
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlatinum(IV) combo prodrugs containing cyclohexane-1R,2R-diamine, valproic acid, and perillic acid as a multiaction chemotherapeutic platform for colon cancer†
Elisabetta
Gabano‡
 a,
Marzia Bruna
Gariboldi‡
b,
Emanuela
Marras
b,
Francesca
Barbato
c and
Mauro
Ravera
*c
a,
Marzia Bruna
Gariboldi‡
b,
Emanuela
Marras
b,
Francesca
Barbato
c and
Mauro
Ravera
*c
aDipartimento per lo Sviluppo Sostenibile e la Transizione Ecologica, Università del Piemonte Orientale, Piazza Sant'Eusebio 5, 13100 Vercelli, Italy
bDipartimento di Biotecnologie e Scienze della Vita (DBSV), Università dell'Insubria, via Dunant 3, Varese, Italy
cDipartimento di Scienze e Innovazione Tecnologica, Università del Piemonte Orientale, Viale Michel 11, 15121 Alessandria, Italy. E-mail: mauro.ravera@uniupo.it
First published on 18th July 2023
Abstract
The complex [PtCl2(cyclohexane-1R,2R-diamine)] has been combined in a Pt(IV) molecule with two different bioactive molecules (i.e., the histone deacetylase inhibitor 2-propylpentanoic acid or valproic acid, VPA, and the potential antimetastatic molecule 4-isopropenylcyclohexene-1-carboxylic acid or perillic acid, PA) in order to obtain a set of multiaction or multitarget antiproliferative agents. In addition to traditional thermal synthetic procedures, microwave-assisted heating was used to speed up their preparation. All Pt(IV) complexes showed antiproliferative activity on four human colon cancer cell lines (namely HCT116, HCT8, RKO and HT29) in the nanomolar range, considerably better than those of [PtCl2(cyclohexane-1R,2R-diamine)], VPA, PA, and the reference drug oxaliplatin. The synthesized complexes showed pro-apoptotic and pro-necrotic effects and the ability to induce cell cycle alterations. Moreover, the downregulation of histone deacetylase activity, leading to an increase in histone H3 and H4 levels, and the antimigratory activity, indicated by the reduction of the levels of matrix metalloproteinases MMP2 and MMP9, demonstrated the multiaction nature of the complexes, which showed biological properties similar to or better than those of VPA and PA, but at lower concentrations, probably due to the lipophilicity of the combo molecule that increases the intracellular concentration of the single components (i.e., [PtCl2(cyclohexane-1R,2R-diamine)], VPA and PA).
Introduction
Although over the years Pt(IV) complexes have not succeeded to be a serious alternative to traditional Pt(II) anticancer agents, they are still widely studied because of their characteristics that make them particularly suitable for the design of metal-based drugs with tailored properties. Actually, octahedral Pt(IV) complexes can reach intact cancer cells and the hypoxic tumor environment can activate them by reductive elimination of their axial ligands, thus releasing the corresponding cytotoxic Pt(II) metabolites (activation by reduction) and increasing the selectivity of the drugs.1–6 In addition, the axial ligands provide that something extra that makes Pt(IV) complexes so interesting. As a matter of fact, the axial ligands that will be released after reduction can be designed to play a role in the overall activity of the Pt(IV) complexes. They can be tumor-targeting molecules (hormones, folates, amino acids, peptides, carbohydrates, etc.), pharmaceutical agents (e.g., enzyme inhibitors), or anticancer drugs with a different mechanism of action, able to synergize with the Pt(II) metabolites. Such multiaction or multitarget prodrugs must contain one or more complementary bioactive molecules.7–13 Apart from the specific role of the ligands, the multiaction Pt(IV) complexes are generally more active than their free components administered separately, as the combination gains in lipophilicity and, thus, in cellular uptake, ensuring high intracellular levels of both the cytotoxic Pt(II) metabolites and the organic active biomolecules (synergistic cellular accumulation).12,14,15In this work, (SP-4-2)-(cyclohexane-1R,2R-diamine)dichloridoplatinum(II) ([PtCl2(dach)] or 1, Scheme 1; dach = cyclohexane-1R,2R-diamine) has been combined with two different bioactive molecules (i.e., 2-propylpentanoic acid or valproic acid, VPA, and 4-isopropenylcyclohexene-1-carboxylic acid or perillic acid, PA) in a set of different Pt(IV) complexes in order to obtain the final multifunctional ‘combo’ molecules (Scheme 1) showing properties better than those of the single components.
 | ||
| Scheme 1 Sketch of the complexes under investigation and the corresponding synthetic pathways: (i) H2O2/CH3COOH; (ii) H2O2/H2O; (iii) acyl chloride of PA, microwave heating, CH3CN, pyridine, (iv) acyl chloride of VPA, microwave heating, CH3CN, pyridine; (v) perillic anhydride, DMSO. The numbering scheme for the assignment of NMR signals is reported in the ESI; PA = perillic acid, VPA = valproic acid. | ||
The dach ligand imparts peculiar characteristics to Pt(II) complexes, being present in the anticancer drug oxaliplatin, (SP-4-2)(cyclohexane-1R,2R-diamine)(ethanedioato)platinum(II). Computer modeling showed that the cyclohexane ring protrudes directly outward into the major groove of the bound DNA, markedly altering the area of the adduct.16,17 The differences in the structure of the adduct produced by cisplatin and oxaliplatin are consistent with the observation that they are differentially recognized by the DNA mismatch repair system, with cisplatin being more easily recognized, making Pt(II)-dach complexes an interesting escape route to bypass cisplatin-resistance.18,19 In particular, 1 was chosen as the equatorial structure based on previous experience acquired by the authors on both the synthesis and the study of the biological properties of this synthon and its Pt(IV) derivatives. Moreover, such Pt(IV) prodrugs show relatively high cellular uptake and good antiproliferative activity, and the presence of dach itself could increase cancer cell aggression by the immune system through immunogenic cell death (ICD).20 In contrast, other authors prefer the use of oxaliplatin, because its Pt(IV) derivatives can be more stable in vitro and more persistent in vivo, even though this depends on the nature of the coordinated ligands.5,21–23
VPA is a well-known histone deacetylase (HDAC) inhibitor. The abnormal enhancement of some members of the HDAC family has been closely related to the appearance and development of various malignant tumors. In addition, changes in the expression and mutation of HDAC-encoding genes have been associated with tumor growth, due to their ability to induce abnormal transcription of genes that regulate key cell functions, such as cell proliferation, cell-cycle regulation, and apoptosis. For this reason, HDAC inhibitors (HDACIs) can induce growth arrest, differentiation, or apoptosis, to a variable extent, in vitro and in vivo. Noticeably, normal cells are usually more resistant than tumor cells to HDACIs.24–27 The combination of HDACIs as chemosensitizers with other cancer therapeutics has shown great potential in preclinical and therefore clinical trials and may represent a way to achieve complete therapeutic benefits.28–32 Different HDAC inhibitors have already been linked in the axial positions of Pt(IV) cores, showing synergistic activity with Pt(II) metabolites.14,15,20,33–40
PA, an active metabolite of limonene, is capable of inhibiting farnesyltransferase (FTase) and geranylgeranyltransferase (GGTase I), the enzymes that catalyze protein prenylation.41 This is necessary for the cellular activity of many oncogenic proteins, including some members of the Ras family. Therefore, prenylation inhibitors are promising for antitumor therapy blocking Ras signaling.42–46 Moreover, PA exhibits good in vivo antimetastatic activity,47 modulates the immune system,48 and acts as a radiosensitizer.49 Recently, a cisplatin-based Pt(IV) complex with two molecules of PA was shown to exhibit high antiproliferative and antimetastatic activity at nanomolar concentrations in lung A-549 cancer cells.50
On the basis of the described activities of VPA and PA, we herein report the design, synthesis, characterization, and biological activities of Pt(IV) derivatives containing different combinations of the two organic ligands. Oxaliplatin in combination with fluorouracil and leucovorin has shown a definite role in the treatment of colorectal cancer (FOLFOX4 regimen). For this reason, the activity of the Pt(IV) derivatives was evaluated in HCT116, HCT8, RKO, and HT29 colon cancer cells, and their effects on HDAC and cell migration were studied to elucidate the mechanism of action of the compounds and reveal the possible multiple effects of some of them.
Results and discussion
Synthesis and characterization
The Pt(IV) complexes under investigation were synthesized by oxidizing [PtCl2(dach)], 1,51,52 with hydrogen peroxide in acetic acid (complex 2) or in water (complex 3) (Scheme 1).53,54 For the subsequent substitution reaction, PA and VPA had to be turned into a more reactive form, that is, acyl chloride or anhydride, using oxalyl chloride in the presence of a catalytic amount of dimethylformamide or the coupling agent N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride, respectively. Reactions of 2 and 3 with acyl chlorides were carried out in acetonitrile with microwave-assisted heating to obtain complexes 4–7.55 The reaction of 3 with perillic anhydride required the use of DMSO as a solvent to favor the formation of monosubstituted complex 8.56 Finally, this intermediate was turned into trifunctional complex 9 upon reaction with VPA acyl chloride as described above.All complexes were characterized by RP-HPLC/ESI-MS and NMR spectroscopy using mono- and bidimensional techniques (see ESI, Fig. S1–S27,† for the most relevant NMR spectra). The dach ligand is supposed to be in a chair configuration, as observed in other similar structures;57,58 but, when the axial ligands are different, couples of corresponding carbon atoms are no longer equivalent and resonate at different frequencies in 13C NMR spectra. As far as the NH2 groups of coordinated dach are concerned, in the 1H spectra in DMSO-d6 of complexes with different axial ligands, four different signals are visible; whereas, when the axial ligands are equal, only two signals are observed. The chemical shift of such signals varies with the change of the other ligands. These features of dach ligands in Pt(IV) complexes have been observed for similar complexes.54 Also, the signals of VPA chains are affected by the other ligands, which results in not-perfectly-equivalent nuclei and the corresponding splitting of most signals. The 195Pt NMR signals of the final dicarboxylato complexes exhibit chemical shifts within the range of 1070–1115 ppm that are consistent with the expected coordination sphere of such Pt(IV) complexes.59–67
The final compounds 4–7 and 9 are supposed to be quite lipophilic and a RP-HPLC technique was used to measure their relative lipophilicity, associated with the ability to cross cell membranes passively.68 This is possible since retention is due to partitioning between the hydrophobic stationary phase (model of cell membranes) and the aqueous mobile phase (model of the environment inside and outside cells).66,69 The retention time (tR) of the complexes was measured on a C18 column employing a 30/70 mixture of 15 mM formic acid/MeOH as an eluent. Data are expressed as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k′ (k′ = (tR − t0)/t0, where t0 is the column dead-time) and are reported in Table 1.
k′ (k′ = (tR − t0)/t0, where t0 is the column dead-time) and are reported in Table 1.
k′) and IC50 values (nM) obtained in HCT116, HCT8, RKO, and HT29 cells by the MTT assay after 72 h of treatment with the studied derivatives and the MTT assay (mean ± S.E. of five independent experiments; #p < 0.001 vs. all others; ***p < 0.001 vs. oxaliplatin, 4, 5 and 7)
| Compound | logk′ (70% MeOH) |
IC50 (nM) HCT116 | IC50 (nM) HCT8 | IC50 (nM) RKO | IC50 (nM) HT29 |
|---|---|---|---|---|---|
| oxaliplatin | −0.48 | 42.82 ± 3.07 | 191.62 ± 16.28 | 217.95 ± 46.00 | 261.18 ± 31.54 |
| 1 | −0.4640 | 343.19 ± 7.38# | 1377.05 ± 187.67# | 1242.68 ± 219.16# | 1659.49 ± 16.40# |
| 4 | 0.43 | 13.90 ± 1.38 | 44.41 ± 2.95 | 29.41 ± 4.38 | 29.41 ± 8.85 |
| 5 | 0.48 | 10.95 ± 3.33 | 34.95 ± 3.50 | 25.85 ± 7.42 | 101.01 ± 20.72 |
| 6 | 1.15 | 1.07 ± 0.30*** | 9.52 ± 1.74*** | 3.78 ± 0.66*** | 4.37 ± 0.32*** |
| 7 | 1.18 | 12.68 ± 4.77 | 34.36 ± 7.22 | 16.19 ± 4.02 | 19.69 ± 6.45 |
| 9 | 1.16 | 0.91 ± 0.19*** | 5.03 ± 0.94*** | 4.55 ± 1.42*** | 8.19 ± 1.75*** |
| PA | 0.5574 | (130.96 ± 22.64) × 103# | (154.87 ± 8.73) × 103# | (161.85 ± 17.3) × 103# | (120.67 ± 7.32) × 103# |
| VPA | 0.56 | (82.91 ± 5.25) × 103# | (139.68 ± 13.83) × 103# | (89.45 ± 9.13) × 103# | (112.09 ± 38.22) × 103# |
Cytotoxicity
The effect of Pt(IV) derivatives on four different human colon cancer cell lines, namely HCT116, RKO, HCT8, and HT29, was evaluated. Cell viability was assessed by the MTT assay upon treatment with increasing concentration of the compounds studied. Compounds 1, oxaliplatin, VPA, and PA were added to the experiments as references. The antiproliferative effect of the compounds was quantitated by calculating the drug concentration that inhibits tumor cell viability by 50% (half-maximal inhibitory concentration, IC50) from the respective dose–response curves (Table 1). Unlike VPA and PA, for which the IC50 values were found to be in the order of hundreds of micromoles per liter, all new Pt(IV) derivatives showed cytotoxic effects at nanomolar concentrations. Taking into account only the new Pt(IV) derivatives, 4, 5, 6, 7 and 9 showed higher potency than their precursor, namely 1. Interestingly, 6 and 9 were shown to be the most potent ones across all cell lines tested. Thus, in agreement with data reported by our group and by other authors,15,35,50 these results indicate that the presence of a perillate or valproate group on the Pt(IV) molecules improved the cytotoxic effect of all progenitor molecules. Actually, as mentioned in the Introduction, a lipophilic Pt(IV) complex is able to increase the intracellular concentration of its (hydrophilic or amphiphilic) fragments by synergistic cellular accumulation when they are combined into a single molecule.In addition, it has been widely reported that the uptake of Pt-based drugs may deeply influence their cytotoxic effects and several membrane transporters have been involved in the cytotoxicity of oxaliplatin and some Pt(IV) derivatives, including organic cation transporters (OCTs).70,71 Notably, recent studies have shown increased expression of OCT1 and OCT2 in several colon cancer cells, including HCT116 and HCT8 cell lines.72,73 The high potencies observed for the compounds under investigation may be explained, at least in part, also considering the potentially increased uptake of the Pt(IV) derivatives due to the increased expression of the OCT transporters.
It has been widely reported that not only Pt(II) derivatives but also PA and VPA can inhibit cancer growth through the induction of cell death, mainly apoptotic or necrotic, and cell cycle alterations.75–77 Thus, to further characterize the mechanisms responsible for the cytotoxic effects of 4, 5, 6, 7 and 9, their ability to induce apoptotic and/or necrotic cell death and/or cell cycle alterations was evaluated by flow cytometric analysis. The results were then compared with those obtained by treating the cells with the reference compounds oxaliplatin, PA, and VPA, in the following experiments.
Due to its better antiproliferative activity compared to 1 (Table 1) and considering that it is one of the most widely used drugs in the treatment of colorectal cancer, oxaliplatin was used as a single reference compound for the subsequent experiments and 1 was no longer investigated.
According to previous reports,75–77PA and VPA induced apoptotic cell death in HCT116, HCT8, RKO and HT29 cell lines. Furthermore, a different degree of apoptosis was observed when cells were treated with equitoxic concentrations of Pt(IV) compounds, corresponding to the respective IC50 values. As shown in Fig. 1, all Pt(IV) derivatives induced apoptotic cell death over controls in HCT116, HCT8, RKO, and HT29 cell lines. However, low percentages of apoptosis were observed in HT29 cells, compared to the other three cell lines. Interestingly, 6, 7 and 9 induced a significantly higher extent of apoptosis compared to PA and VPA in HCT116, HCT8 and RKO cell lines.
 | ||
| Fig. 1 Percentage of apoptotic (A) and necrotic (B) HCT116, HCT8, RKO, HT29 cells after 72 h of treatment with equitoxic concentrations of the studied derivatives and reference compounds (mean ± S.E. of three independent experiments). Statistical analysis: *p < 0.05, **p < 0.01 and ***p < 0.001 vs. control (C). | ||
Analysis of necrotic cell death induction in the four model cell lines under investigation showed that all compounds did not induce necrosis in the HCT116 and RKO cell lines, while a significant increase in the percentage of necrotic cells was observed in HCT8 cells treated with 5, 6, 7, 9, PA and VPA, and in HT29 cells treated with 5, 6, 7, 9 and VPA.
The different responses, in terms of apoptotic cell death, observed in HCT116, HCT8, RKO and HT29 cells upon treatment with the Pt(IV) derivatives could be related to their p53 status, being mutated in the latter and wild type in the other three cell lines. As a matter of fact, it is well known that the status of the tumor suppressor gene p53 may influence the cellular response to anticancer drugs. Specifically, p53 has been related to drug resistance and resistance to apoptotic/necrotic stimuli.78,79 Therefore, the results indicated that the presence of mutated p53 could account for the different apoptotic/necrotic responses to equitoxic concentrations of Pt(IV) observed in HT29 cells.
Considering that a shift in cell cycle phases, an important finding in cell lines undergoing antineoplastic treatment, has previously been demonstrated for both PA and VPA,75,76 the effect of Pt(IV) derivatives included in this study on the distribution of HCT116, HCT8, RKO and HT29 cell lines in the different phases of the cell cycle was evaluated.
Cell-type differences were also observed in the results obtained in this set of experiments (Fig. 2). In particular, none of the Pt(IV) and reference compounds was able to induce cell cycle alterations in HT29 cells, while the cell cycle shift was variable in the other three cell lines. This different behavior could also be related to the status of p53. According to data from other authors,75,76 when cell cycle alterations were present, a shift toward the G0/G1 phase and a corresponding reduction in the S phase was observed.
 | ||
| Fig. 2 Effects on cell cycle progression. Cells were exposed for 72 h to oxaliplatin (oxPt), PA, VPA, and different Pt(IV) derivatives at equitoxic concentrations corresponding to the respective IC50 values (mean ± S.E. of three independent experiments). | ||
Cell cycle progression is finely regulated by the inhibitory activity of a family of proteins capable of binding to specific protein kinase complexes, which are responsible for the passage of cells through the different phases of the cell cycle. In particular, p21waf1/cip1, a member of the Cip/Kip family, is known to play a critical role in the regulation of the cellular transition in the G1/S phase and alteration in its protein levels could explain the results shown in Fig. 2. Thus, the effect of the equitoxic concentrations of 4, 5, 6, 7, 9, PA, VPA, and oxaliplatin on p21waf1/cip1 protein levels was evaluated. Fig. 3 shows that all the compounds studied induced an increase in p21waf1/cip1 levels, although to different extents. In particular, in HCT116 cells, the G1/G0 shift and the consequent lower percentage of apoptotic cells observed following treatment with 7 could be related to a greater increase in p21waf1/cip1 induced by this compound. As a confirmation, the low increase in p21waf1/cip1 protein levels shown in the same cell line after treatment with 5 and 9 did not lead to cell cycle alterations or to a reduced apoptotic response. On the other hand, in RKO cells, a high increase in p21waf1/cip1 following treatment with all Pt(IV) derivatives led to a G0/G1 shift and high levels of apoptosis. Furthermore, HT29 cells increased p21waf1/cip1 levels once treated with 4, 5, 6, 7, 9, PA, VPA, and oxaliplatin; however, no cell cycle alterations were observed. Therefore, p21waf1/cip1 may play a role in the response to Pt(IV) derivatives; however, other factors are probably involved.
 | ||
| Fig. 3 Western blot analysis of p21waf1/cip1 levels in HCT116, HCT8, RKO, and HT29 cell lines treated 72 h with oxaliplatin (oxPt), PA, VPA and the different Pt(IV) derivatives studied at equitoxic concentrations corresponding to the respective IC50 values (mean ± S.E. of three independent experiments). | ||
Effects on histone deacetylases
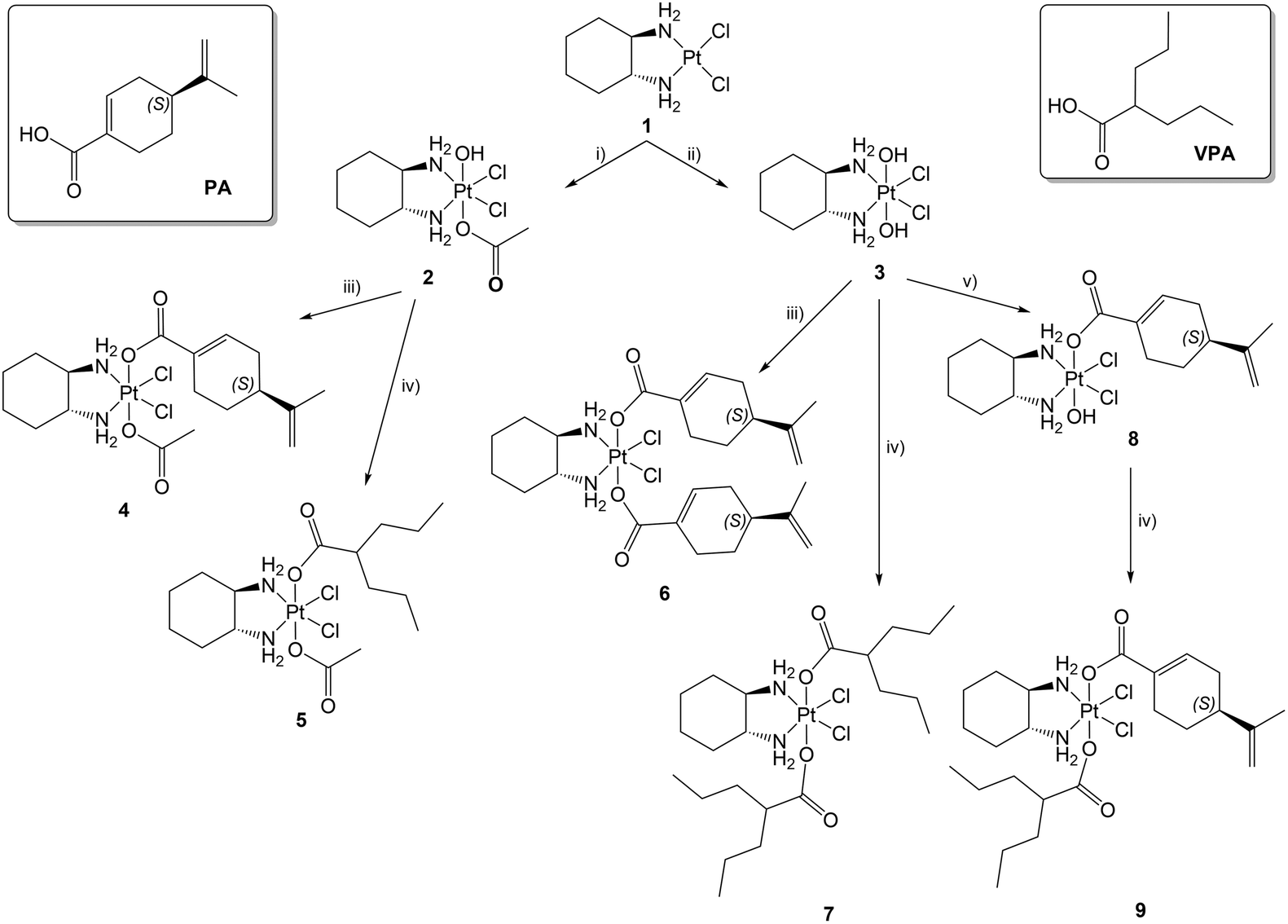
Experimental evidence indicates that VPA-containing Pt(IV) derivatives possess both the DNA binding activity, typical of the Pt(II) derivatives, and the histone deacetylase (HDAC) inhibitory activity of VPA.14,15,34,35,80,81 The latter directly inhibits HDAC, inducing hyperacetylation of histones H3 and H4 in vivo and in vitro82,83 and thereby increasing the accessibility of DNA within chromatin to DNA-binding agents, such as cisplatin and its derivatives.35,84 Furthermore, in addition to their epigenetic effects, HDAC inhibitors are known to play pivotal roles in non-epigenetic regulation, cell cycle arrest, and apoptosis.77To evaluate the possible effects of 4, 5, 6, 7, 9 and reference compounds on histone deacetylase activity, a western blot analysis was performed to evaluate the acetylated histone H3 and H4 proteins in HCT116, HCT8, RKO, and HT29 cell lines after 72 h of treatment with the compounds at concentrations corresponding to the respective IC50 values.
As expected, VPA was able to inhibit HDAC, leading to a significant increase in acetylated H3 and H4 histones (Fig. 4A and B), while PA showed limited effects on HDAC inhibition through a statistically significant increase in histone H4 acetylation, but only in HCT116 cells. Furthermore, in HCT116, HCT8 and RKO cell lines, only the Pt(IV) derivatives containing VPA in their molecule, namely 5, 7, and 9, were able to induce significant increases in the acetylated histones H3 and H4, the index of the inhibition of HDAC, at levels similar to those obtained following treatment with VPA alone, but of course at a considerably lower concentration. Conversely, in HT29 cells only following treatment with 5, it was possible to observe an increase in the acetylated histone protein levels; however, this increase was limited to H3.
 | ||
| Fig. 4 Acetylated histones H3 (A) and H4 (B) in HCT116, HCT8, RKO and HT29 cell lines treated for 72 h with the studied compounds at concentrations corresponding to the respective IC50 values (oxPt = oxaliplatin). The graphs represent the densitometric analysis of three independent western blot experiments. Statistical analysis: *p < 0.05, **p < 0.01 and ***p < 0.001 vs. control (C). | ||
Effects on cellular migration
Degradation of the extracellular matrix and the basal membrane is the first step toward migration and invasion. Matrix metalloproteinases (MMPs) and their tissue inhibitors modulate these processes. Thus, higher expression of different MMPs facilitates invasion and metastasis, as evidenced in a number of human cancers.85,86In this context, a Pt(IV) derivative containing PA has previously been reported to reduce the invasiveness of a lung cancer cell line,50 while other authors have shown that PA administration decreased the metastatic potential of melanoma cells in vivo.47 In both cases, MMP2 (gelatinase-A) and MMP9 (gelatinase-B) were associated with these effects. Interestingly, recently it has been reported that VPA has also shown antimigratory effects in a chicken embryo chorioallantoic membrane model.87 Therefore, to test if the Pt(IV) derivatives could reduce MMP protein levels, thus inhibiting cellular migration, a western blot analysis was performed on total colon cancer lysates to evaluate MMP2 and MMP9 protein levels, after 72 h treatment with the equitoxic concentrations of 4, 5, 6, 7, 9 and reference compounds (Fig. 5).
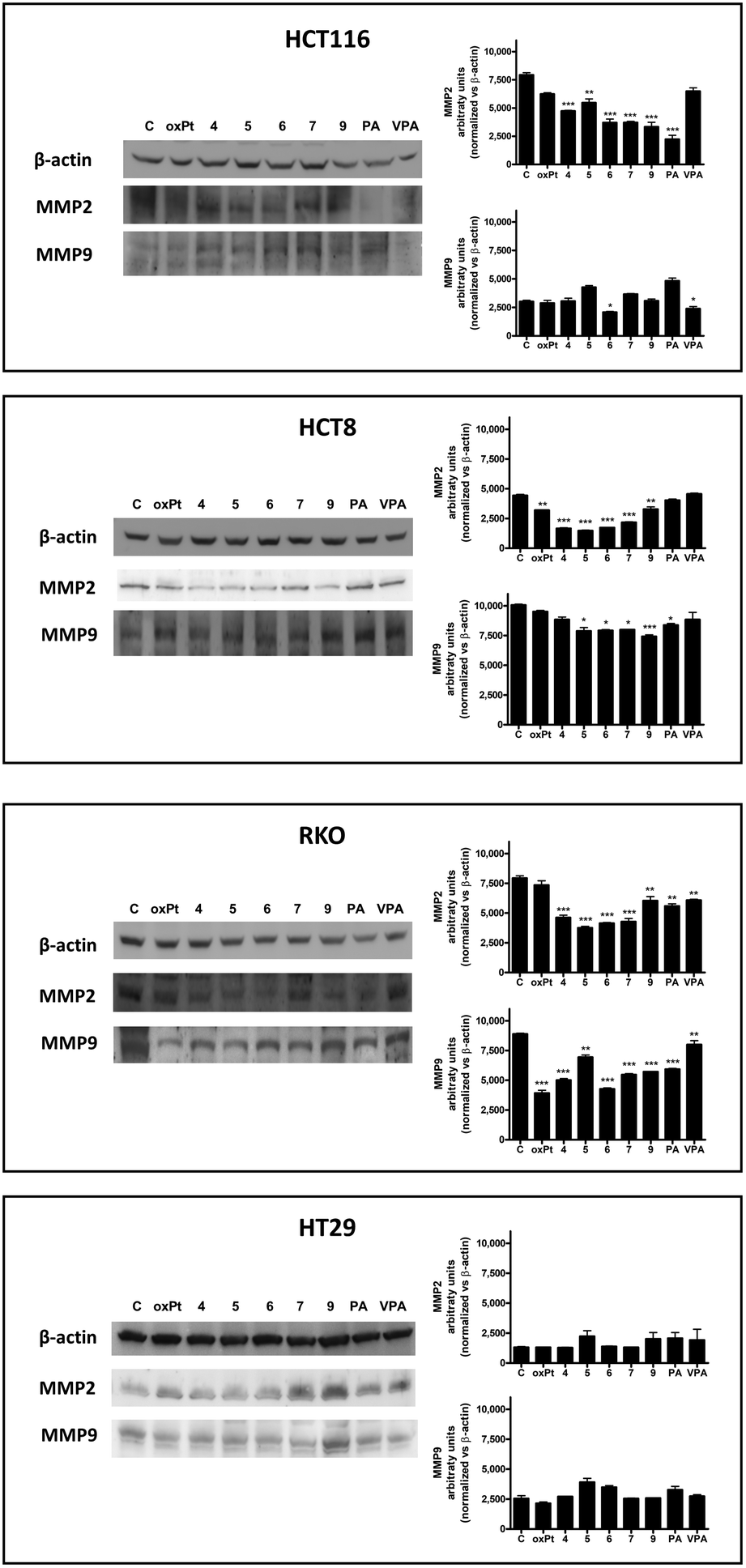 | ||
| Fig. 5 MMP2 and MMP9 protein levels (representative western blot analysis out of three independent experiments with similar results) in HCT116, HCT8, RKO and HT29 cell lines treated 72 h with 4, 5, 6, 7, 9, oxaliplatin (oxPt), PA, and VPA at equitoxic concentrations corresponding to the respective IC50 values and relative densitometric analysis performed on all western blot experiments. Statistical analysis: *p < 0.05; **p < 0.01; ***p < 0.001 vs. control (C). | ||
In general, oxaliplatin, PA and VPA showed variable inhibitory effects on MMP2 and MMP9 levels in the different cell lines. In particular, oxaliplatin statistically reduced MMP2 in HT8, PA in HCT116 and RKO, and VPA in RKO cells. For MMP9, a statistically significant reduction in the protein level was observed for oxaliplatin in RKO, for PA in HT8 and RKO, and for VPA in HCT116 and RKO. No alterations in the protein levels of the considered metalloproteinases were observed in HT29 treated with oxaliplatin, PA, and VPA.
When the effects of the Pt(IV) derivatives on the levels of the MMP2 and MMP9 proteins were evaluated, 4, 5, 6, 7 and 9 significantly reduced the MMP2 levels in all four cell lines. Regarding MMP9, a significant reduction in this metalloproteinase was observed, compared to control levels, in HCT116 treated with 6, HCT8 cells treated with 5, 6, 7 and 9 and RKO cells treated with all compounds. Similar to the case of the reference compounds, no decreases in both MMP2 and MMP9 protein levels were observed in HT29 cells after treatment with Pt(IV) derivatives.
The observed general lack of effects of the Pt(IV) derivatives on the MMP2 and MMP9 protein levels in HT29 could be explained, once again, considering the p53 status of this cell line. Actually, the loss of the p53 function has been reported to enhance invasion and metastasis through MMP production and activation of several survival pathways and it has been reported that the restoration of the p53 function could decrease the activity of MMPs.88,89
Experimental section
General procedures
All chemicals (Sigma Aldrich-Merck or Alfa Aesar-Thermo Fisher Scientific, except where otherwise specified) were used as received and without further purification. Complexes (SP-4-2)-dichlorido(cyclohexane-1R,2R-diamine)platinum(II) ([PtCl2(dach)] (1),51,90 (OC-6-44)-acetatodichlorido(cyclohexane-1R,2R-diamine)hydroxidoplatinum(IV) (2)54 and (OC-6-33)-dichlorido(cyclohexane-1R,2R-diamine)dihydroxidoplatinum(IV) (3),64 and the acyl chloride of perillic acid (PA)74 were prepared according to published procedures.Microwave-assisted synthesis was performed using a CEM Discover® SP System equipped with a focused single-mode and self-tuning cavity, an air-cooling system, and an automated power control based on temperature feedback, supplying power in 1 W increments from 0 to 300 W.
The purity of all the compounds was assessed using an analytical RP-HPLC (≥98%). Chromatographic analysis and purifications were carried out using a C18 Phenomenex PhenoSphere-NEXT (5 μm, 250 × 4.6 mm ID) column on a Waters HPLC-MS instrument (equipped with an Alliance 2695 separation module, a 2487 dual lambda absorbance detector and a 3100 mass detector) with a 70/30 v/v mixture of methanol/15 mM aqueous formic acid as the mobile phase. The flow was set at 0.500 mL min−1 and the wavelength of the UV-vis detector was set at 210 nm.
The MS spectra were recorded using source and desolvation temperatures set at 150 and 250 °C, respectively, with nitrogen used both as a drying gas and as a nebulizing gas. The cone and capillary voltages were usually +30 V (positive ion mode) and 2.70 kV, respectively. Quasi-molecular ion peaks [M + H]+ were assigned on the basis of the m/z values and the simulated isotope distribution patterns.
The NMR spectra were measured on an NMR-Bruker Avance III operating at 500 MHz (1H), 125.7 MHz (13C), and 107.2 MHz (195Pt), respectively. 1H and 13C NMR chemical shifts were referenced to solvent resonances, whereas for 195Pt NMR signals, a solution of K2[PtCl4] in saturated aqueous KCl was used as an external reference (δ = −1628 ppm). The numbering scheme for the assignment of NMR signals is reported in Scheme S1 (see the ESI†).
Microwave-assisted synthesis of complexes 4–7
A solution of the acyl chloride of perillic acid (PA)74 (0.101 g, 0.548 mmol for 4 or 0.056 g, 0.302 mmol for 6) or a solution of the commercially available acyl chloride of VPA (96 μL, 0.548 mmol for 5 or 105 μL, 0.598 mmol for 7) in anhydrous acetonitrile (about 2 mL) was added in a microwave vessel to a suspension of complex 2 (0.050 g, 0.110 mmol for 4 and 5) or complex 3 (0.050 g, 0.121 mmol for 6 and 7) in anhydrous acetonitrile (about 2 mL). Excess pyridine (40 μL) was added dropwise to the reaction mixture under stirring and the vessel was capped and introduced into the microwave cavity. The microwave unit was programmed to heat the mixture to 55 °C over a 5 min ramp period and then maintain it at this temperature for 1 hour under stirring; the power was automatically set at 50 W. After it was heated, the vessel was allowed to cool to room temperature before being removed from the cavity. After filtering the mixture (0.45 μm PTFE filter), the resulting solution was transferred into a 25 mL round-bottom flask and dried with a rotary evaporator. The resulting oil was treated with hexane and then with water at least three times to produce a pale-yellow to pale-brown powder.4. Yield: 0.035 g, 53%. ESI-MS (positive ion mode): 605 m/z; calcd for C18H31Cl2N2O4Pt [M + H]+: 605 m/z. 1H NMR (500.13 MHz, DMSO-d6): δ 1.13 (m, 2H, H16 and H17), 1.32–1.36 (m, 3H, H8, H15 and H18), 1.50–1.52 (m, 2H, H16′ and H17′), 1.71 (m, 3H, H12), 1.76–1.78 (m, 1H, H8′), 1.96 (s, 3H, H1), 2.07 (m, 1H, H6), 2.09 (m, 1H, H7), 2.11 (m, 1H, H9), 2.19–2.21 (m, 2H, H15′ and H18′), 2.24 (m, 1H, H6′), 2.33–2.36 (m, 1H, H9′), 2.50–2.55 (m, 2H, H13 and H14), 4.72 (m, 2H, H11), 6.75 (m, 1H, H5), 8.19–9.53 (m, 4H, –NH2) ppm. 13C NMR (125.76 MHz, DMSO-d6): δ 20.5 (C12), 23.4 (C16 and C17), 23.5 (C1), 24.9 (C9), 26.8 (C8), 30.3 (C6), 31.1–31.2 (C15 and C18), 40.0 (C7), 62.6 (C13 and C14), 109.1 (C11), 132.2 (C4), 137.1 (C5), 148.7 (C10), 176.7 (C3), 180.7 (C2) ppm. 195Pt NMR (107.51 MHz, DMSO-d6): δ 1088 ppm.
5. Yield: 0.014 g, 22%. ESI-MS (positive ion mode): 583 m/z; calcd for C16H33Cl2N2O4Pt [M + H]+: 583 m/z. 1H NMR (500.13 MHz, DMSO-d6): δ 0.84 (m, 6H, H13), 0.97–1.16 (m, 2H, H3 and H4), 1.24–1.34 (m, 8H, H11, H12, H2 and H5), 1.36–1.56 (m, 6H, H11′, H3′ and H4′), 1.95 (2, 3H, H8), 2.18–2.28 (m, 3H, H10, H2′ and H5′), 2.36–2.63 (m, 2H, H1 and H6), 8.15–9.67 (m, 4H, –NH2) ppm. 13C NMR (125.76 MHz, DMSO-d6): δ 13.99–14.04 (C13), 19.9–20.1 (C12), 23.4–23.6–23.7 (C3, C4 and C8), 31.0–31.4 (C2 and C5), 34.6–34.8 (C11), 46.9 (C10), 62.1–63.1 (C1 and C6), 180.8 (C7), 186.0 (C9) ppm. 195Pt NMR (107.51 MHz, DMSO-d6): δ 1113 ppm.
6. Yield: 0.036 g, 42%. ESI-MS (positive ion mode): 711 m/z; calcd for C26H41Cl2N2O4Pt [M + H]+: 711 m/z. 1H NMR (500.13 MHz, DMSO-d6): δ 1.12 (m, 2H, H16 and H17), 1.34–1.36 (m, 4H, H8, H15 and H18), 1.50–1.52 (m, 2H, H16′ and H17′), 1.71 (m, 6H, H12), 1.76–1.78 (m, 2H, H8′), 2.04–2.09 (m, 6H, H6, H7 and H9), 2.20–2.23 (m, 2H, H15′ and H18′), 2.27 (m, 2H, H6′), 2.34–2.37 (m, 2H, H9′), 2.55 (m, 2H, H13 and H14), 4.72 (m, 4H, H11), 6.75 (m, 2H, H5), 8.22–9.56 (m, 4H, –NH2) ppm. 13C NMR (125.76 MHz, DMSO-d6): δ 20.5 (C12), 23.4 (C16 and C17), 24.9 (C9), 26.8 (C8), 30.3 (C6), 31.2 (C15 and C18), 40.0 (C7), 62.7 (C13 and C14), 109.1 (C11), 132.2 (C4), 137.1 (C5), 148.7 (C10), 176.7 (C3) ppm. 195Pt NMR (107.51 MHz, DMSO-d6): δ 1072 ppm.
7. Yield: 0.019 g, 23%. ESI-MS (positive ion mode): 667 m/z; calcd for C22H45Cl2N2O4Pt [M + H]+: 667 m/z. 1H NMR (500.13 MHz, DMSO-d6): δ 0.85 (m, 12H, H11), 1.02 (m, 2H, H3 and H4), 1.27 (m, 14H, H9, H10, H2 and H5), 1.47 (m, 4H, H9′), 1.56 (m, 2H, H3′ and H4′), 2.21–2.30 (m, 4H, H8, H2′ and H5′), 2.45 (m, 2H, H1 and H6), 8.20 and 9.66 (m, 4H, –NH2) ppm. 13C NMR (125.76 MHz, DMSO-d6): δ 14.0–14.1 (C11), 20.0–20.1 (C10), 23.7 (C3 and C4), 31.4 (C2 and C5), 34.6–34.8 (C9), 46.9 (C8), 62.7 (C1 and C6), 186.1 (C7) ppm. 195Pt NMR (107.51 MHz, DMSO-d6): δ 1106 ppm.
Synthesis of complex 8
Complex 8 was prepared by modifying synthetic procedures employed to prepare other unsymmetric Pt(IV) complexes,91 as follows.Microwave-assisted synthesis of complex 9
A solution of the commercial acyl chloride of VPA (62 μL, 0.356 mmol) in anhydrous acetonitrile (2 mL) was added in a microwave vessel to a suspension of complex 8 (0.040 g, 0.071 mmol) in the same solvent (2 mL). Excess pyridine (40 μL) was added dropwise to the stirring reaction mixture and the vessel was capped and introduced into the microwave cavity. The microwave unit was programmed to heat the vessel content to 55 °C over a 5 min ramp period and then maintained it at this temperature for 1 hour under stirring; the power was automatically set at 50 W. After heating, the vessel was allowed to cool to room temperature before removing it from the cavity. The solution obtained was transferred into a round-bottom flask and dried with the rotary evaporator. The resulting oil was treated with hexane and then water to yield a beige powder (yield: 0.026 g, 53%). ESI-MS (positive ion mode): 689 m/z; calcd for C24H43Cl2N2O4Pt [M + H]+: 689 m/z. 1H NMR (500.13 MHz, DMSO-d6): δ 0.85 (m, 6H, H21), 0.98–1.18 (m, 2H, H3 and H4), 1.24–1.41 (m, 9H, H2, H5, H10, H19 and H20), 1.43–1.57 (m, 4H, H19′, H3′ and H4′), 1.71 (s, 3H, H15), 1.73–1.79 (m, 1H, H10′), 2.02–2.11 (m, 3H, H9, H11, H12), 2.19–2.30 (m, 4H, H18, H12, H2′ and H5′), 2.36 (m, 1H, H9′), 2.43–2.63 (m, 2H, H1 and H6), 4.72 (m, 2H, H16), 6.75 (m, 1H, H13), 8.19–9.70 (m, 4H, –NH2) ppm. 13C NMR (125.76 MHz, DMSO-d6): δ 14.5–14.6 (C21), 20.5–20.6 (C20), 21.1 (C15), 23.9–24.3 (C3 and C4), 25.5 (C9), 27.4 (C10), 30.8 (C12), 31.1–31.4 (C2 and C5), 35.1–35.3 (C19), 40.0 (C11), 47.4 (C18), 62.7–63.7 (C1 and C6), 109.7 (C16), 132.8 (C8), 137.5 (C13), 149.2 (C14), 177.2 (C7), 186.5 (C17) ppm. 195Pt NMR (107.51 MHz, DMSO-d6): δ 1097 ppm.Lipophilicity
RP-HPLC was used to measure the capacity factors k′ of the compounds as previously reported.69 Briefly, a chromatogram for each compound was recorded on a C18 column PhenoSphere-NEXT (5 μm, 250 × 4.6 mm internal diameter) with eluent composition of 15 mM formic acid 30% v/v in CH3OH and a flow of 0.5 mL min−1. The corresponding retention time tR was converted into logk′, where k′ = (tR − t0)/t0. The dead time of the column t0 was the tR of reference KCl.
Cell culture and viability assay
Human colon carcinoma cell lines HCT116, RKO, HCT8 and HT29 were obtained from ATCC (American Type Culture Collection, Manassas, VA, USA) and maintained in the DMEM (Euroclone, Milan, Italy), supplemented with 10% fetal calf serum (Euroclone, Milan, Italy), a 1% glutamine and 1% antibiotic mixture, 1% sodium pyruvate and 1% non-essential amino acids (both Sigma-Aldrich, Milan, Italy) at 37 °C under a humidified 5% CO2 atmosphere. Cells were routinely checked for Mycoplasma (Molecular Biology Reagent Set Mycoplasma Species, Euroclone, UK). For all experiments, cells were exposed to the different Pt(IV) derivatives tested and to the reference compounds 1, oxaliplatin, valproic acid, and perillic acid for 72 h. When DMSO was used as a solvent, the final concentration never exceeded 0.1% v/v. This concentration was found to be non-toxic to the cells tested (control). The effect of all derivatives on cellular viability was assessed using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay, according to previously published procedures. Briefly, 3 × 103 cells were seeded in 96-well plates and allowed to attach for 24 h. The cells were then treated for 72 h with growing concentrations (1, 4, 5, 6, 7, 9, and oxaliplatin: from 10 up to 1000 nM; PA and VPA: from 10 to 500 μM) of the compounds studied. At the end of the incubation period, MTT (0.05 mL of a 2 mg mL−1 stock solution in PBS) was added to each well and cells were incubated for 3 h at 37 °C. Cell viability was determined by measuring the absorbance of the DMSO-dissolved blue formazan crystals (λ = 570 nm), formed through MTT reduction by metabolically active cells, in individual wells, using an iMARK Microplate Reader (Bio-RAD). IC50 values were calculated based on the nonlinear regression analysis of dose–response data performed with CalcuSyn software (Biosoft, Cambridge, UK). Differences between IC50 values were statistically evaluated by analysis of variance with the Bonferroni post-test for multiple comparisons.Western blot analysis
The levels of the H3, H4, MMP2 and MMP9, and p21 proteins in whole cell lysates following 72 h treatment with the compounds under investigation at concentrations corresponding to the respective IC50 were detected by western blot analysis. For whole cell lysates, cells were resuspended in lysis buffer (NaCl 120 mM, NaF 25 mM, EDTA 5 mM, EGTA 6 mM, sodium pyrophosphate 25 mM in Tris-buffered saline TBS 20 mM at pH 7.4, phenylmethanesulfonyl fluoride 2 mM, Na3VO4 1 mM, phenylarsine oxide 1 mM, 1% NP-40 and 10% protease inhibitor cocktail) and incubated for 10 min on ice after adding Nonidet P-40 (final concentration 0.1%) and lysates were collected by centrifugation (12800 rpm for 20 min). The protein concentration was determined by a BCA assay (Pierce, Italy) and 50 μg of protein per sample were loaded onto 8% polyacrylamide gels and separated under denaturing conditions. Protein bands were then transferred to Hybond-P membranes (Amersham Biosciences, Italy) and western blot analysis was performed using standard techniques with mouse monoclonal antibodies directed against human MMP2, MMP9, H3, H4 and p21 (Santa Cruz Biotechnology, Inc). Equal loading of the samples was verified by re-probing the blots with a mouse monoclonal anti-β-actin antibody (Santa Cruz Biotechnology, Inc.). Protein bands were visualized using a G-box (Syngene, Chemi-XT4) using peroxidase-conjugated anti-mouse secondary antibodies (Sigma-Aldrich) and a Westar Supernova Substrate (Cyanagen). Densitometric analysis was performed using Image-J software and differences between obtained values were evaluated by analysis of variance with the Bonferroni post-test for multiple comparisons.
Cell death induction and cell cycle analysis
The ability of the Pt(IV) derivatives tested to induce apoptotic and/or necrotic cell death and/or alterations in the distribution of DNA through the cell cycle was evaluated by flow cytometric analysis following 72 h exposure of the cells at concentrations corresponding to the IC50 values. At the end of the treatment, cells were detached, washed in PBS and fixed in 70% ethanol at −20 °C and after a further wash in PBS, DNA was stained with a solution of PI in PBS (50 μg mL−1) in the presence of RNAse A (30 U mL−1) at room temperature for 15 min before analyzing the samples. To determine the percentage of necrotic cells, the fixation step was omitted. The fluorescence emission of PI was collected through a 575 nm band-pass filter using a FACSCalibur (Becton Dickinson) and the percentage of apoptotic cells in each sample was determined based on the sub-G1 peaks detected in mono-parametric histograms acquired in the log mode, while the DNA distribution through the different phases of the cell cycle was determined on peaks acquired in the linear mode. The percentage of necrotic cells was assessed by the increase of PI fluorescence histograms acquired in the log mode.Conclusions
Multitarget Pt(IV) combos represent an interesting (and we dare say not a red herring) chemotherapeutic alternative, allowing the association between the cytotoxic effects of Pt-based drugs and the effects of axial bioactive ligands to improve selectivity and efficacy, and also exploiting the peculiar characteristics of the oxidized Pt(IV) center (activation by reduction).6,9–13,92,93In the present article, a colon-oriented Pt moiety containing the dach ligand, the HDACI properties of valproic acid, and the potential antimetastatic efficacy of perillic acid were combined for the first time. The results indicate that it is relatively easy to obtain Pt(IV) combos operating at the nanomolar scale and hence they are more potent than the reference compounds or the single fragments. However, this was not simply obtained by increasing the lipophilicity of the compounds. Actually, the synthesized complexes possess pro-apoptotic and pro-necrotic properties and the ability to induce cell cycle alterations. Furthermore, they were able to downregulate HDAC activity, leading to an increase in the histone H3 and H4 levels. Finally, they exerted antimigratory activity by reducing the levels of the MMP2 and MMP9 proteins. All these activities, along with the alkylating effect of [PtCl2(dach)], are probably related to a synergistic cellular accumulation of the Pt moiety, VPA and PA, leading to a higher intracellular concentration of these three compounds, compared to the concentrations obtained after co-administration of the three free compounds. Importantly, as a demonstration, the Pt(IV) complexes showed biological properties similar to or better than those of VPA and PA but at lower concentrations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The figure of the table of content was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 Unported license (https://creativecommons.org/licenses/by/3.0/).References
- E. Wexselblatt and D. Gibson, J. Inorg. Biochem., 2012, 117, 220–229 CrossRef CAS PubMed.
- D. Gibson, Dalton Trans., 2016, 45, 12983–12991 RSC.
- D. Gibson, ChemMedChem, 2021, 16, 2188–2191 CrossRef CAS PubMed.
- D. Gibson, J. Inorg. Biochem., 2021, 217, 111353 CrossRef CAS PubMed.
- M. Ravera, E. Gabano, M. J. McGlinchey and D. Osella, Dalton Trans., 2022, 51, 2121–2134 RSC.
- Z. Xu, Z. Wang, Z. Deng and G. Zhu, Coord. Chem. Rev., 2021, 442, 213991 CrossRef CAS.
- X. Y. Wang and Z. J. Guo, Chem. Soc. Rev., 2013, 42, 202–224 RSC.
- J. S. Butler and P. J. Sadler, Curr. Opin. Chem. Biol., 2013, 17, 175–188 CrossRef CAS PubMed.
- E. Gabano, M. Ravera and D. Osella, Dalton Trans., 2014, 43, 9813–9820 RSC.
- R. G. Kenny, S. W. Chuah, A. Crawford and C. J. Marmion, Eur. J. Inorg. Chem., 2017, 1596–1612 CrossRef CAS.
- R. G. Kenny and C. J. Marmion, Chem. Rev., 2019, 119, 1058–1137 CrossRef CAS PubMed.
- M. Ravera, E. Gabano, M. J. McGlinchey and D. Osella, Inorg. Chim. Acta, 2019, 492, 32–47 CrossRef CAS.
- C. Y. Jia, G. B. Deacon, Y. J. Zhang and C. Z. Gao, Coord. Chem. Rev., 2021, 429, 213640 CrossRef CAS.
- R. Raveendran, J. P. Braude, E. Wexselblatt, V. Novohradsky, O. Stuchlikova, V. Brabec, V. Gandin and D. Gibson, Chem. Sci., 2016, 7, 2381–2391 RSC.
- V. Novohradsky, L. Zerzankova, J. Stepankova, O. Vrana, R. Raveendran, D. Gibson, J. Kasparkova and V. Brabec, Biochem. Pharmacol., 2015, 95, 133–144 CrossRef CAS PubMed.
- E. D. Scheeff, J. M. Briggs and S. B. Howell, Mol. Pharmacol., 1999, 56, 633–643 CrossRef CAS PubMed.
- K. Gkionis, S. T. Mutter and J. A. Platts, RSC Adv., 2013, 3, 4066–4073 RSC.
- A. M. Di Francesco, A. Ruggiero and R. Riccardi, Cell. Mol. Life Sci., 2002, 59, 1914–1927 CrossRef CAS PubMed.
- F. Arnesano, A. Pannunzio, M. Coluccia and G. Natile, Coord. Chem. Rev., 2015, 284, 286–297 CrossRef CAS.
- M. Sabbatini, I. Zanellato, M. Ravera, E. Gabano, E. Perin, B. Rangone and D. Osella, J. Med. Chem., 2019, 62, 3395–3406 CrossRef CAS PubMed.
- A. Kastner, T. Mendrina, F. Bachmann, W. Berger, B. K. Keppler, P. Heffeter and C. R. Kowol, Inorg. Chem. Front., 2023, 10, 4126–4138 RSC.
- E. Wexselblatt, E. Yavin and D. Gibson, Angew. Chem., Int. Ed., 2013, 52, 6059–6062 CrossRef CAS PubMed.
- M. Ravera, E. Gabano, S. Bianco, G. Ermondi, G. Caron, M. Vallaro, G. Pelosi, I. Zanellato, I. Bonarrigo, C. Cassino and D. Osella, Inorg. Chim. Acta, 2015, 432, 115–127 CrossRef CAS.
- S. Minucci and P. G. Pelicci, Nat. Rev. Cancer, 2006, 6, 38–51 CrossRef CAS PubMed.
- J. E. Bolden, M. J. Peart and R. W. Johnstone, Nat. Rev. Drug Discovery, 2006, 5, 769–784 CrossRef CAS PubMed.
- D. Morel, D. Jeffery, S. Aspeslagh, G. Almouzni and S. Postel-Vinay, Nat. Rev. Clin. Oncol., 2020, 17, 91–107 CrossRef CAS PubMed.
- R. Roy, T. Ria, D. RoyMahaPatra and U. H. Sk, ACS Omega, 2023, 8, 16532–16544 CrossRef CAS PubMed.
- K. J. Falkenberg and R. W. Johnstone, Nat. Rev. Drug Discovery, 2014, 13, 673–691 CrossRef CAS PubMed.
- J. Roche and P. Bertrand, Eur. J. Med. Chem., 2016, 121, 451–483 CrossRef CAS PubMed.
- T. Eckschlager, J. Plch, M. Stiborova and J. Hrabeta, Int. J. Mol. Sci., 2017, 18, 1414 CrossRef PubMed.
- A. Suraweera, K. J. O'Byrne and D. J. Richard, Front. Oncol., 2018, 8, 15 CrossRef PubMed.
- A. D. Bondarev, M. M. Attwood, J. Jonsson, V. N. Chubarev, V. V. Tarasov and H. B. Schioth, Br. J. Clin. Pharmacol., 2021, 87, 4577–4597 CrossRef PubMed.
- J. Yang, X. Sun, W. Mao, M. Sui, J. Tang and Y. Shen, Mol. Pharm., 2012, 9, 2793–2800 CrossRef CAS PubMed.
- M. Alessio, I. Zanellato, I. Bonarrigo, E. Gabano, M. Ravera and D. Osella, J. Inorg. Biochem., 2013, 129, 52–57 CrossRef CAS PubMed.
- V. Novohradsky, L. Zerzankova, J. Stepankova, O. Vrana, R. Raveendran, D. Gibson, J. Kasparkova and V. Brabec, J. Inorg. Biochem., 2014, 140, 72–79 CrossRef CAS PubMed.
- A. R. Z. Almotairy, V. Gandin, L. Morrison, C. Marzano, D. Montagner and A. Erxleben, J. Inorg. Biochem., 2017, 177, 1–7 CrossRef CAS PubMed.
- M. Ravera, E. Gabano, I. Zanellato, A. Gallina, E. Perin, A. Arrais, S. Cantamessa and D. Osella, Dalton Trans., 2017, 46, 1559–1566 RSC.
- E. Gabano, M. Ravera, I. Zanellato, S. Tinello, A. Gallina, B. Rangone, V. Gandin, C. Marzano, M. G. Bottone and D. Osella, Dalton Trans., 2017, 46, 14174–14185 RSC.
- B. Rangone, B. Ferrari, V. Astesana, I. Masiello, P. Veneroni, I. Zanellato, D. Osella and M. G. Bottone, Life Sci., 2018, 210, 166–176 CrossRef CAS PubMed.
- E. Gabano, B. Rangone, E. Perin, G. Caron, G. Ermondi, M. Vallaro, V. Gandin, C. Marzano, A. Barbanente, N. Margiotta and M. Ravera, Dalton Trans., 2021, 50, 4663–4672 RSC.
- M. H. Gelb, F. Tamanoi, K. Yokoyama, F. Ghomashchi, K. Esson and M. N. Gould, Cancer Lett., 1995, 91, 169–175 CrossRef CAS PubMed.
- Y. Pylayeva-Gupta, E. Grabocka and D. Bar-Sagi, Nat. Rev. Cancer, 2011, 11, 761–774 CrossRef CAS PubMed.
- N. Berndt, A. D. Hamilton and S. M. Sebti, Nat. Rev. Cancer, 2011, 11, 775–791 CrossRef CAS PubMed.
- H. van Hattum and H. Waldmann, Chem. Biol., 2014, 21, 1185–1195 CrossRef CAS PubMed.
- A. D. Cox, C. J. Der and M. R. Philips, Clin. Cancer Res., 2015, 21, 1819–1827 CrossRef CAS PubMed.
- X. D. Zhao and S. Subramanian, Pharmacol. Ther., 2018, 181, 76–84 CrossRef CAS PubMed.
- T. J. Raphael and G. Kuttan, J. Exp. Clin. Cancer Res., 2003, 22, 419–424 CAS.
- T. J. Raphael and G. Kuttan, Immunopharmacol. Immunotoxicol., 2003, 25, 285–294 CrossRef CAS PubMed.
- D. Samaila, B. J. Toy, R. C. Wang and J. A. Elegbede, Anticancer Res., 2004, 24, 3089–3095 CAS.
- M. Ravera, E. Gabano, I. Zanellato, B. Rangone, E. Perin, B. Ferrari, M. G. Bottone and D. Osella, Dalton Trans., 2021, 50, 3161–3177 RSC.
- S. C. Dhara, Indian J. Chem., 1970, 8, 193–194 Search PubMed.
- F. D. Rochon and L. M. Gruia, Inorg. Chim. Acta, 2000, 306, 193–204 CrossRef CAS.
- J. Z. Zhang, P. Bonnitcha, E. Wexselblatt, A. V. Klein, Y. Najajreh, D. Gibson and T. W. Hambley, Chem. – Eur. J., 2013, 19, 1672–1676 CrossRef CAS PubMed.
- E. Gabano, M. Ravera, E. Perin, I. Zanellato, B. Rangone, M. J. McGlinchey and D. Osella, Dalton Trans., 2019, 48, 435–445 RSC.
- E. Gabano and M. Ravera, Molecules, 2022, 27, 4249 CrossRef CAS PubMed.
- C. F. Chin, Q. Tian, M. I. Setyawati, W. Fang, E. S. Q. Tan, D. T. Leong and W. H. Ang, J. Med. Chem., 2012, 55, 7571–7582 CrossRef CAS PubMed.
- M. Ravera, E. Gabano, G. Pelosi, F. Fregonese, S. Tinello and D. Osella, Inorg. Chem., 2014, 53, 9326–9335 CrossRef CAS PubMed.
- V. Gandin, C. Marzano, G. Pelosi, M. Ravera, E. Gabano and D. Osella, ChemMedChem, 2014, 9, 1299–1305 CrossRef CAS PubMed.
- P. S. Pregosin, Coord. Chem. Rev., 1982, 44, 247–291 CrossRef CAS.
- T. G. Appleton, J. R. Hall and S. F. Ralph, Inorg. Chem., 1985, 24, 673–677 CrossRef CAS.
- E. Gabano, E. Marengo, M. Bobba, E. Robotti, C. Cassino, M. Botta and D. Osella, Coord. Chem. Rev., 2006, 250, 2158–2174 CrossRef CAS.
- L. Ronconi and P. J. Sadler, Coord. Chem. Rev., 2008, 252, 2239–2277 CrossRef CAS PubMed.
- S. J. Berners-Price, L. Ronconi and P. J. Sadler, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 49, 65–98 CrossRef CAS.
- P. Gramatica, E. Papa, M. Luini, E. Monti, M. B. Gariboldi, M. Ravera, E. Gabano, L. Gaviglio and D. Osella, J. Biol. Inorg. Chem., 2010, 15, 1157–1169 CrossRef CAS PubMed.
- M. Ravera, E. Gabano, I. Zanellato, F. Fregonese, G. Pelosi, J. A. Platts and D. Osella, Dalton Trans., 2016, 45, 5300–5309 RSC.
- G. Ermondi, G. Caron, M. Ravera, E. Gabano, S. Bianco, J. A. Platts and D. Osella, Dalton Trans., 2013, 42, 3482–3489 RSC.
- A. C. Tsipis and I. N. Karapetsas, Dalton Trans., 2014, 43, 5409–5426 RSC.
- M. Ravera, E. Gabano, I. Zanellato, I. Bonarrigo, M. Alessio, F. Arnesano, A. Galliani, G. Natile and D. Osella, J. Inorg. Biochem., 2015, 150, 1–8 CrossRef CAS PubMed.
- J. A. Platts, G. Ermondi, G. Caron, M. Ravera, E. Gabano, L. Gaviglio, G. Pelosi and D. Osella, J. Biol. Inorg. Chem., 2011, 16, 361–372 CrossRef CAS PubMed.
- S. Z. Zhang, K. S. Lovejoy, J. E. Shima, L. L. Lagpacan, Y. Shu, A. Lapuk, Y. Chen, T. Komori, J. W. Gray, X. Chen, S. J. Lippard and K. M. Giacomini, Cancer Res., 2006, 66, 8847–8857 CrossRef CAS PubMed.
- J. J. Liu, J. Lu and M. J. McKeage, Curr. Cancer Drug Targets, 2012, 12, 962–986 CrossRef CAS PubMed.
- I. Buss, A. Hamacher, N. Sarin, M. U. Kassack and G. V. Kalayda, Metallomics, 2018, 10, 414–425 CrossRef CAS PubMed.
- E. Gabano, I. Zanellato, G. Pinton, L. Moro, M. Ravera and D. Osella, Bioinorg. Chem. Appl., 2022, 2022, 15 Search PubMed.
- M. Ravera, E. Gabano, I. Zanellato, B. Rangone, E. Perin, B. Ferrari, M. G. Bottone and D. Osella, Dalton Trans., 2021, 50, 3161–3177 RSC.
- C. W. Strey, L. Schamell, E. Oppermann, A. Haferkamp, W. O. Bechstein and R. A. Blaheta, Exp. Ther. Med., 2011, 2, 301–307 CrossRef CAS PubMed.
- Y. Laxmi, K. J. Pierre, A. Elegbede, R. C. Wang and S. W. Carper, Cancer Lett., 2007, 257, 216–226 CrossRef PubMed.
- M. Sanaei and F. Kavoosi, Asian Pac. J. Cancer Prev., 2021, 22, 89–95 CrossRef CAS PubMed.
- X. Cao, J. Y. Hou, Q. L. An, Y. G. Assaraf and X. D. Wang, Drug Resist. Updates, 2020, 49, 100671 CrossRef PubMed.
- G. Y. Zhu, C. Y. Pan, J. X. Bei, B. Li, C. Liang, Y. Xu and X. M. Fu, Front. Oncol., 2020, 10, 595187 CrossRef PubMed.
- E. Petruzzella, R. Sirota, I. Solazzo, V. Gandin and D. Gibson, Chem. Sci., 2018, 9, 4299–4307 RSC.
- X. J. Ding, R. Zhang, R. P. Liu, X. Q. Song, X. Qiao, C. Z. Xie, X. H. Zhao and J. Y. Xu, Inorg. Chem. Front., 2020, 7, 1220–1228 RSC.
- M. Gottlicher, S. Minucci, P. Zhu, O. H. Kramer, A. Schimpf, S. Giavara, J. P. Sleeman, F. Lo Coco, C. Nervi, P. G. Pelicci and T. Heinzel, EMBO J., 2001, 20, 6969–6978 CrossRef CAS PubMed.
- N. Gurvich, O. M. Tsygankova, J. L. Meinkoth and P. S. Kein, Cancer Res., 2004, 64, 1079–1086 CrossRef CAS PubMed.
- M. S. Kim, M. Blake, J. H. Baek, G. Kohlhagen, Y. Pommier and F. Carrier, Cancer Res., 2003, 63, 7291–7300 CAS.
- J. Aparna and M. P. Brundha, Indian J. Forensic Med. Toxicol., 2020, 14, 4912–4926 Search PubMed.
- J. A. Eble and S. Niland, Clin. Exp. Metastasis, 2019, 36, 171–198 CrossRef CAS PubMed.
- R. Dirziuviene, L. Slekiene, J. Palubinskiene, I. Balnyte, K. Lasiene, D. Stakisaitis and A. Valanciute, Histol. Histopathol., 2022, 37, 1201–1212 CAS.
- H. Zhu, B. Evans, P. O'Neill, X. C. Ren, Z. D. Xu, W. N. Hait and J. M. Yang, Cancer Biol. Ther., 2009, 8, 1722–1728 CrossRef CAS PubMed.
- B. C. Lewis, D. S. Klimstra, N. D. Socci, S. Xu, J. A. Koutcher and H. E. Varmus, Mol. Cell. Biol., 2005, 25, 1228–1237 CrossRef CAS PubMed.
- F. D. Rochon and L. M. Gruia, Inorg. Chim. Acta, 2000, 306, 193–204 CrossRef CAS.
- R. K. Pathak, S. Marrache, J. H. Choi, T. B. Berding and S. Dhar, Angew. Chem., Int. Ed., 2014, 53, 1963–1967 CrossRef CAS PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- X. J. Li, Y. H. Liu and H. Q. Tian, Bioinorg. Chem. Appl., 2018, 2018, 8276139 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H, 13C, 195Pt, and [1H, 13C] HSQC NMR spectra of complexes 4–9. See DOI: https://doi.org/10.1039/d3dt01876h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |