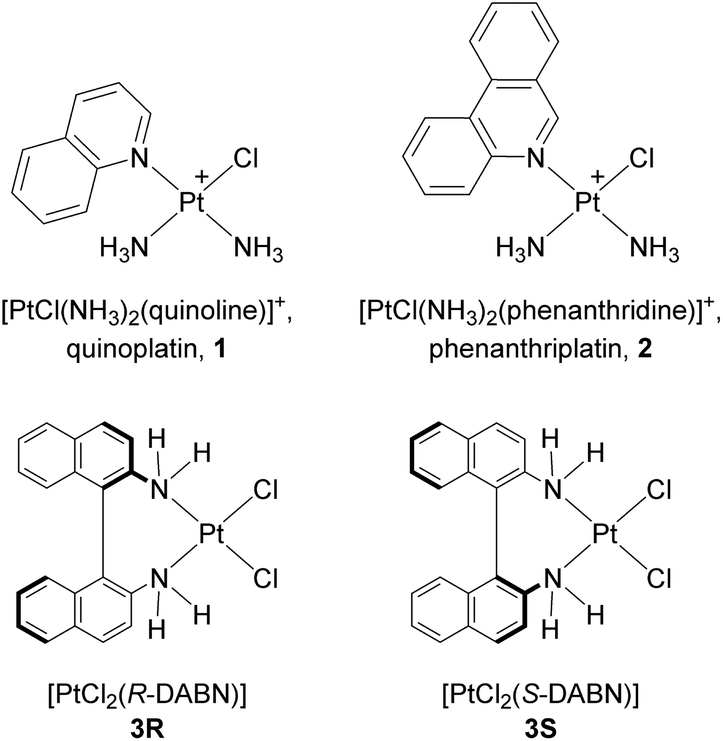
To assess whether observed inhibitory effects of Pt-complexes could be accompanied by variations of ligands’ field around metal center, changes in the UV-vis absorption spectra34–37 were evaluated, upon the addition of NPM1264–277 to aqueous solutions of 1 and 2 at a fixed concentration. These experiments, reported in Fig. 4, confirmed different behaviors for 1 and 2. In fact, the titration with NPM1264–277 of 2 (Fig. 4B) exhibited marked variations of intensity of several absorption bands. In detail the LMCT band at 311 nm (ref. 37) provided saturated values of absorbance at 1:2.5 complex:peptide ratio and an estimation of EC50 (half-maximal effective concentration) of 26 ± 3 μM (inset of Fig. 4B). On the contrary, the same experiments carried out for 1 did not show significant variations of absorbances at 309 nm (Fig. 4A).
 | ||
| Fig. 4 UV-vis titration of compound 1 and 2 with NPM1264–277. Absorption spectra of 1 (A) and 2 (B) upon the addition of increasing amount of NPM1264–277. Arrows indicate the variation of the ratio complex:peptide. As insets UV intensities at indicated wavelengths versus log concentration of NPM1264–277 are reported. | ||
Charged Pt-complexes stabilize the β-conformation of NPM1264–277
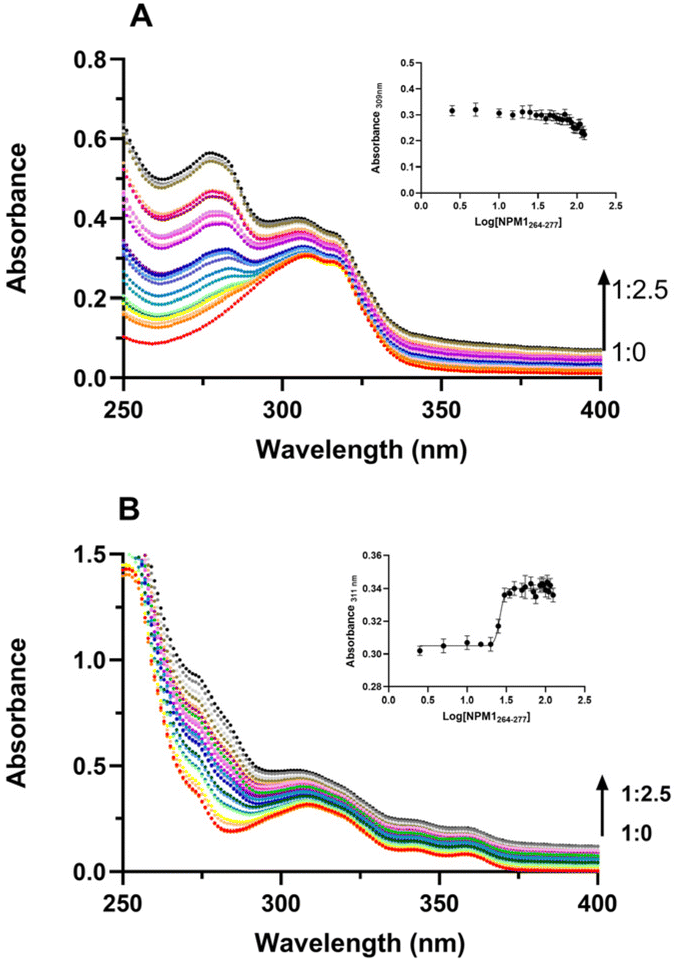
To gain insights into effects of Pt-complexes on the NPM1264–277 conformation, circular dichroism (CD) investigations were carried out. Importantly, the presence of DMSO, even in traces, greatly affects the analysis, and this feature, in addition to the short stabilities of 3R, S, limited the CD experiments to water-soluble 1 and 2 complexes, only. The overlays of CD spectra of NPM1264–277 alone and in the presence of metal complexes, at 1:1 ratio, recorded over a period of 12 h, are reported in Fig. 5. For NPM1264–277 alone, a clear transition from a mixed α-helix and β-structure (Fig. 5A) toward a prevalent β-conformation is observable, as well as a decrease of Cotton effect due to aggregation, as previously reported.38 The presence of 2 complex, already at t = 0 h, stabilized β-conformation leading to the highest β content at 3 h of analysis (Fig. 5C), as confirmed by deconvolution percentages of spectra reported in Table S1.† On the contrary, the sample of NPM1264–277 + 1 exhibited a time-evolution of CD profile more similar to that of the peptide alone (Fig. 5B).
 | ||
| Fig. 5 Overlay of CD spectra of NPM1264–277 (A) alone and incubated with 1 (B) and 2 (C), at 1:1 peptide: Pt(II) compound molar ratio. | ||
Effects of Pt complexes on the morphology of NPM1264–277 fiber
To get insights into the effects of Pt-complexes on the morphology of the fibers derived from NPM1264–277, scanning electron microscopy (SEM) experiments were carried out. Fibers of NPM1264–277 in a previous SEM analysis, exhibited ∼20 μm in diameter and ∼1.3 mm in length.38 Herein, compounds 1, 2 and 3S were added to freshly prepared solutions of NPM1264–277 and after 2 h (for 3S) and 4 h (for 1 and 2) of incubation, the aggregates were analyzed. Compared to peptide alone, both the length and diameter of the aggregates were perturbed in the presence of metal complexes, as shown in Table 2. In detail, the presence of 2 caused a reduction of the diameter of the fiber (from ∼20 μm in NPM1264–277 alone to 8 μm; Fig. 6A–A′′ and Table 2), while the addition of 3S caused a marked reduction of the length dimension (from ∼1.3 mm to ∼0.5 mm; Fig. 6B–B′′ and Table 2), besides to a slight thinning effect. On the contrary, the presence of 1 did not cause significant changes in the fiber, confirming its less modulating effect on the amyloid aggregation (Fig. S2†). | ||
| Fig. 6 Effects of Pt-complexes on amyloid fibers. SEM micrographs of (A–A′′) NPM1264–277 + 2, (B–B′′) NPM1264–277 + 3S at 1:5 ratio, at (A and B) 1 mm, (A′ and B′) 50 μm and (A′′ and B′′) 5 μm. | ||
Charged Pt-complex directly interact with NPM1264–277
On the basis of the greater effect exhibited by 2 with respect to 1 on the aggregation of NPM1-amyloid, the last series of experiments was performed only on 2 complex. NPM1264–277 was incubated with 2 at two different times (0 h and 4 h, respectively) and the resulting mixtures were analyzed by electrospray ionization mass spectrometry (ESI-MS) (Fig. 7At = 0 h; Fig. 7Bt = 4 h). In the presence of 2, the spectra showed the occurrence of peaks due to adducts between one peptide chain and one (#1) or two metal complexes (#2, #3; Table 3). In detail, the peak #1 (m/z 1080.483 u) showed a species corresponding to the adduct with a 1:1 stoichiometry, in which the Pt-complex lost one Cl− and one NH3 ligand. Instead, peaks #2 and #3 are attributed to adducts with a 1:2 stoichiometry, peptide: 2: #2 is related to the adduct among one peptide chain and two 2 moieties lacking 3 × NH3, while #3 corresponds to the #2 carrying one additional acetate ion, which is present in the buffer. As already reported,19 the presence of Cys275, allow the formation of dimeric forms of the amyloid peptide, already at t = 0.
 | ||
| Fig. 7 2 compound interacts with NPM1264–277. ESI-MS spectra of NPM1264–277 peptide incubated with 2 compound at (A) t = 0 h, and (B) t = 4 h. | ||
| Experimental conditions | Experimental m/z | Charge | Theoretical m/z | Description | |
|---|---|---|---|---|---|
| NPM1264–277 + 2, 0 h | 1080.483 | 2 | 1081.63 | NPM1264–277 + 2–1Cl−–1 NH3 | #1 |
| 1305.039 | 2 | 1305.26 | NPM1264–277 + 2 2–3 NH3 | #2 | |
| 1335.087 | 2 | 1335.773 | #2 + acetate | #3 | |
| 1180.955 | 3 | 1180.931 | NPM1264–277 covalent dimer | ||
| NPM1264–277 + 2, 4 h | 1080.483 | 2 | 1081.63 | NPM1264–277 + 2–1Cl−–1 NH3 | #1 |
| 1305.039 | 2 | 1305.26 | NPM1264–277 + 2 2–3 NH3 | #2 | |
| 1335.087 | 2 | 1335.773 | #2 + acetate | #3 | |
| 1180.955 | 3 | 1180.931 | NPM1264–277 covalent dimer | ||
Modelling studies of interaction complex 2 with NPM1264–277
With the aim of identifying the most favorable metal-binding modes between NPM1264–277 and the Pt complexes computational modelling studies were performed. Initial tests at the semi-empirical GFN2-xTB level39 indicate that compound 2 binds preferentially to the deprotonated thiol side chain of Cys275, with a binding energy more stable of ∼30 kJ mol−1 than any other available site. Possible cation–π interactions between the extended π-system of the ligand with Lys273 and Arg277 were evident. However, the flexible nature of the peptide means that such interactions may not persist, so we used molecular dynamics (MD) simulations to predict the conformations of the peptide with and without 2 bound. Over the course of 3 independent 100 ns simulations, metal-free monomeric NPM1264–277 folds quickly and consistently into α-helical fold (Fig. S2 and S3†). Helices start to form within 10 ns and persist over the course of each simulation, albeit with some fluctuations to partially folded forms. Clustering procedure finds that the most populated cluster is a fully-folded α-helix, consisting of 19% of all frames recorded (Fig. 8A), and other clusters consisting of partially-folded helices (14 and 11%), all in agreement with the native secondary and tertiary structures of NPM1 protein.40 Conformations adopted by a non-covalent dimer of NPM1264–277 strongly depend on starting conformation: in parallel or crossed construction no clear secondary structure forms, and in some cases, the dimer dissociates. However, when constructed in antiparallel form from extended monomer conformation, MD simulation, quickly and consistently, brings to β-strand conformations (Fig. 8B).41 These strands form almost instantly and encompass the entire length of each peptide chain, this self-interaction is at the basis of observed amyloid aggregation of the sequence NPM1264–277.16 | ||
| Fig. 8 Most populated clusters from 3 independent 100 ns simulations. (A) metal-free monomer; (B) metal-free non-covalent anti-parallel dimer; (C) monomer peptide:2; (D) dimer peptide:2 adducts. Monomer is colored from N-terminus (blue) to C-terminus (red), while dimers are colored according to secondary structure (yellow = β-strand, blue = turn). | ||
The presence of 2 bound to Cys275 drastically changes the conformational preferences of both monomer and dimer. The Pt-bound monomer (Fig. 8C) exhibits almost no helical character indeed the dictionary of protein secondary structure (DSSP) algorithm identifying only turn and bend elements42 suggesting that the presence of the large Pt complex disrupts the helical organization of the isolated peptide. Similarly, the anti-parallel dimer appears strongly inhibited by the binding of 2 (Fig. 8D). In this case, some recorded frames contain β-strands, but these are smaller and shorter-lived than in the metal-free case. The maximum strand content is 22% of all recorded frames and is found for Phe268 of chain A and Phe276 of chain B. Closer analysis indicates the occurrence of several non-covalent interactions between 2 and the peptide chain in addition to the coordination bond to Cys275. These connections include C–H⋯π interactions between Lys and the aromatic ligand of 2, as well as hydrogen bonds from ammine ligands to backbone nitrogens of Val, Glu and Arg. The lack of a precise agreement between the ESI† data and the MD data is due to different phases, gas and aqueous buffer, and ratios of peptide to 2 complex.
Conclusions
Platinum-based therapeutics are widely used in a variety of chemotherapy regimens and studies aimed to deepen their MOAs demonstrated that some of them are able to induce ribosome biogenesis stress pathway. In detail phenanthriplatin, which is a monofunctional Pt(II) compound, demonstrated the ability to relocalize NPM143 indicating that the spatial orientation and/or hydrophobicity of aromatic ring around metal ion can finely tune the cytotoxic effects of this important class of anticancer agents.44 Much more recent is the interest in the ability of Pt-anticancers to interact with amyloid proteins and peptides45 mainly through a coordinative mechanism often with concomitant interactions as electrostatic attraction, hydrogen bonding and van der Waals force46 as well as π–π stacking among amino acids side chains and planar aromatic ligands. In the present study we have investigated the abilities of several Pt-complexes to impede fibril formation of amyloidogenic peptides reported in Table 1. Although the interaction between Pt complexes and amyloids is relatively weak47 the coordination of these compounds to peptides remarkably changes the aggregation behavior of the amyloid peptides and among studied Pt-complexes phenanthriplatin, compound 2 (Fig. 1), demonstrated the most active. We propose that this difference may be due to a greater π-stacking contribution to the inhibitory mechanism of NPM1264–277 exerted by 2, which bears a phenanthridine ligand. Indeed, the aromatic core constituted by Phe268,276 and Tyr271 residues in NPM1 plays a crucial role in the self-aggregation mechanism of this amyloid model.41 ThT-time courses, registered both upon the addition of the Pt compounds to freshly prepared samples or to soluble amyloid aggregates, indicated that 2 is very efficient to inhibit the fibrillation and to alter the particle sizes of peptide aggregates as clearly demonstrated, by means of SEM, on the morphology of aggregates. Other investigations demonstrated that the ligand configuration is a crucial factor in modulatory effect on amyloid fibrillation, which severely affects the binding patterns and sites of Pt-inhibitors. The MS analysis provided evidences for the formation of different types of adducts, all implying the binding of compound 2 to NPM1264–277 through a common ligands’ release process, with the retention of phenanthroline and the loss of the chlorine and ammonia, even if with different stoichiometries. CD studies indicated that 2 increased and stabilized the β-content of the amyloid peptide in early stages of aggregation and MD simulations indicated in the Cys residue the most involved in the coordination of metal center, with additional π–cation interactions and that the binding to 2 strongly affects the conformation of the peptide chain.In conclusion this research provides critical information on the inhibition and disaggregation of amyloid fibrillation by phenanthroline-based metal complexes with impact on the biomedical value of clinical platinum drugs against amyloid diseases.
Author contributions
S. L. M. synthesized the peptide and performed fluorescence, UV and CD studies; V. M. and M. M. performed ESI-experiments;† V. R. did SEM assays; M. R. and E. G. synthesized and characterized the metal complexes; J. A. P. performed MD experiments; D. M. designed the concept, supervised the experiments and wrote the manuscript. All authors have read and approved the final version of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC) grant IG 2022, Rif. 27378 (D. M.) and by #NEXTGENERATIONEU (NGEU), Ministry of University and Research (MUR), National Recovery and Resilience Plan (NRRP), project MNESYS (PE0000006) – A Multiscale integrated approach to the study of the nervous system in health and disease (DN. 1553 11.10.2022).References
- G. Son, B. I. Lee, Y. J. Chung and C. B. Park, Acta Biomater., 2018, 67, 147–155 CrossRef CAS PubMed.
- I. Yousuf, M. Bashir, F. Arjmand and S. Tabassum, Coord. Chem. Rev., 2021, 445, 214104 CrossRef CAS.
- M. A. Telpoukhovskaia and C. Orvig, Chem. Soc. Rev., 2013, 42, 1836–1846 RSC.
- C. Soto and S. Pritzkow, Nat. Neurosci., 2018, 21, 1332–1340 CrossRef CAS PubMed.
- T. D. Samdin, A. G. Kreutzer and J. S. Nowick, Curr. Opin. Chem. Biol., 2021, 64, 106–115 CrossRef CAS PubMed.
- K. J. Barnham, V. B. Kenche, G. D. Ciccotosto, D. P. Smith, D. J. Tew, X. Liu, K. Perez, G. A. Cranston, T. J. Johanssen and I. Volitakis, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6813–6818 CrossRef CAS PubMed.
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- M. Van Beusichem and N. Farrell, Inorg. Chem., 1992, 31, 634–639 CrossRef CAS.
- F. Gumus, G. k. e. Eren, L. Açık, A. Celebi, F. Ozturk, S. u. k. Yılmaz, R. I. Sagkan, S. Gur, A. Ozkul and A. Elmalı, J. Med. Chem., 2009, 52, 1345–1357 CrossRef CAS PubMed.
- N. A. Al-Masoudi, B. H. Abdullah, A. H. Essa, R. Loddo and P. LaColla, Arch. Pharm., 2010, 343, 222–227 CAS.
- I. Sasaki, C. Bijani, S. Ladeira, V. Bourdon, P. Faller and C. Hureau, Dalton Trans., 2012, 41, 6404–6407 RSC.
- F. Collin, I. Sasaki, H. Eury, P. Faller and C. Hureau, Chem. Commun., 2013, 49, 2130–2132 RSC.
- T. G. Chan, C. L. Ruehl, S. V. Morse, M. Simon, V. Rakers, H. Watts, F. A. Aprile, J. J. Choi and R. Vilar, Chem. Sci., 2021, 12, 9485–9493 RSC.
- G. Gong, J. Xu, X. Huang and W. Du, JBIC, J. Biol. Inorg. Chem., 2019, 24, 179–189 CrossRef CAS PubMed.
- R. Cukalevski, B. Boland, B. Frohm, E. Thulin, D. Walsh and S. Linse, ACS Chem. Neurosci., 2012, 3, 1008–1016 CrossRef CAS PubMed.
- S. La Manna, M. Leone, I. Iacobucci, A. Annuziata, C. Di Natale, E. Lagreca, A. M. Malfitano, F. Ruffo, A. Merlino, M. Monti and D. Marasco, Inorg Chem, 2022, 61, 3540–3552 CrossRef CAS PubMed.
- C. Di Natale, P. L. Scognamiglio, R. Cascella, C. Cecchi, A. Russo, M. Leone, A. Penco, A. Relini, L. Federici, A. Di Matteo, F. Chiti, L. Vitagliano and D. Marasco, FASEB J., 2015, 29, 3689–3701 CrossRef CAS PubMed.
- D. Florio, M. Cuomo, I. Iacobucci, G. Ferraro, A. M. Mansour, M. Monti, A. Merlino and D. Marasco, Pharmaceuticals, 2020, 13, 171 CrossRef CAS PubMed.
- S. La Manna, V. Roviello, F. Napolitano, A. M. Malfitano, V. Monaco, A. Merlino, M. Monti, K. Kowalski, Ł. Szczupak and D. Marasco, Inorg. Chem., 2023, 62, 10470–10480 CrossRef CAS PubMed.
- M. M. Patino, J.-J. Liu, J. R. Glover and S. Lindquist, Science, 1996, 273, 622–626 CrossRef CAS PubMed.
- J. R. Glover, A. S. Kowal, E. C. Schirmer, M. M. Patino, J.-J. Liu and S. Lindquist, Cell, 1997, 89, 811–819 CrossRef CAS PubMed.
- M. Balbirnie, R. Grothe and D. S. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2375–2380 CrossRef CAS PubMed.
- S. La Manna, D. Florio, I. Iacobucci, F. Napolitano, I. D. Benedictis, A. M. Malfitano, M. Monti, M. Ravera, E. Gabano and D. Marasco, Int. J. Mol. Sci., 2021, 22, 3015 CrossRef CAS PubMed.
- G. Y. Park, J. J. Wilson, Y. Song and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11987–11992 CrossRef CAS PubMed.
- D. R. Roe and T. E. Cheatham III, J. Chem. Theory Comput., 2013, 9, 3084–3095 CrossRef CAS PubMed.
- S. Bombard, M. B. Gariboldi, E. Monti, E. Gabano, L. Gaviglio, M. Ravera and D. Osella, JBIC, J. Biol. Inorg. Chem., 2010, 15, 841–850 CrossRef CAS PubMed.
- E. Gabano, S. Gama, F. Mendes, M. B. Gariboldi, E. Monti, S. Bombard, S. Bianco and M. Ravera, JBIC, J. Biol. Inorg. Chem., 2013, 18, 791–801 CrossRef CAS PubMed.
- D. Case, D. Cerutti, T. Cheatham, T. Darden, R. Duke, T. Giese, H. Gohlke, A. Goetz, D. Greene and N. Homeyer, AMBER 2016, University of California, San Francisco Search PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- P. Li and K. M. Merz Jr., J. Chem. Inf. Model., 2016, 599–604 CrossRef CAS PubMed.
- J. M. Seminario, Int. J. Quantum Chem., 1996, 60, 1271–1277 CrossRef.
- C. Di Natale, S. La Manna, A. M. Malfitano, S. Di Somma, D. Florio, P. L. Scognamiglio, E. Novellino, P. A. Netti and D. Marasco, Biochim. Biophys. Acta, Proteins Proteomics, 2019, 1867, 637–644 CrossRef CAS PubMed.
- T. Tanaka, V. V. Betkekar, K. Ohmori, K. Suzuki and H. Shigemori, Pharmaceuticals, 2021, 14, 1118 CrossRef CAS PubMed.
- G. Arena, G. Calogero, S. Campagna, L. Monsù Scolaro, V. Ricevuto and R. Romeo, Inorg. Chem., 1998, 37, 2763–2769 CrossRef CAS PubMed.
- E. L. McInnes, R. Farley, C. Rowlands, A. Welch and L. Yellowlees, J. Chem. Soc., Dalton Trans., 1999, 4203–4208 RSC.
- V. X. Jin and J. D. Ranford, Inorg. Chim. Acta, 2000, 304, 38–44 CrossRef CAS.
- M. Imran, Z. ur Rehman, G. Hogarth, D. A. Tocher, I. S. Butler, F. Bélanger-Gariepy and T. Kondratyuk, Dalton Trans., 2020, 49, 15385–15396 RSC.
- D. Florio, V. Roviello, S. La Manna, F. Napolitano, A. M. Malfitano and D. Marasco, Bioorg. Chem., 2022, 127, 106001 CrossRef CAS PubMed.
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed.
- C. G. Grummitt, F. M. Townsley, C. M. Johnson, A. J. Warren and M. Bycroft, J. Biol. Chem., 2008, 283, 23326–23332 CrossRef CAS PubMed.
- A. Russo, C. Diaferia, S. La Manna, C. Giannini, T. Sibillano, A. Accardo, G. Morelli, E. Novellino and D. Marasco, Biochim. Biophys. Acta, Proteins Proteomics, 2017, 1865, 176–185 CrossRef CAS PubMed.
- W. G. Touw, C. Baakman, J. Black, T. A. Te Beek, E. Krieger, R. P. Joosten and G. Vriend, Nucleic Acids Res., 2015, 43, D364–D368 CrossRef CAS PubMed.
- C. E. McDevitt, M. V. Yglesias, A. M. Mroz, E. C. Sutton, M. C. Yang, C. H. Hendon and V. J. DeRose, JBIC, J. Biol. Inorg. Chem., 2019, 24, 899–908 CrossRef CAS PubMed.
- Z. Deng and G. Zhu, Curr. Opin. Chem. Biol., 2023, 74, 102303 CrossRef CAS PubMed.
- T. Zheng, Y. Huo, Y. Wang and W. Du, J. Inorg. Biochem., 2022, 237, 111989 CrossRef CAS PubMed.
- J. Zhao, K. Li, K. Wan, T. Sun, N. Zheng, F. Zhu, J. Ma, J. Jiao, T. Li and J. Ni, Angew. Chem., Int. Ed., 2019, 58, 18032–18039 CrossRef CAS PubMed.
- G. Ma, E. Wang, H. Wei, K. Wei, P. Zhu and Y. Liu, Metallomics, 2013, 5, 879–887 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM micrographs of NPM1264–277 + 1, at 1:5 ratio, at 1 mm, and 300 μm; time evolution of RMSD relative to initial extended structure; time evolution of secondary structure in metal-free monomer and dimer MD simulation; deconvolution of CD spectra; bonded parameters from MCPB.py for 2 bound to Cys12. See DOI: https://doi.org/10.1039/d3dt02187d |
| This journal is © The Royal Society of Chemistry 2023 |