
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Switchable cyclopalladation of substrates containing two directing groups: on the way to non-symmetrical [2.2]-dipalladaparacyclophanes†
Tereza
Korábková
a,
Jan
Bartáček
a,
Lukáš
Marek
 a,
Jiří
Hanusek
a,
Aleš
Růžička
b and
Jiří
Váňa
*a
a,
Jiří
Hanusek
a,
Aleš
Růžička
b and
Jiří
Váňa
*a
aInstitute of Organic Chemistry and Technology, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 53210 Pardubice, The Czech Republic. E-mail: jiri.vana@upce.cz
bDepartment of General and Inorganic Chemistry, Faculty of Chemical Technology, University of Pardubice, Studentská 573, 53210 Pardubice, The Czech Republic
First published on 13th July 2023
Abstract
Simple switching of the site-selectivity of C–H activation reactions of substrates containing multiple directing groups is particularly important for the so-called late stage functionalization synthetic approach. In this work, we verified the possibility of achieving this by adding acids of different strengths. Using a substrate containing two differently strong (and basic) directing groups, the influence of the addition of acids on the regioselectivity of the C–H activation step of the reaction with palladium acetate was thoroughly studied. The addition of no or weak acids results in cyclopalladation being controlled by a stronger directing group. However, the addition of a strong acid causes protonation of this group and the reaction is then controlled by a weaker directing group. Finally, this approach enables double C–H activation leading to a unique class of compounds: “non-symmetrical” [2.2]-dipalladaparacyclophanes.
Introduction
Directing group (DG) assisted C–H activation is important not only in the synthesis of palladacycles1 but also especially as a key step of the intensively studied C–H functionalization reactions.2 During the past few decades, a number of DGs were introduced.3 Despite progress in this area, understanding and control of site-selectivity in complex substrates containing multiple DGs remains a challenge.4 The basic pillar for the prediction of regioselectivity is the determination of relative DG abilities. A few DG ability scales were introduced and their validity was verified on substrates containing two DGs.5 In simplicity, C–H activation is preferably assisted by a stronger DG. However, the possibility of controlling the reaction using a weaker DG can be very useful for purposes of organic synthesis, especially during the decoration of complex molecules in the late stages of their synthesis.6 Most probably the simplest and the most predictable way is based on changing the acid–base properties of the reaction mixture. This way, suggested e.g. by Sanford5b or us7 and verified on one example of C–H functionalization by Norrby et al.,5a takes advantage of the fact that the stronger DG tends to be more basic.5b Therefore, the addition of an acid to the reaction mixture causes its protonation and thus the loss of DG ability.8 This allows the weaker DG to direct the reaction (Scheme 1).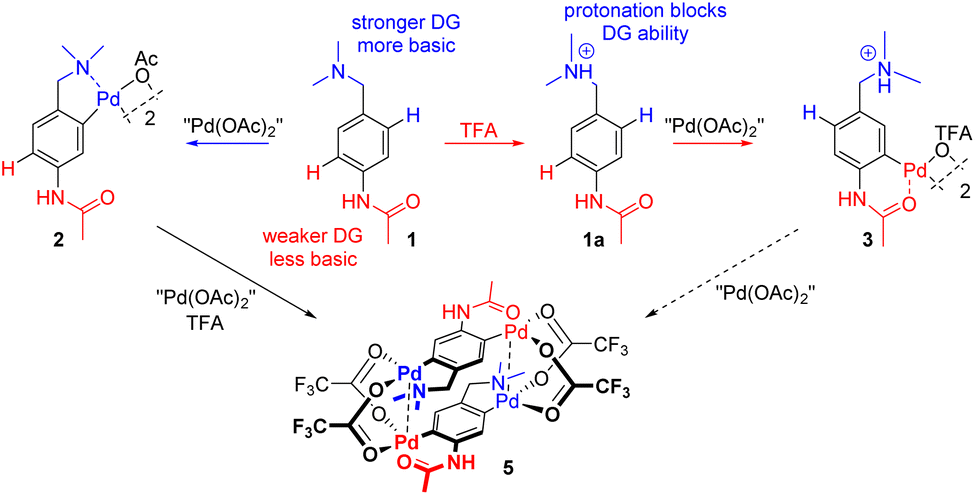 | ||
| Scheme 1 General principle of site selectivity switch controlled by protonation of directing groups. | ||
In addition, the occurrence of two different DGs offers an opportunity for gradual double C–H activation leading to unique “non-symmetrical” [2.2]-dipalladaparacyclophanes. Such doubly cyclopalladated compounds are known to be only “symmetrical”, i.e. containing two identical DGs, prepared by the reaction of substrates with two equivalents of palladium salts.9 In the case of substrates containing two different DGs, the reaction requires two steps (Scheme 1). The first stoichiometric C–H activation directed by a stronger DG is followed by the addition of a second equivalent of palladium acetate and a strong acid additive.
The aim of our study was to verify the viability of this approach for synthesizing (di)palladacycles and to examine the factors that influence the formation of individual regioisomers. By investigating these factors, we hope to enhance the understanding of DG-assisted C–H activation and contribute to the development of efficient synthetic strategies in organic chemistry.
Results and discussion
Protonation of substrates
We synthesized N-(4-((dimethylamino)methyl)phenyl) acetamide 1 as the model substrate, i.e. a substrate containing two DGs differing in DG abilities and basicity. The N,N-dimethylamino group is a stronger and more basic DG (pKa ≈ 9).10 Therefore, it is expected to direct C–H activation under ‘neutral’ conditions (without or with the addition of a weak acid). On the other hand, this group was protonated to give 1a after the addition of a stronger acid (e.g. trifluoroacetic acid (TFA)) or a higher amount of a weaker acid to the reaction mixture. The successful protonation was documented by changes in the chemical shifts especially of CH2 and N-(CH3)2 to higher values (Fig. 1a and b) and above that in the case of addition of strong TFA by the splitting of signals of the CH2 and N-(CH3)2 groups in proton NMR (Fig. 1c). Thus, in more acidic media the weaker acetamido group (pKa ≈ −4) should direct cyclopalladation. | ||
| Fig. 1 Influence of additional acids (AcOH and TFA) on the 1H NMR spectra of compounds 1 and 2. | ||
Cyclopalladation in the media without additional acids
Initially, we examined the equimolar reaction of 1 with Pd3(OAc)6 (hereafter referred to as “Pd(OAc)2”) in dichloromethane (DCM) at room temperature. The reaction gave a satisfactory yield (68%) of yellow precipitate 2. The 1H NMR spectrum (Fig. 1d) shows features analogous to the spectra of known C2-symmetric (clamshell) dinuclear palladacycles derived from N,N-dimethylbenzylamines,11 specifically, the diastereotopic CH2 group appears as two AB doublets (CH2-a and CH2-b) and two different N-CH3 groups are assigned to the axial and equatorial positions of the five-membered ring. Single-crystal X-ray diffraction confirmed the predicted structure palladated in the ortho position proximal to the stronger N,N-dimethylamino group (Fig. 2).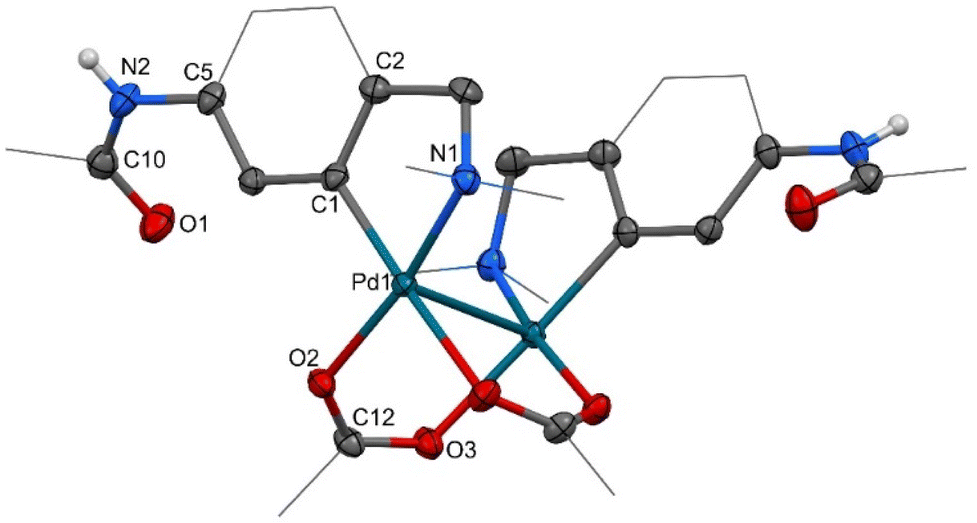 | ||
| Fig. 2 The ORTEP structures of dinuclear complex 2. Thermal ellipsoids are set at 50% probability and peripheral atoms are shown as wireframes; most of the hydrogen atoms are omitted for clarity. Selected interatomic distances (Å) and angles (°): Pd1–C1 1.966(2), Pd1–O2 2.0603(17), Pd1–N1 2.065(2), Pd1–O3i 2.1500(17), Pd1–Pd1i 2.9304(4), O1–C10 1.226(3), N1–C9 1.486(3), C1–Pd1–O2 93.59(9), C1–Pd1–N1 82.40(9), O2–Pd1–N1 170.10(7), C1–Pd1–O3i 176.12(9), O2–Pd1–O3i 90.17(7), N1–Pd1–O3i 93.74(8), C1–Pd1–Pd1i 102.33(7), O2–Pd1–Pd1i 81.09(5), N1–Pd1–Pd1i 108.58(6), O3i–Pd1–Pd1i 79.18(5); symmetry code: (i) −x + 1, y, −z + 3/2. | ||
The structure of centrosymmetric dinuclear palladium complex 2 consists of two palladium centers bridged by two acetate ligands in an isobidentate-bridging fashion. The oxygen atoms of these ligands are mutually cis oriented at the base of square pyramidal coordination polyhedra of each palladium atom. Substituted phenyl with a pendant dimethylaminomethyl moiety chelates palladium atoms in the rest of the base positions, while a weak palladium–palladium bonding interaction12 is responsible for the presence of the second palladium atom on the top of the pyramid. The mutual orientation of both chelating phenyl ligands is anti-C2 symmetry. Such a coordination geometry is similar to ca. 30 examples of doubly carboxylate bridged dinuclear palladium complexes bearing adjacent monoanionic C,N-chelates with two five-membered palladacycles found within the Cambridge Crystal Structure Database.13 Most of the parameters characterizing the coordination polyhedra, interatomic distances and angles are similar, but significant differences are found in cases of Pd–Pd separations. While these distances are usually a weak bond of ∼2.9 Å (also for 2), the same type of parameter could be much longer for complexes with sterically demanding substituents on the amino groups (∼3.2 Å),14 but also much shorter as, for example, for the dinuclear PdIII complex (∼2.5 Å).15
Cyclopalladation in the presence of weak acids
Very often, the C–H functionalization protocols involve the presence of additional acetic or other carboxylic acids. Therefore, we examined their influence on cyclopalladation using NMR spectroscopic (time-conversion) experiments. CD3CN was selected as a solvent to avoid precipitation of the product 2. While cyclopalladation without additional acid gave signals of dinuclear palladacycle 2 as an exclusive product, the addition of acetic acid (5 and 10 equivalents) to the reaction mixture caused the appearance of signals of a new compound 2a/b (Fig. 1e), whose abundance increased with increasing amount of acetic acid present in the reaction mixture. The 1H NMR spectrum showed signals of three aromatic protons with chemical shifts and coupling constants very close to the signals of 2. On the other hand, signals in the aliphatic region resonated as singlets. After the addition of a high excess of AcOH (more than 50 equivalents) the signals of 2a/b are solely present (Fig. 1f). To examine the nature of this behavior and the structure of 2a/b, we took the resulting reaction mixture from the experiment without additional acid and containing only product 2 and added 10 equivalents of AcOH. The spectra recorded immediately after the addition were identical to the spectra recorded at the end of the kinetics in the presence of 10 equivalents of AcOH (Fig. S1†). This shows that 2a/b is a different form of 1 and the ratio of the individual forms is dependent exclusively on the amount of added acetic acid. Evaporation of volatiles from the NMR tube, where exclusively signals of 2a/b were recorded, to dryness and subsequent redissolution of the residue in CD3CN or acetone-d6 followed by immediate recording of the 1H NMR spectrum gave rise to a sole set of signals attributable to 2 (Fig. 1g). This indicates that 2a/b exists only in the solution containing AcOH and that the evaporation of solvents leads to the release of AcOH and the restoration of dinuclear palladacyclic complex 2.Two possible structures of compound 2a/b can be proposed (Scheme 2). First, the clamshell dinuclear complex 2 is fragmented by adjacent acid to unprotonated monopalladium species 2b. In this case it can be expected that aliphatic signals are not split (or only very little) analogously to the ‘open-shell’ shaped chloride bridged dinuclear complexes.11 Second, confirmed by other evidence, there is a possibility of protonation of the basic N,N-dimethylamino group giving acyclic palladium species 2a, as is known e.g. from the chemistry of organostannanes.16 The validity of the protonation is confirmed not only by the chemical shift of the CH2 and N-(CH3)2 groups (Fig. 1fvs.Fig. 1a) but also by the observation that the amount of additional acid necessary for full conversion of 2 to 2a decreases with its strength or concentration (Fig. 1f and Fig. S2†). Thus, full conversion requires the addition of more than 50 equivalents of AcOH but less than 2 equivalents of much stronger TFA. Furthermore, the evaporation of 2a-TFA formed in the presence of stronger trifluoroacetic acid does not lead to the restoration of dinuclear complex 2 (Fig. 1h).
 | ||
| Scheme 2 Possible structures of compounds 2a/b. | ||
From the NMR and FTIR data it is not possible to distinguish the exact structure of 2a and whether it is mononuclear or dinuclear. The NMR analysis of 2a-TFA reveals the presence of a signal set corresponding to one trifluoroacetate anion. However, the exact stoichiometry of the “substrate vs. OTFA” cannot be determined. Furthermore, FTIR analysis of the powder does not show a band typical of nitriles in the region 2200–2300 cm−1 (Fig. S23†).
In any case, the occurrence of such an acyclic species 2a in acidic media may be interesting especially from a mechanistic point of view in studies of C–H functionalization, where its occurrence has not yet been considered and at the same time it can be an important reactive intermediate or catalyst resting state.
Finally, the time–conversion experiments (Fig. S3†) show that the addition of acetic acid (5 equiv.) causes a decrease in the reaction rate. However, further increasing the amount of acid has no longer such a significant effect. The addition of stronger acids (methoxyacetic or dichloroacetic) leads to a further reaction rate slowdown and full cyclopalladation is achieved in a few days.
Cyclopalladation in the presence of strong acids
This part of our work examines the influence of additional strong acid (TFA) on the regioselectivity and cyclopalladation outcome. First, the treatment of substrate 1 with 5 equivalents of TFA in DCM for 15 minutes (giving 1a) was followed by the addition of 1 equivalent of “Pd(OAc)2”. In this case, it can be assumed that the reactive palladium containing species are [Pd3(OAc)6−x(OTFA)x] (x = 1–6) formed by a sequential ligand exchange from [Pd3(OAc)6] prior to the reaction with 1a.17 The resulting (rapidly formed) oil solidified after trituration in hexane, giving yellow powder 4. After its dissolution in CD3CN the 1H NMR spectrum contains signals of three aromatic protons (two doublets and one singlet) indicating successful cyclopalladation. The palladation directed by the acetamido group is confirmed by e.g. the 1H and 13C-HMBC NMR spectra showing the correlation of the benzylic carbon atom with two aromatic protons (Fig. S20†). Furthermore, chemical shifts of N-CH3 and CH2 protons are almost identical to those of the protonated substrate 1a, whereas the signals of acetamido CH3 groups are downfield-shifted (Fig. S4†). However, the spectrum slowly changed in time (Fig. 3). Within a few hours signals of a new compound 3 appeared. These are signals of two aromatic protons accompanied by signals of the CH2 and N-(CH3)2 groups. After three days the change was complete reaching a ratio of prime to new signals of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.7. The analogous behavior results in a 19F NMR spectrum containing one broad signal whose intensity increased in time and four narrow signals in the region 74.5–75.5 ppm that slowly disappeared.
:0.7. The analogous behavior results in a 19F NMR spectrum containing one broad signal whose intensity increased in time and four narrow signals in the region 74.5–75.5 ppm that slowly disappeared.
 | ||
| Fig. 3 Changes with time of the important areas of the 19F and 1H NMR spectra of the solution of complex 4 in CD3CN. | ||
The probable explanation (Scheme 3) is that the original yellow complex formed in DCM is trinuclear palladacyclic complex 4,18 which is also indicated by elemental analysis. Its dissolution in CD3CN led to the rapid expulsion of “Pd(OTFA)2”, giving a dinuclear palladacyclic complex 3 (major signals in NMR). Such an instability of the trinuclear species in coordinating solvents and in the presence of nucleophiles was observed in our previous work.17 The formed palladacycle 3 slowly underwent a second C–H activation with liberated “Pd(OTFA)2(AN)2” (narrow signals in 19F NMR) directed by the N,N-dimethylamino group (there was no additional free acid present in this case), giving doubly activated compound 5. The structure of the complex 5 was determined using single-crystal X-ray diffraction (Scheme 4.). The results show its existence in the [2.2]-dipalladaparacyclophane form containing two doubly activated C–H units connected by four bridging acetate ligands. The addition of a higher amount of TFA to the reaction mixture caused a slowdown of the reaction rate.
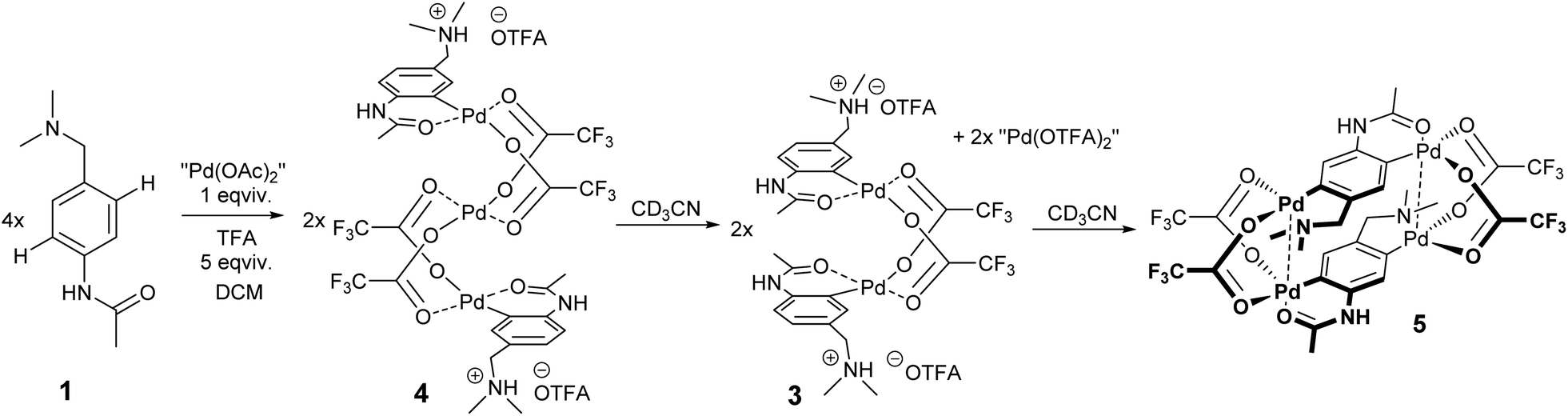 | ||
| Scheme 3 Probable structure of complex 4 and its transformation in CD3CN. | ||
 | ||
| Scheme 4 Scheme of one pot double functionalization (top). The ORTEP structures of double activated complex 5. Thermal ellipsoids are set at 40% probability; most of the hydrogen atoms and minor disordered parts of CF3 groups are omitted for clarity. Selected interatomic distances (Å) and angles (°): Pd1–C1 1.956 (5), Pd1–O1 1.986(4), Pd1–O5i 2.073(3), Pd1–O2 2.169(3), Pd1–Pd2i 3.0074(5), Pd2–C4 1.960(5), Pd2–O4 2.058(4), Pd2–N2 2.064(5), Pd2–O3i 2.197(4), C1–Pd1–O1 91.91(18), C1–Pd1–O5i 95.46(18), O1–Pd1–O5i 170.47(14), C1–Pd1–O2 178.55(18), O1 –Pd1–O2 86.68(14), O5i–Pd1–O2 85.98(13), C1–Pd1–Pd2i 99.53(14), O1–Pd1–Pd2i 106.01(12), O5i–Pd1–Pd2i 78.75(10), O2–Pd1–Pd2i 80.56(9), C4–Pd2–O4 95.00(18), C4–Pd2–N2 81.6(2), O4–Pd2–N2 174.40(16), C4–Pd2–O3i 176.60(18), O4–Pd2–O3i 86.09(15), N2–Pd2–O3i 97.54(17), C4–Pd2–Pd1i 97.62(14), O4–Pd2–Pd1i 79.89(10), N2–Pd2–Pd1i 104.92(14), O3i–Pd2–Pd1i 79.38(10); Symmetry code: (i) −x + 1, −y + 1, −z + 1. | ||
Interestingly, the change of the solvent from DCM to acetonitrile caused a dramatic effect on the reaction course. First, the reaction slowed down significantly. At the beginning, only 1H NMR signals of protonated substrate 1a-TFA were present (Fig. S5†). Surprisingly, within a few days, signals of the complex 2a-TFA began to appear. Thus, in acetonitrile the C–H activation is directed solely by the N,N-dimethylamino group even in acidic media. Over time (weeks), the signals of 2a-TFA were accompanied by signals from substance 5.
We currently have no clear explanation for this. The result of the NMR titration speaks against the possibility of a fundamentally different basicity of the individual DGs of both solvents (Fig. S6†); however, it cannot be fully excluded. Titration of substrate 1 with TFA in both solvents leads to similar changes in the chemical shifts of individual NMR signals. Another possible explanation is the different structure (nuclearity) and thus the reactivity of palladium trifluoroacetate in dichloromethane and acetonitrile. While we have confirmed16 the retention of the trinuclear cyclic structure in dichloromethane, this may not be the case in acetonitrile. However, a detailed explanation will require further work.
The double C–H activation giving complex 5 can be achieved by an alternative way (Scheme 4). First, an equimolar amount of 1 was reacted with “Pd(OAc)2” in DCM. After 30 minutes the second equivalent of “Pd(OAc)2” dissolved in DCM containing 5 equivalents of TFA was added. The 1H NMR spectrum of the formed yellow precipitate 5 (70% yield) shows signals of only two aromatic protons and the chemical shifts are analogous/identical to the chemical shifts shown in Fig. 3. Furthermore, a small number of broad signals appear in the 1H NMR spectra, probably belonging to the polymer structure of the doubly activated species.
With respect to the structure of 2, 5 is also centrosymmetric but tetranuclear with the doubly deprotonated (palladated) ligands in para positions. These two ligands with an opposite orientation of the N,N-dimethylamino and acetamide groups bridge the dinuclear units. The major difference from the published structures of a similar kind is the fact the C,N- and C,O-chelates have the same orientation of donors within the dinuclear moiety, while the donors in the rest of forty-five dinuclear species containing two carboxylates and C,N- or C,O-chelating ligands are in opposite positions. Nevertheless, the most interesting fact is that the ligands form a doubly bridged structure, which resembles the structural motif of [2.2]paracyclophane. Such a prototype of a unique tetrametalla [2.2]paracyclophane structure was described only for one tetragermanium compound19 and a very few9 analogues of palladium species containing e.g. the 1,4-bis-(benzothiazol-2-yl)phenylene ligand,9e whereas all of these are based on symmetric ligands. There is a direct comparison between 5 and the former, where the Pd–Pd separation is shorter by ∼0.2 Å, and the π–π stacking interaction between ligands of 3.613 Å in 5 is a bit longer than the 3.418 Å in the published structure as a result of flexible donor parts versus completely flat ones, which allow the planes to get closer.
DFT calculations
DFT calculations were employed to gain further insights into the observed trends. Therefore, we calculated20 and compared simplified energy profiles for C–H activation directed by both directing groups (Fig. 4). For simplicity, the reaction profiles contain only pre-complexes (pre), intermediates (int) and transition states (TS).21 Furthermore, it can be assumed that a relative comparison of profiles is not strongly affected by the nuclearity of the reaction species;22 therefore, only mononuclear species are involved in the calculations. | ||
| Fig. 4 Comparison of simplified reaction profiles (relative Gibbs energies in DCM at 298 K in kcal mol−1) of the C–H activation of 1 directed by N,N-dimethylamino (blue) and acetamido (red) groups. | ||
The comparison of relative energies of pre-complexes (pre) shows a strong preference for the formation of the N,N-dimethylamino coordinated one. On the other hand, the energy differences between pre-complexes (pre) and transition states (TS) in both profiles are almost identical (15.2 vs. 14.6 kcal mol−1). It follows that preferential coordination of palladium by the DG and not C–H-activation itself is responsible for the different directing group abilities.5 This offers a complement to the work of Norrby et al.,5a who used relative stabilities of formed palladacycles for the determination of DG abilities, or to the work of Lapkin.23
Conclusions
This work demonstrates that it is possible to switch the site-selectivity of the cyclopalladation reaction of substrates containing two different directing groups using the acidity of the environment. However, it depends on factors such as solvent or nuclearity. Furthermore, the work proves the existence of a protonated palladacyclic compound 2a and its possible application as a reaction intermediate in C–H functionalization reactions. Last but not least, directed stepwise double cyclopalladation enables the synthesis of “non-symmetrical” [2.2]-dipalladaparacyclophanes.Author contributions
T. K., J. B. and L. M.: investigation; A. R:. crystallography and writing – review & editing; J. H.: writing – review & editing; and J. V.: conceptualization, investigation and writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The University of Pardubice is greatly acknowledged for financial support.References
- Palladacycles, ed. J. Dupont and M. Pfeffer, 2008WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, ISBN: 978-3-527-31781-3 Search PubMed.
- (a) A. Kapdi and D. Maiti, Strategies for Palladium-Catalyzed Non-directed and Directed C-H Bond Functionalization, Elsevier 2017, Paperback ISBN: 9780128052549 Search PubMed; (b) T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, C-H activation, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef CAS.
- (a) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Transition metal-catalyzed C–H bond functionalizations by the use of diverse directing groups, Org. Chem. Front., 2015, 2, 1107 RSC; (b) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry, Chem. Soc. Rev., 2018, 47, 6603 RSC.
- S. R. Neufeldt and M. S. Sanford, Controlling Site Selectivity in Palladium-Catalyzed C–H Bond Functionalization, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed.
- (a) A. Tomberg, M. E. Muratore, M. J. Johansson, I. Terstiege, C. Sköld and P.-O. Norrby, Relative Strength of Common Directing Groups in Palladium-Catalyzed Aromatic C-H Activation, iScience, 2019, 20, 373 CrossRef CAS PubMed; (b) L. V. Desai, K. J. Stowers and M. S. Sanford, Insights into Directing Group Ability in Palladium-Catalyzed C−H Bond Functionalization, J. Am. Chem. Soc., 2008, 130, 13285 CrossRef CAS PubMed; (c) X. Sun, G. Shan, Y. Sun and Y. Rao, Regio- and Chemoselective C-H Chlorination/Bromination of Electron-Deficient Arenes by Weak Coordination and Study of Relative Directing-Group Abilities, Angew. Chem., Int. Ed., 2013, 52, 4440 CrossRef CAS PubMed; (d) D. S. Timofeeva, D. M. Lindsay, W. J. Kerr and D. J. Nelson, A quantitative empirical directing group scale for selectivity in iridium-catalysed hydrogen isotope exchange reactions, Catal. Sci. Technol., 2020, 10, 7249 RSC; (e) J. McIntyre, I. Mayoral-Soler, P. Salvador, A. Poater and D. J. Nelson, Insights into mechanism and selectivity in ruthenium(II)-catalysed ortho-arylation reactions directed by Lewis basic groups, Catal. Sci. Technol., 2018, 8, 3174 RSC.
- (a) L. Zhang and T. Ritter, A Perspective on Late-Stage Aromatic C-H Bond Functionalization, J. Am. Chem. Soc., 2022, 144, 2399 CrossRef CAS PubMed; (b) J. Börgel and T. Ritter, Late-Stage Functionalization, Chem, 2020, 1877 CrossRef.
- J. Váňa, J. Bartáček, J. Hanusek, J. Roithová and M. Sedlák, C–H Functionalizations by Palladium Carboxylates: The Acid Effect, J. Org. Chem., 2019, 84, 12746 CrossRef PubMed.
- G. Cai, Y. Fu, Y. Li, X. Wan and Z. Shi, Indirect ortho Functionalization of Substituted Toluenes through ortho Olefination of N,N-Dimethylbenzylamines Tuned by the Acidity of Reaction Conditions, J. Am. Chem. Soc., 2007, 129, 7666 CrossRef CAS PubMed.
- (a) B. J. O'Keefe and P. J. Steel, Cyclometalated Compounds. 10.1 Preparation and Crystal Structure of a Nonpolymeric, Acetate-Bridged, Multiply Cyclopalladated Compound, Organometallics, 1998, 17, 3621 CrossRef; (b) B. J. O'Keefe and P. J. Steel, Cyclometalated Compounds. XVI.1 Double Cyclopalladations of Bis(2-pyridyloxy)naphthalenes. Kinetic versus Thermodynamic Control of Regiospecificity, Organometallics, 2003, 22, 1281 CrossRef; (c) A. G. Panova, K. A. Radyushin and K. P. Balashev, Cyclopalladated complexes based on 2-phenylbenzothiazole and 1,4-(benzothiazol-2-yl)benzene with acetate ligands and ethylenediamine, Russ. J. Gen. Chem., 2011, 81, 743 CrossRef CAS; (d) D. J. de Geest, B. J. O'Keefe and P. J. Steel, Cyclometallated compounds. XIII. Cyclopalladation of 2-phenoxypyridine and structurally-related compounds, J. Organomet. Chem., 1999, 79, 97 CrossRef; (e) A. Bjelopetrović, M. Robić, I. Halasz, D. Babić, M. J. Kulcsár and M. Ćurić, Facile Mechanochemical Anion Substitution in Cyclopalladated Azo-Benzenes, Organometallics, 2019, 38(22), 4479 CrossRef.
- Predicted by https://chemaxon.com/products/marvin.
- J. Milani, N. E. Pridmore, A. C. Whitwood, I. J. S. Fairlamb and R. N. Perutz, The Role of Fluorine Substituents in the Regioselectivity of Intramolecular C–H Bond Functionalization of Benzylamines at Palladium(II), Organometallics, 2015, 34, 4376 CrossRef CAS.
- J. E. Bercaw, A. C. Durrell, H. B. Gray, J. C. Green, N. Hazari, J. A. Labinger and J. R. Winkler, Electronic Structures of PdII Dimers, Inorg. Chem., 2010, 49, 1801 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed.
- J. Albert, J. Granell, R. Qadir, J. Quirante, C. Calvis, R. Messeguer, J. Badia, L. Baldoma, M. Font-Bardia and T. Calvet, Cyclopalladated Benzophenone Imines: Synthesis, Antitumor Activity, Cell Accumulation, DNA Interaction, and Cathepsin B Inhibition, Organometallics, 2014, 33, 7284 CrossRef CAS.
- D. Powers and T. R. Ritter, Bimetallic Pd(III) complexes in palladium-catalysed carbon–heteroatom bond formation, Nat. Chem., 2009, 1, 302 CrossRef CAS PubMed.
- P. Švec, E. Černošková, Z. Padělková, A. Růžička and J. Holeček, Tri- and diorganostannates containing 2-(N,N-dimethylaminomethyl)phenyl ligand, J. Organomet. Chem., 2010, 695, 2475 CrossRef.
- J. Váňa, J. Lang, M. Šoltésová, J. Hanusek, A. Růžička, M. Sedlák and J. Roithová, The role of trinuclear species in a palladium acetate/trifluoroacetic acid catalytic system, Dalton Trans., 2017, 46, 16269 RSC.
- I. J. S. Fairlamb, J. Lang, A. Růžička, M. Sedlák and J. Váňa, Direct Cyclopalladation of Fluorinated Benzyl Amines by Pd3(OAc)6: The Coexistence of Multinuclear Pdn Reaction Pathways Highlights the Importance of Pd Speciation in C–H Bond Activation, Organometallics, 2023 DOI:10.1021/acs.organomet.3c00178.
- A. Sekiguchi, T. Yatabe, C. Kabuto and H. Sakurai, 1,1,2,2,9,9,10,10-Octamethyl-1,2,9,10-tetragerma[2.2] paracyclophane: the first [2.2]paracyclophane bridged by two GeGe bonds, J. Organomet. Chem., 1990, 390, c27 CrossRef CAS.
- Initial geometries of the reaction centers were taken from: (a) J. Váňa, V. Petrović, T. Terencio, O. Tischler, Z. Novák and J. Roithová, Palladium-Catalyzed C−H Activation: Mass Spectrometric Approach to Reaction Kinetics in Solution, Organometallics, 2017, 36, 2072 CrossRef; (b) D. L. Davies, S. M. A. Donald and S. A. Macgregor, Computational Study of the Mechanism of Cyclometalation by Palladium Acetate, J. Am. Chem. Soc., 2005, 127, 13754 CrossRef CAS PubMed.
- There is still a lively discussion about the classification of reaction mechanisms (AMLA/CMD/BIES) and the nomenclature of intermediates (agostic, Wheeland, σ-, π-, and σ-π continuum complexes) of C-H activation reactions (a) D. L. Davies, S. A. Macgregor and C. L. McMullin, Computational Studies of Carboxylate-Assisted C−H Activation and Functionalization at Group 8–10 Transition Metal Centers, Chem. Rev., 2017, 117, 8649 CrossRef CAS PubMed; (b) K.-M. Altus and J. A. Love, The continuum of carbon–hydrogen (C–H) activation mechanisms and terminology, Commun. Chem., 2021, 4, 173 CrossRef PubMed; (c) T. Rogge, J. C. A. Oliveira, R. Kuniyil, L. Hu and L. Ackermann, Reactivity-Controlling Factors in Carboxylate-Assisted C–H Activation under 4d and 3d Transition Metal Catalysis, ACS Catal., 2020, 10(18), 10551 CrossRef CAS.
- J. Váňa, J. Hanusek and M. Sedlák, Bi and trinuclear complexes in palladium carboxylate-assisted C–H activation reactions, Dalton Trans., 2018, 47, 1378 RSC.
- L. Cao, M. Kabeshov, S. V. Ley and A. A. Lapkin, In silico rationalisation of selectivity and reactivity in Pd-catalysed C–H activation reactions, Beilstein J. Org. Chem., 2020, 16, 1465 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra and DFT calculation results. CCDC 2241934 and 2241935. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02019c |
| This journal is © The Royal Society of Chemistry 2023 |