
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Robust and efficient transfer hydrogenation of carbonyl compounds catalyzed by NN-Mn(I) complexes†
Zheng
Wang
 *abc,
Ning
Ma
a,
Xiaochi
Lu
a,
Ming
Liu
b,
Tian
Liu
b,
Qingbin
Liu
*c,
Gregory A.
Solan
*bd and
Wen-Hua
Sun
*b
*abc,
Ning
Ma
a,
Xiaochi
Lu
a,
Ming
Liu
b,
Tian
Liu
b,
Qingbin
Liu
*c,
Gregory A.
Solan
*bd and
Wen-Hua
Sun
*b
aCollege of Science, Hebei Agricultural University, Baoding 071001, China
bKey Laboratory of Engineering Plastics and Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: wangzheng@iccas.ac.cn; whsun@iccas.ac.cn
cHebei Key Laboratory of Organic Functional Molecules, College of Chemistry and Material Science, Hebei Normal University, Shijiazhuang 050024, China. E-mail: liuqingb@sina.com
dDepartment of Chemistry, University of Leicester, University Road, Leicester LE1 7RH, UK. E-mail: gas8@leicester.ac.uk
First published on 11th July 2023
Abstract
A series of manganese(I) carbonyl complexes bearing structurally related NN- and NNN-chelating ligands have been synthesized and assessed as catalysts for transfer hydrogenation (TH). Notably, the NN-systems based on N-R functionalized 5,6,7,8-tetrahydroquinoline-8-amines, proved the most effective in the manganese-promoted conversion of acetophenone to 1-phenylethanol. In particular, the N-isopropyl derivative, Mn1, when conducted in combination with t-BuONa, was the standout performer mediating not only the reduction of acetophenone but also a range of carbonyl substrates including (hetero)aromatic-, aliphatic- and cycloalkyl-containing ketones and aldehydes with especially high values of TON (up to 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200; TOF of 3550 h−1). These findings, obtained through a systematic variation of the N-R group of the NN ligand, are consistent with an outer-sphere mechanism for the hydrogen transfer. As a more general point, this Mn-based catalytic TH protocol offers an attractive and sustainable alternative for producing alcoholic products from carbonyl substrates.
200; TOF of 3550 h−1). These findings, obtained through a systematic variation of the N-R group of the NN ligand, are consistent with an outer-sphere mechanism for the hydrogen transfer. As a more general point, this Mn-based catalytic TH protocol offers an attractive and sustainable alternative for producing alcoholic products from carbonyl substrates.
Introduction
The conversion of ketones and aldehydes to their corresponding alcohols constitutes one of the most fundamental reactions and is central to the synthesis of numerous important derivatives such as fragrances, flavors, and fine-chemical intermediates as well as pharmaceuticals.1 Indeed, a wide range of approaches have been applied over the years to realize this transformation.1a Conventionally, stoichiometric amounts of metal hydrides (e.g. NaBH4 or LiAlH4) or boranes are employed (Scheme 1a), but these reagents can be problematic on account of their hazardous or toxic properties and moreover can afford large amounts of noxious by-products.1,2 As an alternative to these stoichiometric approaches, catalytic transfer hydrogenation (TH) protocols offer an attractive and sustainable strategy.1b,c,3 To this end, precious metal-based catalysts based on Ru,4 Rh,5 Ir,6 or Os7 have paved the way (Scheme 1b). However, the high cost of some of these complexes and their limited availability of their metal centers poses problems with their long-term application.3b,8 Therefore, there is a considerable impetus to develop catalysts based on inexpensive and earth-abundant 3d transition metals in combination with robust and synthetically accessible ligand frameworks.9 As a consequence, a great deal of reports have emerged on the development of iron-10 and cobalt-11 based catalysts for TH (Scheme 1b), whereas the use of manganese, the third most abundant metal in the Earth's crust, is rather more limited. Indeed, manganese has traditionally been considered as a metal for promoting oxidation reactions.12 | ||
| Scheme 1 Strategies for the reduction of ketones or aldehydes to give alcohols. | ||
Nonetheless, the last six years or so have seen some key advances in Mn-catalyzed TH, particularly with regard to the conversion of ketones to secondary alcohols.13–15 In 2017, Beller first showed that dipicolylamine-manganese NNN-pincer complexes can promote the reduction of ketones using isopropanol as the hydrogen source.13a Later, Sortais's group reported the use of Mn(I) complexes bearing bidentate 2-picolylamine ligands13b that could facilitate turnover frequencies (TOF) as high as 3600 h−1. Since then, much attention has been dedicated to developing simple air-stable bidentate ligand systems that can facilitate the TH process.13c–i However, most of these NN-chelated Mn(I)-complexes, including aminotriazole,13c benzimidazole13d or bipyridine ligands,13e,f require high catalyst loading (0.2–3.0 mol%) to achieve good conversions.13c–g Elsewhere, Pidko demonstrated that the replacement of one N-donor unit in the NN-ligand by an N-heterocyclic carbene resulted in a significant improvement in the Mn-catalyzed TH of ketones. Indeed, an outstanding TON of 17300 was achieved using 25 ppm of this NHC–Mn(I) complex in the reduction of acetophenone.13i As a key observation, the reaction efficiency was found to be strongly dependent on temperature especially for low catalyst loadings where higher temperatures resulted in faster catalyst deactivation.
Up until now, several challenges remain to be confronted before earth-abundant transition metal catalytic systems can be practically utilized in (industrial) organic synthesis. For example, when compared to their noble metal counterparts, catalyst loadings remain up to four orders of magnitude higher at several thousands of ppm (i.e. 0.1–1.0 mol%).12 Moreover, in order to reduce operational costs, it is desirable to replace phosphine-based ligands that are a feature of precious metal catalysts for simpler and more scalable alternatives.
As part of our ongoing program, ligand design has been at the cornerstone of our efforts in developing transition metal complexes that can promote transformations such as ethylene polymerization16 and the (de)hydrogenation of biomass-carbonyl compounds17,18 for the production of fine chemicals and new materials. In particular, we have found the 8-amino-5,6,7,8-tetrahydroquinoline and 8-imino-5,6,7-trihydroquinoline skeletons as useful structural motifs that can be used as NN-chelating ligands or derivatized to expand the denticity of the coordinating ligand.19,20 Based on this design strategy, we herein disclose five new NN-manganese(I) carbonyl complexes bearing either an N-R substituted (R = i-Pr, n-Bu, Bn, Ph) 5,6,7,8-tetrahydroquinoline-8-amine (Scheme 1c) or a N-NHPh-substituted 8-imino-5,6,7-trihydroquinoline; a related NNN-manganese(I) complex incorporating an N-ethyl-1H-benzimazole-functionalized 5,6,7,8-tetrahydroquinoline-8-amine is also reported. All six complexes are then explored initially as precatalysts for the TH of acetophenone to determine their relative effectiveness before extending the most promising system to the TH of a wide range of alkyl- and aryl-containing ketones and aldehydes. Besides the in-depth catalytic evaluation, full details are presented for the synthesis of both the ligands, complexes and attempts to characterize intermediates involved in the catalysis.
Results and discussion
Synthesis and characterization of manganese(I) complexes (Mn1–Mn6)
Treatment of Mn(CO)5Br with N-R-8-N(H)C9H10N (R = i-Pr L1, n-Bu L2, Bn L3, Ph L4) or imine-containing 8-(PhHN-N)C9H9N (L5) in tetrahydrofuran at 60 °C for 12 h gave on work-up, [(NN)Mn(CO)3]Br [NN = L1 (Mn1), L2 (Mn2), L3 (Mn3), L4 (Mn4), L5 (Mn5)], in good yield (Scheme 2). On the other hand, the preparation of the N-ethyl-1H-benzimazole-functionalized [(NNN)Mn(CO)2]Br (Mn6) required more forcing conditions involving the reaction of N-((CH2)2CHN2HC6H4)-8-N(H)C9H10N (L6) with Mn(CO)5Br in toluene at 110 °C for 24 h. The ligands themselves, L1–L6, were synthesized in good overall yield via the interaction of 5,6,7-trihydroquinolin-8-one7b with the corresponding amine under reductive amination (L1–L4, L6) or condensation (L5) conditions (see ESI†).18,19 All six manganese complexes were found to be stable under atmospheric conditions in the solid state but slowly decomposed under exposure to light. Complex characterization was achieved using 1H/13C NMR, FT-IR spectroscopy and elemental analysis (Fig. S1–S18, see ESI†). | ||
| Scheme 2 Synthetic routes to manganese(I) complexes, Mn1–Mn6. | ||
In the 13C NMR spectra of Mn1–Mn6, the metal-carbonyls were seen as three low intensity downfield signals in the range δ 218–223 ppm.13–15,18 By contrast, three strong absorption peaks for the CO groups were visible between 1904 to 2027 cm−1 in their IR spectra (Table 1). Notably the average carbonyl stretching frequencies for Mn1–Mn4 follow the order Mn4 (1960) > Mn3 (1953) > Mn2 (1950) > Mn1 (1948), which support the donating properties of the N-R substituent being: i-Pr > n-Bu > Bn > Ph. The elemental analyses of all six complexes were in support of the proposed compositions.
| Complex | Chemical shift of the Mn-COsa (ppm) | CO stretching frequenciesb (cm−1) |
|---|---|---|
| a 13C NMR spectra were recorded in DMSO-d6. b FT-IR spectra have been recorded in the solid state. | ||
| Mn1 | 222.2, 222.8, 223.0 | 1906, 1922, 2017 |
| Mn2 | 222.0, 222.5, 223.1 | 1901, 1926, 2023 |
| Mn3 | 220.0, 220.8, 222.4 | 1904, 1936, 2019 |
| Mn4 | 220.8, 222.6, 223.2 | 1907, 1949, 2023 |
| Mn5 | 219.0, 221.3, 223.3 | 1919, 1942, 2027 |
| Mn6 | 218.9, 219.8, 220.2 | 1912, 1930, 2027 |
Crystals of Mn1 as well as the bromide salt of L1, [L1H]Br, suitable for single crystal X-ray determinations were grown by the slow diffusion of hexane into a dichloromethane solution of the corresponding compound. ORTEP views of the resulting structures are shown in Fig. 1, while selected bond distances and angles for Mn1 are given in the caption. Inspection of the structure of Mn1 reveals the manganese center to be surrounded by an NN-chelating L1, three carbonyl ligands and a terminal bromide so as to complete a distorted octahedral coordination geometry. The CO ligands are disposed in a fac arrangement with the two nitrogen donors belonging to L1 and bromide occupying the trans sites. Some variation in the Mn–N distances was evident with that involving the tetrahydroquinoline nitrogen slightly shorter than that for the amine nitrogen [Mn–N1 2.046(2) Å vs. Mn–N2 2.116(2) Å] in line with the better donor properties of the former. Similar observations have been noted with structurally related NNN- and NNS-manganese complexes (see Mn7 and Mn10, Fig. 2),18 though the variation was less distinct. Within the carbocyclic section of the tetrahydroquinoline, some puckering of the ring is observed on account of the sp3-hybridized carbons, C5, C6, C5 and C8. In turn this leads to some deviation of the N–C–C–N coordination plane from planarity [N1–C9–C8–N2 torsion = −30.8°]. There were no intermolecular contacts of note.
 | ||
| Fig. 1 ORTEP representations of [L1H]Br (left) and Mn1 (right) with the thermal ellipsoids shown at the 30% probability level; the hydrogen atoms have been omitted for clarity. Selected bond distances (Å) for Mn1: Br1–Mn1 2.5275(4), Mn1–N1 2.046(2), Mn–N2 2.116(2); Selected bond angles (°) for Mn-1: N1–Mn1–Br 86.42(6), N1–Mn1–N2 78.74(8), C13–Mn1–Br1 178.75(8), C13–Mn1–N1 92.79(10), C13–Mn1–C14 90.76(12), C13–Mn1–N2 95.38(10), N2–Mn1–Br1 85.43(6). | ||
 | ||
| Fig. 2 Evaluation of Mn1–Mn10 as catalysts for transfer hydrogenation of acetophenone (a1). Conditions: 5.0 mmol a1, 10 μmol (0.2 mol%) [Mn], 0.5 mmol t-BuOK (10 mol%), 5 mL i-PrOH, 90 °C (oil bath temperature), 2 h. The conversions were determined by GC with dodecane as the internal standard. | ||
Catalytic studies
:[Mn]:t-BuOK set at 5:0.01:0.5; the % conversions to 1-phenylethanol (b1) over 2 h are collected in Fig. 2.
Pleasingly, all ten manganese complexes, under these conditions stipulated, proved to be active catalysts for the reduction of a1 with conversions to b1 in the range of 41% to 97%. With respect to the NN-chelated manganese complexes Mn1–Mn5, the relative level of catalytic performance fell in the order: Mn1 > Mn2 > Mn3 > Mn5 > Mn4. Evidently, the nature of the N-R substituent has some effect on the catalytic performance with i-Pr-containing Mn1 the most productive; imine-containing Mn5 was notably at the lower end of the conversion range (69% conversion). The reason behind the enhanced performance for Mn1 is uncertain but may be due to the N-i-Pr group providing the most suitable steric protection to inhibit deactivation pathways at high temperature.13i By comparison, the N-Ph-containing Mn4 showed much lower conversions (58%) than seen with Mn1–Mn3 and Mn5. This finding could plausibly be due to the rigidity of the N-Ph group leading to a barrier in the formation of active Mn–H species.13c,d Nevertheless, electronic factors are also likely influential and indeed the order of activity for Mn1–Mn4 matches the trend in electron donating capacity of the N-R substituents seen in the IR data (vide supra). On the other hand, NNN-chelated Mn6 incorporating the N-ethyl-1H-benzimidazole unit displayed the lowest conversion (41%) among this series of complexes. This observation can plausibly be accredited to the difficulty in the dissociation of either the benzimidazole-nitrogen donor or a carbon monoxide ligand from the metal center to generate a vacant coordination site to allow the formation of the active Mn–H species; this observation is in accordance with that mentioned in Kundu's report.13d By contrast, the NNN- and NNS-type ligands in Mn7–Mn10 were less influential on the conversion (range: 78% to 85%), with their relative performance being: Mn7 > Mn8 > Mn10 > Mn9.
Overall, these results suggest that the NN-chelated Mn1 is the most effective TH catalyst of this series of complexes outperforming its NN-counterparts as well as its NNN or NNS derivatives. As a control, we also tested the reaction in the absence of any manganese complex with the conditions otherwise identical. Significantly, only trace conversion to b1 was observed highlighting the importance of the 3d metal complex in the TH process. Therefore, on the basis of the superior performance characteristics of Mn1, this complex was employed for all further studies.
With the aim to optimize the catalyst system, a selection of different bases, namely t-BuOK, t-BuONa, i-PrONa, EtONa, MeONa, NaBHEt3, KHMDS (potassium bis(trimethylsilyl)amide), NaHMDS (sodium bis(trimethylsilyl)amide), Cs2CO3, Na2CO3, LiOH·H2O, NaOH, KOH, Ca(OH)2 and Ba(OH)2, was assessed with the loading of Mn1 fixed at 0.2 mol% and base at 10 mol% (conditions A, Chart 1 and Table S3†). Of these fifteen bases screened, t-BuONa proved the most compatible allowing the greatest conversion of a1 to b1 of 99%. Nonetheless, more than half of the bases used displayed good performance characteristics for the TH of a1 with conversions in the range of 86–97%. Furthermore, it was evident that the bases used in which sodium provided the counter ion were more efficient than their potassium counterparts: t-BuONa (99%) vs. t-BuOK (97%), NaHMDS (91%) vs. KHMDS (86%) and NaOH (94%) vs. KOH (91%) (Chart 1 and Table S3†). Similar findings have been reported elsewhere.19b Control experiments performed in the absence of either base or Mn1 provided only trace amounts of product after 2 h at 90 °C (entries 16 and 17, Table S3†). Similarly, use of MnBr(CO)5 in place of Mn1, under the same reaction conditions, allowed a conversion of only 6% (entry 18, Table S3†).
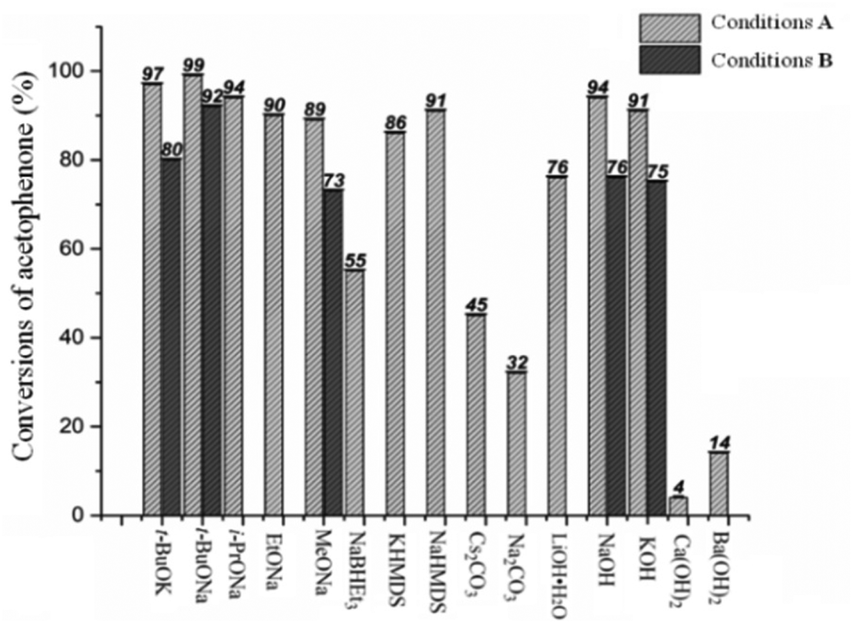 | ||
| Chart 1 Transfer hydrogenation of acetophenone (a1) as a function of the base. Conditions A: 5.0 mmol a1, 10 μmol (0.2 mol%) Mn1, 0.5 mmol base (10 mol%), 5 mL i-PrOH, 90 °C (oil bath temperature), 2 h; Conditions B: 25 mmol a1, 5 μmol (0.02 mol%) Mn1, 1.25 mol base (5 mol%), 10 mL i-PrOH (2.5 M), 90 °C (oil bath temperature), 6 h. Notes: the conversions (%) were determined by GC with dodecane as the internal standard. | ||
Next, we set about exploring the effect of varying the loading of t-BuONa on the TH of a1 (Table S4†). As the base loading was decreased from 25 to 1.25 mol%, a clear reduction in conversion (down to 38%) was observed. Interestingly, most cases resulted in the conversions to b1 being over 50% with the exception of loadings of less than 2.5 mol% of t-BuONa. Furthermore, by prolonging the reaction time to 6 h with 5 mol% t-BuONa, a high conversion of 92% could be obtained (entry 9, Table S4†). On the basis of this latter finding, five selected bases, namely t-BuOK, t-BuONa, MeONa, NaOH, KOH, were additionally investigated for the reduction of a1 with the loading set at 5 mol%, the loading of Mn1 fixed at 0.02 mol% and the run time extended to 6 h (conditions B, Chart 1). Once again, t-BuONa proved the optimal base with the conversions of a1 to b1 up to 92%, while the remaining four bases also provided good conversions in the range 73 to 80%.
With the intention to explore the effect of catalyst loading on the conversion, Mn1 was deployed at levels between 1.0 to 0.0050 mol% for the reduction of a1 with the mol% of t-BuONa set at 10 mol% (Table 2). With either 1 mol% or 0.5 mol% of Mn1 and the run temperature and duration kept at 90 °C for 20 min, respectively, essentially full conversion to b1 was observed (entries 1 and 2, Table 2). Moreover, by extending the reaction time to 2 h, excellent conversions to b1 could again be achieved when the catalyst loading was lowered to 0.2 or 0.1 mol%. However, further reduction of the loading from 0.050 mol% (500 ppm) to 0.010 mol% (100 ppm) saw the conversion drop from 86% to 78%, while TONs and TOFs increased up to 7100 and 3550 h−1, respectively. Notably, with an exceptionally low catalyst loading of 0.0050 mol% (50 ppm), the highest TON of 17200 was achieved with the run time extended to 360 min (6 h).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Mn1 [n mol%, (x ppm)] | t (min) | Conv.b (%) | TON | TOF (h−1) |
| a Conditions: 0.5–100 mmol a1, 5 μmol Mn1, 0.1 eq. (for a1) of t-BuONa (10 mol%), 5–100 mL i-PrOH (1 M), 90 °C (oil bath temperature), 20–360 min. b Determined by GC with dodecane as the internal standard, isolated yield in parentheses. Notes: TON = turnover number, TOF = Turnover frequency (mol of substrate per mol of complex per hour). | |||||
| 1 | 1.0 | 20 | 99 (92) | 99 | 294 |
| 2 | 0.50 | 20 | 99 | 198 | 594 |
| 3 | 0.20 | 120 | 99 | 495 | 247 |
| 4 | 0.10 | 120 | 97 | 970 | 485 |
| 5 | 0.050 (500) | 120 | 86 | 1720 | 860 |
| 6 | 0.025 (250) | 120 | 82 | 3280 | 1640 |
| 7 | 0.020 (200) | 120 | 78 | 3900 | 1950 |
| 8 | 0.010 (100) | 120 | 71 | 7100 | 3550 |
| 9 | 0.0050 (50) | 360 | 86 | 17200 |
2867 |

In terms of the mechanism of this TH catalysis, we propose as one possibility that Mn1 firstly undergoes the loss of H+ and Br−, under the action of the base t-BuONa, forming amide complex A along with the elimination of KBr and t-BuOH (Scheme 3). Subsequently, the HOCH unit in i-PrOH adds across the Mn![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in A, to form the active hydride complex B. Then, the NH proton and the Mn–H hydride can transfer by an outer-sphere pathway to a1 (via transition state C), leading to the reduction of a1 to b1 and the re-formation of A. This proton transfer may be assisted by the conjugate acid of t-BuONa, which may explain its influence on conversion.21 Finally, species A can dehydrogenate i-PrOH (via transition state D), to give acetone, thereby regenerating the active species B.
N bond in A, to form the active hydride complex B. Then, the NH proton and the Mn–H hydride can transfer by an outer-sphere pathway to a1 (via transition state C), leading to the reduction of a1 to b1 and the re-formation of A. This proton transfer may be assisted by the conjugate acid of t-BuONa, which may explain its influence on conversion.21 Finally, species A can dehydrogenate i-PrOH (via transition state D), to give acetone, thereby regenerating the active species B.
 | ||
| Scheme 3 One possible mechanism for the TH of a1 promoted by Mn1. | ||
To lend some support for the proposed mechanism, we set about studying the process of precatalyst activation by performing a series of 1H NMR experiments (Fig. S19–S21†). On the basis of these experiments, the amido complex [(NN)Mn(CO)3] (A in Scheme 3) is formed when Mn1 was treated with t-BuONa in the presence of i-PrOH at 30 °C. Indeed, related monometallic complexes have been reported as effective catalysts for the TH of ketones by Leitner13c and Sun.14f By contrast, our findings are unlike that reported in Sortais’ work in which a dimeric manganese complex has been identified.13b Furthermore, isolated A displayed high activity for the TH of a1 with good conversions (80%) observed under conditions A. On the other hand, treatment of Mn1 with t-BuONa in the presence of i-PrOH at 90 °C gave what we tentatively ascribe as the active species B (see ESI†). Unfortunately, B proved very sensitive and attempts to obtain a crystal structure of this complex were unsuccessful. Nevertheless, the ESI-MS mass spectrum of B provided evidence for a M–H parent ion peak at m/z 329.25 [B–H]+ (see Fig. S23†).
To gain a broader understanding of the scope of Mn1, we extended the TH evaluation to cover a wide variety of (hetero)aromatic ketones using two sets of conditions namely A (0.5 mol% Mn1, 10 mol% t-BuONa, 90 °C, 20 min) and B (0.02 mol% Mn1, 5 mol% t-BuONa, 90 °C, 6 h). As shown in Table 3, a wide variety of (hetero)aromatic ketones could be reduced to their corresponding alcohols. Indeed, various substituted acetophenones (a2–a16, Table 3) containing both electron withdrawing and donating groups on the aryl group were effectively reduced under conditions A. Halide-containing substrates produced the desired alcohols with conversions up to 99%. Interestingly, the acetophenone derivatives (a2–a4, a7–a9, a12–a14, a17, a18 and a20) bearing electron-withdrawing substituents (R = F, Cl and Br) were more effectively converted to their corresponding alcohols (b2–b4, b7–b9 and b12–b14) than that observed by their analogues containing electron-donating groups (a5, a6, a10, a11, a15 and a16, R = Me and OMe) under conditions B. Evidently, these halide-containing substrates can be reduced without the formation of dehalogenated products. It was also found that acetophenones bearing meta or para substituents typically display higher conversions than those bearing ortho substituents (a2–a11vs.a12–a16, Table 3). These differences may be the result of greater steric hindrance induced by the ortho substituents.
| a Reaction conditions A: substrate (2.0 mmol), Mn1 (10 μmol, 0.5 mol%), t-BuONa (0.2 mmol, 10 mol%), in i-PrOH (5 mL) at 90 °C (oil bath temperature) for 20 min. Reaction conditions B: substrate (25.0 mmol), Mn1 (5 μmol, 0.02 mol%), t-BuONa (2.5 mmol, 10 mol%), in i-PrOH (25 mL) at 90 °C (oil bath temperature) for 6 h. Notes: the conversions (%) were measured using GC (n-dodecane was used as an internal standard). b Isolated % yield. |
|---|
|
With the goal to explore the effect of electronic properties and steric hindrance on catalytic performance, acetophenones containing chloride substituents (a17–a19) at two different positions on the aryl group were firstly investigated (Table 3). The 2,4- and 3,4-substituted derivatives a17 and a18 could be smoothly converted to the corresponding alcohols, b17 and b18, while 2′,6′-dichloroacetophenone (b19) was more sluggish when compared to its analogues, a17 and a18. A variety of propiophenone derivatives bearing functional groups, such as Me and Cl (a21–a22) were reduced to give the expected alcohols with nearly quantitative conversions (conditions A, Table 3). With regard to benzophenones and acetyl-naphthalenes, the conversions to the corresponding alcohols were in general high, with benzophenone, phenyl(p-tolyl)methanone and 1-/2-acetylnaphthalenes amenable to 97%, 96%, 93% and 96% conversion (under conditions A), respectively. Benzocyclones (a30–a32) with carbocyclic ring sizes of between six and eight gave moderate to high conversions to their corresponding alcohols, with 9,10-dihydroanthracen-9-ol, 1,2,3,4-tetrahydronaphthalen-1-ol and 6,7,8,9-tetrahydro-5H-benzo[7] annulen-5-ol being obtained with percentages of 54%, 72% and 94% (under conditions A, Table 3), respectively. When compared to 3,4-dihydronaphthalen-1(2H)-one (a31), 6,7-dihydroquinolin-8(5H)-one (a33) underwent higher conversion, with 5,6,7,8-tetrahydroquinolin-8-ol (b33) being obtained in 88% isolated yield. With respect to heteroaryl ketones, a34–a38, the conversions to the corresponding alcohols, b34–a38, were slightly lower than that observed by the aryl ketones (Table 3). Notably, the 2-acetylfuran derivatives, a35 and a36, gave only low conversions to their alcohols which could be due to the presence of a strongly coordinating oxygen heteroatom.
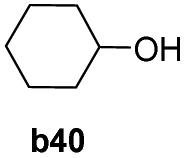
To further examine the substrate scope of Mn1, cyclic alkyl and acyclic dialkyl ketones were also assessed using reaction conditions A (Table 4). For the cyclic alkyl ketones, it was found that the ring size was influential on the conversion with the smallest ring ketone, cyclopentanone (a39, n = 5) affording the lowest conversion of 69% (entry 1, Table 4). Conversely, the medium size cyclic ketones (a40–a42, 6 ≤ n ≤ 8) gave higher conversions, with cyclohexanol, cycloheptanol and cyclooctenone formed between 84% and 94%. On the other hand, the largest ring ketone cyclododecanone (a43, n = 12) again led to a lower conversion to alcohol b43 (entry 5, Table 4).
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Conv.b (%) |
| a Reaction conditions A: substrate (2.0 mmol), Mn1 (10 μmol, 0.5 mol%), t-BuONa (0.2 mmol, 10 mol%), i-PrOH (5 mL) at 90 °C (oil bath temperature) for 20 min. b Measured by GC with n-dodecane used as an internal standard. Isolated yields are given in parentheses. | |||
| 1 |

|

|
69 |
| 2 |
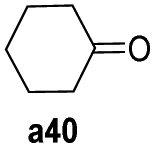
|
|
94 |
| 3 |
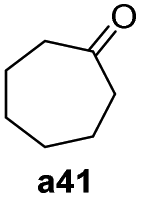
|

|
90 |
| 4 |

|

|
84 |
| 5 |
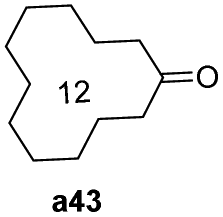
|
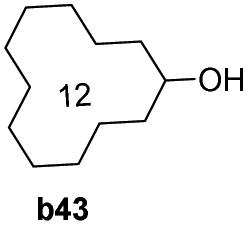
|
72 |
| 6 |

|

|
90 (85) |
| 7 |

|

|
81 |
| 8 |

|

|
91 |
| 9 |

|

|
60 |
| 10 |

|

|
65 (60) |
| 11 |

|

|
99 |
| 12 |

|

|
93 (90) |

With the ring size of the cyclic alkyl ketone maintained at six, the 4-substituted cyclohexan-1-ones, a44–a47, all gave high conversions (81–91%) to the corresponding cyclic alcohols (b44–b47, entries 6–8, Table 4). By contrast, 2-substituted cyclohexan-1-one, a47, gave a lower conversion to b47 (60%) which likely reflects the steric properties of the 2-isopropyl substituent (entry 7, Table 4). Similarly, the N-heterocyclic containing ketone, quinuclidin-3-one a48, gave lower conversion (65%) to b48 (entry 10, Table 4). By comparison, the conversions for the long chain dialkyl-containing ketones, 4-phenylbutan-2-one (a49) and 1-phenylhexan-3-one (a50), were higher at 99% and 93% in 20 min, respectively (entries 11 and 12, Table 4).
This manganese-mediated TH is not just limited to ketonic substrates. Indeed, a number of substituted aldehydes (c1–c11, entries 1–10, Table 5) were shown to undergo TH using reaction conditions A. Indeed, most of the aldehydes employed were efficiently reduced to the corresponding primary alcohol and in some cases quantitatively. The aryl-containing aldehydes, c1–c5, with either electron-rich or electron-poor groups gave good to high conversions to d1 (90%), d2 (99%), d3 (85%), d4 (99%) and d5 (99%), respectively (entries 1–5, Table 5). Heteroatom-containing aldehydes, picolinaldehyde (c6), thiophene-2-carboxaldehyde (c7) and furan-2-carbaldehyde (c8) also worked effectively (entries 6–8, Table 5). Finally, our catalytic system also promoted the reduction of the long chain alkyl-containing aldehydes, heptaldehyde (c9) and octanal (c10) affording excellent conversions (entries 9 and 10, Table 5).
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Conv.b (%) |
| a Reaction conditions A: aldehyde (c) (2.0 mmol), Mn1 (10 umol, 0.5 mol%), t-BuONa (0.2 mmol, 10 mol%), i-PrOH (5 mL) at 90 °C (oil bath temperature) for 30 min. b Measured by GC with n-dodecane used as an internal standard. Isolated yields are given in parentheses. | |||
| 1 |
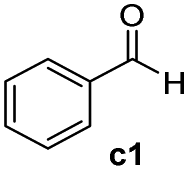
|
|
90 (85) |
| 2 |

|

|
99 (95) |
| 3 |
|
|
85 (81) |
| 4 |

|

|
99 (94) |
| 5 |
|
|
99 (95) |
| 6 |

|

|
46 (41) |
| 7 |

|

|
98 (93) |
| 8 |

|

|
89 (82) |
| 9 |

|
|
99 |
| 10 |

|

|
99 |

Conclusions
In summary, a new set of NN-manganese carbonyl complexes (Mn1–Mn5) along with the NNN-containing Mn6, all incorporating either an 8-amino-5,6,7,8-tetrahydroquinoline or an 8-imino-5,6,7-trihydroquinoline backbone, have been synthesized and characterized. All six complexes, along with previously reported NNN- and NNS-complexes, Mn7–Mn10 (Fig. 2), have been evaluated as catalysts for the transfer hydrogenation of acetophenone. Notably, isopropyl containing NN-Mn1, in the presence of t-BuONa, proved the most effective and was able to operate efficiently with a catalyst loading of just 0.005 mol%. Indeed, using Mn1/t-BuONa as catalyst, a wide range of (hetero)aromatic-, aliphatic- and cycloalkyl-containing ketones (50 examples) and aldehydes (10 examples) could be reduced to give their corresponding alcohols with conversions in the range 39–99%. Furthermore, the catalytic results suggest an outer-sphere hydrogen transfer involving the amino NH functionality in Mn1 as the proton source. Further mechanistic studies are currently ongoing to elucidate the detailed catalytic cycle.Experimental section
General information
All manipulations were carried out under a nitrogen atmosphere using standard Schlenk techniques. All solvents were dried and distilled under nitrogen prior to use. All substrates, both liquid and solid, were used directly without further purification. Other reagents were purchased from Aldrich, Acros or local suppliers. 1H and 13C NMR spectra were recorded on Bruker AV-400 NMR and Bruker AV-500 NMR spectrometers. Chemical shift values in the 1H and 13C NMR spectra were referenced internally to the residual solvent resonances. Infrared spectroscopy was performed in the solid state on a Bruker ALPHA. Elemental analysis was performed with a Vario EL III CHN microanalyzer. GC was performed using a FuLi GC-9790Plus instrument (Zhe Jiang FuLi Analytical Instrument) using a PB-FFAP column (30 m × 0.32 mm × 0.25 μm, Wuhan Puli Technology Co., Ltd) or RB-WAX column (30 m × 0.25 mm × 0.25 μm, Wuhan Puli Technology Co., Ltd): injector temp. 300 °C, detector temp. 300 °C, column temp. 120 °C, withdraw time 2 min, then 20 °C min−1 to 180 °C for 5 min, then 20 °C min−1 to 250 °C, withdraw time for 5 min. Synthetic procedures for L1–L6 and Mn1–Mn6 are given in the ESI†.Transfer hydrogenation studies
Under an atmosphere of nitrogen, a 25–250 mL dried Schlenk tube, containing a stirrer bar, was charged with the selected ketonic substrate (2.5–25.0 mmol), manganese complex (Mn1–Mn10, 5.0–10.0 μmol), the desired amount of base (e.g. t-BuOK, t-BuONa, i-PrONa, NaOEt, NaOMe KOH, NaOH, CsCO3, Na2CO3, LiOH·H2O, LiOH, Ca(OH)2, K2CO3, Na2CO3, NaBHEt3, NaHMDS, KHMDS) (0.5–4.0 mmol) and i-PrOH (5–100 mL). The vessel was sealed and the contents then stirred at 90 °C (oil bath temperature). After the desired reaction time, the mixture was allowed to cool to room temperature and the pressure slowly released. The reaction mixture was filtered through a plug of silica gel and then analyzed by GC; the composition of the reaction mixture was confirmed by running a separate GC on a mixture of pure ketone, alcohol and dodecane. All conversions were determined by GC using dodecane as internal standard. Where indicated, the crude products were purified by flash gel chromatography (eluent: petroleum ether/ethyl acetate = 200:1 to 10:1) and an isolated yield determined.
X-ray structure determination
The single-crystal X-ray diffraction studies of [L1H]Br and Mn1 were conducted on a Rigaku Sealed Tube CCD (Saturn 724+) diffractometer with graphite-mono chromated Cu-Kα radiation (λ = 1.54184) at 169(8) K and the cell parameters obtained by global refinement of the positions of all collected reflections. Intensities were corrected for Lorentz and polarization effects and empirical absorption. The structures were solved by direct methods and refined by full-matrix least-squares on F2. All non-hydrogen atoms were refined anisotropically and all hydrogen atoms were placed in calculated positions. Structure solution was performed using SHELXL (2015) and structure refinement performed using SHELXL.22 Crystal data and processing parameters for [L1H]Br and Mn1 are summarized in Table S6.†Conflicts of interest
Acknowledgements
We acknowledge the support from the Nature Science Foundation of Hebei Province (B2022205020, B2022204020) and Talent Introduction Foundation of Hebei Agricultural University (YJ201931). Research Project of Fundamental Scientific Research Business Expenses of Provincial Colleges and Universities in Hebei Province (KY2021028); Opening Fund of CAS Key Laboratory of Engineering Plastics, Institute of Chemistry. G.A.S. is grateful to the Chinese Academy of Sciences for a President's International Fellowship for Visiting Scientists.References
- (a) P. G. Andersson and I. J. Munslo, in Modern Reduction Methods, Wiley, New York, 2008 CrossRef; (b) P. A. Dub and T. Ikariya, ACS Catal., 2012, 2, 1718–1741 CrossRef CAS; (c) J. Ito and H. Nishiyama, Tetrahedron Lett., 2014, 55, 3133–3146 CrossRef; (d) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS PubMed; (e) L. Shi, Y. Liu, Q. Liu, B. Wei and G. Zhang, Green Chem., 2012, 14, 1372–1375 RSC.
- (a) E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551–5553 CrossRef CAS; (b) L. Deloux and M. Srebnik, Chem. Rev., 2002, 93, 763–784 CrossRef; (c) D. Lunic, N. Sanosa, I. Funes-Ardoiz and C. J. Teskey, Angew. Chem., Int. Ed., 2022, 61, e202207647 CrossRef CAS PubMed.
- (a) A. Matsunami and Y. Kayaki, Tetrahedron Lett., 2018, 59, 504–513 CrossRef CAS; (b) D. Baidilov, D. Hayrapetyan and A. Y. Khalimon, Tetrahedron, 2021, 98, 132435 CrossRef CAS.
- Selected examples for TH ruthenium catalysts, see. (a) A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS; (b) A. Del Zotto, W. Baratta, M. Ballico, E. Herdtweck and P. Rigo, Organometallics, 2007, 26, 5636–5642 CrossRef CAS; (c) W. Baratta, G. Chelucci, S. Magnolia, K. Siega and P. Rigo, Chem. – Eur. J., 2009, 15, 726–732 CrossRef CAS PubMed; (d) W. Du, P. Wu, Q. Wang and Z. Yu, Organometallics, 2013, 32, 3083–3090 CrossRef CAS; (e) H. Chai, T. Liu, Q. Wang and Z. Yu, Organometallics, 2015, 34, 5278–5284 CrossRef CAS; (f) J. M. Botubol-Ares, S. Cordón-Ouahhabi, Z. Moutaoukil, I. G. Collado, M. Jiménez-Tenorio, M. C. Puerta and P. Valerga, Organometallics, 2021, 40, 792–803 CrossRef CAS; (g) A. Dubey and E. Khaskin, ACS Catal., 2016, 6, 3998–4002 CrossRef CAS.
- (a) K. Murata, T. Ikariya and R. Noyori, J. Org. Chem., 1999, 64, 2186–2187 CrossRef CAS; (b) K. Farrell, H. Müller-Bunz and M. Albrecht, Organometallics, 2015, 34, 5723–5733 CrossRef CAS.
- Selected examples for TH iridium catalysts, see. (a) M. Albrecht, J. R. Miecznikowski, A. Samuel, J. W. Faller and R. H. Crabtree, Organometallics, 2002, 21, 3596–3604 CrossRef CAS; (b) U. Hintermair, J. Campos, T. P. Brewster, L. M. Pratt, N. D. Schley and R. H. Crabtree, ACS Catal., 2014, 4, 99–108 CrossRef CAS; (c) R. Wang, X. Han, J. Xu, P. Liu and F. Li, J. Org. Chem., 2020, 85, 2242–2249 CrossRef CAS PubMed.
- Selected examples for TH osmium catalysts, see. (a) W. Baratta, M. Ballico, G. Chelucci, K. Siega and P. Rigo, Angew. Chem., Int. Ed., 2008, 47, 4362–4365 CrossRef CAS PubMed; (b) W. Baratta, F. Benedetti, A. Del Zotto, L. Fanfoni, F. Felluga, S. Magnolia, E. Putignano and P. Rigo, Organometallics, 2010, 29, 35639 CrossRef; (c) G. Chelucci, S. Baldino and W. Baratta, Acc. Chem. Res., 2015, 48, 363–379 CrossRef CAS PubMed; (d) J. P. C. Coverdale, I. Romero-Canelón, C. Sanchez-Cano, G. J. Clarkson, A. Habtemariam, M. Wills and P. J. Sadler, Nat. Chem., 2018, 10, 347–354 CrossRef CAS PubMed.
- T. Zell and R. Langer, ChemCatChem, 2018, 10, 1930–1940 CrossRef CAS.
- F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed.
- (a) T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS PubMed; (b) Y.-Y. Li, S.-L. Yu, W.-Y. Shen and J.-X. Gao, Acc. Chem. Res., 2015, 48, 2587–2598 CrossRef CAS PubMed; (c) D. Wei and C. Darcel, Chem. Rev., 2019, 119, 2550–2610 CrossRef CAS PubMed; (d) P. O. Lagaditis, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2011, 133, 9662–9665 CrossRef CAS PubMed; (e) R. Bigler, R. Huber and A. Mezzetti, Angew. Chem., Int. Ed., 2015, 54, 5171–5174 CrossRef CAS PubMed; (f) R. A. Farrar–Tobar, B. Wozniak, A. Savini, S. Hinze, S. Tin and J. G. de Vries, Angew. Chem., Int. Ed., 2019, 58, 1129–1133 CrossRef PubMed; (g) X. Liu, J. G. de Vries and T. Werner, Green Chem., 2019, 21, 5248–5255 RSC; (h) S. Zhang, Z. Wang, Q. Cao, E. Yue, Q. Liu, Y. Ma, T. Liang and W.-H. Sun, Dalton Trans., 2020, 49, 15821–15827 RSC.
- (a) W. Liu, B. Sahoo, K. Junge and M. Beller, Acc. Chem. Res., 2018, 51, 1858–1869 CrossRef CAS PubMed; (b) A. Mukherjee and D. Milstein, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS; (c) W. Ai, R. Zhong, X. Liu and Q. Liu, Chem. Rev., 2018, 119, 2876–2953 CrossRef PubMed; (d) G. Zhang and S. K. Hanson, Chem. Commun., 2013, 49, 10151–10153 RSC; (e) C. Hou, J. Jiang, Y. Li, Z. Zhang, C. Zhao and Z. Ke, Dalton Trans., 2015, 44, 16573–16585 RSC.
- (a) E. S. Gulyaeva, E. S. Osipova, R. Buhaibeh, Y. Canac, J.-B. Sortais and D. A. Valyaev, Coord. Chem. Rev., 2022, 458, 214421 CrossRef CAS; (b) K. Azouzi, D. A. Valyaev, S. Bastin and J.-B. Sortais, Curr. Opin. Green Sustainable Chem., 2021, 31, 100511 CrossRef CAS; (c) K. Das, S. Waiba, A. Jana and B. Maji, Chem. Soc. Rev., 2022, 51, 4386–4464 RSC; (d) Y. Wang, M. Wang, Y. Li and Q. Liu, Chem, 2021, 7, 1180–1223 CrossRef CAS.
- (a) M. Perez, S. Elangovan, A. Spannenberg, K. Junge and M. Beller, ChemSusChem, 2017, 10, 83–86 CrossRef CAS PubMed; (b) A. Bruneau-Voisine, D. Wang, V. Dorcet, T. Roisnel, C. Darcel and J.-B. Sortais, Org. Lett., 2017, 19, 3656–3659 CrossRef CAS PubMed; (c) O. Martínez-Ferraté, C. Werlé, G. Franciò and W. Leitner, ChemCatChem, 2018, 10, 4514–4518 CrossRef PubMed; (d) K. Ganguli, S. Shee, D. Panja and S. Kundu, Dalton Trans., 2019, 48, 7358–7366 RSC; (e) A. Dubey, S. M. W. Rahaman, R. R. Fayzullin and J. R. Khusnutdinova, ChemCatChem, 2019, 11, 3844–3852 CrossRef CAS; (f) C. Zhang, B. Hu, D. Chen and H. Xia, Organometallics, 2019, 38, 3218–3226 CrossRef CAS; (g) V. Vigneswaran, S. N. MacMillan and D. C. Lacy, Organometallics, 2019, 38, 4387–4391 CrossRef CAS; (h) N. V. Shvydkiy, O. Vyhivskyi, Y. V. Nelyubina and D. S. Perekalin, ChemCatChem, 2019, 11, 1602–1605 CrossRef CAS; (i) R. Putten, J. Benschop, V. J. Munck, M. Weber, C. Müller, G. A. Filonenko and E. A. Pidko, ChemCatChem, 2019, 11, 5232–5235 CrossRef PubMed; (j) D. Wang, A. Bruneau-Voisine and J.-B. Sortais, Catal. Commun., 2018, 105, 31–36 CrossRef CAS; (k) S. Budweg, K. Junge and M. Beller, Chem. Commun., 2019, 55, 14143–14146 RSC.
- Selected examples for ATH manganese catalysts:. (a) A. Zirakzadeh, S. R. M. M. Aguiar, B. Stöger, M. Widhalm and K. Kirchner, ChemCatChem, 2017, 9, 1744–1748 CrossRef CAS; (b) D. Wang, A. Bruneau-Voisine and J.-B. Sortais, Catal. Commun., 2018, 105, 31–36 CrossRef CAS; (c) R. van Putten, G. A. Filonenko, A. Gonzalez de Castro, C. Liu, M. Weber, C. Müller, L. Lefort and E. Pidko, Organometallics, 2019, 38, 3187–3196 CrossRef CAS PubMed; (d) G.-Y. Zhang, S.-H. Ruan, Y.-Y. Li and J.-X. Gao, Chin. Chem. Lett., 2021, 32, 1415–1418 CrossRef CAS; (e) K. Azouzi, A. Bruneau-Voisine, L. Vendier, J.-B. Sortais and S. Bastin, Catal. Commun., 2020, 142, 106040 CrossRef CAS; (f) L. Wang, J. Lin, Q. Sun, C. Xia and W. Sun, ACS Catal., 2021, 11, 8033–8041 CrossRef CAS; (g) J. Yang, L. Yao, Z. Wang, Z. Zuo, S. Liu, P. Gao, M. Han, Q. Liu, G. A. Solan and W.-H. Sun, J. Catal., 2023, 418, 40–50 CrossRef CAS; (h) K. Z. Demmans, M. E. Olson and R. H. Morris, Organometallics, 2018, 37, 4608–4618 CrossRef CAS; (i) H. Jayaprakash, Dalton Trans., 2021, 50, 14115–14119 RSC; (j) H. Jayaprakash, P. Coburger, M. Worle, A. Togni and H. Grutzmacher, Chem. – Eur. J., 2022, 28, e202201522 CrossRef CAS PubMed; (k) F. Li, L. Long, Y. M. He, Z. Li, H. Chen and Q. H. Fan, Angew. Chem., Int. Ed., 2022, 61, e202202972 CAS; (l) C. L. Oates, A. S. Goodfellow, M. Buhl and M. L. Clarke, Angew. Chem., Int. Ed., 2023, 62, e202212479 CrossRef CAS PubMed.
- (a) J. A. Garduño, M. Flores–Alamo and J. J. García, ChemCatChem, 2019, 11, 5330–5338 CrossRef; (b) D. Wei, A. Bruneau–Voisine, M. Dubois, S. Bastin and J. B. Sortais, ChemCatChem, 2019, 11, 5256–5259 CrossRef CAS; (c) C. L. Oates, M. B. Widegren and M. L. Clarke, Chem. Commun., 2020, 56, 8635–8638 RSC; (d) X. Liu and T. Werner, Chem. Sci., 2021, 12, 10590–10597 RSC; (e) K. Sarkar, K. Das, A. Kundu, D. Adhikari and B. Maji, ACS Catal., 2021, 11, 2786–2794 CrossRef CAS.
- (a) Z. Wang, Q. Liu, G. A. Solan and W.-H. Sun, Coord. Chem. Rev., 2017, 350, 68–83 CrossRef CAS; (b) Z. Wang, G. A. Solan, W. Zhang and W.-H. Sun, Coord. Chem. Rev., 2018, 363, 92–108 CrossRef CAS; (c) Q. Mahmood, X. Li, L. Qin, L. Wang and W.-H. Sun, Dalton Trans., 2022, 51, 14375–14407 RSC.
- (a) B. Pan, B. Liu, E. Yue, Q. Liu, X. Yang, Z. Wang and W.-H. Sun, ACS Catal., 2016, 6, 1247–1253 CrossRef CAS; (b) Z. Wang, X. Chen, B. Liu, Q. Liu, G. A. Solan, X. Yang and W.-H. Sun, Catal. Sci. Technol., 2017, 7, 1297–1304 RSC; (c) Z. Wang, B. Pan, Q. Liu, E. Yue, G. A. Solan, Y. Ma and W.-H. Sun, Catal. Sci. Technol., 2017, 7, 1654–1661 RSC; (d) Z. Wang, Y. Li, Q. Liu, G. A. Solan, Y. Ma and W.-H. Sun, ChemCatChem, 2017, 9, 4275–4281 CrossRef CAS.
- Z. Wang, Q. Lin, N. Ma, S. Liu, M. Han, X. Yan, Q. Liu, G. A. Solan and W.-H. Sun, Catal. Sci. Technol., 2021, 11, 8026–8036 RSC.
- (a) J. Li, Y. Ma, Z. Wang, Q. Liu, G. A. Solan, Y. Ma and W.-H. Sun, Dalton Trans., 2018, 47, 8738–8745 RSC; (b) Z. Wang, Y. Liu, M. Han, N. Ma, Q. Lyu, Q. Liu and W.-H. Sun, Dalton Trans., 2022, 51, 10983–10991 RSC; (c) F. Cao, Y. Wang, X. Wang, W. Zhang, G. A. Solan, R. Wang, Y. Ma, X. Hao and W.-H. Sun, Catal. Sci. Technol., 2022, 12, 6687–6703 RSC.
- (a) D. Zerla, G. Facchetti, M. Fusè, M. Pellizzoni, C. Castellano, E. Cesarotti, R. Gandolfi and I. Rimoldi, Tetrahedron: Asymmetry, 2014, 25, 1031–1037 CrossRef CAS; (b) D. S. Zerla, I. Rimoldi, E. Cesarotti, G. Facchetti, M. Pellizzoni and M. Fusè, J. Organomet. Chem., 2014, 771, 2–8 CrossRef CAS.
- (a) R. van Putten, E. A. Uslamin, M. Garbe, C. Liu, A. Gonzalez-de-Castro, M. Lutz, K. Junge, E. J. M. Hensen, M. Beller, L. Lefort and E. A. Pidko, Angew. Chem., Int. Ed., 2017, 56, 7531–7534 CrossRef CAS PubMed; (b) Y. Wang, S. Liu, H. Yang, H. Li, Y. Lan and Q. Liu, Nat. Chem., 2022, 14, 1233–1241 CrossRef CAS PubMed.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, A71, 3–8 CrossRef PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, spectra (NMR), Fig. S1–S53, Tables S1–S6 and X-ray crystallographic data. CCDC 2248092 ([L1H]Br) and 2248093 (Mn1). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02022c |
| This journal is © The Royal Society of Chemistry 2023 |