
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of the unique magnetic behaviours of isomers in a 1,2-dithiooxalato-bridged diiron(II) complex†
Takuya
Kanetomo
 *a,
Koki
Yokoyama
a,
Yudai
Suzuki
a,
Hiromichi
Ida
a,
Atsushi
Okazawa
b and
Masaya
Enomoto
*a
*a,
Koki
Yokoyama
a,
Yudai
Suzuki
a,
Hiromichi
Ida
a,
Atsushi
Okazawa
b and
Masaya
Enomoto
*a
aTokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: kanetomo@rs.tus.ac.jp; enomoto.masaya@rs.tus.ac.jp
bWaseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
First published on 11th August 2023
Abstract
1,2-Dithiooxalate (dto) can be employed as a bridging ligand and it exhibits symmetric (O,S-chelation) or asymmetric (O,O- and S,S-chelation) coordination forms. In this study, we prepared a novel dto-bridged diiron(II) complex, [{Fe(TPA)}2(μ-dto)](ClO4)2 (1), where TPA is tris(2-pyridylmethyl)amine. Interestingly, the bridging dto ligand exhibited not only the asymmetric form but also a linkage isomer and a diastereomer within the same crystal. Notably, the three isomers of 1 exhibited different magnetic properties, resulting in a multi-step spin crossover behaviour.
Introduction
1,2-Dithiooxalate (dto, C2O2S2) has O,O,S,S-donor atoms and can adopt various coordination forms. For instance, it can exhibit O,O- and S,S-chelation forms or two O,S-chelation forms, as illustrated in Fig. 1a and b, respectively. Note that the two configurations depicted in Fig. 1a and b are denoted in this paper as asymmetric and symmetric configurations, respectively, based on the centrosymmetry of dto. The choice of coordinating donor atoms to a metal centre is determined by the hard and soft acids and bases (HSAB) rule.1 In the case of heteronuclear metal complexes, asymmetric coordination environments are commonly observed (Fig. 1a).2–23 This arrangement often leads to desirable physical properties such as ferromagnetism,11–13 single-molecule magnets (SMMs),14 charge-transfer (CT) phase transition15–21 and ferroelectrics.22 On the other hand, the studies of homonuclear complexes with dto are relatively rare. For example, [{NiII(en)2}NiII(dto)2] (en = ethylenediamine) exhibits an asymmetric configuration (Fig. 1a),24 while homonuclear complexes of indium(III), silver(I) and zirconium(IV) with dto exhibit the symmetric configuration of dto (Fig. 1b).25–27 | ||
| Fig. 1 Linkage isomers: (a) asymmetric and (b) symmetric conformations. Green filled circles represent metal centres. | ||
An iron(II) ion with a 3d6 electron configuration in an octahedral environment can exhibit two spin states, namely high-spin (hs, S = 2) and low-spin (ls, S = 0) states. These spin states depend on the coordination field in the presence of an appropriate ligand field. There is a spin transition between the hs and ls states triggered by external stimuli. This phenomenon is called a spin crossover (SCO).28–32 It has been intensively studied using similar 3d-transition metal complexes such as iron(III),33–35 cobalt(II)36–40 and manganese(III) ions.41,42 SCO materials have garnered significant attention and investigation due to their potential applications: for example, building blocks for switchable coordination polymers43,44 and multifunctional magnetic materials with conductivity,45–47 fluorescence48–50 and redox ability.51–53
In this study, we synthesized a novel homonuclear diiron(II) complex [{Fe(TPA)}2(μ-dto)](ClO4)2 (1). When both Fe2+ centres have the same or different spin states, the dto bridging mode corresponds to a symmetric or asymmetric configuration, respectively. Notably, our spectroscopic and physical investigations revealed that 1 exhibits both symmetric and asymmetric dto bridging modes within the same crystal. This intriguing finding suggests the coexistence of different coordination fields, namely {N4OS}, {N4O2} and {N4S2}, around the Fe2+ centre. As a result of this unique situation, 1 exhibited a multi-step SCO and undergoes an irreversible structural and magnetic change at about 400 K. Our investigation aims to determine the relative proportions of symmetric and asymmetric dto bridging modes within 1 and to understand the factors that influence these distributions. In addition, we aim to gain insight into the mechanisms governing the magnetic properties exhibited by the compound.
Results and discussion
Synthesis and characterization
The dto-bridged diiron(II) complex 1 was prepared as green block crystals from FeII(ClO4)2·6H2O (1 eq.), TPA (1 eq.), potassium dithiooxalate (0.5 eq.) and a small amount of ascorbic acid in methanol. The product was characterized by elemental analysis, mass spectroscopy and X-ray crystallographic analysis. The charge and spin states of the iron centres were determined by 57Mössbauer spectroscopy (for details, see below). The thermal properties of 1 were evaluated by thermogravimetric analysis (TGA) and differential thermal analysis (DTA). The experimental curves, shown in Fig. 2, demonstrate that 1 exhibits a thermal stability of up to 440 K and does not contain any crystal solvents. | ||
| Fig. 2 TG (red line) and DTA curves (blue line) of 1. The sweep rate of temperature is 2 K min−1. | ||
Crystal structure
Crystal structures of 1 were obtained at five different temperatures: initial 90, 293, 400, 223 and final 90 K. The corresponding crystallographic parameters are summarized in Table 1. Here, we describe the structure at an initial temperature of 90 K, while the temperature dependence of structural parameters will be discussed later. Compound 1 crystallizes in the monoclinic P21/c space group. The asymmetric unit consists of the Fe2+ ion, the TPA capping ligand, half of the dto bridging ligand and the perchlorate anion. The centrosymmetric dinuclear complex comprises two Fe2+ centres with TPA bridged by dto shown in Fig. 3a. During the structural solution process, a non-negligible residual negative electron density was observed in the vicinity of the S1 atom. To solve this issue, it was necessary to consider a disordered state associated with different coordination forms, 1-SymA, 1-Asym (linkage isomer) and 1-SymB (diastereomer), as illustrated in Fig. 3b–d. Note that 1-SymA means that the N4 atom is positioned opposite to the S atom, whereas the diastereomer (1-SymB) means that the N4 atom is positioned opposite to the O atom. However, the simultaneous analysis of three isomers in the disorder model posed a significant challenge. To overcome this difficulty, we employed a mixed occupancy model, which is used in the analysis of inorganic compounds.54,55 This model assumes that the S1 and O1 sites have mixed occupancy by the S1/O2 and O1/S2 atoms, respectively, with occupancy factors denoted as p1 and p2 (where p1 + p2 = 1). The analysis revealed that at an initial temperature of 90 K, the p1 and p2 factors were determined to be 0.672(9) and 0.328(9), respectively. Furthermore, the estimated ratios of the isomers 1-SymA, 1-SymB and 1-Asym were found to be 0.45(1), 0.108(6) and 0.44(2), respectively.‡ Four Fe–N distances (Fe1–N1 to Fe1–N4) are 1.993(3)–2.060(4) Å, which are close to the typical values observed for the ls-Fe2+ ion (1.95–2.00 Å).28 | ||
| Fig. 3 (a) Crystal structure of 1 at 90 K (1st). Symmetric code: a = 1 − x, –y, 1 − z. The perchlorate anion molecules and H atoms are omitted for clarity. (b–d) Schematic isomers of dto in 1. (b) 1-SymA, (c) 1-Asym and (d) 1-SymB. | ||
| T/K | 90 (1st) | 293 | 400 | 223 | 90 (2nd) |
|---|---|---|---|---|---|
| a R = ∑||Fo| − |Fc||/∑|Fo|. b R w = [∑w|Fo2 − Fc2|2/∑w(Fo2)2]1/2. | |||||
| Formula | C38H36Cl2Fe2N8O10S2 | ||||
| Formula weight | 1011.47 | ||||
| Crystal system | Monoclinic | ||||
| Space group | P21/c | ||||
| a/Å | 9.3104(10) | 9.4639(6) | 9.5293(15) | 9.4258(9) | 9.2720(18) |
| b/Å | 13.5947(15) | 13.8477(10) | 13.955(2) | 13.8090(15) | 13.600(3) |
| c/Å | 16.6268(17) | 16.8880(11) | 17.003(3) | 16.7478(16) | 16.515(3) |
| β/° | 102.270(3) | 104.177(2) | 104.716(4) | 104.095(3) | 101.876(6) |
| V/Å3 | 2056.4(4) | 2145.8(2) | 2187.0(6) | 2114.3(4) | 2037.9(7) |
| Z | 2 | 2 | 2 | 2 | 2 |
| d calcd/g cm−3 | 1.634 | 1.565 | 1.536 | 1.589 | 1.648 |
| μ(Mo Kα)/mm−1 | 1.006 | 0.964 | 0.946 | 0.978 | 1.015 |
| R(F)a (I > 2σ(I)) | 0.0658 | 0.0506 | 0.0566 | 0.0523 | 0.0563 |
| R w (F2)b (all data) | 0.1372 | 0.1252 | 0.1490 | 0.1401 | 0.1279 |
| Goodness of fit | 1.103 | 1.081 | 1.051 | 1.056 | 1.043 |
| No. of unique reflns | 4329 | 3262 | 3402 | 4706 | 4438 |
| Occupancy factor (p1) | 0.672(9) | 0.668(8) | 0.748(9) | 0.765(7) | 0.728(9) |
The selected structural parameters of the dto moiety are summarized in Table 2. The C1–C1, C1–O1/S2 and C1–S1/O2 bond lengths are 1.520(7), 1.324(6) and 1.582(5) Å, respectively. Compared to the mean of the reported values for the C–C, C–O and C–S bond lengths (1.53(3), 1.24(4) and 1.70(4) Å, respectively),1,10,13–17,25–27 the values in this work show only a negligible difference in the C–C bond length. However, the experimental C–O bond length is longer than the referential value, while the experimental C–S bond length is shorter. These findings suggest that the values of C1–O1 and C1–S1 are influenced by the presence of C1–S2 and C1–O2 bonds, respectively.
| T/K | 90 (1st) | 293 | 400 | 223 | 90 (2nd) |
|---|---|---|---|---|---|
| a 1 − x, –y, 1 − z. b 2 − x, –y, 1 − z. c x, −1/2 − y, 1/2 + z. d 1 − x, 1/2 + y, 1/2 − z. | |||||
| Coordination bond length | |||||
| Fe1–N1/Å | 2.015(4) | 2.121(4) | 2.153(4) | 2.114(3) | 2.017(3) |
| Fe1–N2/Å | 1.993(3) | 2.110(3) | 2.138(3) | 2.097(3) | 1.993(3) |
| Fe1–N3/Å | 2.017(3) | 2.130(4) | 2.169(4) | 2.126(3) | 2.025(3) |
| Fe1–N4/Å | 2.060(4) | 2.192(3) | 2.232(4) | 2.185(3) | 2.061(3) |
| Fe1–O1/Å | 2.101(3) | 2.175(3) | 2.174(3) | 2.131(2) | 2.036(3) |
| Fe1–S1/Å | 2.296(2) | 2.287(2) | 2.368(2) | 2.330(1) | 2.311(2) |
| Bond length in the dto ligand | |||||
| C1–C1a/Å | 1.520(7) | 1.528(8) | 1.540(6) | 1.522(5) | 1.516(6) |
| C1–O1/Å | 1.324(6) | 1.351(5) | 1.246(6) | 1.258(5) | 1.272(6) |
| C1–S1/Å | 1.582(5) | 1.585(4) | 1.634(5) | 1.635(2) | 1.624(4) |
| Intra- and intermolecular distances | |||||
| S1⋯C8a/Å | 3.209(5) | 3.414(5) | 3.531(6) | 3.439(4) | 3.244(4) |
| O1⋯C7/Å | 3.159(6) | 3.353(6) | 3.442(8) | 3.353(5) | 3.146(6) |
| O1⋯C19/Å | 3.262(5) | 3.471(6) | 3.582(7) | 3.469(5) | 3.239(6) |
| S1⋯C5b/Å | 3.714(5) | 3.689(6) | 3.643(7) | 3.622(5) | 3.656(5) |
| S1⋯C11c/Å | 3.534(6) | 3.550(6) | 3.535(7) | 3.493(4) | 3.498(6) |
| O1⋯C5b/Å | 3.443(5) | 3.457(6) | 3.569(8) | 3.513(5) | 3.535(5) |
| O1⋯C11c/Å | 3.391(6) | 3.373(6) | 3.415(8) | 3.358(5) | 3.390(6) |
| O4⋯C16d/Å | 3.078(7) | 3.169(9) | 3.22(1) | 3.125(7) | 3.063(6) |
Both intramolecular and intermolecular interactions between the dto and TPA ligands were observed in the crystal structure, as shown in Fig. 4. First, the intramolecular distance S1/O2⋯C8 is 3.209(5) Å, while the O1/S2⋯C7 and O1/S2⋯C19 distances are 3.159(6) and 3.262(5) Å, respectively (green dashed lines in Fig. 4). These values are smaller or comparable to the sum of the van der Waals (vdW) radii (C/S: 3.50 Å; C/O: 3.22 Å),56 even considering the influence of the disordered analysis. On the other hand, the intermolecular distances of O1/S2⋯C11, O1/S2⋯C5, S1/O2⋯C11 and S1/O2⋯C5 are 3.391(6), 3.443(5), 3.534(6) and 3.714(5) Å, respectively (blue dashed lines in Fig. 4). Although these values were found to be slightly larger than the sum of the vdW radii, it is important to consider the actual positions of the O and S atoms in dto. These positions may allow the formation of intermolecular contacts. The ClO4 anions are located surrounding the [{FeII(TPA)}2(μ-dto)] cation units, resulting in the anions being sufficiently isolated from each other.
 | ||
| Fig. 4 Intra- and intermolecular hydrogen bonds of 1 at 1st 90 K. Symmetric code: a = 1 − x, −y, 1 − z; b = 2 − x, −y, 1 − z; and c = x, −1/2 − y, 1/2 + z. The perchlorate anions and H atoms are omitted for clarity. Green and blue dashed lines represent the intra- and intermolecular contacts, respectively. | ||
Magnetic properties
The magnetic properties of the polycrystalline 1 were investigated through heating and cooling processes. During the first heating (5–400 K; red open circles in Fig. 5), the values exhibited a plateau at 0.6–0.9 cm3 K mol−1 between 10 K and 100 K. These values were larger than the theoretical values expected for two ls-Fe2+ ions (S = 0; 0 cm3 K mol−1). The residual magnetic momentum indicated the presence of a small amount of hs-Fe2+, which was confirmed by 57Fe Mössbauer spectroscopy (for details, see below). Upon further heating from 100 K, the χmT values exhibited three-step SCO behaviours: 1st, 150–200 K; 2nd, 200–350 K; and 3rd, 350–400 K. In the Δ(χmT)/ΔT vs. T plot (Fig. 5), two distinct peaks corresponding to the abrupt 1st and 3rd SCO behaviours were observed, giving the transition temperatures Tc1 = 184 K and Tc3 = 371 K, respectively. Considering the presence of three isomers in 1 as shown in Fig. 3b–d, we must take into account four Fe2+ centres with different coordination fields: namely, two species of N4OS (α and β) from 1-SymA and 1-SymB, respectively, and N4O2 and N4S2 from 1-Asym. Comparing the energy levels and the magnitude of the orbital lobes for the O and S atoms, the coordination field for N4O2 is weaker than that of N4S2. Therefore, the 1st, 2nd and 3rd SCO behaviours can be attributed to α-N4OS, β-N4OS and N4S2, respectively, based on the order of coordination fields and the corresponding changes in χmT values. There could also be slight variation in the exact coordination field between 1-SymA and 1-SymB, leading to differences in the SCO temperature. After the 3rd SCO behaviour, the χmT value reached 6.71 cm3 K mol−1 at 400 K. Although this value suggests that all iron(II) centres are in the hs state, it exceeds the expected value of 6.0 cm3 K mol−1, which is derived from two hs-Fe2+ ions (S = 2 and g = 2.0; 3.0 cm3 K mol−1). This discrepancy indicates that the g value is greater than 2, due to the distorted coordination environment around the hs-Fe2+ centres.28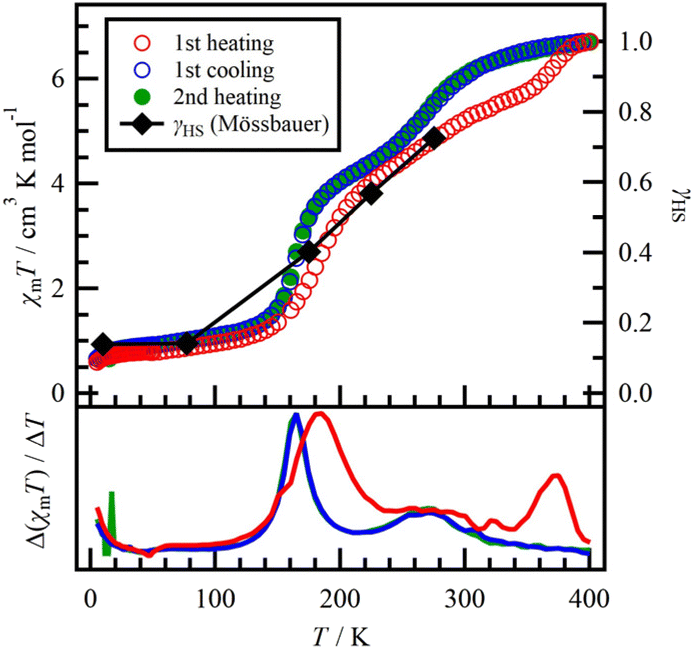 | ||
| Fig. 5 Temperature dependence of the product χmT and the HS fraction γHS of 1 measured at 0.5 T. Black diamonds represent γHS of 1 obtained from the results of Mössbauer spectra (for details, see the text). | ||
During the cooling process from 400 K (blue open circles in Fig. 5), χmT showed a clear two-step SCO behaviour (1st, 100–200 K; 2nd, 200–300 K), compared to the 1st heating process. The Δ(χmT)/ΔT vs. T plot gave the transition temperatures T′c1 = 165 K and T′c2 = 268 K. The 1st SCO behaviour observed during the cooling process is comparable to that observed during the 1st heating process. However, the 2nd SCO behaviour appears to be a cooperative occurrence of the 2nd and 3rd SCO behaviours observed in the 1st heating process. When heated again from 5 K (2nd heating process, green filled circles in Fig. 5), the χmT values followed the same route as the cooling process. This observation indicates an irreversible change between the 1st heating and cooling processes. While the desorption of the crystalline solvent could be a potential cause of this irreversible change, such a scenario was ruled out by TGA and structural studies. In this study, we propose that the as-grown crystal of 1 is initially metastable and undergoes a transition to a stable state during the 1st heating process. The stable state could have a lower ΔHtrs, which is a transition enthalpy defined as ΔG = ΔHtrs − TcΔStrs = 0, compared to the metastable state due to the lowered transition temperatures after the irreversible change. The decrease in ΔHtrs is often explained by a change in the structural factor corresponding to the crystal lattice and the coordination field around the Fe2+ centres.
57Fe Mössbauer spectroscopy
Variable temperature 57Fe Mössbauer spectra were utilized to determine the spin state of the iron centres in 1. The spectra were recorded during the 1st heating process and the results are presented in Table 3 and Fig. 6. | ||
| Fig. 6 57Fe Mössbauer spectra of 1 recorded at 10, 77, 175, 225 and 275 K. The blue pattern represents the area of ls-A, and the green and yellow patterns represent the areas of hs-A and hs-B, respectively. | ||
| T/K | Spin state | Area/% | δ /mm s−1 | ΔEQ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b/mm s−1 b/mm s−1 |
Γ /mm s−1 |
|---|---|---|---|---|---|
| a δ [mm s−1] = isomer shift. b ΔEQ [mm s−1] = quadrupole splitting. c Γ [mm s−1] = full width at the half-maximum (FWHM) of the line. | |||||
| 10 | hs-A | 9.4 | 1.327(10) | 1.92(2) | 0.42(4) |
| hs-B | 4.5 | 1.063(8) | 2.97(2) | 0.24(3) | |
| ls-A | 86.1 | 0.4669(10) | 0.574(2) | 0.399(3) | |
| 77 | hs-A | 9.5 | 1.32(2) | 1.96(3) | 0.51(6) |
| hs-B | 4.7 | 1.058(11) | 3.01(2) | 0.27(4) | |
| ls-A | 85.8 | 0.462(1) | 0.569(3) | 0.444(4) | |
| 175 | hs-A | 9.5 (fixed) | 1.155(10) | 1.83(2) | 0.32(2) |
| hs-B | 30.7 | 0.962(4) | 2.958(8) | 0.433(13) | |
| ls-A | 59.8 | 0.411(2) | 0.607(4) | 0.413(7) | |
| 225 | hs-A | 9.5 (fixed) | 1.084(11) | 1.75(2) | 0.31(2) |
| hs-B | 47.3 | 0.948(4) | 2.821(9) | 0.54(2) | |
| ls-A | 43.2 | 0.392(5) | 0.621(9) | 0.46(2) | |
| 275 | hs-A | 9.5 (fixed) | 0.87(2) | 1.67(4) | 0.34(4) |
| hs-B | 63.1 | 0.904(6) | 2.600(12) | 0.61(2) | |
| ls-A | 27.4 | 0.341(2) | 0.44(3) | 0.58(6) | |
At 10 K, the Mössbauer spectrum exhibited three distinct doublets. The major doublet (blue filled area) showed a quadrupole splitting (ΔEQ) of 0.574(2) mm s−1 and an isomer shift (δ) of 0.4669(10) mm s−1, indicating the presence of Fe2+ atoms in the ls state (ls-A).1 Two minor doublets (green and yellow areas) exhibited ΔEQ and δ values of 1.92(2) and 1.327(10) mm s−1 and 2.97(2) and 1.063(8) mm s−1, respectively. These minor doublets were assigned to hs-Fe2+ sites,1 namely, hs-A (green area) and hs-B (yellow area).
Upon heating from 10 K to 275 K, the intensity of the ls-A doublet decreased to about 60% of the total area, while the hs-B signal increased (Table 3). This result provides evidence for the SCO behaviour of the Fe2+ centres, corresponding to the ls and hs states for α-N4OS and a part of β-N4OS, as discussed in the magnetic studies. In addition, the hs-A area remained almost constant across all temperatures and was utilized as the fixed parameter for analysis above 175 K. This temperature-independent component can be attributed to N4O2, which exhibits the weakest coordination field. Furthermore, the ls-A component is attributed to the N4S2 environment, which has the same area as N4O2 (hs-A).
The sum of the areas of hs-A and hs-B at each temperature was superimposed as shown in Fig. 5. These data agree well with the fraction of the hs state (γHS) determined from the corresponding χmT values. Overall, the 57Fe Mössbauer spectra directly support the SCO phenomenon at the Fe2+ sites in 1.
Variable-temperature dependence of single-crystal X-ray diffraction
We performed variable-temperature single-crystal X-ray diffraction of 1 upon heating (90, 293 and 400 K) and then cooling (223 and 90 K), as shown in Table 1 and 2. Compound 1 exhibited in the monoclinic P21/c space group at all temperatures. Fig. 7a shows the temperature dependence of the cell parameters, a, b, c and V. There was a slight difference in cell parameters (<1%) between the initial and final 90 K, indicating the irreversible structural change. | ||
| Fig. 7 Temperature dependence of selected structural parameters. (a) Cell parameters: a (red), b (yellow), c (green) and V (blue). (b) Bond lengths in the dto moiety; C1–C1 (red), C1–O1 (green) and C1–S1 (blue). (c) Coordination bonds around the Fe2+ centre: Fe1–O1 (red) and Fe1–S1 (blue). (d) Intramolecular contacts: O1⋯C7 (red), O1⋯C19 (green) and S1⋯C8 (blue). The arrows represent the direction of the temperature sweep. | ||
Fig. 7b shows the temperature dependence of the bond lengths in dto. Although there are negligible differences in the C–C bond lengths across all temperatures, the C–O and C–S bond lengths undergo noticeable changes from 293 K to 400 K. The occupancy value p1 also increases from the initial 90 K (0.672) to the final 90 K (0.728).
At the initial 90 K, the four Fe–N distances indicate the ls state of the Fe2+ centre. Upon heating to 293 K, these lengths increase and fall within the typical range of 2.12–2.18 Å observed for hs-Fe2+ complexes.28 These structural changes are consistent with the SCO behaviour of the Fe2+ centres. Fig. 7c shows that the Fe1–O1 bond length decreases while the Fe1–S1 one increases, between the initial and final 90 K. These findings suggest potential strengthening or weakening, respectively, of the coordination field around the Fe2+ centre. Based on the magnetic studies conducted, it is hypothesized that the change in the Fe–S bond plays a dominant role and contributes to a lower SCO temperature during the cooling process compared to the 1st heating process.
The temperature dependence of intramolecular contacts between dto and TPA moieties is shown in Fig. 7d. At the initial 90 K, the O1/S2⋯C7, O1/S2⋯C19 and S1/O2⋯C8 distances are 3.159(6), 3.262(5) and 3.209(5) Å, respectively. Upon heating to 400 K, these distances increased to 3.442(8), 3.582(7) and 3.531(6) Å, respectively, which are larger than the sum of vdW radii (C/S: 3.50 Å; C/O: 3.22 Å).56 This change in intramolecular distances indicates the weakening of the contacts between the dto and TPA moieties, leading to increased freedom of motion for dto. As a result of this increased freedom, 1 exhibits an irreversible change from metastable to stable phases. It is likely that the formation of the metastable phase is related to the synthetic conditions during the mixing of the three isomers (Fig. 3). Before the crystallization of 1, the reaction solution was held at 300 K (reaction) and 255 K (crystallization). Based on magnetic studies conducted in the solid state (Fig. 5), 1 exhibits SCO behaviour. It is important to note that the transition temperature in solution can be lower than that in the solid state due to a reduction in ΔHtrs. This lower Tc in solution allows 1 to exist as a mixture of hs and ls states. In this particular condition, the bridging conformation of the dto ligand could be confused with either the symmetric or asymmetric form. This conformation confusion leads to incomplete crystallization, resulting in the formation of a metastable phase.
Conclusions
We have synthesized a novel dto-bridged diiron(II) complex 1. The bridging ligand (dto) adopts a symmetric S,O-chelation form, while 1 also encompasses the linkage isomer (S,S- and O,O-chelation) and the diastereomer within the same crystal. The bridging modes of dto in 1 are influenced by the spin state (hs or ls) of the two Fe2+ centres. During complex formation, the modes are determined by the Fe centres, resulting in a mixture of the three isomeric forms due to the presence of both hs and ls states. These isomers exhibit distinct temperature-dependent magnetic properties, and 1 displays a multi-step SCO behaviour. Interestingly, magnetic studies revealed differences in SCO behaviour between the first heating and cooling processes. This discrepancy can be attributed to an irreversible structural change occurring within the complex. Our observations indicate that the transition from the metastable phase to the stable phase is triggered by the thermal expansion of the crystal and the stretching of bond lengths due to the SCO phenomenon.Experimental
Materials and methods
Iron(II) perchlorate hexahydrate, TPA, ascorbic acid and methanol were purchased. Methanol was used as the solvent without further purification. Potassium 1,2-dithiooxalate K2(dto) was prepared using the previously reported procedure.57 Infrared (IR) spectra were recorded using a JASCO FT/IR-4600 spectrometer by the diamond attenuated total reflectance (ATR) method. The spectral data were reported in the form of major peaks in wavenumbers (cm−1). The peaks were recorded in a spectral window of 4000–400 cm−1. Elemental analyses were carried out using a PerkinElmer Series II CHNS/O 2400 analyser. Thermogravimetry and differential thermal analysis (TG and DTA, respectively) of 1 were carried out using a Bruker AXS TG2000SA. The temperature scan rate was 2 K min−1 in the range of 296–481 K. Melting point measurement was performed using an ATM-02 (AS ONE). Mass spectra (MS) were recorded in the electrospray ionization (ESI) mode using an AccuTOF-JMS-T100LP (JEOL) spectrometer. The specimen was dissolved in methanol.Synthesis of [{Fe(TPA)}2(μ-dto)](ClO4)2 (1)
Under an Ar atmosphere, iron(II) perchlorate hexahydrate (51.0 mg, 0.141 mmol), TPA (43.1 mg, 0.148 mmol) and ascorbic acid (7.9 mg) were dissolved in methanol (30 mL). K2(dto) powder (8.30 mg, 0.0420 mmol) was added to the solution. The reaction mixture was stored in a refrigerator (<−18 °C). The precipitated polycrystals were separated on a filter and washed with distilled water. The yield of 1 was 13.1 mg (0.0130 mmol, 31%). Melting point: 185 °C (dec.). Analytical calculations for C38H36N8S2O10Fe2Cl2: C, 45.12; H, 3.59; N, 11.08%. Result: C, 44.77; H, 3.37; N, 10.81%. IR (ATR): 1530, 1478, 1439, 1075, 1053, 1020, 870, 763, 620 and 596 cm−1. MS (ESI+): m/z 911.2 and 913.2 [1-ClO4].Single crystal X-ray diffraction (SXRD)
X-ray diffraction data of 1 were collected on a Bruker D8 Quest diffractometer (Mo Kα radiation: λ = 0.71073 Å). X-ray data analysis was carried out using SHELXT58 and SHELXL,59 which were operated with Olex2 software.60 Numerical absorption correction was used. All hydrogen atoms were refined using the riding model. The thermal displacement parameters of the nonhydrogen atoms were refined anisotropically. The Cambridge Crystallographic Data Centre (CCDC) numbers of 1 are 2108397–2108401† measured at 90 (1st), 90 (2nd), 223, 400 and 293 K, respectively.Magnetic measurements
The direct current magnetic properties of the polycrystalline specimens of 1 in wrap were measured using a Quantum Design MPMS-XL7AC SQUID magnetometer equipped with a 7 T coil in the temperature range of 5–400 K. The experimental data were corrected using the measured diamagnetic blank data of the sample holder. The diamagnetic contribution of the sample was estimated using Pascal's constants.61Mössbauer spectroscopy
57Fe Mössbauer spectra of 1 were recorded on a constant acceleration spectrometer with a γ-ray source of 57Co/Rh in the transmission mode. The measurements were performed using a closed-cycle helium refrigerator (Iwatani Industrial Gases Corp.) and a conventional Mössbauer spectrometer (Topologic Systems). All isomer shifts were obtained relative to α-Fe at room temperature. The Mössbauer spectra were fitted using the least-squares fitting program MossWinn 4.0.62Conflicts of interest
There are no conflicts to declare.Acknowledgements
The structural solution of 1 was prepared as advised by Prof. Nobuyuki Matsushita at Rikkyo University, Japan. Mr Daigo Matsunaga and Ms Sayaka Ono (Tokyo University of Science) collected the MS data for 1.Notes and references
- W. Dietzsch, P. Strauch and E. Hoyer, Coord. Chem. Rev., 1992, 121, 43 CrossRef CAS.
- M. Mitsumi, H. Okawa, H. Sakiyama, M. Ohba, N. Matsumoto, T. Kurisaki and H. Wakita, J. Chem. Soc., Dalton Trans., 1993, 2991 RSC.
- S. Decurtins, H. W. Schmalle, R. Pellaux, P. Schneuwly and A. Hauser, Inorg. Chem., 1996, 35, 1451 CrossRef CAS PubMed.
- M. Siebold, S. Eidner, A. Kelling, M. U. Kumke, U. Schilde and P. Strauch, Z. Anorg. Allg. Chem., 2006, 632, 1963 CrossRef CAS.
- M. Nowotny, S. Foro, S. Heinschke, R. C. Hoffmann and J. J. Schneider, Eur. J. Inorg. Chem., 2015, 512 CrossRef CAS.
- J. König, A. Kelling, U. Schilde and P. Strauch, Molbank, 2016, 2016, M895 CrossRef.
- A. Hijazi, J. C. Kemmegne-Mbouguen, S. Floquet, J. Marrot, C. R. Mayer, V. Artero and E. Cadot, Inorg. Chem., 2011, 50, 9031 CrossRef CAS PubMed.
- J. McGuire, B. Wilson, J. McAllister, H. N. Miras, C. Wilson, S. Sproules and J. H. Farnaby, Dalton Trans., 2019, 48, 5491 RSC.
- M. Nakayama, T. Kanetomo and M. Enomoto, Chem. Lett., 2020, 49, 1050 CrossRef CAS.
- C. J. Adams, J. Chem. Soc., Dalton Trans., 2002, 1545 RSC.
- H. Ōkawa, M. Mitsumi, M. Ohba, M. Kodera and N. Matsumoto, Bull. Chem. Soc. Jpn., 1994, 67, 2139 CrossRef.
- J. M. Bradley, S. G. Carling, D. Visser, P. Day, D. Hautot and G. J. Long, Inorg. Chem., 2003, 42, 986 CrossRef CAS PubMed.
- Y. Ono, M. Okubo and N. Kojima, Solid State Commun., 2003, 126, 291 CrossRef CAS.
- G.-F. Xu, P. Gamez, J. Tang, R. Clérac, Y.-N. Guo and Y. Guo, Inorg. Chem., 2012, 51, 5693 CrossRef CAS PubMed.
- M. Itoi, Y. Ono, N. Kojima, K. Kato, K. Osaka and M. Takata, Eur. J. Inorg. Chem., 2006, 1198 CrossRef CAS.
- M. Itoi, A. Okazawa, J. Yamaura, S. Maki, T. Komatsu, I. Maurin, E. Codjovi, K. Boukheddaden and N. Kojima, Inorg. Chem., 2018, 57, 13728 CrossRef CAS PubMed.
- K. Nomura, T. Kanetomo and M. Enomoto, Cryst. Growth Des., 2022, 22, 2139 CrossRef CAS.
- T. Nakamoto, Y. Miyazaki, M. Itoi, Y. Ono, N. Kojima and M. Sorai, Angew. Chem., Int. Ed., 2001, 40, 4716 CrossRef CAS PubMed.
- Y. Kobayashi, M. Itoi, N. Kojima and K. Asai, J. Phys. Soc. Jpn., 2002, 71, 3016 CrossRef CAS.
- N. Kida, M. Enomoto, I. Watanabe, T. Suzuki and N. Kojima, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 144427 CrossRef.
- N. Kida, M. Hikita, I. Kashima, M. Okubo, M. Itoi, M. Enomoto, K. Kato, M. Takata and N. Kojima, J. Am. Chem. Soc., 2009, 131, 212 CrossRef CAS PubMed.
- X. Liu, B. Wang, X. Huang, X. Dong, Y. Ren, H. Zhao, L. Long and L. Zheng, J. Am. Chem. Soc., 2021, 143, 5779 CrossRef CAS PubMed.
- P. D. W. Boyd, J. Hope, C. L. Raston and A. H. White, Aust. J. Chem., 1990, 43, 601 CrossRef CAS.
- D. Coucouvanis and D. Piltingsrud, J. Am. Chem. Soc., 1973, 95, 5556 CrossRef CAS.
- L. Golič, N. Bulc and W. Dietzsch, Inorg. Chem., 1982, 21, 3560 CrossRef.
- L. Golič, N. Bulc and W. Dietzsch, Polyhedron, 1983, 2, 1201 CrossRef.
- C. A. Hester, M. Draganjac and A. W. Cordes, Inorg. Chim. Acta, 1991, 184, 137 CrossRef CAS.
- Spin Crossover in Transition Metal Compounds I, II, and III, ed. P. Gütlich and H. A. Goodwin, Springer, Berlin, 2004 Search PubMed.
- P. Gütlich, Y. Garcia and H. A. Goodwin, Chem. Soc. Rev., 2000, 29, 419 RSC.
- R. W. Hogue, S. Singh and S. Brooker, Chem. Soc. Rev., 2018, 47, 7303 RSC.
- H. Hagiwara, R. Minoura, T. Udagawa, K. Mibu and J. Okabayashi, Inorg. Chem., 2020, 59, 9866 CrossRef CAS PubMed.
- M. J. H. Ojea, J. M. van Raden, S. Louie, R. Collins, D. Pividori, J. Cirera, K. Meyer, R. Jasti and R. A. Layfield, Angew. Chem., Int. Ed., 2021, 60, 3515 CrossRef PubMed.
- S. Mossin, B. L. Tran, D. Adhikari, M. Pink, F. W. Heinemann, J. Sutter, R. K. Szilagyi, K. Meyer and D. J. A. Mindiola, J. Am. Chem. Soc., 2012, 134, 13651 CrossRef CAS PubMed.
- Z.-Y. Li, H. Ohtsu, T. Kojima, J.-W. Dai, T. Yoshida, B. K. Breedlove, W.-X. Zhang, H. Iguchi, O. Sato, M. Kawano and M. Yamashita, Angew. Chem., Int. Ed., 2016, 55, 5184 CrossRef CAS PubMed.
- S. K. Karuppannan, A. Martín-Rodríguez, E. Ruiz, P. Harding, D. J. Harding, X. Yu, A. Tadich, B. Cowie, D. Qi and C. A. Nijhuis, Chem. Sci., 2021, 12, 2381 RSC.
- S. Hayami, Y. Komatsu, T. Shimizu, H. Kamihata and Y. H. Lee, Coord. Chem. Rev., 2011, 255, 1981 CrossRef CAS.
- O. Drath and C. Boskovic, Coord. Chem. Rev., 2018, 375, 256 CrossRef CAS.
- S. Ghosh, S. Selvamani, S. Mehta and A. Mondal, Dalton Trans., 2020, 49, 9208 RSC.
- R. Akiyoshi, Y. Komatsumaru, M. Donoshita, S. Dekura, Y. Yoshida, H. Kitagawa, Y. Kitagawa, L. F. Lindoy and S. Hayami, Angew. Chem., Int. Ed., 2021, 60, 12717 CrossRef CAS PubMed.
- T. Kanetomo, Z. Ni and M. Enomoto, Dalton Trans., 2022, 51, 3034 RSC.
- G. G. Morgan, K. D. Murnaghan, H. Müller-Bunz, V. McKee and C. J. A. Harding, Angew. Chem., Int. Ed., 2006, 45, 7192 CrossRef CAS PubMed.
- S. Wang, M. Ferbinteanu, C. Marinescu, A. Dobrinescu, Q.-D. Ling and W. Huang, Inorg. Chem., 2010, 49, 9839 CrossRef CAS.
- Molecular Magnetic Materials, ed. B. Sieklucka and D. Pinkowich, Wiley-VCH, Weinheim, 2017 Search PubMed.
- F. Bigdeli, C. T. Lollar, A. Morsali and H.-C. Zhou, Angew. Chem., Int. Ed., 2020, 59, 4652 CrossRef CAS PubMed.
- K. Takahashi, H.-B. Cui, Y. Okano, H. Kobayashi, H. Mori, H. Tajima, Y. Einaga and O. Sato, J. Am. Chem. Soc., 2008, 130, 6688 CrossRef CAS PubMed.
- H. Phan, S. M. Benjamin, E. Steven, J. S. Brooks and M. Shatruk, Angew. Chem., 2015, 127, 837 CrossRef.
- R. Ishikawa, S. Ueno, S. Nifuku, Y. Horii, H. Iguchi, Y. Miyazaki, M. Nakano, S. Hayami, S. Kumagai, K. Katoh, Z.-Y. Li, M. Yamashita and S. Kawata, Chem. – Eur. J., 2020, 26, 1278 CrossRef CAS PubMed.
- J. Yuan, S.-Q. Wu, M.-J. Liu, O. Sato and H.-Z. Kou, J. Am. Chem. Soc., 2018, 140, 9426 CrossRef CAS.
- C.-F. Wang, G.-Y. Yang, Z.-S. Yao and J. Tao, Chem. – Eur. J., 2018, 24, 3218 CrossRef CAS PubMed.
- S. Ghosh, S. Kamilya, T. Pramanik, M. Rouzières, R. Herchel, S. Mehta and A. Mondal, Inorg. Chem., 2020, 59, 13009 CrossRef CAS PubMed.
- B. Schneider, S. Demeshko, S. Dechert and F. Meyer, Angew. Chem., Int. Ed., 2010, 49, 9274 CrossRef CAS PubMed.
- I. A. Gural'skiy, S. I. Shylin, V. Ksenofontov and W. Tremel, Eur. J. Inorg. Chem., 2017, 3125 CrossRef.
- L. Zappe, S. Schönfeld, G. Hörner, K. A. Zenere, C. F. Leong, C. J. Kepert, D. M. D'Alessandro, B. Weber and S. M. Neville, Chem. Commun., 2020, 56, 10469 RSC.
- K. Mereiter, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 1085 CrossRef CAS PubMed.
- S. Funahashi, Y. Michiue, T. Takeda, R.-J. Xie and N. Hirosaki, Acta Crystallogr., Sect. C: Struct. Chem., 2014, 70, 452 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- H. S. Tasker and H. O. Jones, J. Chem. Soc., Trans., 1909, 95, 1910 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59 CrossRef CAS PubMed.
- O. Kahn, Molecular Magnetism, VCH-Verlag, Weinheim, New York, 1993 Search PubMed.
- Z. Klencsár, E. Kuzmann and A. Vértes, J. Radioanal. Nucl. Chem., 1996, 210, 105–118 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2108397–2108401. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01992f |
| ‡ The ratios of the isomers 1-symA, 1-symB and 1-Asym were determined using the formulas, p1 × p1, (1 − p1) × (1 − p1) and 2 × {p1 × (1 − p1)}, respectively, where p1 represents the occupancy factor of the predominant atoms (O1 and S1); for example, a p1 value of 0.672(9) at an initial temperature of 90 K gives 0.672 × 0.672, 0.328 × 0.328 and 2 × (0.672 × 0.328) for 1-SymA (0.45(1)), 1-SymB (0.108(6)) and 1-Asym (0.44(2)), respectively. |
| This journal is © The Royal Society of Chemistry 2023 |