
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDicationic copper(I) complexes bearing ENE (E = S, Se) pincer ligands; catalytic applications in regioselective cyclization of 1,6-diynes†
Bhagyashree
Das
a,
Amiya Kumar
Sahoo
a,
Shyam Kumar
Banjare
b,
Subhra Jyoti
Panda
b,
Chandra Shekhar
Purohit
 b and
Adinarayana
Doddi
*a
b and
Adinarayana
Doddi
*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research Berhampur, Transit Campus, Industrial Training Institute (ITI), Engineering School Road, Ganjam, 760010, Odisha, India. E-mail: adoddi@iiserbpr.ac.in
bSchool of Chemical Sciences, National Institute of Science Education and Research (NISER), Bhubaneswar, 752050, India
First published on 12th August 2023
Abstract
Two novel dicationic binuclear Cu(I) complexes of the type [{(BPPP)E2}Cu]2[BF4]2 (E = S (3a); Se (3b)) bearing (BPPP)E2 (BPPP = bis(diphenylphosphino)pyridine) pincer systems were isolated, and structurally characterized. The solid-state structures of 3a/3b display the presence of intermolecular cuprophilic (Cu⋯Cu) interactions between the two monocationic species, and consist of weak Cu⋯S bonding between the two cations. Besides, complex 3a was introduced as a molecular copper(I) catalyst in cyclization reactions, and new protocols were developed for the synthesis of a series of new oxazole and triazole derivatives bearing alkyne-phenyl propargylic ether substituents. 3a was also found to be active in achieving these two classes of heterocyclic compounds by the mechanical grinding method. One of the key intermediate copper-azide species was detected by the high-resolution mass spectrometry technique, which supports the proposed catalytic pathway. All the reported transformations were accomplished sustainably by employing a well-defined, earth-abundant, and cheap copper(I) catalytic system.
Introduction
Oxazole heterocycles are an important class of structural motifs found in numerous natural products and pharmacologically active synthetic drug molecules due to their biological activities.1 A vast variety of synthetic protocols have been developed for the construction of differently substituted oxazole derivatives through metal-mediated and metal-free catalytic approaches2 by using different substrates. Click chemistry is one of the widely employed techniques for the synthesis of oxazoles and triazoles. In this regard, coinage metal salts have been widely employed as catalysts in click chemistry.3 Moses et al.,4 and Zhan et al.5 independently developed protocols using silver-based catalysts. In 2012, Jiang and coworkers6 employed copper(II) salts for the preparation of oxazoles by using internal alkynes and nitriles.Similarly, Jiao and co-workers7 used copper(II) salts for the preparation of such systems starting from aldehydes and amines under aerobic conditions. Pan and co-workers have also reported 2,5-disubstituted oxazoles by employing copper(I) chloride aerobic dehydrogenative cyclization methods from alkene and azides.8 To date, there are only three reports available describing various oxazoles derived from the terminal alkynes and acyl azides catalyzed by earth-abundant, and cheap copper-based catalysts. In 1995, Abraham and co-workers first time showed the photochemical formation of oxazoles from azides and alkynes.9 Latter, Pérez, and co-workers described the isolation of various oxazoles using sterically bulky amine ligands bearing Cu(I) catalysts such as [(Tpm*Br)Cu(CH3CN)]BF4 (Tpm*Br = tris(3,5-dimethyl-4-bromopyrazolyl)methane), and [(Tpa)*Cu]PF6 (Scheme 1).10–12 In contrast to triazoles,13 synthesis of oxazole derivatives requires relatively more reaction times. Moreover, their synthesis involving cheap and earth-abundant molecular metal-based homogeneous catalysis is still in its infancy. Therefore, high-yielding, atom-economic, and green catalytic approaches are desirable.
 | ||
| Scheme 1 Copper(I) mediated synthesis of various oxazoles (L′ = (BPPP)E2; E = S, and Se). | ||
In recent times, synthesis and applications of various pincer ligands have been widely used as potential systems as ancillary ligands in homogeneous catalysis.14 It can be envisioned that stable pincer ligands supported cationic copper(I) based complexes would be valuable candidates for the construction of such heterocyclic compounds due to the fact that the alkyne bonds can easily be activated formally by the Lewis acidic d10 copper(I) systems. We presumed that pincer ligands consisting of S, Se as coordinating centers would be suitable species,15 thus rarely explored ENE coordinating ligands of the type (BPPP)E2 (BPPP = bis(diphenylphosphino)pyridine; E = S, Se) can offer potential ancillary ligand properties for the effective stabilization of Cu(I) centers in the pincer pocket (Scheme 1). To the best of our knowledge, metal complexes of (BPPP)E2 ligands have never been utilized in any catalytic reactions. It should be noted that the coordination chemistry of (BPPP)E2 was already explored to some extent only with Sn,16 Re,17 and Cr18 metals. In the current study, structurally characterized robust dicationic copper(I) complex bearing (BPPP)E2 was developed, and used as a new catalyst for the facile synthesis of various oxazoles from ethereal-based terminal internal alkynes and azides (Scheme 1). This catalyst was also employed for the preparation of various triazoles.
Results and discussion
The BPPP (1)16,17 was prepared by a slightly modified procedure,19 and subsequently used for the isolation of sulfide and selenide derivatives 2a/2b.16 The treatment of 2a/2b with [Cu(CH3CN)4]BF4 in CH2Cl2 readily afforded [(2a)Cu]2[BF4]2 (E = S (3a), Se (3b)) in 82%, and 53% isolated yields as orange solids (Scheme 2). 3a/3b are highly soluble in polar solvents but insoluble in non-polar solvents such as n-hexane, ether, and pentane.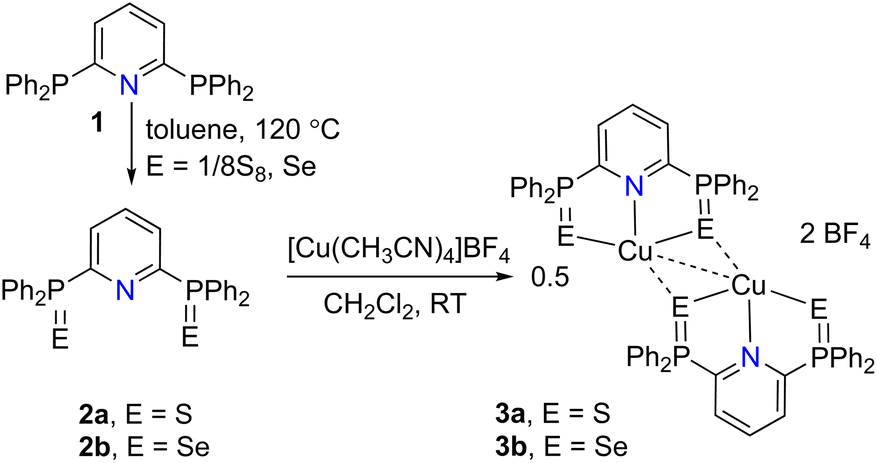 | ||
| Scheme 2 Preparation of dicationic copper(I) complexes with (BPPP)E2 systems. | ||
The spectral data of 3a/3b differed from the corresponding free ligands 2a/2b, indicating metal coordination. When compared with the free ligands, the 1H NMR (in CDCl3) signals of 3a and 3b of p-pyridyl protons at δ = 8.54, and 8.47 ppm respectively are downfield shifted, whereas the meta protons are high field shifted.16 The 31P NMR signals observed at δ = 60.9 (3a), 54.4 (3b) ppm indicated the metal coordination as these shifts are significantly downfield shifted compared with the corresponding free ligands (δ = 37.7 (2a), and 31.8 ppm (2b)). The ESI HRMS data showed the m/z values at 574.0049 (calcd 574.0043) and 669.8940 (calcd 669.8936) for 3a and 3b which can be unambiguously assigned for the presence of stable monocationic fragments [(2a/2b)Cu]+ in acetonitrile (ESI Fig. S9 and S15†).
Single crystals suitable for X-ray diffraction analysis were grown at RT by the layering of hexane over the CH2Cl2 solutions of 3a/3b, and these crystalize in the monoclinic space group P21/c, and P21/n respectively. The molecular structures of 3a (Fig. 1) and 3b (Fig. S125†) show that the distorted tetrahedral copper center is stabilized by three sulfur and one nitrogen donor, and the presence of ENE coordination mode of B(PPP)E2. Both structures show the presence of strong cuprophilic interactions (Cu⋯Cu) between the cationic moieties in an intermolecular fashion. The Cu1–Cu2 distance of 2.5941(6) Å in 3a is shorter than the same observed in 3b (2.7291 Å), however, these contacts fall in the range for reported examples.20 Furthermore, the copper centers in both monocationic moieties are also connected by one of the sulfur atoms of the other unit; for instance the Cu1–S3 and Cu2–S2. In addition, the reactivity of 2a/2b with neutral copper(I) source CuCl was also studied. Reactions in CH2Cl2 afforded 4a (45%) and 4b (40%) respectively (Scheme S1†). In contrast to 3a/3b, the spectral data indicated the paramagnetic nature of 4a/4b, however, the CHNS analysis and HRMS spectral analysis unambiguously supported the presence of similar dicationic moiety but with different counter anion (Fig. S17/S19†). Single crystal X-ray structures of 4a/4b could not be established, and our attempts yielded amorphous insoluble solids.
 | ||
| Fig. 1 ORTEP view with 30% ellipsoid probability of the cationic part of the complex [(2a)Cu]2[BF4]2 (3a). Anionic moiety and hydrogen atoms are omitted for clarity. Important bond lengths [Å] and bond angles [°]. Cu1–Cu2 2.5941(6), Cu1–S1 2.3286(8), Cu1–S3 2.4146(9), Cu1–S4 2.3135(8), Cu1–N1 2.069(3), Cu2–S4 2.4731(9), Cu2–S3 2.3358(9), Cu2–S2 2.3034(9), Cu2–N2 2.092(3), S1–P2 2.0055(11), S3–P3 2.0023(12), S4–P1 1.9811(11), P4–S2 1.9820(13), S2–Cu2–S3 138.04(4), and S4–Cu1–S1 136.39(3). | ||
Catalytic studies of 3a and 3b
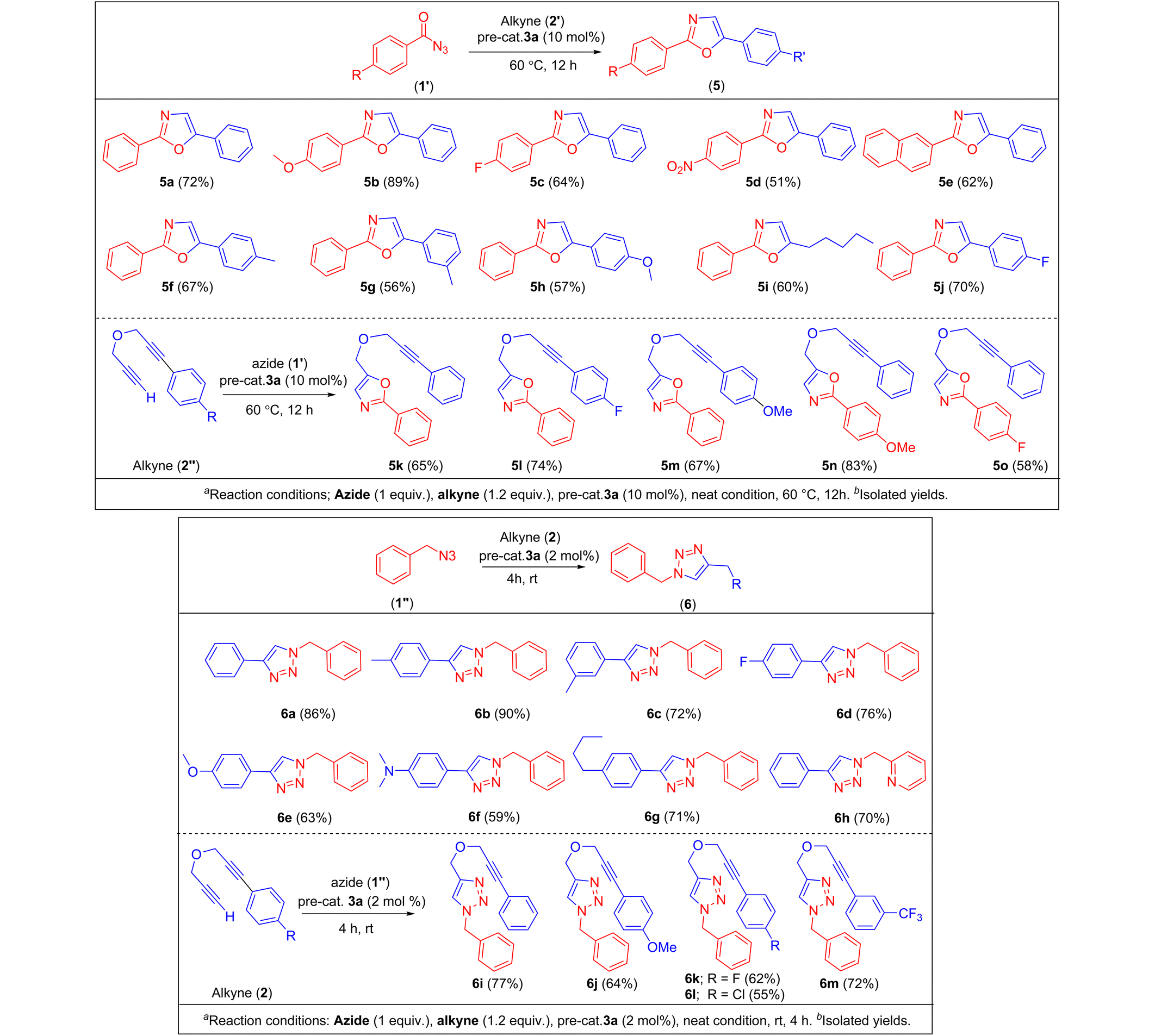
NMR experiments showed that both 3a/3b are stable to moisture and air in solution and solid state, however, in the case of 3b signs of decomposition were observed after several days. Their stability can be attributed to the strong cuprophilic interactions and is another advantage to handle these complexes at higher temperatures. To the best of our knowledge, copper-based catalysts have not been explored in oxazole synthesis as extensively as in the case of triazole synthesis (vide supra). Pérez and coworkers10,11 reported oxazole synthesis using benzoyl azides and terminal alkynes with copper catalyst [(Tpm*Br)Cu(NCMe)]BF4 [(Tpm*Br) = tris(3,5-dimethyl-4-bromopyrazolyl)methane], and showed to yield a mixture of 2,5-disubstituted, and 2,3,5-trisubstituted oxazoles in the solvent media, however, when performed under solvent-free conditions using cationic copper complex [TpaCu]PF6 (Tpa* = tris(3,5-dimethyl-pyrazolylmethyl)amine), 2,5-disubstituted oxazoles were obtained selectively.We first established the best experimental conditions by screening both 3a/3b for the synthesis of various oxazoles. In order to study the solvent effects, a series of solvents such as CH3CN, CHCl3, CH2Cl2, and dichlorobenzene were employed, however, better yields could not be obtained, and in some cases, no product formation was observed (Table 1). Under the solvent-free neat condition (Table 1, entry 1), the reaction of benzoyl azide with phenyl acetylene and 10 mol% 3a at 60 °C temperature afforded the corresponding oxazole (5). Notably, the pre-catalyst 3a yielded very good conversions (50–90%) within 12 h than the 3b, which indicates, the phosphorus sulfide–copper complex is the best choice over the selenium derivative. A gradual increase in the reaction temperature showed more efficiency in the overall oxazole formations with complex 3a. Furthermore, reactions performed at different temperatures for instance at 25 °C (13%), 40 °C (31%), 60 °C (77%) and 80 °C (60%) afforded different yields, indicating the best performance of the pre-catalyst at 60 °C.
|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions | Yield of productb (%) |
| a Reaction conditions: azide (1 equiv.), alkyne (1.2 equiv.), pre-cat. 3a (10 mol%), neat condition, 60 °C, 12 h. b Isolated yields. | ||
| 1 | None | 77 |
| 2 | DCM (0.2 M) instead of neat reaction | 52 |
| 3 | MeCN as a solvent instead of neat reaction | 0 |
| 4 | DCB as a solvent instead of neat reaction | 33 |
| 5 | CHCl3 as a solvent instead of neat reaction | 45 |
| 6 | 0.4 equiv. of Li2CO3 as additive | 29 |
| 7 | 2 mol% Cu-complex instead of 10 mol% | 36 |
| 8 | 5 mol% Cu-complex instead of 10 mol% | 51 |
| 9 | Temperature rt instead of 60 °C | 13 |
| 10 | Temperature 40 °C instead of 60 °C | 31 |
| 11 | Temperature 80 °C instead of 60 °C | 60 |
| 12 | 3b instead of 3a complex | 41 |
| 13 | Alkyne 1 equiv. instead of 1.2 equiv. | 43 |
| 14 | Alkyne 1.5 equiv. instead of 1.2 equiv. | 65 |
| 15 | CuCl & CuI as catalyst | 0 |
| 16 | [Cu(CH3CN)4]BF4 as catalyst | 20 |
| 17 | Without catalyst | 0 |
| 18 | Copper powder | 0 |
| 19 | 2a alone | 0 |
| 20 | 2a/[Cu(CH3CN)4]BF4 | 42 |

Furthermore, lithium carbonate as an additive also did not improve the yield instead it decreased the product formation significantly (Table 1, entry 6). Attempts with 2 mol% and 5 mol% catalyst loadings resulted in the formation of the target compound in 36 and 51% yields respectively (Table 1, entries 7 and 8), indicating more catalyst loading is required to achieve better conversions. Similarly, an equimolar reaction of azide and terminal alkyne, and in another case slight excess of the alkyne (1.5 equiv.) did not show yield enhancement. It is worth mentioning that when commercially available copper(I) salts such as CuCl and CuI were employed as catalysts, no reaction was observed, indicating the crucial role of cationic copper centers for such transformation (Table 1, entry 15). This indicates the requirement of an ancillary ligand attached to the copper atoms which accelerates the reaction dynamics as observed in various click reactions.21,22 Furthermore, we have also tested the same substrates with cationic copper(I) complex [Cu(CH3CN)4]BF4 under similar conditions, and only 20% yield was obtained; however, it indicates the necessity of the cationic copper centre for this set of reactions (Table 1, entry 16). In addition, the same reaction was also performed without the pre-catalyst 3a, and no reaction was observed (Table 1, entry 17); in a similar manner, we have also tested the same reaction in the presence of copper powder, and the ligand 2a; however, both reactions did not show product formations (entries 18 and 19). However, the ligand/metal precursor combination (entry 20; 2a/[Cu(CH3CN)4]BF4) showed a 42% yield, which indicates better yields can be achieved by the molecular catalytic approach (entry 1) over the ligand–metal salt combination.
As shown in Table 2, a series of two different oxazole derivatives were prepared. Under the optimal neat condition by using 10% catalyst, a variety of 2,5-disubstituted oxazoles 5a–5j were prepared as sole products in 50–90% isolated yields. All these derivatives were achieved with complete control of the regioselectivity without any side products. These results are comparable with the similar conversions reported earlier by using sterically bulky ligand-supported cationic copper(I) complex [Tpa*Cu]PF6.11 Furthermore, to expand the derivatives in terms of the directing groups, we examined electron-rich, and electron-poor substituted benzoyl azides by maintaining the same terminal alkyne. The electron-rich azide furnished a higher yield of oxazole 5b, than those with electron-withdrawing groups on the phenyl rings such as 5c, and 5d. In contrast, when we employed electron-withdrawing aryl acetylenes only a slight enhancement in the yield of corresponding oxazole was observed. This confirms the marginal influence of the electronic effect of the substituents. We have further explored the applicability of 3a in the coupling of benzoyl azides with ethereal-based internal-terminal alkynes. For these reactions, three varieties of 1,6-diynes (2′, Table 2) were used. In this set of substrates, five different varieties of 2,5-disubstituted oxazoles featuring ethereal-based alkynes were obtained in moderate to good yields. Similar to the other oxazoles, electron-rich azides afforded the corresponding 2,5-disubstituted oxazoles (5n) in good yields when compared with the electron-withdrawing groups (5o). It should be noted that the pre-catalyst 3a selectively promotes cyclization reactions only at the terminal alkyne positions rather than internal alkyne. By the present protocol, a series of oxazoles bearing ethereal-based alkynes were achieved selectively, and to the best of our knowledge such internal-terminal alkynes have never been employed as substrates in the oxazole synthesis.
|
Furthermore, the catalytic efficiency of 3a was also studied in the synthesis of two different classes of triazoles (Table 2; 6a–6m). Pre-cat. 3a was found to be highly active in this set of reactions as well, and the best conversions were obtained under the solvent-free neat conditions by using 2 mol% catalyst. In this case, both simple and internal-terminal alkynes were coupled with benzyl azide. Under the optimal condition, eight different triazoles 6a–6h were prepared selectively in 59–90% yields. In a similar fashion, another five different para-substituted phenyl groups containing propargylic ether groups were screened for the isolation of 6i–6m in 55–77% yields (Table 2). It should be noted that 1,6-diynes have rarely been employed in the triazoles synthesis, and to the best of our knowledge, only one publication reported where phenyl group substituted bis(propargylic) ether was used in the synthesis of bis(triazole) by employing copper(II) salts and expensive iridium(I) catalysts.23 Noteworthy to mention that a few oxazole and triazole derivatives could be achieved by mechanical grinding and shaking methods (ESI Fig. S126–S142†) by using cat. 3a. However, these results require more in-depth studies which will be reported in our future works.
Studies towards the mechanistic understanding
In order to establish a plausible mechanism, a series of reactions in different conditions were performed as illustrated in Scheme 3. We first studied a radical scavenger reaction (Scheme 3a) by employing one equiv. of TEMPO (2,2,6,6-tetramethylpiperidin-1-yl)oxyl and BHT separately under the optimized conditions with the combination of phenylacetylene and benzoyl azide. From these reactions, 5a was isolated in 29 and 27% yields respectively, and radical scavengers did not prevent the expected product, indicating a preferential approach through the non-radical pathway.6,10 | ||
| Scheme 3 Mechanistic understanding and control experiments in oxazole synthesis. | ||
To further gain more insight into the substituent electronic effects, a competition experiment was performed between the phenylacetylene, and 4-methoxy/4-fluoro benzoyl azides under the established conditions (Scheme 3b). Consequently, 5b and 5c were isolated in 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Fig. S22 and S23†) respectively in pure forms, showing that there is almost negligible impact of the electronic effects in oxazole synthesis under the optimized conditions. Additionally, we have also performed a stoichiometric reaction between alkyne, benzoyl azide, and pre-cat. 3a in an equimolar reaction under the same conditions (Scheme 3c). Mass spectral analysis showed the formation of copper-azide species (A) (detected as M + K; HRMS; 790.0199; calcd 790.0219; Fig. S24†), which is in line with the previously proposed intermediate species in 1,3-dipolar cycloaddition reactions. This species upon releasing the N2 can generate the copper-acyl nitrene intermediate.
:1 ratio (Fig. S22 and S23†) respectively in pure forms, showing that there is almost negligible impact of the electronic effects in oxazole synthesis under the optimized conditions. Additionally, we have also performed a stoichiometric reaction between alkyne, benzoyl azide, and pre-cat. 3a in an equimolar reaction under the same conditions (Scheme 3c). Mass spectral analysis showed the formation of copper-azide species (A) (detected as M + K; HRMS; 790.0199; calcd 790.0219; Fig. S24†), which is in line with the previously proposed intermediate species in 1,3-dipolar cycloaddition reactions. This species upon releasing the N2 can generate the copper-acyl nitrene intermediate.
Furthermore, in order to rule out the possibility of formation of copper nanoparticles and better understanding of the homogeneity of the reactions, mercury drop experiments were performed (ESI Fig. S145–S150†). To prove the proof of concept excess mercury metal was used in the ratio of 10:100 mol% (cat. Cu/Hg). Reactions were performed by using three different substrates, and isolated the corresponding oxazoles 5a (69%), 5e (55%), and 5f (63%) in almost same yields as established in the reactions without Hg metal as clean products. Additionally, the mass spectral analysis of the reaction mixture in the preparation of 5b after 12 h at 60 °C clearly showed the formation of oxazole 5b (m/z = 252.0083) and the presence of cationic pre-catalyst [(2a)Cu]+ (m/z = 574.0087). In addition, the 31P NMR (in CDCl3) of the reaction mixture after 12 h reaction course also indicated the absence of free ligand (δ = 37.73 ppm), indicating the pincer ligand bound cationic copper species. These results clearly demonstrate the robustness of the catalyst, and also support the homogeneous catalytic pathway.
A detailed plausible mechanism for the transformation of alkynes and azides to the corresponding oxazoles is illustrated in Scheme 4. The copper catalyst first reacts with the benzoyl azide to form A (Fig. S24†), this could leave N2 (detected by GC; Fig. S25†) to form the copper-acyl nitrene intermediate B which further reacts with terminal alkyne to form C. This intermediate can transfer into ketinimide D. Subsequent protonation followed by rearrangement of D triggers the formation of copper(I) bound cyclized intermediate F. It can be presumed that 1,2-hydrogen shift would further liberate the end product oxazole by releasing the active pre-catalytic species L′Cu+.10 Noteworthy, similar mechanistic investigations have been proposed for such a class of compounds by computational methods involving other copper(I) sources.11 In a similar manner, a plausible mechanism can be derived for the triazole synthesis catalyzed by pre-cat. 3a (Scheme S2†).22,24
 | ||
| Scheme 4 Proposed catalytic cycle for the synthesis of 2,5-disubstituted oxazole derivatives by pre-cat. 3a. | ||
Conclusions
In conclusion, (BPPP)E2 pincer ligands supported robust dicationic bi-nuclear Cu(I) complexes that were structurally characterized, and they display intermolecular cuprophilic interactions between two monocationic units. The newly designed copper(I) system serves as a potential catalyst in the synthesis of various di-substituted oxazoles starting from rarely explored bis(propargylic) ethers without additives. The pre-cat. 3a selectively cyclizes the terminal alkyne group by keeping the internal alkyne intact, and mechanistic studies indicate the formation of copper-acyl azide species.11 All the reported conversions were achieved under neat solvent-free conditions. The triazole derivatives bearing alkyne-phenyl propargylic ether substituents synthesized in this study can serve as potential starting materials for the preparation of various fused polycyclic systems.25 Further applications of (BPPP)E2 ligands in organometallic chemistry are currently under consideration in our laboratory.Experimental section
Materials and methods
All the reagents were purchased from commercially available sources (Loba Chemie, Spectrochem and Sigma-Aldrich, TCI, and Avra). The copper powder was purchased from Spectrochem. Internal terminal alkyne, 1-phenyl-4-oxahepta-1,6-diyne, and its substituted derivatives were prepared according to the reported synthetic protocols.26 Starting materials were prepared by following the reported procedures. Dichloromethane was dried using a distillation setup over calcium hydride in an argon/nitrogen atmosphere. THF and n-hexanes were dried using a distillation setup over sodium metal and benzophenone in an atmosphere of dry N2. All the catalytic reactions were carried out in a Borosil-sealed tube in an N2 atmosphere. The TLC plates were detected using UV light. The NMR spectra (1H, 13C, 31P, 19F, and 11B NMR) were measured using the Bruker Avance 400 and 700 MHz NMR spectrometers. Chemical shifts (δ) are given in ppm referenced to tetramethylsilane (TMS), using the residual solvent as an internal standard (CDCl3, 1H; 7.26 ppm, and 13C; 77.16 ppm; DMSO-d6; 1H; 2.50 ppm, and 13C; 39.52 ppm). Elemental analysis was measured on the Elementar Unicube instrument. The ESI mass spectral data was measured using Waters Xevo G2-XS QToF instrument using CH3OH or CH3CN solvents.Crystallography
The single crystal X-ray analysis was performed using a Rigaku SmartLab X-ray diffractometer. Crystals were mounted on the nylon loop under the microscope using a paraffin bar, and X-ray data were collected on the Rigaku Super Nova fine-focused dual diffractometer, with CuKα radiation (λ = 1.54178 Å) and MoKα radiation (0.71073 Å) equipped with a PILATUS200K. Using Olex2, the structures were solved with the ShelXS structure solution program using direct methods and refined with the ShelXL refinement package. The CCDC numbers 2257376 and 2257375 for compounds 3a and 3b respectively.†Synthesis of reported compounds
Catalytic reactions
Synthesis of 5a–5j. In a 50 mL sealed tube charged with a stirring bar was added azide (1.0 equiv.), alkyne (1.2 equiv.), and pre-catalyst 3a (10 mol%) under nitrogen atmosphere. Then, the reaction mixture was allowed to stir for 12 h at 60 °C. After cooling to room temperature, the crude reaction mixture was purified by silica gel column chromatography (10% EtOAc/hexane mixture) to afford the corresponding substituted oxazoles. Specific yields and spectral data are given in the ESI† accordingly.
Synthesis of 5k–5o. In a 50 mL sealed tube charged with a stirring bar was added azide (1 equiv.), bis(propargylic) ethers (1.2 equiv.), and pre-catalyst 3a (10 mol%) under N2 atmosphere. Then, the reaction mixture was allowed to stir for 12 h at 60 °C. After cooling to room temperature, the crude reaction mixture was purified by silica gel column chromatography (10% EtOAc/hexane mixture) to afford the corresponding substituted oxazoles. Specific yields and spectral data of 5k–5o are given in the ESI† accordingly.
Synthesis of 6a–6h. In a 50 mL sealed tube charged with a stirring bar was added benzyl azide (1 equiv.), alkyne (1.2 equiv.), and pre-catalyst 3a (2 mol%) under N2 atmosphere. The reaction mixture was allowed to stir for 4 h at room temperature. Then the crude mixture was purified by silica gel column chromatography (30% EtOAc/hexane mixture) to afford the corresponding triazole derivatives. Specific yields and spectral data of 6a–6h are given in the ESI† accordingly.
Synthesis of 6i–6m. In a 50 mL seal tube charged with a magnetic bar was added benzyl azide (1 equiv.), propargylic ethers (1.2 equiv.), and pre-catalyst 3a (2 mol%) under the nitrogen atmosphere. The reaction mixture was allowed to stir for 4 h at room temperature. Then the crude reaction mixture was purified by silica gel column chromatography (30% EtOAc/hexane mixture) to afford the corresponding triazole derivatives. Specific yields and spectral data of 6i–6m are given in the ESI† accordingly.
Data availability
All the other specific experimental procedures, spectral and crystallographic details of the compounds are given in the ESI.† Crystallographic data for compounds 3a/3b has been deposited as CCDC 2257376 and 2257375 respectively.†Author contributions
BD and AKS prepared metal complexes, and substrates, and worked on the manuscript. SKB assisted in the synthesis of a series of substrates. SJP and CP performed the X-ray diffraction studies. AD supervised the whole project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AD thanks IISER Berhampur for the CAIF facility and SERB for the Start-Up Research Grant. BD and AKS are thankful for the institute fellowship. We are also thankful to Dr Ponneri C. Ravikumar, Department of Chemical Sciences at NISER Bhubaneswar for suggestions related to the project.References
- (a) P. Wipf, Chem. Rev., 1995, 95, 2115–2134 CrossRef CAS; (b) V. S. Yeh, Tetrahedron, 2004, 60, 11995–12042 CrossRef CAS; (c) A. C. Giddens, H. I. Boshoff, S. G. Franzblau, C. E. Barry and B. R. Copp, Tetrahedron Lett., 2005, 46, 7355–7357 CrossRef CAS; (d) S. Kakkar and B. Narasimhan, BMC Chem., 2019, 13, 16 CrossRef PubMed; (e) Y. Hamada and T. Shioiri, Chem. Rev., 2005, 105, 4441–4482 CrossRef CAS PubMed; (f) Z. Jin, Nat. Prod. Rep., 2006, 23, 464–496 RSC; (g) A. Thakur, M. Verma, R. Bharti and R. Sharma, Tetrahedron, 2022, 119, 132813 CrossRef CAS; (h) P. Garg, R. S. Rawat, H. Bhatt, S. Kumar and S. R. Reddy, ChemistrySelect, 2022, 7, e202201706 CrossRef CAS.
- (a) T. Ibata and R. Sato, Chem. Lett., 1978, 7, 1129–1130 CrossRef; (b) M. Sun, L. Zhao and M.-W. Ding, J. Org. Chem., 2019, 84, 14313–14319 CrossRef CAS PubMed; (c) T. Chatterjee, J. Y. Cho and E. J. Cho, J. Org. Chem., 2016, 81, 6995–7000 CrossRef CAS PubMed; (d) W. Xu, U. Kloeckner and B. J. Nachtsheim, J. Org. Chem., 2013, 78, 6065–6074 CrossRef CAS PubMed; (e) I. Romero-Estudillo, V. R. Batchu and A. Boto, Adv. Synth. Catal., 2014, 356, 3742–3748 CrossRef CAS.
- (a) W. Yang, R. Zhang, F. Yi and M. Cai, J. Org. Chem., 2017, 82, 5204–5211 CrossRef CAS PubMed; (b) A. D. Gillie, R. Jannapu Reddy and P. W. Davies, Adv. Synth. Catal., 2016, 358, 226–239 CrossRef CAS.
- D. J. Ritson, C. Spiteri and J. E. Moses, J. Org. Chem., 2011, 76, 3519–3522 CrossRef CAS PubMed.
- Y. Pan, F. Zheng, H. Lin and Z. Zhan, J. Org. Chem., 2009, 74, 3148–3151 CrossRef CAS.
- X. Li, L. Huang, H. Chen, W. Wu, H. Huang and H. Jiang, Chem. Sci., 2012, 3, 3463 RSC.
- Z. Xu, C. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 11367–11370 CrossRef CAS.
- J. Li, Y. Wang, W. Li, H. Wang, D. Mo and Y. Pan, Chem. Commun., 2015, 51, 17772–17774 RSC.
- K.-U. Clauss, K. Buck and W. Abraham, Tetrahedron, 1995, 51, 7181–7192 CrossRef CAS.
- I. Cano, E. Álvarez, M. C. Nicasio and P. J. Pérez, J. Am. Chem. Soc., 2011, 133, 191–193 CrossRef CAS PubMed.
- E. Haldón, M. Besora, I. Cano, X. C. Cambeiro, M. A. Pericàs, F. Maseras, M. C. Nicasio and P. J. Pérez, Chem. – Eur. J., 2014, 20, 3463–3474 CrossRef PubMed.
- E. Haldón, M. Delgado-Rebollo, A. Prieto, E. Álvarez, C. Maya, M. C. Nicasio and P. J. Pérez, Inorg. Chem., 2014, 53, 4192–4201 CrossRef.
- E. Haldón, M. C. Nicasio and P. J. Pérez, Org. Biomol. Chem., 2015, 13, 9528–9550 RSC.
- (a) D. Morales-Morales, Pincer compounds. Chemistry and applications, ed. D. Morales-Morales, Elsevier, Amsterdam, 2018 Search PubMed; (b) G. van Koten and R. A. Gossage, The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications, Springer International Publishing, Cham, 2016, vol. 54 Search PubMed; (c) H. Valdés, M. A. García-Eleno, D. Canseco-Gonzalez and D. Morales-Morales, ChemCatChem, 2018, 10, 3136–3172 CrossRef; (d) G. van Koten and D. Milstein, Organometallic Pincer Chemistry, Springer Berlin Heidelberg, Berlin, Heidelberg, 2013, vol. 40 Search PubMed; (e) J. Dupont, C. S. Consorti and J. Spencer, in The Chemistry of Pincer Compounds, ed. D. Morales-Morales and C. M. Jensen, Elsevier Science B.V., Amsterdam, 2007, pp. 1–24 Search PubMed.
- N. Mézailles and P. Le Floch, The Chemistry of Pincer Compounds, Elsevier, 2007, pp. 235–271 Search PubMed.
- R. Sevcik, M. Necas and J. Novosad, Polyhedron, 2003, 22, 1585–1593 CrossRef CAS.
- P. J. Heard and A. E. Aliev, Polyhedron, 1998, 17, 3981–3987 CrossRef CAS.
- H. Christina, E. McFarlane, W. McFarlane and A. S. Muir, Polyhedron, 1990, 9, 1757–1764 CrossRef.
- J. Ye, J.-Q. Zhang, Y. Saga, S. Onozawa, S. Kobayashi, K. Sato, N. Fukaya and L.-B. Han, Organometallics, 2020, 39, 2682–2694 CrossRef CAS.
- (a) N. V. S. Harisomayajula, S. Makovetskyi and Y.-C. Tsai, Chem. – Eur. J., 2019, 25, 8936–8954 CrossRef CAS PubMed; (b) F. A. Cotton, X. Feng and D. J. Timmons, Inorg. Chem., 1998, 37, 4066–4069 CrossRef CAS PubMed; (c) T. C. Davenport and T. D. Tilley, Angew. Chem., Int. Ed., 2011, 50, 12205–12208 CrossRef CAS PubMed; (d) A. Doddi, D. Bockfeld, A. Nasr, T. Bannenberg, P. G. Jones and M. Tamm, Chem. – Eur. J., 2015, 21, 16178–16189 CrossRef CAS PubMed.
- (a) V. O. Rodionov, S. I. Presolski, D. D. Díaz, V. V. Fokin and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12705–12712 CrossRef CAS PubMed; (b) V. O. Rodionov, S. I. Presolski, S. Gardinier, Y.-H. Lim and M. G. Finn, J. Am. Chem. Soc., 2007, 129, 12696–12704 CrossRef CAS PubMed; (c) Y.-C. Lin, Y.-J. Chen, T.-Y. Shih, Y.-H. Chen, Y.-C. Lai, M. Y. Chiang, G. C. Senadi, H.-Y. Chen and H.-Y. Chen, Organometallics, 2019, 38, 223–230 CrossRef CAS.
- C. Nolte, P. Mayer and B. F. Straub, Angew. Chem., Int. Ed., 2007, 46, 2101–2103 CrossRef CAS PubMed.
- X. Duan, N. Zheng, M. Li, X. Sun, Z. Lin, P. Qiu and W. Song, Chin. Chem. Lett., 2021, 32, 4019–4023 CrossRef CAS.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460 CrossRef CAS PubMed.
- Y. Shi and V. Gevorgyan, Chem. Commun., 2015, 51, 17166–17169 RSC.
- T. Kudoh, T. Mori, M. Shirahama, M. Yamada, T. Ishikawa, S. Saito and H. Kobayashi, J. Am. Chem. Soc., 2007, 129, 4939–4947 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, spectral data, experimental procedures and full characterization data. CCDC 2257375 (3b) and 2257376 (3a). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01989f |
| This journal is © The Royal Society of Chemistry 2023 |