
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVapochromic behaviour of a gold(I)–lead(II) complex as a VOC sensor†
Sonia
Moreno
,
David
Royo
,
Abdel G.
El-Hachimi
 ,
María
Rodríguez-Castillo
,
Miguel
Monge
,
M. Elena
Olmos
* and
José M.
López-de-Luzuriaga
*
,
María
Rodríguez-Castillo
,
Miguel
Monge
,
M. Elena
Olmos
* and
José M.
López-de-Luzuriaga
*
Departamento de Química, Universidad de La Rioja, Centro de Investigación en Síntesis Química (CISQ), Complejo Científico-Tecnológico, 26006 – Logroño, Spain. E-mail: josemaria.lopez@unirioja.es; m-elena.olmos@unirioja.es
First published on 21st September 2023
Abstract
The reaction among [Au2Ag2(C6F5)4(OEt2)2]n, PbCl2 and terpyridine leads to the polymeric complex [{Au(C6F5)2}2{Pb(terpy)}]n (1). Its crystal structure reveals potential voids close to the lead centres large enough to hold different molecules. The availability of these free sites allows complex 1 to act as a VOC sensor. Thus, when 1 is exposed to different solvent vapours such as acetonitrile, toluene or THF, variations in its solid appearance and its photophysical properties are observed as a consequence of the formation of the new polymorphs [{Au(C6F5)2}2{Pb(terpy)(CH3CN)2}]n (2), [{Au(C6F5)2}2{Pb(terpy)}]n·Tol (3) and [{Au(C6F5)2}2{Pb(terpy)(THF)}]n·THF (4). Each polymorph displays a different emission energy depending on its structure and the presence of metallophilic interactions. In addition, the reversible solvent molecule exchange allows the tuning of the luminescence emissions in the greenish yellow–red range. DFT and TD-DFT calculations were performed to explain the origin of the luminescence of all these complexes.
Introduction
During the last few years, several studies in several laboratories1 have shown that heterometallic complexes containing Au(I) and other closed shell metal ions are responsible for interesting optical properties.2–6 In them, it has been observed that the luminescent behaviour of the complexes is associated with the presence of different Au⋯M or/and Au⋯Au interactions in their solid state structures and also serves as an indication of the presence of these interactions even in solution.7,8 Additionally, the number and type of ligands also influence the geometry and electronics of the metal centres and, therefore, the emissive character of the complexes.On the other hand, McCrone defined “a solid crystalline phase of a given compound resulting from the possibility of at least two different arrangements of the molecules of that compound in the solid state” as polymorphism.9 This situation can obviously affect the optical properties of the materials depending on the different arrangements of metals or on the interactions between them in each polymorph. One special class of polymorphism called solvatopolymorphism is present in systems where the crystal structures are defined by different unit cells that differ in their elemental composition by means of the inclusion of one or more solvent molecules.10
The presence of solvent molecules in the crystal packing should not affect the optical behaviour whether there is no metal⋯solvent interaction (neither increasing their coordination numbers nor varying their geometries) or even if they exist when they do not influence the strength of the intermetallic interactions or other weak noncovalent bonds, including hydrogen bonds, π-stacking or van der Waals interactions;11 nevertheless, the introduction of these molecules into the network as solvation molecules in solution or as vapours frequently leads to important modifications in the crystal packing, producing changes in these interactions and, as a consequence, in their colours, which are often perceptible to the human eye, and even deeper under UV light. This situation is particularly interesting since these can find application as smart materials such as gas sensors and optical fibre sensors for volatile organic compound detection.
Although in the last few years there has been an increasing number of metallic complexes and materials dealing with this topic, especially in the case of gold-containing compounds, they offer a poorly explored, but fertile area of work, based on the facility of drawing together gold atoms to sub-van der Waals distances in their complexes, interactions that are strongly influenced by the environment and that greatly modify the photophysical properties of the aggregates. This was, for instance, the case of the pioneering well-known Eisenberg's dithiocarbamate gold(I) complex [Au(S2CN(C5H11)2)]2, in which the presence of polar aprotic solvents causes its polymerization through Au⋯Au contacts between dimers, being completely reversible by drying.12 Since then, a number of reports on this topic have been published,13,14 but most of the complexes described show this behaviour in an irreversible way, or exclusively for one specific solvent/vapour. A few reports described complexes sensitive to several vapours and these, at the same time, show reversibility under mild conditions. This is the case of the heteronuclear gold–copper complex described by Koshevoy et al. in which up to four different crystalline forms are obtained when the alkynyl complex [Au2Cu2(C2OHC5H8)4]n is crystallized from methanol; ethanol, acetone or chloroform; toluene; and diethyl ether or ethyl acetate solutions, each polymorph showing different photoluminescence characteristics and some of them being interconvertible upon exposure to vapours of other solvents.13a
In spite of the interest that this research is attracting, there are still many questions to respond and more examples are needed to try to rationalize the mechanisms of this behaviour. In fact, the exhaustive control of polymorph formation has been recognized as an important aim for the production of functional solid materials.
With regard to our ongoing studies on the optical properties associated with heteronuclear gold–heterometal materials, one of the main goals is the analysis of the influence of external stimuli on their optical behaviour. In our case, we have contributed with several examples of this type of behaviour in heteronuclear complexes. Thus, for instance, the mixed gold(I)–thallium(I) complex [AuTl(C6Cl5)2]n, reported by some of us,13b,c reacts with different volatile organic compounds that covalently bond to the thallium centres, which produces visible changes that are completely reversible by moderate heating at 100 °C. Other examples are those of a series of gold(I)–silver(I) derivatives of stoichiometry [Au2Ag2(C6X5)2L2]n (X = halogen; L = neutral ligand),13d–f in which reversible or irreversible conversions were dependent on the donor affinity of the different vapours to silver and the nature of the perhalophenyl ligands.
In addition, we have recently contributed a number of works in which we analyse the optical properties of heterometallic complexes showing Au(I)⋯Pb(II) interactions.15 The number of reported complexes that present Au(I)⋯Pb(II) interactions is very scarce and, in particular, studies on the influence of external stimuli, such as vapours, solvents, temperature or pressure, on these derivatives have not been published so far.
Therefore, we focused our efforts on this last field of research, and in this paper we report the synthesis, structural characterization, reversible response to solvents and their vapours as well as a computational study of the complex [{Au(C6F5)2}2{Pb(terpy)}]n (1) and its four solvatopolymorphs containing acetonitrile (2), toluene (3), tetrahydrofuran (4) or benzonitrile (5).
Results and discussion
Synthesis and characterization
Complex [{Au(C6F5)2}2{Pb(terpy)}]n (1) was prepared according to the literature procedure.16 Complex 1 was obtained by reaction of the heterometallic polymer [Au2Ag2(C6F5)4(OEt2)2]n with PbCl2 and terpyridine in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 (Au:Pb:Terpy) molar ratio, using methanol as the solvent. Complex 1 is stable to air and moisture for long periods. It is soluble in MeOH and slightly soluble in most common organic solvents such as dichloromethane, acetonitrile, toluene, THF or benzonitrile, but insoluble in diethyl ether or n-hexane.
:1:1 (Au:Pb:Terpy) molar ratio, using methanol as the solvent. Complex 1 is stable to air and moisture for long periods. It is soluble in MeOH and slightly soluble in most common organic solvents such as dichloromethane, acetonitrile, toluene, THF or benzonitrile, but insoluble in diethyl ether or n-hexane.
Treatment of complex 1 with solvents with different donor abilities leads to new heterometallic Au(I)–Pb(II) complexes displaying the same Au–Pb–Au metallic sequence, but a new coordination environment for the Pb(II) atom. Thus, the addition of acetonitrile, toluene, tetrahydrofuran or benzonitrile gives rise to polymorphs 2–5, respectively (Scheme 1), the latter recently published by us.17 By contrast, solvents and vapours such as diethyl ether and n-hexane, wherein the complex is not soluble, do not provoke detectable changes in it.
 | ||
| Scheme 1 Syntheses of complexes 2–5. | ||
All the analytical and spectroscopic data of the new complexes agree with the proposed stoichiometries. Thus, for all complexes, the IR spectra show, among others, the absorption bands related to the presence of the [Au(C6F5)2]− units close to 1505, 955 and 770 cm−1 and those due to the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) stretching vibrations arising from the terpy ligand, appearing around 1590 cm−1. In the case of polymorph 2, an additional band associated with the ν(C
N) stretching vibrations arising from the terpy ligand, appearing around 1590 cm−1. In the case of polymorph 2, an additional band associated with the ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) stretching vibrations from the acetonitrile molecules is observed at 2247 (2) cm−1 (Fig. S2–S4†).
N) stretching vibrations from the acetonitrile molecules is observed at 2247 (2) cm−1 (Fig. S2–S4†).
The molar conductivity measurements of complexes 2–4 in acetone show values corresponding to the 2:1 electrolyte, suggesting complete dissociation of their ionic counterparts, two [Au(C6F5)2]− anions and a [Pb(terpy)]2+ cation, and the loss of the metallophilic interactions in solution, in a similar way that has been described for 116 and 5.17
Regarding their MALDI(−) mass spectra, they show a peak corresponding to the [Au(C6F5)2]− unit at m/z = 530, and in their MALDI(+) mass spectra a peak corresponding to the [{Au(C6F5)2}{Pb(terpy)}]+ fragment is detected at m/z = 972.
The 1H NMR spectra of all complexes in [D6]-DMSO show six signals corresponding to the terpyridine ligand in the range 8.76–7.47 ppm. In addition, the signals related to solvent molecules are also observed (Fig. S5–S7†).
Finally, the 19F NMR spectra of complexes 2–4 (Fig. S8–S10†) display signals corresponding to the C6F5 groups bonded to gold(I) at −114.6 (m, Fo), −161.5/−161.7 (t, Fp) and −162.8/−163.0 (m, Fm) ppm, with chemical shifts very similar to those of the precursor NBu4[Au(C6F5)2], suggesting the rupture of the Au⋯Pb metallophilic interactions in solution.
X-ray crystal structure determination
The crystal structure of complex 1 has been previously described in the literature.16 However, it will be also commented on in this work for comparative purposes. An in-depth study of the 3D structural arrangement of complex 1 revealed the presence of empty channels that run parallel to the crystallographic y axis with interatom distances of around 3.765 Å (Fig. 1) and diameters close to 17.6 Å, which are large enough to accommodate small molecules. In fact, the slow evaporation of acetonitrile, toluene or tetrahydrofuran solutions of complex 1 leads to the growth of single crystals adequate for their study by X-ray diffraction techniques revealing the filling of the empty channels by solvent molecules. | ||
| Fig. 1 Crystal structure of 1 viewed down the crystallographic y axis. | ||
Compounds 2–5 display a similar structural arrangement concerning the trinuclear [{Au(C6F5)2}{Pb(terpy)}{Au(C6F5)2}] building blocks. The X-ray structure of complex 5 has been previously reported;17 but it is briefly discussed for comparative purposes. Both gold(I) atoms within the trinuclear Au2Pb unit are linearly coordinated to two pentafluorophenyl groups with an additional (and rather uncommon) Au⋯Pb metallophilic interaction. These heterometallic interactions are quite similar for all the complexes with Au–Pb distances of 2.8964(5)–2.9670(5) (1), 2.9740(5)–2.9825(5) (2), 2.8470(14) and 2.8856(9) (3), 2.9262(3) and 2.9645(3) Å (4), and 2.8349(4) and 2.8646(4) Å (5). These Au–Pb distances compare well with those found in the decanuclear Au6Pb4 complex [{Au(C6F5)2}6{PbCl(terpy)}2{Pb(terpy)}2] where the atom trans to gold is another gold centre (2.8776(9)–2.9332(9) Å),16 and to those observed for [Au2Pb(CH2P(S)Ph2)4] (2.896(1) and 2.963(2) Å),18 in which the Au⋯Pb contacts are supported by the P,S-donor bridging ligands. In contrast, the Au–Pb distances are longer when the atom trans to gold is chlorine, as observed in the related compounds [{Au(C6F5)2}6{PbCl(terpy)}2{Pb(terpy)}2] (3.2719(9); 3.2183(9) Å) and [{Au(C6F5)2}{PbCl(terpy)}] (3.2957(6) Å).16
With the only exception of compound 4, this trinuclear Au2Pb units are held together through aurophilic interactions of different strengths [Au–Au distances of 3.1315(5) and 3.3060(5) Å in 1; 3.3626(7), 3.4345(7) and 3.5413(7) Å in 2; 2.9309(13) Å in 3, and 2.8920(5) Å in 5], thus forming polymeric chains (Fig. 2). In contrast, in the case of 4, the shortest Au–Au distance (5.818 Å) is too long to consider any kind of aurophilic interaction;19 therefore, the crystal structure of complex 4 can be considered as formed by discrete molecules (Fig. 3).
 | ||
| Fig. 2 Crystal structures of complexes 2 (left) and 3 (right). Selected bond lengths (Å) and angles (°) of complex 2: Au(1)–Pb = 2.9827(5), Au(2)–Pb = 2.9781(5), Au(1)–Au(1)#1 = 3.4343(5), Au(2)–Au(2)#1 = 3.3620(7), Pb–N(3) = 3.314, Au(1)–Pb–Au(2) = 177.251(15), Pb–Au(2)–Au(2)#1 = 158.693(19). Selected bond lengths (Å) and angles (°) of complex 3: Au(1)–Pb = 2.8446(15), Au(2)–Pb = 2.8878(11), Au(1)–Au(2) = 2.9315(15), Au(1)–Pb–Au(2) = 160.21(4), Pb–Au(2)–Au(1) = 176.98(5). | ||
 | ||
| Fig. 3 Crystal structure of complex 4. Selected bond lengths (Å) and angles (°): Au(1)–Pb = 2.9645(3), Au(2)–Pb = 2.9263(3), Pb–O = 2.787, and Au(1)–Pb–Au(2) = 163.965(7). | ||
In all the cases, the intermetallic contacts are reinforced by π–π interactions of different strengths that appear between pentafluorophenyl groups and/or between aromatic rings of perhalophenyl and terpyridine ligands, all of them with centroid–centroid distances within the range of π-stacking interactions.20
The lead atom within each trinuclear Au2Pb unit, in addition to displaying two Pb⋯Au metallophilic interactions, is symmetrically coordinated to the three nitrogen atoms of a terpy ligand with Pb–N bond lengths in the ranges of 2.490(7)–2.548(7) Å in 2, 2.457(14)–2.488(19) Å in 3, and 2.514(3)–2.566(3) Å in 4, with longer distances found in the non-polymeric complex 4. All these values are similar to those found in 1 (2.451(6)–2.526(6) Å) and 5 (2.450(7)–2.532(11) Å), and lie within the usual values found for hemidirected Pb(II) complexes21 containing terpy or terpyridine-type ligands.22
One of the main structural differences among the polymorphs described in this study is found in the inclusion of solvent molecules to 1, which can either be incorporated into the coordination sphere of Pb(II) or stay in its immediate surroundings. Thus, for complex 2, two acetonitrile molecules are weakly connected to each lead(II) centre with dissimilar Pb–N distances of 3.256(1) and 3.538(1) Å, generating a distorted pentagonal bipyramid environment for the lead atom, in which the inert pair is stereochemically inactive, with the gold centres in the apical positions (Fig. 2 left and S11†). On the other hand, complex 3 incorporates a toluene molecule per trinuclear unit, which is only interconnected through C–H⋯F hydrogen bonds (Fig. S12†), while no π–π interactions are found between the solvent and the aromatic rings present in the heterometallic compound. In the structure of polymorph 4, two THF molecules are present per trinuclear unit, but while one of them is bonded to the lead(II) atom (Pb–O = 2.786(3) Å), the second one just acts as a crystallization solvent molecule (Fig. 3 and S13†). Finally, complex 5, as previously discussed in detail in the literature,17 displays one disordered benzonitrile molecule per trinuclear fragment with a different percentage of coordinated and non-coordinated solvent molecules depending on the external pressure or temperature.
On the other hand, the differences found in the Au–Pb–Au and Au–Au–Pb angles within the heterometallic chains of the solvate-containing compounds 2–5 are also worth noting (Fig. 4). Thus, the Au–Au–Pb angles in 3 and 5 are approximately linear (168.07(4) and 176.93(4)° in 3, and 170.13(1) and 175.66(1)° in 5), while the Au–Pb–Au angles in the structures of compounds 3–5 are narrower (160.26(3)° in 3, 164.0(1)° in 4, and 156.87(2)° in 5). In contrast, an opposite trend is found in the crystal structure of complex 2, which shows Au–Au–Pb angles in the range 155.64(2)–158.70(2)°, and values of 177.26(1) and 177.53(1)° for the Au–Pb–Au angles, that is, the lead atoms are closer to linearity than the gold ones in the case of 2.
 | ||
| Fig. 4 Metallic chain skeleton of compounds 2 (up), 3 (centre) and 5 (bottom). Colour codes: gold: yellow; lead: dark grey. | ||
Optical properties
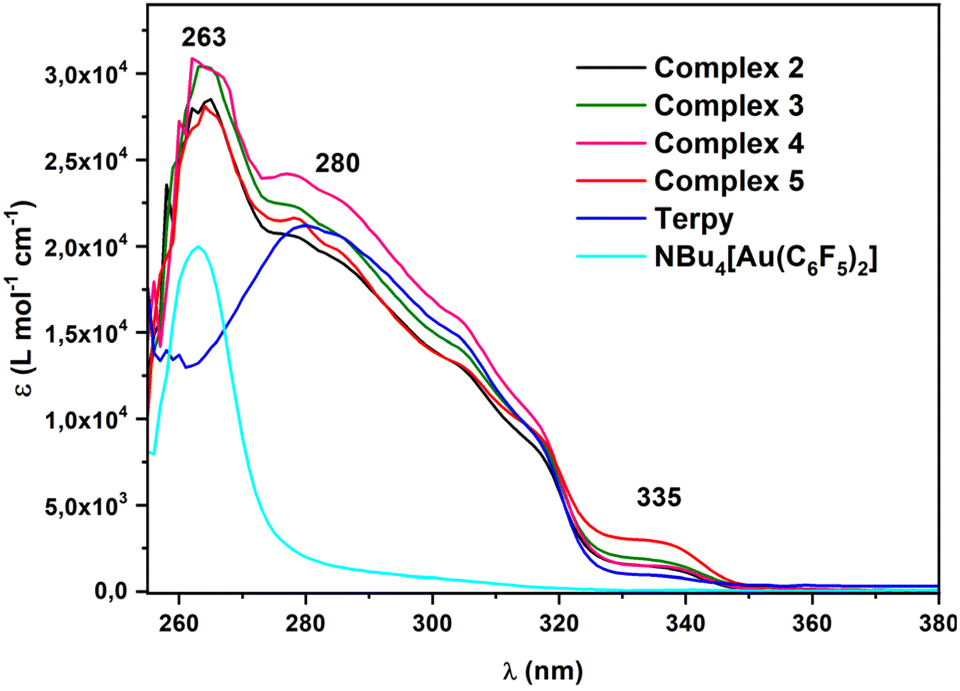 | ||
| Fig. 5 UV-vis absorption spectra of compounds 2–5, the gold(I) precursor NBu4[Au(C6F5)2] and the terpyridine ligand in DMSO. | ||
In summary, due to dissociation in solution, the spectra of all the complexes are similar and can be considered the sum of the Au precursor and the terpy ligand.
On the other hand, their solid-state absorption spectra (Fig. 6) display interesting features, because, in addition to a strong and wide absorption band between 260 and 350 nm, a lower energy absorption also appears. Besides, the absorption spectra corresponding to the gold(I) precursor NBu4[Au(C6F5)2] and to the terpyridine ligand show no absorption at similar values, which seems to indicate that these transitions are influenced by the gold–gold or gold–lead interactions.
 | ||
| Fig. 6 UV-vis absorption spectra of compounds 2–5, the gold(I) precursor NBu4[Au(C6F5)2] and the terpyridine ligand in the solid state. | ||
This is in accordance with the shift of the lower energy band depending on the strength of the metallophilic interactions observed in their crystal structures. Thus, in the spectrum of 5, which shows the shortest Au–Au and Au–Pb distances, this absorption is located at the longest wavelength.
 | ||
| Fig. 7 Photographs of the solid colour (up) and luminescence (bottom) of compounds 1–5. | ||
 | ||
| Fig. 8 Excitation and emission spectra in the solid state at room temperature (black) and 77 K (red) for complexes 2 (up, left), 3 (up, right), 4 (down, left) and 5 (down, right). | ||
By contrast, the excitation spectra resemble those of the absorption spectra in the solid state, indicating that these absorptions give rise to the observed emissions. Thus, in the solid state at room temperature, complex 2 shows a greenish yellow luminescence with an emission band maximum at 550 nm upon excitation at 460 nm. Complexes 3 and 5 exhibit a dark red luminescence almost imperceptible to the human eye because it is at the limit of the UV region, due to emission maximum bands at 770 (3) and 780 (5) nm (excitation at 670 (3) and 725 (5) nm). The remaining complex with the O-donor solvent (4) shows the greatest displacement with respect to compound 1, with very intense green luminescence with an emission band at 530 nm upon excitation at 320 or 400 nm.
The large Stokes shifts and emission lifetime measurements both at room temperature and at 77 K in the microsecond range allow us to suggest that these transitions are based on phosphorescence processes (Fig. S29–S35†). As can be seen in Table 1, the values obtained for lifetimes at 77 K are higher than those obtained at room temperature for all compounds. Quantum yield measurements of complexes 2–5 in the solid state at room temperature exhibit different emission quantum yields with values of 43 (2), 26 (3), 98 (4) and 25% (5).
| Complex | 2 | 3 | 4 | 5 |
|---|---|---|---|---|
| τ (μs) (RT) | 0.853 | 0.226 | 2.592 | 0.286 |
| τ (μs) (77 K) | 1.214 | 1.734 | 4.021 | — |
| Φ (%, RT) | 43 | 26 | 98 | 25 |
Finally, at 77 K, the emission bands shift to lower energies in all the cases. The red emission shifts observed at 77 K are common in heteronuclear derivatives displaying unsupported intermetallic interactions, in which the metallic centres are involved in the transitions responsible for the emissive properties. This has been repeatedly justified as a consequence of the thermal contraction of the structure at 77 K that reduces the metal–metal distances and, hence, the HOMO–LUMO gap. Thus, complex 2 exhibits a shift in its emission band from 550 nm (excitation at 460 nm) to 580 nm (excitation at 480 nm); complexes 3 and 5 show an infrared emission band at 882 or 940 nm (770 or 780 nm at RT), respectively, upon excitation at 760 (3) or 800 nm (5). Finally, the emission band in complex 4 shifts from 530 (excitation at 320 or 400 nm) to 550 nm (excitation at 325 or 425 nm) when lowering the temperature.
The powder diffraction patterns of derivatives 2–5 differ from that obtained for starting compound 1 (Fig. S15–S19†), indicating the complete insertion of the VOCs into the complex, whether coordinated or not, unequivocally confirming the ability of 1 to react in the solid–gas phase with these solvents when acting as VOCs, and demonstrating that a change in the structure of the complex occurs.
Therefore, we carried out an X-ray powder diffraction study in order to analyse the reversibility of the exchange process of five different solvents (methanol, acetonitrile, toluene, tetrahydrofuran and benzonitrile) when compound 1 is subjected to solvent vapours (Fig. S20–S28†).
Thus, upon subjecting any of these compounds to methanol vapours for a few seconds, all of them change to a reddish colour and exhibit intense red luminescence, which indicates that complete transformation into compound 1 occurs. To confirm this transformation and that it is complete, the powder X-ray diffractograms of pure compounds 2–5 were recorded and their diffraction patterns were compared with those of the same derivatives after subjecting them to methanol vapours, as well as with the diffraction pattern of compound 1 (Fig. S21–S24†).
In addition, exposure of compounds 2–5 to air for a not very long period of time leads to the loss of the previously incorporated solvents, all of them regenerating the starting derivative 1. This process can be accelerated by the application of manual pressure in a mortar, achieving the total change in a few seconds, as confirmed by the X-ray powder diffractograms of these three compounds before and after being subjected to pressure and their comparison with that of the precursor 1.
Finally, it should be noted that any of these compounds can be interconverted into one another without going through compound 1. Thus, if any of compounds 2–5 are subjected to acetonitrile, toluene, THF or benzonitrile vapours, they displace the previous solvent, becoming another pseudopolymorph, thus demonstrating in all cases the possibility of interconversion that these derivatives have. In summary, with a single solid of any of them, the rest of the compounds can be obtained by simple exposition to vapours of the adequate solvent.
In the case of exposure to acetonitrile vapours, a mixture of 2 and precursor compound 1 is observed in all cases since, as discussed above, this compound is not stable in air for long periods of time, gradually losing solvent and reverting to the starting complex 1.
Computational studies
DFT and TD-DFT computational calculations were carried out to elucidate the origin of the photophysical properties displayed by all the complexes in the solid state. Models 2a–4a were chosen based on the crystal structures of complexes 2–4, representing the most important interactions. Model systems were built up considering two equal Au2Pb units linked through an unsupported gold(I)⋯gold(I) contact to study this type of interaction, except for complex 4, in which this intermetallic distance is too large (Fig. S36†).The electronic structures of the three models (2a–4a) were computed through dispersion-corrected single point DFT calculations. For the models displaying the same Au2Pb trinuclear core disposition (2a and 3a), the HOMOs are mainly placed at the metal centres, largely at gold atoms and to a lesser extent at the lead atoms, with a minor contribution from the perhalophenyl groups (Fig. 9 and 10 and Tables S2 and S3†). On the other hand, the LUMOs calculated for all models appear to be mostly located at the terpy ligand and display a π* character, indicating that terpy has a highly relevant role in the luminescent behavior of these complexes, being involved in metal to ligand charge transfer (MLCT) transitions.
 | ||
| Fig. 9 Molecular orbitals involved in the most important transitions calculated for model [{Au(C6F5)2}2{Pb(terpy)(NCMe)2}]22a. | ||
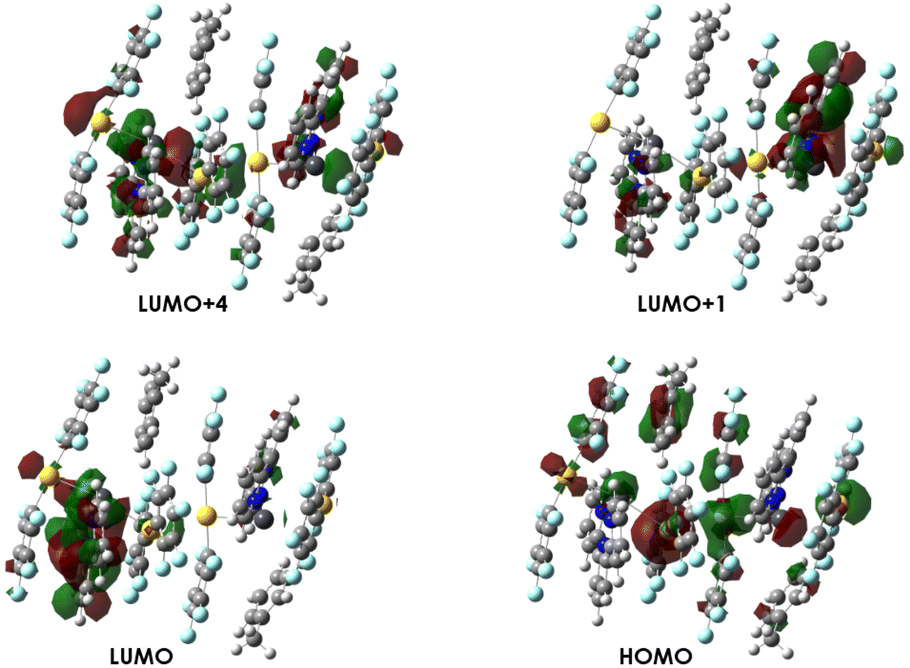 | ||
| Fig. 10 Molecular orbitals involved in the most important transitions calculated for model {[{Au(C6F5)2}2{Pb(terpy)}]·toluene}23a. | ||
In the case of model 4a, in which aurophilic interactions are not present, the computed electronic structure provides a different arrangement of molecular orbitals. The HOMO is now placed at the neighbouring perhalophenyl groups, and the LUMO is located at the terpy ligand (Fig. 11 and Table S4†). Therefore, for this complex, ligand-to-ligand charge transfer (LL′CT) is likely favoured upon excitation.
 | ||
| Fig. 11 Molecular orbitals involved in the most important transitions calculated for model {[{Au(C6F5)2}2{Pb(terpy)(THF)}]·THF}24a. | ||
To confirm the origin of electronic transitions that could be related to the absorption spectra and luminescent emissions observed experimentally for the reported complexes, we performed an analysis of the energy of the most intense singlet–singlet electronic excitations computed using the TD-DFT approach. Thus, we compared these allowed transitions with the experimental UV-vis solid-state spectra of the complexes, obtaining a good match between the predicted excitations and the experimental absorption profile. Moreover, since the emissive behaviour of these complexes could arise from phosphorescence processes in view of their lifetimes, in the microsecond range, we also computed the lowest singlet–triplet excitation at the TD-DFT level for all models, and we compared the corresponding predicted energies with the experimental excitation spectra.
The computed first few singlet–singlet excitations for models 2a–3a provide the predicted absorption spectra, which can be divided into two types of excitations: one corresponding to the HOMO–LUMO transition (at lower energy) and the rest of excitations at higher energies. The excitation calculated at 364 nm for model 2a and at 354 nm for 3a takes place from the HOMO to LUMO+4. The latter is located at the fragment [Pb(terpy)]2+. For model 2a, the electronic transition associated with singlet–singlet excitation at 351 nm occurs between HOMO−14 and LUMO+1, being quite similar to the HOMO and LUMO, centered at the metal centers and terpy ligands, respectively. In the case of model 3a, the transition at 458 nm takes place between the HOMO and LUMO+1, centered at the terpy ligand, as well as the LUMO.
Regarding model 4a, the lowest energy excitation is a combination of two electronic transitions: the HOMO → LUMO+1 (50%) and the HOMO−1 → LUMO (23%) transitions. On the other hand, there is another more energetic excitation at 334 nm that takes place between two electronic transitions: HOMO−9 → LUMO+3 (37%) and HOMO−19 → LUMO+1 (31%). The HOMO−19 orbital is mainly located at the terpy ligand with a minor contribution of the perhalophenyl rings. However, HOMO−9 is inverted, being mainly centered at the perhalophenyl groups. LUMO+1 and LUMO+3, as well as the LUMO, are located at the terpy ligand.
If we analyze the lowest energy excitations, the first singlet–triplet transition arises as a whole for all model systems from the HOMO to the LUMO. These computed results seem to indicate that the electronic transition responsible for the phosphorescence emission observed for all complexes could have its origin in metal to ligand (terpy) charge transfer (MLCT) transition. Besides, in the case of 4a, there is another transition with a high contribution, which is from HOMO−1 to the LUMO. HOMO−1 is located at neighbouring perhalophenyl groups, not only at the perhalophenyl groups with π–π stacking, but also at the other perhalophenyl groups joined to the same gold(I) centres, with a small contribution from the metal centers (Fig. 11).
We also carried out periodic DFT calculations (computational details) to explore the electronic structure and the complex⋯solvent interaction energies (IE) of the experimentally described Au(I)–Pb(II) heterometallic complexes, bearing four different solvents namely acetonitrile (2), toluene (3), THF (4) or benzonitrile (5).
We performed an interaction energy (IE) calculation of the Au–Pb molecules using periodic boundaries. Complexes 2–5 display an explicit solvation that, as is shown experimentally, leads to a reversible process, complex 1 being the most stable species from a thermodynamic point of view. Therefore, the calculation of the IE could explain the tendency for the displacement of the solvate molecules from each supramolecular arrangement. To evaluate the IE, eqn (1) in Fig. 12 is used, where E(solvent-complex), E(complex), and E(solvent) correspond to the energy of the supramolecular arrangements of complexes 2–5 interacting with the solvents, the complexes without solvent, and the explicit solvent, respectively. This qualitative approach highlights the interaction between the solid-state systems and the solvent molecules acetonitrile (2), toluene (3), tetrahydrofuran (4) or benzonitrile (5). Fig. 12 also depicts the computed periodic crystal structure of complex 2 with and without solvent molecules and the isolated explicit acetonitrile solvent. The interpretation of the solvent–complex interaction is based on two main parts: (i) the observed experimental tendency compared to the calculation results, as well as the effect caused by this interaction to make stress in the cells of the four materials, and (ii) the high tendency to lose the solvent spontaneously during the synthesis.
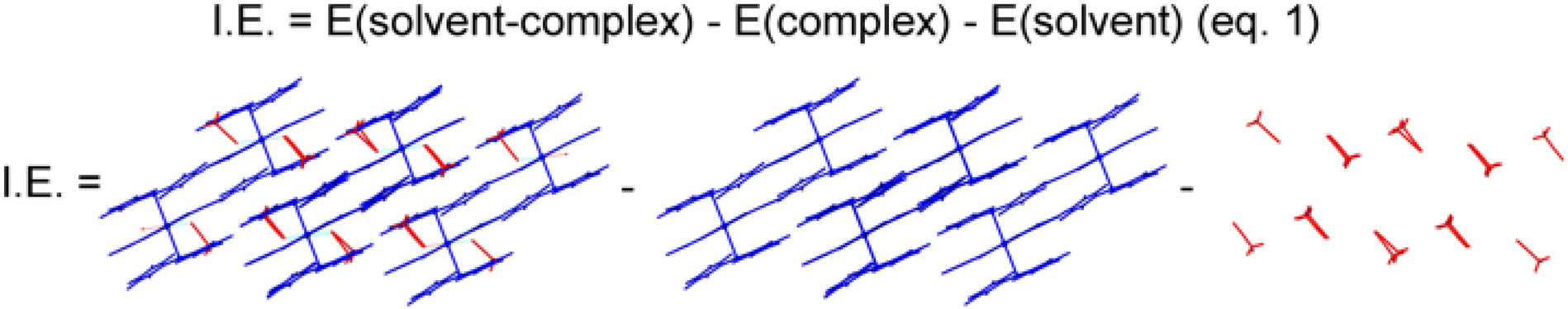 | ||
| Fig. 12 Representation of the model systems used for the estimation of the interaction energy for complex 2. | ||
The computed results show that complex 2, bearing acetonitrile molecules, displays a repulsive (positive) interaction energy value of +8.23 eV. This value reflects a clear tendency of losing solvent molecules, as observed experimentally, leading to a more thermodynamically stable empty supramolecular arrangement and free solvent molecules. In the case of the acetonitrile solvate, this is the highest interaction energy found for all systems. In addition, complex 4 bearing THF has also a high tendency to lose the solvent molecules and we found an interaction energy value of +2.28 eV, followed by complex 5 with benzonitrile (IE = +0.33 eV) and then complex 3 with toluene molecules (IE = +0.09 eV). The latter presents the smallest interaction energy value of the four solvates. It is noteworthy that the value of the interaction energy given could be overestimated for complex 2, probably due to the complex electronic interactions and the limitlessness of the pseudopotential approach.
In view of these results, it could be argued that the starting supramolecular complex 1 displays a high tendency to interact with solvent molecules due to the opened structural arrangement with large voids but, at the same time, this also facilitates the spontaneous loss of the solvent molecules under ambient conditions, leading to facile recovery of complex 1.
Conclusions
In view of the results obtained throughout this work, it can be concluded that compound 1 presents a vapochromic behaviour by which it lodges small organic solvent molecules in the holes in its crystalline structure, with an evident change in both the colour of the compound and its luminescence. These changes also affect the homo- and heterometallic distances presented by its crystal structures. Moreover, the theoretical study carried out on all of them reveals that the origin of the emission is not affected in any case by the solvent molecules inside them, but by charge transfers from gold centres and/or perhalophenyl ligands to the neutral terpyridine ligand.Experimental section
General
[Au2Ag2(C6F5)4(Et2O)2]n was prepared according to literature methods.13dMaterials and physical measurements
Infrared spectra were recorded in the 3500–450 cm−1 range on a PerkinElmer FT-IR Spectrum Two with a UATR (Single Reflection Diamond) accessory. 1H and 19F NMR spectra were recorded on a Bruker Avance 300 instrument in deuterated DMSO solutions at room temperature. Chemical shifts are quoted relative to SiMe4 (1H external) and CFCl3 (19F external). C, H, and N analyses were carried out with a C.E. Instrument EA-1110 CHNSO microanalyser. The MALDI mass spectra were registered on a Microflex Bruker spectrometer using DIT (dithranol) and DCTB (T-2-(3-(4-t-butylphenyl)-2-methyl-2-propenylidene)-malononitrile) as the matrix. The m/z values are obtained for the higher peak in the isotopic pattern. Absorption spectra in solution were recorded on a Hewlett-Packard 8453 diode array UV–visible spectrophotometer. Diffuse reflectance UV–vis spectra of pressed powder samples diluted with silica were recorded on a Shimadzu UV-3600 spectrophotometer with a Harrick Praying Mantis accessory and recalculated following the Kubelka–Munk function. Excitation and emission spectra in the solid state were recorded with an Edinburgh FLS 1000 fluorescence spectrometer. Luminescence lifetime was measured on an Edinburgh FLS 1000 fluorescence spectrometer. For measurements at RT, this equipment was used with a supercontinuum White Light Laser SuperK Extreme NKT Photonics. Time-resolved phosphorescence decay measurements at 77 K were analysed using numerical reconvolution. To do this, the corresponding instrumental response function (IRF) is necessary to detect the scattered light from the sample. Quantum yields were measured in the solid state using an N-M01 integrating sphere with excitation at 560 nm on an Edinburgh FLS 1000 fluorescence spectrometer.Synthesis
N) (Fig. 8), ν = 2247 cm−1 (acetronitrile). MALDI(+): m/z (%): 972 (100) [AuPb(C6F5)2(terpy)]+; MALDI(−): m/z (%): 530 (100) [Au(C6F5)2]−; elemental analysis calcd (%) for C43H17Au2F20N5Pb: C, 32.59; H, 1.08; N, 4.42. Found: C, 32.22; H, 1.01; N, 4.31. ΛM (acetone): 226.5 K−1 cm2 mol−1.
N). MALDI(+): m/z (%): 972 (100) [AuPb(C6F5)2(terpy)]+; MALDI(−): m/z (%): 530 (100) [Au(C6F5)2]−; elemental analysis calcd (%) for C46H19Au2F20N3Pb: C, 34.64; H, 1.20; N, 2.64. Found: C, 34.41; H, 1.04; N, 2.83. ΛM (acetone): 215.7 K−1 cm2 mol−1.
N). MALDI(+): m/z (%): 972 (100) [AuPb(C6F5)2(terpy)]+; MALDI(−): m/z (%): 530 (100) [Au(C6F5)2]−; elemental analysis calcd (%) for C47H27Au2O2F20N3Pb: C, 34.28; H, 1.65; N, 2.55. Found: C, 34.45; H, 1.71; N, 2.66. ΛM (acetone): 214.8 K−1 cm2 mol−1.
Crystallography
Crystals were mounted in inert oil on a MiteGen MicroMounts™ and transferred to the cold gas stream of a Nonius Kappa CCD (3) or in a Bruker APEX-II CCD diffractometer (2 and 4) equipped with an Oxford Instruments low-temperature attachment. Data were collected using monochromated Mo Kα radiation (λ = 0.71073 Å). Scan type: ω and ϕ. Absorption corrections: empirical (2 and 3) or semiempirical (4). In spite of having tested several absorption correction types in order to choose the best one to minimize them, the crystal structure of complex 2 contains residual peaks and holes larger than desired, although they are close to the heavy atoms. This fact is probably due to the number of heavy atoms and the high value of the absorption coefficient in these structures. The structures were solved with the XT structure solution program using Intrinsic Phasing and refined with the ShelXL refinement package using Least Squares minimization (2 and 4) or by direct methods (3) and refined on F2 using the program SHELXL-97.23 The non-coordinated THF molecule in 4 is disordered over two different positions (60:40). All non-hydrogen atoms, except those of the non-coordinated THF molecule in 4, were refined anisotropically. Hydrogen atoms were included using a riding model. CCDC 2266131–2266133† contain the supplementary crystallographic data for this paper.
Computational details
All calculations were performed using the Gaussian 09 suite of programs24 using DFT-BP86 level of theory.25We have performed single-point calculations on all model systems at the DFT-D3/BP86 level, including the empirical dispersion correction by Grimme et al.26 This level of theory has been proven to represent noncovalent interactions at lower computational cost. For these calculations, the corresponding def2-TZVP basis sets were used.27
Periodic DFT calculations were based on the plane augmented wave (PAW) method with pseudopotentials.28 This methodology is implemented in the Quantum Espresso package.29 Plane waves and basis sets were expanded with an energy cut-off equal to 70 Ry. The periodic cell boundaries and the lattice constants were taken from the experimental data and are depicted in Table 1. For single point energy calculations, a 4 × 4 x 4 k-point Monkhorst Pack-Mesh is used in the first Brillouin zone. For all systems, the energy convergence should reach a value equal to 10−7 eV, and the convergence in the forces should reach a threshold of 0.01 eV A−1. In all the calculations, GGA-PBE was used to treat the exchange–correlation potential.30
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge DGI MICINN/FEDER (project number PID2022-139739NB-I00 (AEI/FEDER, UE)) and “ERDF A way of making Europe”. S. M. also acknowledges MINECO for an FPI grant. A. G. E.-H. thanks the MU and the EU-NextGeneration program for a Maria Zambrano post-doctoral scholarship.References
- V. W.-W. Yam, V. K.-M. Au and S. Yu-Lut, Leung. Light-Emitting Self-Assembled Materials Based on d8 and d10 Transition Metal Complexes, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- Modern Supramolecular Gold Chemistry: Gold−Metal Interactions and Applications, ed. A. Laguna, Wiley-VCH, Weinheim, Germany, 2008, p. 181 Search PubMed.
- See for example: (a) M. Gil-Moles, M. C. Gimeno, J. M. López-de-Luzuriaga, M. Monge and D. Pascual, Tailor Made Luminescent Polymers through Unusual Metallophilic Interaction Arrays Au⋯Au⋯Ag⋯Ag, Inorg. Chem., 2017, 56, 9281–9290 CrossRef CAS PubMed; (b) M. E. Olmos, A. Schier and H. Schmidbaur, 2-(Diphenylphosphino)pyridine as an Ambidentate Ligand in Homo- and Hetero-binuclear Complexes of Copper, Silver, and Gold, Z. Naturforsch., B: J. Chem. Sci., 1997, 52, 203–208 CrossRef CAS; (c) V. J. Catalano and A. L. Moore, Mono-, Di-, and Trinuclear Luminescent Silver(I) and Gold(I) N-Heterocyclic Carbene Complexes Derived from the Picolyl-Substituted Methylimidazolium Salt: 1-Methyl-3-(2-pyridinylmethyl)-1H-imidazolium Tetrafluoroborate, Inorg. Chem., 2005, 44, 6558–6566 CrossRef CAS PubMed; (d) Q. M. Wang, Y. A. Lee, O. Crespo, J. Deaton, C. Tang, H. J. Gysling, M. C. Gimeno, C. Larraz, M. D. Villacampa, A. Laguna and R. Eisenberg, Intensely Luminescent Gold(I)-Silver(I) Cluster Complexes with Tunable Structural Features, J. Am. Chem. Soc., 2004, 126, 9488–9489 CrossRef CAS PubMed.
- See for example: (a) R. Donamaría, E. J. Fernández, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, D. Pascual and M. Rodríguez-Castillo, New Au(I)–Cu(I) heterometallic complexes: the role of bridging pyridazine ligands in the presence of unsupported metallophilic interactions, Dalton Trans., 2017, 46, 10941–10949 RSC; (b) M. J. Calhorda, C. Ceamanos, O. Crespo, M. C. Gimeno, A. Laguna, C. Larraz, P. D. Vaz and M. D. Villacampa, Heteropolynuclear Gold Complexes with Metallophilic Interactions: Modulation of the Luminescent Properties, Inorg. Chem., 2010, 49, 8255–8269 CrossRef CAS PubMed; (c) E. J. Fernández, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, M. Montiel, M. E. Olmos and M. Rodríguez-Castillo, Unsupported Au(I)⋯Cu(I) interactions: influence of nitrile ligands and aurophilicity on the structure and luminescence, Dalton Trans., 2009, 36, 7509–7518 RSC.
- See for example: (a) A. Burini, J. P. Fackler, Jr., R. Galassi, T. A. Grant, M. A. Omary, M. A. Rawashdeh-Omary, B. R. Pietroni and R. J. Staples, Supramolecular Chain Assemblies Formed by Interaction of a π Molecular Acid Complex of Mercury with π-Base Trinuclear Gold Complexes, J. Am. Chem. Soc., 2000, 122, 11264–11265 CrossRef CAS; (b) J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos and D. Pascual, Experi-mental and Theoretical Comparison of the Metallophilicity between d10–d10 AuI–HgII and d8−d10 AuIII–HgII Interactions, Inorg. Chem., 2014, 53, 1275–1277 CrossRef PubMed; (c) T. Lasanta, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos and D. Pascual, Synthesis of the molecular amalgam [{AuHg2(o,-C6F4)3}{Hg3(o-C6F4)3}]−: a rare example of a heterometallic homoleptic metallacycle, Dalton Trans., 2016, 45, 6334–6338 RSC.
- See for example: (a) R. Donamaría, M. C. Gimeno, V. Lippolis, J. M. López-de-Luzuriaga, M. Monge and M. E. Olmos, Tuning Au(I)⋯Tl(I) Interactions via Mixed ThiaAza Macrocyclic Ligands: Effects on the Structural and Luminescence Properties, Inorg. Chem., 2017, 56, 12551–12563 CrossRef PubMed; (b) V. J. Catalano, B. L. Bennett, H. M. Kar and B. C. Noll, Synthesis and Characterization of Trigonal Gold(I) Cage Complexes: Luminescent Metallocryptates Encapsulating Tl(I) and Na+ Ions, J. Am. Chem. Soc., 1999, 121, 10235–10236 CrossRef CAS.
- (a) S. H. Lim, M. M. Olmstead, J. C. Fettinger and A. L. Balch, Polymorphs and Aurophilic Interactions in Colourless Crystals of Au2(μ-1,2 bis-(diphenylarsino)ethane)X2, (X = Cl, Br, I), Inorg. Chem., 2012, 51, 1925–1932 CrossRef CAS PubMed; (b) A. L. Balch, Polymorphism and luminescent behaviour of linear, two-coordinate gold(I) complexes, Gold Bull., 2004, 37, 45–50 CrossRef CAS.
- E. J. Fernández, M. C. Gimeno, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, P. Pyykkö and D. Sundholm, Luminescent Characterization of Solution Oligomerization Process Mediated Gold-Gold Interactions. DFT Calculations on [Au2Ag2R4L2]n Moieties, J. Am. Chem. Soc., 2000, 122, 7287–7293 CrossRef.
- W. C. McCrone, Physics and Chemistry of the Organic Solid State, ed. D. Fox, M. M. Labels and A. Weissberger, Interscience, London, 1965, vol. 2, pp. 725–767 Search PubMed.
- H. G. Brittain, Polymorphism and solvatomorphism 2006, J. Pharm. Sci., 2008, 97, 3611–3636 CrossRef CAS PubMed.
- M. Asaoka, Y. Kitagawa, R. Teramoto, K. Miyagi, Y. Natori and M. Nakano, Origin of Solvent-independent Optical Property of Unsubstituted BODIPY Revisited, Chem. Lett., 2017, 46, 1–3 CrossRef.
- M. A. Mansour, W. B. Connick, R. J. Lachicotte, H. J. Gysling and R. Eisenberg, Linear Chain Au(I) Dimer Compounds as Environmental Sensors: A Luminescent Switch for the Detection of Volatile Organic Compounds, J. Am. Chem. Soc., 1998, 120, 1329–1330 CrossRef CAS.
- (a) I. O. Koshevoy, Y.-C. Chang, A. J. Karttunen, M. Haukka, T. Pakkanen and P.-T. Chou, Modulation of Metallophilic Bonds: Solvent-Induced Isomerization and Luminescence Vapochromism of a Polymorphic Au-Cu Cluster, J. Am. Chem. Soc., 2012, 134, 6564–6567 CrossRef CAS PubMed; (b) E. J. Fernández, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, J. Pérez, A. Laguna, A. A. Mohamed and J. P. Facker, Jr., {Tl[Au(C6Cl5)2]}n: A Vapochromic Complex, J. Am. Chem. Soc., 2003, 125, 2022–2023 CrossRef PubMed; (c) E. J. Fernández, J. M. López-de-Luzuriaga, M. Monge, M. Montiel, A. A. Mohamed and J. P. Facker, A Detailed Study of the Vapochromic Behaviour of {Tl[Au(C6Cl5)2]}n, Inorg. Chem., 2004, 43, 3573–3581 CrossRef PubMed; (d) E. J. Fernández, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, R. C. Puelles, A. Laguna, A. A. Mohamed and J. P. Facker, Jr., Inorg. Chem., 2008, 47, 8069–8076 CrossRef PubMed; (e) E. J. Fernández, A. Laguna, J. M. López-de-Luzuriaga, M. E. Olmos and R. C. Puelles, Z. Naturforsch., B: J. Chem. Sci., 2009, 64, 1500–1512 CrossRef; (f) T. Lasanta, M. E. Olmos, A. Laguna, J. M. López-de-Luzuriaga and P. Naumov, J. Am. Chem. Soc., 2011, 133, 16358–16361 CrossRef CAS PubMed; (g) J. Lefebvre, R. J. Batchelor and D. B. Leznoff, Cu[Au-(CN)2]2(DMSO)2: Golden Polymorphs That Exhibit Vapochromic Behaviour, J. Am. Chem. Soc., 2004, 126, 16117–16125 CrossRef CAS PubMed; (h) J. Lefebvre, J. L. Korčok, M. J. Katz and D. B. Lenznoff, Vapochromic Behaviour of M[Au(CN)2]2-Based Coordination Polymers (M = Co, Ni), Sensors, 2012, 12, 3669–3692 CrossRef CAS PubMed; (i) J. Lefebvre, F. Callaghan, M. J. Katz, J. E. Sonier and D. B. Leznoff, A New Basic Motif in Cyanometallate Coordination Polymers: Structure and Magnetic Behaviour of M(μ-OH2)2[Au(CN)2]2 (M = Cu, Ni), Chem. – Eur. J., 2006, 12, 6748–6761 CrossRef CAS PubMed; (j) J. Lefebvre, P. Tyagi, S. Trudel, V. Pacradouni, C. Kaiser, J. E. Sonier and D. B. Leznoff, Magnetic Frustration and Spin Disorder in Isostructural M(μ-OH2)2[Au(CN)2]2 (M = Mn, Fe, Co) Coordination Polymers Containing Double Aqua-Bridged Chains: SQUID and μSR Studies, Inorg. Chem., 2009, 48, 55–67 CrossRef CAS PubMed; (k) M. J. Katz, T. Ramnial, H. Z. Yu and D. B. Leznoff, Polymorphism of Zn[Au(CN)2]2 and Its Luminescent Sensory Response to NH3 Vapour, J. Am. Chem. Soc., 2008, 130, 10662–10673 CrossRef CAS PubMed; (l) B. R. Varju, J. S. Ovens and D. B. Leznoff, Mixed Cu(I)/Au(I) Coordination Polymers as Reversible Turn-on Vapoluminescent Sensors for Volatile Thioethers, Chem. Commun., 2017, 53, 6500–6503 RSC; (m) A. Luquin, C. Bariáin, E. Vergara, E. Cerrada, J. Garrido, I. R. Matias and M. Laguna, New Preparation of Gold−Silver Complexes and Optical Fibre Environmental Sensors Based on Vapochromic [Au2Ag2(C6F5)4(phen)2]n, Appl. Organomet. Chem., 2005, 19, 1232–1238 CrossRef CAS; (n) R. Usón, A. Laguna, M. Laguna, P. G. Jones and G. M. Sheldrick, Synthesis and Reactivity of Bimetallic Au−Ag Complexes. X-Ray Structure of a Chain Polymer Containing the Moiety ⋯(F5C6)2Au(μ-AgSC4H8)2Au(C6F5)2⋯, J. Chem. Soc., Chem. Commun., 1981, 1097–1098 RSC; (o) R. Usón, A. Laguna, M. Laguna, B. R. Manzano, P. G. Jones and G. M. Sheldrick, Synthesis and Reactivity of Bimetallic Au-Ag Polyfluorophenyl Complexes; Crystal and Molecular Structures of [{AuAg(C6F5)2(SC4H8)}n] and [{AuAg(C6F5)2(C6H6)}n], Dalton Trans., 1984, 285–292 RSC; (p) E. J. Fernández, M. C. Gimeno, A. Laguna, J. M. López-de-Luzuriaga, M. Monge, P. Pyykkö and D. Sundholm, Luminescent Characterization of Solution Oligomerization Process Mediated Gold-Gold Interactions. DFT Calculations on [Au2Ag2R4L2]n Moieties, J. Am. Chem. Soc., 2000, 122, 7287–7293 CrossRef; (q) M. Laguna, C. Bariáin, J. Garrido, I. R. Matías and I. Romeo, Spanish Patent, E-99-02862, 1999 Search PubMed; C. Bariáin, J. Garrido, I. R. Matias and I. Romeo, European Patent, PCT/ES00/00485, 2000 Search PubMed; (r) M. Laguna, J. Garrido and I. Romeo, Spanish Patent, 2151424, 2001 Search PubMed; (s) C. Bariáin, I. R. Matías, I. Romeo, J. Garrido and M. Laguna, Detection of Volatile Organic Compound Vapours by Using a Vapochromic Material on a Tapered Optical Fiber, Appl. Phys. Lett., 2000, 77, 2274–2276 CrossRef; (t) C. Bariáin, I. R. Matías, I. Romeo, J. Garrido and M. Laguna, Behavioural Experimental Studies of a Novel Vapochromic Material Towards Development of Optical Fiber Organic Compounds Sensor, Sens. Actuators, B, 2001, 76, 25–31 CrossRef; (u) C. Bariáin, I. R. Matías, C. Fdez-Valdivielso, C. Elosúa, A. Luquin, J. Garrido and M. Laguna, Optical Fibre Sensors Based on Vapochromic Gold Complexes for Environmental Applications, Sens. Actuators, B, 2005, 108, 535–541 CrossRef; (v) S. C. Terrones, C. E. Aguado, C. Bariáin, A. S. Carretero, I. R. M. Maestro, A. F. Gutiérrez, A. Luquin, J. Garrido and M. Laguna, Volatile-Organic-Compound Optic Fiber Sensor Using a Gold-Silver Vapochromic Complex, Opt. Eng., 2006, 45, 044401–044407 CrossRef; (w) C. Elosua, I. Matías, C. Bariain and F. Arregui, Development of an In-Fiber Nanocavity Towards Detection of Volatile Organic Gases, Sensors, 2006, 6, 578–592 CrossRef CAS; (x) Z. Lei, S. S. Chang and Q. W. Wang, Vapochromic Gold(I)-Silver(I) Cluster Protected by Alkynyl and Phosphine Ligands, Eur. J. Inorg. Chem., 2017, 2017, 5098–5102 CrossRef CAS; (y) L. Q. Mo, J. H. Jia, L. J. Sun and Q. M. Wang, Solvent-Induced Intercluster Rearrangements and the Reversible Luminescence Responses in Sulfide Bridged Gold(I)-Silver(I) Clusters, Chem. Commun., 2012, 48, 8691–8693 RSC; (z) S. H. Lim, M. M. Olmstead and A. L. Balch, Molecular Accordion: Vapoluminescence and Molecular Flexibility in the Orange and Green Luminescent Crystals of the Dimer, Au2(μ-bis-(diphenylphosphino)ethane)2Br2, J. Am. Chem. Soc., 2011, 133, 10229–10238 CrossRef CAS PubMed.
- (a) K. R. England, S. H. Lim, L. M. C. Luong, M. M. Olmstead and A. L. Balch, Vapoluminescent Behaviour and the Single-Crystal-to-Single-Crystal Transformations of Chloroform Solvates of [Au2(μ-1,2-bis(diphenylarsino)ethane)2](AsF6)2, Chem. – Eur. J., 2019, 25, 874–878 CrossRef CAS PubMed; (b) M. S. Jiang, Y. H. Tao, Y. W. Wang, C. Lu, D. J. Young, J. P. Lang and Z. G. Ren, Reversible Solid-State Phase Transitions Between Au-P Complexes Accompanied by Switchable Fluorescence, Inorg. Chem., 2020, 59, 3072–3078 CrossRef CAS PubMed; (c) A. Chu, F. K. W. Hau, L. Y. Yao and V. W. W. Yam, Decanuclear Gold(I) Sulfido Pseudopolymorphs Displaying Stimuli-Responsive RGBY Luminescence Changes, ACS Mater. Lett., 2019, 1, 277–284 CrossRef CAS; (d) Y. F. Hsu, T. W. Wu, Y. H. Kang, C. Y. Wu, Y. H. Liu, S. M. Peng, K. V. Kong and J. S. Yang, Porous Supramolecular Assembly of Pentiptycene-Containing Gold(I) Complexes: Persistent Excited-State Aurophilicity and Inclusion-Induced Emission Enhancement, Inorg. Chem., 2022, 61, 11981–11991 CrossRef CAS PubMed; (e) J. A. Pells, D. Guan and D. B. Leznoff, Heterobimetallic Ln(III)-Containing Materials Based on One-Dimensional Aurophilic Chains of Gold(I) Dithiolate Dimers and Their Vapochromic Response to DMF, Eur. J. Inorg. Chem., 2022, e202200049 CrossRef CAS; (f) Y. F. Hsu, S. Y. Chen, S. Maity, Y. H. Liu, S. M. Peng and J. S. Yang, A polymorphic pentiptycene-containing gold(I) isocyanide complex: solvent- and conformation-dependent supramolecular luminescence, Dalton Trans., 2020, 49, 15602–15606 RSC; (g) L. M. C. Luong, C. D. Lowe, A. V. Adams, V. Moshayedi, M. M. Olmstead and A. L. Balch, Seeing luminescence appear as crystals crumble. Isolation and subsequent self-association of individual [(C6H11NC)2Au ]+ ions in crystals, Chem. Sci., 2020, 11, 11705–11713 RSC; (h) L. M. C. Luong, M. A. Malwitz, V. Moshayedi, M. M. Olmstead and A. L. Balch, Role of Anions and Mixtures of Anions on the Thermochromism, Vapochromism, and Polymorph Formation of Luminescent Crystals of a Single Cation, [(C6H11NC)2Au ]+, J. Am. Chem. Soc., 2020, 142, 5689–5701 CrossRef CAS PubMed; (i) S. Nayeri, S. Jamali, A. Jamjah and H. Samouei, Tetranuclear Au2Cu2 Clusters with Butterfly- and Planar-Shaped Metal Cores: Strong Rigidochromism Induced by Jahn–Teller Distortion in Two-Coordinated Gold(I) Centres, Inorg. Chem., 2019, 58, 12122–12131 CrossRef CAS PubMed; (j) S. Ibáñez and E. Peris, Chemically Tunable Formation of Different Discrete, Oligomeric, and Polymeric Self-Assembled Structures from Digold Metallotweezers, Chem. – Eur. J., 2018, 24, 8424–8431 CrossRef PubMed.
- (a) R. Echeverría, J. M. López-de-Luzuriaga, M. Monge and M. E. Olmos, The gold(I)⋯lead(II) interaction: a relativistic connection, Chem. Sci., 2015, 6, 2022–2026 RSC; (b) R. Echeverría, J. M. López-de-Luzuriaga, M. Monge, S. Moreno and M. E. Olmos, New Insights into the Au(I)⋯Pb(II) Closed-Shell Interaction: Tuning of the Emissive Properties with the Intermetallic Distance, Inorg. Chem., 2016, 20, 10523–10534 CrossRef PubMed; (c) R. Echeverría, J. M. López-de-Luzuriaga, M. Monge, S. Moreno, M. E. Olmos and M. Rodríguez-Castillo, Lead encapsulation by a golden clamp through multiple electrostatic, metallophilic, hydrogen bonding and weak interactions, Chem. Commun., 2018, 3, 295–298 RSC.
- J. M. López-de-Luzuriaga, M. Monge, S. Moreno, M. E. Olmos and M. Rodríguez-Castillo, Rational Assembly of Metallophilic Gold(I)–Lead(II) and Gold(I)–Gold(I) Puzzle Pieces, Angew. Chem., Int. Ed., 2021, 60, 640–644 CrossRef PubMed.
- S. Moreno, N. Casati, M. Rodríguez-Castillo, M. Monge, M. E. Olmos and J. M. López-de-Luzuriaga, Switching on/off of a solvent coordination in a Au(I)-Pb(II) complex: high-pressure and temperature as external stimuli, Inorg. Chem., 2023, 62, 10307–10316 CrossRef CAS PubMed.
- S. Wang, G. Garzon, C. King, J.-C. Wang and J. P. Fackler, Jr., Luminescent extended one-dimensional heterobimetallic chain compounds with relativistic metal-metal bonds. Synthesis, crystal structures, and spectroscopic studies of AuTl(MTP)2 a nd Au2Pb(MTP)4 (MTP = [CH2P(S)Ph2]−, Inorg. Chem., 1989, 28, 4623–4629 CrossRef CAS.
- https://www.webelements.com/ .
- S. Alvarez, A cartography of the van der Waals territories, Dalton Trans., 2013, 42, 8617–8636 RSC.
- L. Shimoni-Livny, J.-P. Glusker and C. W. Bock, Lone Pair Func-tionality in Divalent Lead Compounds, Inorg. Chem., 1998, 37, 1853–1867 CrossRef CAS.
- (a) A. Morsali, Syntheses and Characterization of Two New Lead(II) Acetate Complexes, Pb(L)(CH3COO)2, L = 2,2′: 6′,2′′-Terpyridine (tpy) and 2,4,6-Tris(2-pyridyl)-1,3,5-Triazine (trz), Crystal Structure of Pb(tpy)(CH3COO)2, Z. Naturforsch., B: J. Chem. Sci., 2004, 59, 1039–1044 CrossRef CAS; (b) N. Noshiranzadeh, A. Ramazani, A. Morsali, A. D. Hunter and M. Zeller, 4′-(4-Pyridyl)-2,2′: 6′,2′′-terpyridine as ligand in the lead(II) complexes, [Pb(pyterpy)(MeOH)I2]·MeOH and [Pb(pyterpy)(μ-AcO)]2(ClO4)2, Inorg. Chim. Acta, 2007, 360, 3603–3609 CrossRef CAS.
- G. M. Sheldrick, SHELXL97, Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) A. D. Becke, Density–functional thermochemistry. I. The effect of the exchange–only gradient correction, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS; (b) A. D. Becke, Density–functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. Lett., 1998, B37, 785–789 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 1–19 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. D. Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio Jr., A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H.-V. Nguyen, A. Otero-de-la-Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, Advanced capabilities for materials modelling with Quantum ESPRESSO, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Perdew, Burke, and Ernzerhof reply, Phys. Rev. Lett., 1998, 80, 891 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic characterization, structural characterization, powder X-ray diffraction analysis, and computational studies. CCDC 2266131–2266133. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt02000b |
| This journal is © The Royal Society of Chemistry 2023 |