
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNot the usual suspect – an unexpected organometallic product during the synthesis of cytotoxic platinum(II) complexes†‡
Thomas
Maier
ab,
Judith
Wutschitz
a,
Natalie
Gajic
a,
Michaela
Hejl
a,
Klaudia
Cseh
a,
Sebastian
Mai
 c,
Michael A.
Jakupec
ad,
Mathea S.
Galanski
*a and
Bernhard K.
Keppler
*ad
c,
Michael A.
Jakupec
ad,
Mathea S.
Galanski
*a and
Bernhard K.
Keppler
*ad
aUniversity of Vienna, Faculty of Chemistry, Department of Inorganic Chemistry, Waehringer Strasse 42, 1090 Vienna, Austria. E-mail: mathea.galanski@univie.ac.at; bernhard.keppler@univie.ac.at
bUniversity of Vienna, Doctoral School in Chemistry (DoSChem), Waehringer Strasse 42, 1090 Vienna, Austria
cUniversity of Vienna, Faculty of Chemistry, Department of Theoretical Chemistry, Waehringer Strasse 17, 1090 Vienna, Austria
dResearch Cluster “Translational Cancer Therapy Research”, University of Vienna, Waehringer Strasse 42, 1090 Vienna, Austria
First published on 6th October 2023
Abstract
The reaction of (1R,2R)-(cyclohexane-1,2-diamine)dichloridoplatinum(II) with maleic acid unexpectedly resulted in the formation of an organometallic platinum(II) complex featuring a C,O-coordinating ligand. Additionally, a small series of close derivatives with increasing lipophilicity was synthesized. All complexes were fully characterized by multinuclear one- and two-dimensional (1H, 13C, 15N, and 195Pt) NMR spectroscopy, high resolution mass spectrometry, and in one case by X-ray diffraction. The lipophilicity and the impact on the DNA secondary structure as well as the cytotoxic properties in three human cancer cell lines (A549, SW480, and CH1/PA-1) were investigated. Unexpectedly, no clear-cut trend in cytotoxicity was observed with increasing lipophilicity. Also unexpectedly, the complexes showed only a low potential to inhibit cancer cell growth and no sign of interaction with DNA, in sharp contrast to the parent drug oxaliplatin, which seems to be caused by the low reactivity of the investigated compounds.
Introduction
Since Barnett Rosenberg's discovery of the antitumor activity of cisplatin,1–4 platinum compounds have been shown to be a major force in the fight against various cancers, leading to the worldwide approval of cisplatin, carboplatin, and oxaliplatin.5 Despite their huge success, platinum-based chemotherapy is accompanied by unfortunate drawbacks. A prominent issue is the strong side effects of these compounds, which are in some cases dose limiting. This diminishes the outcome of therapy and thus overall the rate of successful treatment. Additionally, cancers can show intrinsic or acquired resistance to platinum compounds.6 Consequently, tuning platinum compounds towards higher activity and/or reduced side effects is still of utmost importance.Oxaliplatin (Scheme 1) is mainly used in the case of colorectal cancer (stage II–IV), which is one of the most common causes of cancer related deaths in women and men.7 Neurotoxicity is known to be the dose limiting toxicity. It is believed that the oxalate as leaving ligand is at least partially responsible for this toxicity. Thus, we were interested in synthesizing novel oxaliplatin derivatives, leaving the (cyclohexane-1,2-diamine)platinum(II) fragment, which is responsible for the cytotoxic properties, intact. As one of the potential dicarboxylato ligands, we have chosen maleate.
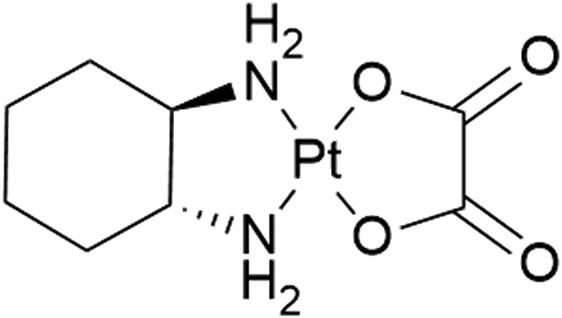 | ||
| Scheme 1 Chemical structure of oxaliplatin. | ||
In order to synthesize the target maleato complex 2 (Scheme S1, ESI†), a standard reaction procedure was used. But to our surprise, the main product turned out to be the pair of organometallic diastereoisomers 3a, featuring a C,O- instead of an O,O-chelating ligand as shown in Scheme 2. To the best of our knowledge, complex 3a is mentioned only once in the literature in a patent.8 Similar complexes with the (cyclohexane-1,2-diamine)platinum(II) moiety and a C,O-chelating ligand have been reported only scarcely. These focus mainly on ascorbato complexes,9–19 but are not limited thereto.20,21 Furthermore, complexes with up to two coordinating nitrogen atoms and at least one coordinating carbon atom are known as well.22–30
 | ||
| Scheme 2 Synthesis of complex 3a and close derivatives 3b–3f. | ||
With the aim of learning more about the cytotoxic properties of this type of organometallic platinum(II) complex, we additionally synthesized a small series of close derivatives with increasing lipophilicity (complexes 3b–3f, Scheme 2). The target complexes were fully characterized by one- and two-dimensional multinuclear (1H, 13C, 15N, 195Pt) NMR spectroscopy and high-resolution mass spectrometry and were investigated with respect to their cytotoxic properties in three human cancer cell lines and for their impact on the DNA secondary structure in a cell-free assay.
Results and discussion
Synthesis and characterization
Dichloridoplatinum(II) complex 1 was suspended in water and mixed with maleic acid in the presence of Ag2CO3 (Scheme 2). The silver salt thereby has a double function. (i) It reacts efficiently with chloride ions of 1, producing the soluble diaquaplatinum(II) complex and insoluble silver chloride. (ii) The reaction of maleic acid with the activated platinum species is accelerated, since acidic protons of the chelating ligand react with carbonate, releasing CO2 from the solution. As delineated above, the organometallic species 3a was isolated and not the expected dicarboxylato species 2. X-ray diffraction quality single crystals of 3a were obtained directly from the reaction solution (Fig. 1).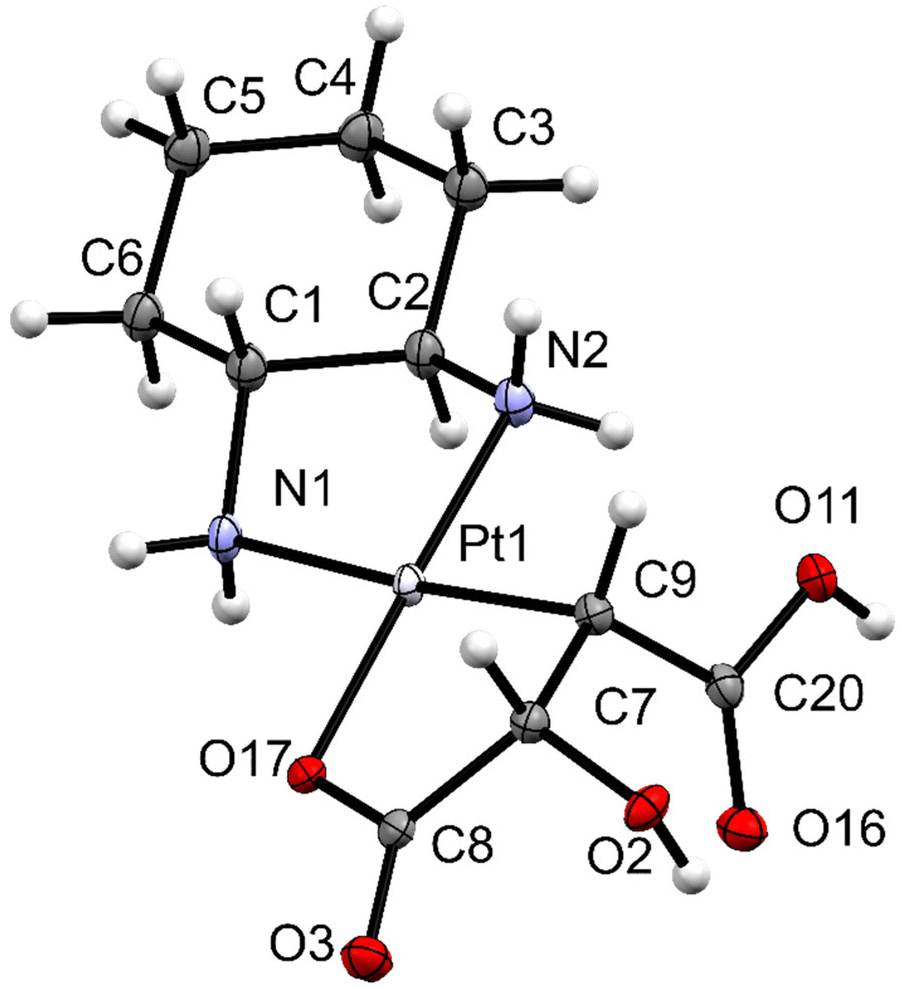 | ||
| Fig. 1 ORTEP view of complex 3a. One of the two diastereoisomers in the asymmetric unit of 3a is drawn with 50% displacement ellipsoids. Solvent molecules are omitted for clarity. Selected bond lengths (of the shown diastereoisomer) [Å]: Pt1–N2: 2.032(12), Pt1–N1: 2.118(13), Pt1–O17: 2.053(10), Pt1–C9: 2.054(14), and C9–C7: 1.512(14) and selected angles [°]: N2–Pt1–N1: 82.1(5), N1–Pt1–O17: 98.4(4), O17–Pt1–C9: 85.5(4), and C9–Pt1–N2: 94.1(5). | ||
For brevity, the following discussion references only the values of one diastereoisomer of 3a (Fig. 1). The same correlations are found for the second diastereoisomer, albeit with small deviations in absolute numbers. The platinum(II) ion has a square-planar PtN2CO coordination geometry and is chelated by two bidentate ligands: (1R,2R)-trans-(cyclohexane-1,2-diamine) and 2-hydroxybutanedioic acid. The diamine ligand acts as a neutral ligand coordinating to the platinum center via nitrogen atoms N1 and N2. The second ligand is doubly negatively charged and bound to platinum(II) through oxygen atom O17 and carbon atom C9. The cyclohexane ring adopts a chair conformation and both amine groups are in the equatorial position. A distorted envelope conformation is found in both five-membered chelate rings. The torsion angle serves as a measure of deviation from the planarity of the chelate ring, which was found to be −56(1)° (ΘN1–C1–C2–N2), and −30(2)° (ΘO17–C8–C7–C9), respectively. The two diastereoisomers differ in the orientation of the substituents of the C,O-chelating ligand (compare Scheme 2). Either the carboxyl group, the hydroxyl group and the neighboring N–CH hydrogen atom point in one direction relative to the PtN2CO coordination plane, or the COOH and OH both point to one side and the respective N–CH hydrogen atom to the opposite side.
The hydroxyl and the carboxyl substituents at C7 and C9 are placed in the cis configuration. The C7–C9 bond length of 1.512(14) Å is in the typical range of a C–C single bond; the vicinal protons H(C7) and H(C9) feature a torsion angle ΘH–C7–C9–H of 32°.
The Pt–C bond [2.054(14) Å] is shorter than the corresponding bond of an ascorbatoplatinum(II) complex, Pt(en)(C2,O5-Asc) [2.126(4) Å].19 The Pt–N bond length is dependent on the coordinated atom in the trans position. Due to the higher trans effect of the bound σ-carbon atom compared to coordinated oxygen, the Pt–N bond trans to the carbon atom [Pt1–N1, 2.118(13) Å] is longer than that trans to oxygen [Pt1–N2, 2.032(12) Å]. This observation is in accordance with those values reported for Pt(en)(C2,O5-Asc) [2.083(3) versus 2.052(3) Å].19
Synthesis of complex 3a was reproducible and has subsequently been optimized. Recrystallization from water by treatment with activated charcoal improved the appearance of the product. However, recrystallization was omitted for further batches and only activated charcoal was added approximately 5 min before filtering off the precipitated AgCl. This led to a drastic increase in yield, from 20% to 59%, while no significant decrease in purity was observed.
The reaction of dichlorido(trans-1R,2R-cyclohexane-1,2-diamine)platinum(II), 1, with maleic acid results in the formation of two diastereoisomers with either 10S,11R- or 10R,11S-configuration in the C11,O-chelating ligand (the NMR numbering scheme for all synthesized compounds is shown in Fig. S1 in the ESI†). The isomers could not be separated by preparative HPLC. Consequently, a doubled set of NMR signals is found in 1H, 13C, 15N, and 195Pt NMR spectra, making complete peak assignment more complicated. Most indicative of the PtN2CO coordination sphere, besides the absence of the double bond, in diastereoisomers of 3a are the 195Pt chemical shifts at −1280.2 and −1282.4 ppm, respectively. In contrast, a 195Pt resonance at around −400 ppm was expected for complex 2 exhibiting a PtN2O2 coordination. These values and differences in 195Pt chemical shifts are well comparable with those found in the literature for ascorbatoplatinum(II) complexes in the Pt(en)(C2,O5-Asc) or Pt(en)(O2,O3-Asc) coordination mode at −1060 and −96 ppm, respectively.19
Finally, peak and signal assignments to both diastereoisomers were performed on the basis of two-dimensional homonuclear and heteronuclear NMR spectroscopy. Most useful was recording the [1H,195Pt] HSQC spectrum (Fig. 2). As a result, specific protons of the isolated spin systems of the cyclohexanediamine (NH protons), as well as those of the C,O-chelating ligand (protons H11), show shift correlation signals with the 195Pt atom of the respective diastereoisomer. From these anchor points, diastereoisomer A and B were defined and peaks were further assigned with the help of [1H,1H] COSY, [1H,13C] HSQC, [1H,13C] HMBC, and [1H,15N] HSQC NMR spectroscopy.
 | ||
| Fig. 2 [1H,195Pt] HSQC spectrum of 3a showing shift correlation signals between 195Pt resonances of both diastereoisomers and NH protons (4.1–5.8 ppm) as well as protons H11 at 3.23 and 3.35 ppm. | ||
However, the complete assignment of every single signal was limited in the case of overlapping signals, especially that of protons in the cyclohexane ring or of 13C resonances, featuring small differences in their chemical shifts due to the diastereoisomeric nature of 3a (Fig. 3). Additionally, complex 3a was further characterized by high resolution mass spectrometry and elemental analysis.
 | ||
| Fig. 3 13C NMR spectrum of complex 3a. The zoomed double peaks show the diastereomeric nature of the product. | ||
Complex 3a has good water solubility. Since the cytotoxic properties of compounds are in part dependent on their lipophilicity, close analogues of 3a have additionally been synthesized (Scheme 2). For that purpose, first the respective mono esters of maleic acid were prepared by adjusting a procedure from the literature31 from maleic anhydride and the respective alcohol. Target complexes 3b–3f were synthesized and purified via preparative HPLC in an isocratic fashion using acetonitrile and MilliQ water with 0.1% formic acid as the additive. Separation of the diastereoisomers was not possible with the utilized chromatography setup. Compounds 3b–3f were fully characterized by multinuclear NMR spectroscopy, high resolution mass spectrometry and elemental analysis. Again, in accordance with complex 3a, novel platinum(II) compounds showed for nearly every single atom a double set of signals in NMR spectra due to the presence of two diastereoisomers. For example, two 195Pt chemical shifts for complexes 3b–3f were found in the region between −1273 and −1285 ppm, which were separated by 2–9 ppm. As expected, these signals are well comparable with 195Pt resonances of −1280.2 and −1282.8 ppm in the case of compound 3a.
Cytotoxicity
Cytotoxicity was investigated in three human cancer cell lines differing in their general chemosensitivity: the multidrug-resistant non-small cell lung cancer cell line A549, the colon cancer cell line SW480 with mostly intermediate sensitivity, and the quite broadly sensitive ovarian teratocarcinoma cell line CH1/PA-1. The IC50 values were interpolated from the concentration–effect curves (Fig. S2, ESI†) and are listed in Table 1. Overall, complexes 3a–3f have low cytotoxic potencies, which was unexpected for organometallic platinum(II) complexes at first sight. This observation is even more unexpected taking into consideration that 3a–3f, upon release of the chelating ligands, are expected to form the same bisadducts with DNA as oxaliplatin. Consequently, the high IC50 values most probably reflect the low reactivity of the compounds. One further contribution to the low cytotoxicity of compound 3a could be its deprotonation under physiological conditions. The attack of the resulting negatively charged species on DNA would be hampered, since DNA is also negatively charged. This assumption would only hold true if complex 3a arrives intact at the DNA without the ligand exchange reaction.| A549 | SW480 | CH1/PA-1 | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kw kw |
|
|---|---|---|---|---|
| a Crystals dissolved in DMSO and then diluted in the medium; all other samples were prepared from lyophilized compounds and directly dissolved in the medium. b As previously reported in ref. 32. | ||||
|
3aa |
>200 | 91 ± 17 | 42 ± 6 | 0.17 |
| 3a | >200 | 32 ± 5 | 20 ± 4 | |
| 3b | >200 | 166 ± 9 | 102 ± 13 | 0.64 |
| 3c | >200 | 147 ± 4 | 105 ± 22 | 1.15 |
| 3d | 164 ± 25 | 42 ± 2 | 25 ± 9 | 1.20 |
| 3e | >200 | 96 ± 11 | 47 ± 8 | 2.01 |
| 3f | >200 | 136 ± 34 | 37 ± 4 | 2.38 |
| Oxaliplatin | 0.98 ± 0.21b | 0.29 ± 0.05b | 0.18 ± 0.01b | 0.24 |
With regard to cell line dependency, the IC50 values of 3a–3f follow the expected trend for oxaliplatin derivatives: A549 > SW480 > CH1/PA-1, but they are by factors of ∼100 to >500 less potent than the parent drug oxaliplatin. This observation is difficult to explain and warrants further investigation. The literature data for reasonable comparison are scarce: a shikimate based DACH Pt(II) complex bearing a C,O-coordination motif has been reported to yield an IC50 value of 3.2 μM (after 72 h of exposure for L1210 murine leukemia cells), which is 80 times higher than that of [Pt(DACH)SO4] (with oxaliplatin not included in this study); nevertheless, in vivo activity has been claimed for this compound.20 An only remotely related dinuclear Pt(II) complex containing a bridging C,N-chelating benzohydroxamate ligand has been found to be five times less potent after 72 h of exposure of A2780 ovarian cancer cells than the mononuclear [Pt(DACH)Cl2] (with oxaliplatin not included either).21 Even if seemingly more pronounced, the reduced cytotoxicity of complexes described here is in line with these reports.
DNA-interaction assay
A dsDNA plasmid assay was performed to check whether DNA could be the main target of the tested compounds like for the conventional platinum drugs.33 Additionally, this assay is useful in judging the reactivity of compounds towards DNA. The native plasmid is mainly present in the negatively supercoiled (sc) form, but upon interaction with compounds, it may, depending on the kind of interaction, adopt an open circular (oc), linear or cross-linked form. As has been shown recently, cisplatin interacts with plasmid DNA relatively fast.33 Total untwisting of the supercoiled form to the open circular form was already found after 30 min. In the case of oxaliplatin, featuring a bidentate oxalate ligand, pronounced interaction with dsDNA plasmid starts after 4 hours. However, complexes 3a and 3d showed no ability to untwist or break the supercoiled form towards the open circular or linear forms within 6 hours of incubation. Therefore, compounds 3a and 3d do not show any signs of DNA cross-linking or strand breaking under these conditions (Fig. S3, ESI†).Lipophilicity
Due to the low cytotoxicity and the absence of interaction with isolated DNA, we investigated the lipophilicity of the synthesized complexes in comparison with oxaliplatin in order to shed more light on their modes of action. Lipophilicity is a crucial parameter in drug discovery, since it has a great influence on the diffusion of drugs through cell membranes, or it affects how efficiently drugs are distributed in body tissue rather than plasma. The lipophilic properties of complexes 3a–3f in comparison with oxaliplatin were determined via an HPLC-based method as reported previously.34 The change in the retention time on a C18 HPLC column with varying contents of the organic modifier was used to determine the logkw values. As expected, the lipophilicity increases from 3a to 3f (Table 1), while the lipophilic properties of complex 3a are well comparable with those of oxaliplatin.
Unexpectedly, there was no clear-cut dependency between the lipophilicity and cytotoxicity of complexes 3a to 3f. Usually, the cellular accumulation of compounds is facilitated by increasing lipophilicity, resulting in decreasing IC50 values, within homologous series of complexes. This deviation might be explained by two opposing effects: increasing lipophilicity on the one hand, but a concurrently increasing steric demand affecting cytotoxicity on the other.
Reactivity towards 5′-GMP
The cytotoxicity of 3a–3f is remarkably low and interaction with DNA was not observed for compounds 3a and 3d. In contrast, the lipophilic properties of complexes are comparable with (3a) or better (3b–3f) than those of clinically established oxaliplatin, suggesting improved antiproliferative activity. Accordingly, it seems to be obvious that reactivity plays a crucial role in their mode of action. Since guanine in cellular DNA is known to be the most important target of platinum based drugs,35 the reactivity of 3a in comparison with oxaliplatin towards 5′-GMP as a model nucleotide was investigated. Thus, oxaliplatin and complex 3a, respectively, were incubated with a 10-fold excess of 5′-GMP. The corresponding HPLC traces are depicted in Fig. S4–S6 of the ESI.†First, the stability of 5′-GMP, oxaliplatin and complex 3a was investigated in PBS buffered solution at pH 7.4 over 194 h. 5′-GMP is stable in PBS under the chosen conditions (Fig. S6, ESI†). In contrast, oxaliplatin is not fully stable in PBS. This can be seen in the decrease of the oxaliplatin peak at rt = 0.8 min over time accompanied by an increase in the peak at rt = 0.35 min. Furthermore, deposition of yellow crystals in the HPLC vial was observed. Presumably, oxaliplatin reacts with Cl− ions from the buffered solution (PBS), yielding complex 1, which is sparsely soluble in water. Complex 3a is fairly stable in PBS over the course of the assay. A very slow increase in the peak at rt = 0.45 accompanied by a very slow decrease in the compound peak at rt = 0.9 min shows only minor decomposition (Fig. S5, ESI†).
In the presence of 5′-GMP, the oxaliplatin containing sample shows a fast reaction of oxaliplatin with the nucleotide (Fig. S4, ESI†). In contrast, in comparison with oxaliplatin, complex 3a reacts very slowly with 5′-GMP (compare Fig. S4 and S5 of the ESI†).
Reactivity assessment via DFT calculations
In order to exclude possible differences in reactivity due to protonation or deprotonation of the carboxylic acid moiety of complex 3a, DFT calculations were performed. The starting point for these optimizations was the geometry provided by crystal structure measurements. Comparison of the optimized geometries with the data obtained from X-ray crystallography shows only minor differences (Fig. S28 of the ESI†).Since the Pt–C bond length of the protonated complex 3a is well comparable with that of the deprotonated complex (2.061 vs. 2.044 Å for molecule A and 2.019 vs. 2.062 Å for molecule B), the thermodynamic stability is not significantly different (Fig. S29 of the ESI†).
Conclusions
Six platinum(II) complexes related to oxaliplatin, but with C,O-chelating maleic acid derived ligands, were prepared, five of them, to the best of our knowledge, for the first time. Overall, the cytotoxic potency of this class of organometallic compounds was unexpectedly low, and in contrast to oxaliplatin (as shown previously), no evidence for cross-linking was obtained upon cell-free interaction with a dsDNA plasmid. In addition, the reaction with the model nucleotide 5′-GMP was also very slow compared to oxaliplatin. It can be concluded that such types of organometallic platinum complexes feature very low reactivity. Remarkably, cytotoxic activity could not be steadily improved by increasing the lipophilicity. Additionally, it seems that the steric bulk in the C,O-chelating ligand has an opposing effect. All these observations warrant further synthesis of close analogues and comparative studies of selected representatives of this remarkable class of platinum-based complexes.Methods and experimental
General
All solvents and chemicals were obtained from commercial suppliers and used as received. K2[PtCl4] was bought from Johnson Matthey (Switzerland). Water was purified with a MilliQ system (Milli-Q, 18.2 MΩ cm−1, Milli-Q Advantage A10, Darmstadt, German). Although all products seem to be bench stable, reactions involving platinum complexes were conducted under protection from light. Unless stated otherwise, all reactions were performed in 20 mL scintillation vials and stirred with PTFE coated stirring bars. Vials were heated with an aluminum heating block.Analytical HPLC measurements were performed on a Thermo Fisher HPLC with a DAD detector and an Acquity UPLC BEH C18 1.7 μm column (3.0 × 50 mm) or an Agilent HPLC-MS with the same column and a dual wavelength detector. For all analyses, the 220 nm channel was used; additionally, MS data from HPLC-MS were used for the identification of product peaks. Lipophilicity measurements were conducted on the same Thermo Fisher device using the same Acquity UPLC BEH C18 1.7 μm column (3.0 × 50 mm). Data collection for analytical HPLC was done using Chromeleon 7.2 SR5 from Thermo Scientific. Preparative HPLC was performed on an Agilent preparative HPLC system with an Xbridge Prep C18 10 μm column (19 × 250 mm). Procedures for purification via preparative HPLC were developed via optimizing isocratic methods on the analytical setup.
NMR experiments were performed with a Bruker Avance NEO 500 MHz spectrometer at 500.32 (1H), 125.81 (13C), 107.55 (195Pt) and 50.70 (15N) MHz in DMF-d7, D2O or CDCl3 at 298 K. The (residual) solvent resonances were used as the internal reference for 1H (DMF-d7, 2.93 ppm, low-field methyl signal;34 CDCl3, 7.26 ppm (ref. 36)) and 13C (DMF-d7, 34.6 ppm, low-field methyl signal;34 CDCl3 77.16 ppm (ref. 36)) chemical shifts. 195Pt and 15N signals were referenced relative to external K2[PtCl4]34 or NH4Cl, respectively. The NMR numbering scheme for all synthesized compounds is shown in Fig. S1 in the ESI.†
Elemental analyses were performed with a PerkinElmer 2400 CHN Elemental Analyzer or an Eurovector EA 3000 CHNS-O elemental analyzer at the Microanalytical Laboratory of the University of Vienna.
ESI-MS was performed with a Bruker maXis UHR-TOF spectrometer in the positive and negative mode using ACN/MeOH 1/1 with 1% water as the solvent (ACN = acetonitrile).
Single crystal X-ray diffraction data were collected with a Stadivari diffractometer (STOE & Cie GmbH, Germany) equipped with an EIGER2 R500 detector (Dectris Ltd, Switzerland). Data were processed and scaled with the STOE software suite X-Area (STOE & Cie GmbH). Structures were solved using SHELXT37 and refined with SHELXL38 or Olex2.39 Model building was carried out using Olex2 or ShelXle.40 Structures were validated using CHECKCIF (https://checkcif.iucr.org/). Please see the respective CIF files for exact versions and more details. An overview of the sample and crystal data, data collection and structure refinement, as well as an overview of bond lengths and angles for complex 3a can be found in Tables S1–S3 in the ESI.† The ORTEP view of complex 3a drawn with 50% displacement ellipsoids is shown in Fig. S30 in the ESI.†
Determination of lipophilicity via RP-HPLC
Analyte solutions with a concentration of 0.5 mM were prepared in an ACN/MilliQ water mixture consisting of 90% (v/v) MilliQ water and 10% (v/v) ACN. This solvent was acidified with 0.1% (v/v) formic acid. The retention time was determined for three different, isocratic eluent mixtures on a Thermo Fisher HPLC system. Evaluation of the 220 nm channel via an established method34,41 gave the listed logkw values.
Cytotoxicity test
Adherent CH1/PA-1 ovarian teratocarcinoma cells (a donation from Lloyd R. Kelland, CRC Centre for Cancer Therapeutics, Institute of Cancer Research, Sutton, UK), as well as SW480 colon and A549 lung adenocarcinoma cells (both kindly provided by the Institute of Cancer Research, Department of Medicine I, Medical University of Vienna, Austria) were grown in minimal essential medium supplemented with 10% v/v heat-inactivated fetal bovine serum (Biowest, Nuaillé, France), 4 mM L-glutamine, 1 mM sodium pyruvate and 1% v/v non-essential amino acids (from a ready-to-use solution) at 37 °C under 5% CO2. All culture media, supplements and reagents were purchased from Sigma Aldrich (St Louis, MO, USA) and all plasticware from Starlab (Hamburg, Germany), unless stated otherwise.Cytotoxicity of the compounds was determined by using the colorimetric MTT assay (MTT = 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide). CH1/PA-1, SW480 and A549 cells were harvested by trypsinization and seeded in 96-well flat-bottom microculture plates at densities of 1 × 103, 2 × 103 and 3 × 103 cells (100 μL per well), respectively. Cells were allowed to settle for 24 h to resume exponential adherent growth. Test compounds were dissolved and serially diluted in a supplemented medium and then added in aliquots of 100 μL to each well. After continuous exposure for 96 h, the drug-containing medium was replaced with 100 μL of an RPMI 1640 medium/MTT mixture [6 parts of the RPMI 1640 medium (supplemented with 10% heat-inactivated fetal bovine serum and 2 mM L-glutamine) and 1 part of MTT in phosphate-buffered saline (5 mg mL−1)] per well. After incubation for 4 h, the mixtures were removed, and the formazan crystals formed by viable cells were dissolved in 150 μL of DMSO per well. Optical densities at 550 nm were measured with a microplate reader (ELx808, Bio-Tek, Winooski, VT, USA), using a reference wavelength of 690 nm to correct for unspecific absorption. 50% inhibitory concentrations (IC50) were interpolated from concentration–effect curves based on quantities of viable cells relative to untreated controls. Data are means from at least three independent experiments, each with triplicates per concentration.
DNA-interaction assay
Stock solutions of 3a and 3d (5 mM) were prepared in MilliQ water and respective volumes were added to reaction solutions to obtain 50 μM of the test compounds. 0.1 μg μL−1 pUC19 dsDNA (2686 bp) plasmid (New England BioLabs) was exposed to the compounds for different time intervals (15 min to 6 h) at 37 °C under continuous shaking. 20 μL of the samples was added to 4 μl of 6x DNA loading dye (ThermoFisher Scientific) and loaded onto a 1% agarose gel in 1× TBE buffer. Electrophoresis was performed at 60 V for 5 min, followed by 120 V for 90 min. Ethidium bromide (Serva) staining was carried out in 1× TBE (0.75 μg ml−1) for 20 min. Images were taken using the GelDoc-It Imaging System Fusion Fx7 (Vilber Lourmat, Germany).Reaction towards 5′-GMP
The following three stock solutions were prepared in phosphate buffered saline (PBS; commercially available powder yielding 0.2 g L−1 KCl, 0.2 g L−1 KH2PO4, 8.0 g L−1 NaCl, and 2.26 g L−1 Na2HPO4 anhydrous) at a pH value of 7.4: 0.005 mM oxaliplatin (stock A), 0.005 mM complex 3a (stock B) and 0.05 mM 5′-guanosine monophosphate disodium salt (5′-GMP) (stock C).From these solutions, five samples (1 mL each) were prepared: stock A diluted 1:1 with PBS, stock B diluted 1:1 with PBS, stock C diluted 1:1 with PBS, stock A combined 1:1 with stock C and stock B combined 1:1 with stock C.
After thorough mixing and subsequent filtration through a Minisart® RC4 Syringe filter, the samples were measured immediately on a Thermo Fisher HPLC and kept at 36.9 °C in the autosampler for 26 h. Subsequently, they were transferred to an aluminium heating block at 37 °C. During the first 7 hours, each sample was measured approximately every 48 min. Further measurements were performed at 26 h, 100 h and 194 h. The obtained chromatograms were compared in a qualitative fashion. The peaks were identified via their respective m/z values, measured via injecting the same samples into an Agilent HPLC-MS under the same chromatographic conditions.
DFT calculations
Syntheses
Yield: 4.09 g (89%). EA: (C6H14N2Cl2Pt): C 18.95% H 3.71% N 7.37%; found: C 18.83% H 3.68% N 7.24%. 1H NMR (DMF-d7, 500.32 MHz): δ = 1.16 (m, 2H, H4/H5 ax), 1.49 (m, 2H, H3/H6 ax), 1.56 (m, 2H, H4/H5 eq.), 2.10 (m, 2H, H3/H6 eq.), 2.60 (m, 2H, H1/H2), 5.06 (m, 2H, H7/H8 ax), 5.62 (m, 2H, H7/H8 eq.) ppm. 13C NMR (DMF-d7, 125.81 MHz): δ = 24.46 (C4/C5), 31.96 (C3/C6), 63.32 (C1/C2) ppm. 15N NMR (DMF-d7, 50.70 MHz): δ = −20.1 ppm. 195Pt NMR (DMF-d7, 107.57 MHz): δ = −654.5 ppm. HR-MS: m/z = 403.0048 g mol−1vs. calc. 403.0055 g mol−1 [M + Na+].
Yield: 14.05 g (88%). 1H NMR (CDCl3, 500.32 MHz): δ = 0.97 (t, J(1H,1H) = 7.4 Hz, 3H, H7), 1.74 (sex, J(1H,1H) = 7.1 Hz, 2H, H6), 4.23 (t, J(1H,1H) = 6.6 Hz, 2H, H5), 6.37 (d, J(1H,1H) = 12.8 Hz, 1H, H3), 6.43 (d, J(1H,1H) = 12.6 Hz, 1H, H2), 10.73 (s(b), 1H, H1) ppm. 13C NMR (CDCl3, 125.81 MHz): δ = 10.34 (C7), 21.73 (C6), 68.63 (C5), 129.85 (C3), 135.64 (C2), 165.40 (C4), 167.75 (C1) ppm. HR-MS: m/z = 181.0471 g mol−1vs. calc. 181.0471 g mol−1 [M + Na+].
Yield: 14.23 g (83%). 1H NMR (CDCl3, 500.32 MHz): δ = 0.94 (t, J(1H,1H) = 7.3 Hz, 3H, H8), 1.40 (sex, J(1H,1H) = 7.5 Hz, 2H, H7), 1.68 (p, J(1H,1H) = 7.1 Hz, 2H, H6), 4.26 (t, J(1H,1H) = 6.6 Hz, 2H, H5), 6.36 (d, J(1H,1H) = 12.8 Hz, 1 h, H3), 6.42 (d, J(1H,1H) = 13.0 Hz, 1H, H2), 11.17 (s(b), 1H, H1) ppm. 13C NMR (CDCl3, 125.81 MHz): δ = 13.67 (C8), 19.07 (C7), 30.29 (C6), 66.91 (C5), 129.93 (C3), 135.40 (C2), 165.54 (C4), 167.68 (C1) ppm. HR-MS: m/z = 173.0808 g mol−1vs. calc. 173.0808 g mol−1 [M + H+].
Yield: 16.42 g (88%). 1H NMR (CDCl3, 500.32 MHz): δ = 0.90 (m, 3H, H9), 1.34 (m, 4H, H7/H8), 1.70 (m, 2H, H6), 4.26 (t, J(1H,1H) = 6.8 Hz, 2H, H5), 6.36 (d, J(1H,1H) = 12.6 Hz, 1H, H3), 6.42 (d, J(1H,1H) = 12.6 Hz, 1H, H2), 10.29 (s(b), 1H, H1) ppm. 13C NMR (CDCl3, 125.81 MHz): δ = 13.98 (C9), 22.31 (C8), 27.95 (C7), 27.99 (C6), 67.24 (C5), 129.82 (C3), 135.68 (C2), 165.33 (C4), 167.75 (C1) ppm. HR-MS: m/z = 209.0783 g mol−1vs. calc. 209.0784 g mol−1 [M + Na+].
Yield: 99 mg (41%). 1H NMR (DMF-d7, 500.32 MHz): δ = 1.15 (m, 4H, H4/H5 ax, isomer A/B), 1.33–1.53 (m, 4H, H3/H6 ax, isomer A/B), 1.57 (m, 4H, H4/H5 eq. + H3/H6 ax, isomer A/B), 2.05 (m, 3H, H3/H6 eq., isomer A/B), 2.15 (m, 1H, H3/H6 eq., isomer A/B), 2.33 (m, 4H, H1/H2, isomer A/B), 3.22 (d, J(1H,1H) = 6.9 Hz, 1H, H11, isomer A/B), 3.36 (d, J(1H,1H) = 6.9 Hz, 1H, H11, isomer A/B), 3.43 (s, 3H, H15, isomer A/B), 3.47 (s, 2H, H15, isomer A/B), 3.81 (d, J(1H,1H) = 7.0 Hz, 1H, H10, isomer A/B), 3.83 (d, J(1H,1H) = 7.0 Hz, 1H, H10, isomer A/B), 4.24 (t, J(1H,1H) = 10.0 Hz, 1H, H7/8 ax, isomer A/B), 4.38 (t, J(1H,1H) = 10.5 Hz, 1H, H7/8 ax, isomer A/B), 4.57 (t, J(1H,1H) = 9.7 Hz, 1H, H7/8 ax, isomer A/B), 4.84 (d, J(1H,1H) = 9.0 Hz, 1H, H7/8 eq., isomer A/B), 5.09 (t, J(1H,1H) = 10.3 Hz, 1H, H7/8 ax, isomer A/B), 5.15 (d, J(1H,1H) = 10.3 Hz, 1H, H7/8 eq., isomer A/B), 5.22 (d, J(1H,1H) = 8.9 Hz, 1H, H7/8 eq., isomer A/B), 5.69 (d, J(1H,1H) = 9.4 Hz, 1H, H7/8 eq., isomer A/B) ppm. 13C NMR (DMF-d7, 125.81 MHz): δ = 19.07 (C11, isomer A/B), 19.19 (C11, isomer A/B), 24.48 (C4/C5, isomer A/B), 24.59 (C4/C5, isomer A/B), 24.84 (C4/C5, isomer A/B), 24.98 (C4/C5, isomer A/B), 32.44 (C3/C6, isomer A/B), 32.53 (C3/C6, isomer A/B), 33.62 (C3/C6, isomer A/B), 33.78 (C3/C6, isomer A/B), 49.21 (C15, isomer A/B), 49.31 (C15, isomer A/B), 58.35 (C1, isomer A/B), 59.44 (C1, isomer A/B), 63.40 (C2, isomer A/B), 63.70 (C2, isomer A/B), 71.23 (C10, isomer A/B), 71.36 (C10, isomer A/B), 176.85 (C9, isomer A/B), 176.89 (C9, isomer A/B), 186.46 (C12, isomer A/B), 186.55 (C12, isomer A/B) ppm. 15N NMR (DMF-d7, 50.70 MHz): δ = −39.4 (N7/8, isomer A/B), 2.6 (N7/8, isomer A/B) ppm. 195Pt NMR (DMF-d7, 107.57 MHz): δ = −1274.5 (b, isomer A/B), −1276.5 (b, isomer A/B) ppm. EA: (C11H20N2O5Pt)(H2O)0.5: C 28.80% H 4.57% N 5.96%; found: C 28.45% H 4.56% N 6.03%. HR-MS: m/z = 478.0917 g mol−1vs. calc. 478.0913 g mol−1 [M + Na+].
Yield: 107 mg (43%). 1H NMR (DMF-d7, 500.32 MHz): δ = 1.15 (m, 10H, H4/H5 ax + H16, isomer A/B), 1.34–1.66 (m, 8H, H3/H6 ax + H4/H5 eq., isomer A/B), 2.05 (m, 3H, H3/H6 eq., isomer A/B), 2.16 (m, 1H, H3/H6 eq., isomer A/B), 2.34 (m, 4H, H1/H2, isomer A/B), 3.20 (d, J(1H,1H) = 6.9 Hz, 1H, H11, isomer A/B), 3.23 (d, J(1H,1H) = 5.5 Hz, 2H, H14, isomer A/B), 3.34 (d, J(1H,1H) = 7.0 Hz, 1H, H11, isomer A/B), 3.76–3.87 (m, 3H, H10 + H15, isomer A/B), 3.91–4.05 (m, 3H, H15, isomer A/B), 4.24 (t, J(1H,1H) = 10.2 Hz, 1H, H7/H8 ax, isomer A/B), 4.43 (m, 2H, H7/H8 ax(A) + H7/H8 eq./ax(B), isomer A/B), 4.65 (d, J(1H,1H) = 9.5 Hz, 1H, H7/H8 eq., isomer A/B), 5.15 (m, 2H, H7/H8 eq./ax + H7/H8 ax, isomer A/B), 5.22 (d, J(1H,1H) = 8.9 Hz, 1H, H7/H8 eq., isomer A/B), 5.74 (d, J(1H,1H) = 9.5 Hz, 1H, H7/H8 eq., isomer A/B) ppm. 13C NMR (DMF-d7, 125.81 MHz): δ = 14.09 (C16, isomer A/B), 14.15 (C16, isomer A/B), 19.28 (C11, isomer A/B), 19.49 (C11, isomer A/B), 24.51 (C4/C5, isomer A/B), 24.59 (C4/C5, isomer A/B), 24.90 (C4/C5, isomer A/B), 25.02 (C4/C5, isomer A/B), 32.53 (C3/C6, isomer A/B), 32.64 (C3/C6, isomer A/B), 33.67 (C3/C6, isomer A/B), 33.82 (C3/C6, isomer A/B), 57.71 (C15, isomer A/B), 57.75 (C15, isomer A/B), 58.34 (C1, isomer A/B), 59.40 (C1, isomer A/B), 63.45 (C2, isomer A/B), 63.79 (C2, isomer A/B), 71.24 (C10, isomer A/B), 71.41 (C10, isomer A/B), 176.22 (C9, isomer A/B), 176.52 (C9, isomer A/B), 186.47 (C12, isomer A/B), 186.59 (C12, isomer A/B) ppm. 15N NMR (DMF-d7, 50.70 MHz): δ = −39.0 (N7/8, isomer A/B), 2.7 (N7/8, isomer A/B) ppm. 195Pt NMR (DMF-d7, 107.57 MHz): δ = −1277.0 (b, isomer A/B), −1285.2 (b, isomer A/B) ppm. EA: (C12H22N2O5Pt)(H2O)0.5: C 30.12% H 4.85% N 5.86%; found: C 30.19% H 4.76% N 5.98%. HR-MS: m/z = 492.1070 g mol−1vs. calc. 492.1070 g mol−1 [M + Na+].
Yield: 118 mg (46%). 1H NMR (DMF-d7, 500.32 MHz): δ = 0.88 (t, J(1H,1H) = 7.3 Hz, 3H, H17, isomer A/B), 0.91 (t, J(1H,1H) = 7.5 Hz, 3H, H17, isomer A/B),1.13 (m, 4H, H4/H5 ax, isomer A/B), 1.31–1.63 (m, 12H, H3/H6 ax + H4/H5 eq. + H16, isomer A/B), 2.05 (m, 4H, H3/H6 eq., isomer A/B), 2.32 (m, 4H, H1/H2, isomer A/B), 3.24 (d, J(1H,1H) = 7.00 Hz, 1H, H11, isomer A/B), 3.36 (d, J(1H,1H) = 6.76 Hz, 1H, H11, isomer A/B), 3.76 (m, 1H, H10, isomer A/B), 3.85 (H15, isomer A/B, taken from HSQC), 3.91 (H10, isomer A/B, taken from HSQC), 4.19 (m, 1H, H7/H8 ax, isomer A/B), 4.36 (m, 2H, H7/H8 ax + H7/H8, isomer A/B), 4.76 (d, J(1H,1H) = 10.0 Hz, 1H, H7/H8 eq., isomer A/B), 5.08 (m, 1H, H7/H8 eq., isomer A/B), 5.14 (m, 2H, H7/H8 ax + H7/H8, isomer A/B), 5.78 (d, J(1H,1H) = 10.0 Hz, 1H, H7/H8 eq., isomer A/B) ppm. 13C NMR (DMF-d7, 125.81 MHz): δ = 9.88 (C17, isomer A/B), 9.91 (C17, isomer A/B), 19.32 (C11, isomer A/B), 19.52 (C11, isomer A/B), 21.75 (C16, isomer A/B), 21.80 (C16, isomer A/B), 24.17 (C4/C5, isomer A/B), 24.21 (C4/C5, isomer A/B), 24.53 (C4/C5, isomer A/B), 24.61 (C4/C5, isomer A/B), 32.24 (C3/C6, isomer A/B), 32.34 (C3/C6, isomer A/B), 33.28 (C3/C6, isomer A/B), 33.36 (C3/C6, isomer A/B), 58.17 (C1, isomer A/B), 59.01 (C1, isomer A/B), 63.08 (C2, isomer A/B), 63.31 (C2, isomer A/B), 63.70 (C15, isomer A/B), 63.74 (C15, isomer A/B), 71.08 (C10, isomer A/B), 71.20 (C10, isomer A/B), 176.90 (C9, isomer A/B), 176.94 (C9, isomer A/B), 186.54 (C12, isomer A/B), 186.71 (C12, isomer A/B) ppm. 195Pt NMR (DMF-d7, 107.57 MHz): δ = −1273.4 (b, isomer A/B), −1279.9 (b, isomer A/B) ppm. EA: (C13H24N2O5Pt)(H2O)0.5: C 31.71% H 5.12% N 5.69%; found: C 31.77% H 5.01% N 5.96%. HR-MS: m/z = 506.1223 g mol−1vs. calc. 506.1227 g mol−1 [M + Na+].
Yield: 159 mg (61%). 1H NMR (DMF-d7, 500.32 MHz): δ = 0.89 (t, J(1H,1H) = 7.3 Hz, 3H, H18, isomer A/B), 0.90 (t, J(1H,1H) = 7.3 Hz, 3H, H18, isomer A/B), 1.15 (m, 4H, H4/H5 ax, isomer A/B), 1.31–1.68 (m, 16H, H4/H5 eq. + H3/H6 ax + H16 + H17, isomer A/B), 2.0–2.17 (m, 4H, H3/H6 eq., isomer A/B), 2.34 (m, 4H, H1/H2, isomer A/B), 3.21 (d, J(1H,1H) = 6.8 Hz, 2H, H11, isomer A/B), 3.24 (m, 2H, H14, isomer A/B), 3.34 (d, J(1H,1H) = 6.9 Hz, 1H, H11, isomer A/B), 3.76–3.85 (m, 3H, H10 + H15, isomer A/B), 3.87–4.03 (m, 3H, H15, isomer A/B), 4.23 (t, J(1H,1H) = 10.3 Hz, 1H, H7/H8 ax, isomer A/B), 4.37 (t, J(1H,1H) = 10.5 Hz, 1H, H7/H8 ax, isomer A/B), 4.44 (t, J(1H,1H) = 10.5 Hz, 1H, H7/H8 ax, isomer A/B), 4.62 (d, J(1H,1H) = 9.2 Hz, 1H, H7/H8 eq., isomer A/B), 5.15 (m, 2H, H7/H8 ax + H7/H8 eq., isomer A/B), 5.23 (m, 1H, H7/H8 eq., isomer A/B), 5.76 (d, J(1H,1H) = 9.1 Hz, 1H, H7/H8 eq., isomer A/B) ppm. 13C NMR (DMF-d7, 125.81 MHz): δ = 13.36 (C18, isomer A/B), 13.39 (C18, isomer A/B), 19.05 (C17, isomer A/B), 19.08 (C17, isomer A/B), 19.35 (C11, isomer A/B), 19.48 (C11, isomer A/B), 24.47 (C4/C5, isomer A/B), 24.54 (C4/C5, isomer A/B), 24.84 (C4/C5, isomer A/B), 24.95 (C4/C5, isomer A/B), 30.97 (C16, isomer A/B), 31.03 (C16, isomer A/B), 32.53 (C3/C6, isomer A/B), 32.62 (C3/C6, isomer A/B), 33.61 (C3/C6, isomer A/B), 33.73 (C3/C6, isomer A/B), 58.38 (C1, isomer A/B), 59.25 (C1, isomer A/B), 61.72 (C15, isomer A/B), 61.80 (C15, isomer A/B), 63.47 (C2, isomer A/B), 63.66 (C2, isomer A/B), 71.23 (C10, isomer A/B), 71.37 (C10, isomer A/B), 176.47 (C9, isomer A/B), 176.66 (C9, isomer A/B), 186.49 (C12, isomer A/B), 186.67 (C12, isomer A/B) ppm. 15N NMR (DMF-d7, 50.70 MHz): δ = −39.12 (N7/8, isomer A/B), −38.01 (N7/8, isomer A/B), 2.4 (N7/8, isomer A/B) ppm. 195Pt NMR (DMF-d7, 107.57 MHz): δ = −1276.3 (b, isomer A/B), −1284.6 (b, isomer A/B) ppm. EA: (C14H26N2O5Pt)(H2O): C 31.62% H 5.48% N 5.44%; found: C 32.70% H 5.38% N 5.38%. HR-MS: m/z = 520.1384 g mol−1vs. calc. 520.1383 g mol−1 [M + Na+].
Yield: 117 mg (43%). 1H NMR (DMF-d7, 500.32 MHz): δ = 0.89 (t, J(1H,1H) = 7.1 Hz, 3H, H19, isomer A/B), 0.90 (t, J(1H,1H) = 6.9 Hz, 3H, H19, isomer A/B), 1.08–1.24 (m, 4H, H4/H5 ax, isomer A/B), 1.26–1.65 (m, 20H, H4/H5 eq. + H3/H6 ax + H16 + H17 + H18, isomer A/B), 2.0–2.17 (m, 4H, H3/H6 eq., isomer A/B), 2.34 (m, 4H, H1/H2, isomer A/B), 3.22 (d, J(1H,1H) = 6.8 Hz, 1H, H11, isomer A/B), 3.24 (m, 1H, H14, isomer A/B), 3.35 (d, J(1H,1H) = 7.0 Hz, 1H, H11, isomer A/B), 3.75–3.85 (m, 3H, H10 + H15, isomer A/B), 3.86–4.03 (m, 3H, H15, isomer A/B), 4.24 (t, J(1H,1H) = 10.22 Hz, 1H, H7/H8 ax, isomer A/B), 4.36 (t, J(1H,1H) = 10.4 Hz, 1H, H7/H8 ax, isomer A/B), 4.45 (t, J(1H,1H) = 10.4 Hz, 1H, H7/H8 ax, isomer A/B), 4.61 (d, J(1H,1H) = 9.5 Hz, 1H, H7/H8 eq., isomer A/B), 5.16 (m, 2H, H7/H8 eq. + H7/H8 ax, isomer A/B), 5.23 (m, 1H, H7/H8 eq., isomer A/B), 5.77 (d, J(1H,1H) = 9.5 Hz, 1H, H7/H8 eq., isomer A/B) ppm. 13C NMR (DMF-d7, 125.81 MHz): δ = 13.563 (C19, isomer A/B), 13.572 (C19, isomer A/B), 19.32 (C11, isomer A/B), 19.46 (C11, isomer A/B), 22.243 (C18, isomer A/B), 22.255 (C18, isomer A/B), 24.48 (C4/C5, isomer A/B), 24.55 (C4/C5, isomer A/B), 24.85 (C4/C5, isomer A/B), 24.96 (C4/C5, isomer A/B), 28.10 (C17, isomer A/B), 28.14 (C17, isomer A/B), 28.56 (C16, isomer A/B), 28.64 (C16, isomer A/B), 32.56 (C3/C6, isomer A/B), 32.64 (C3/C6, isomer A/B), 33.68 (C3/C6, isomer A/B), 33.76 (C3/C6, isomer A/B), 58.39 (C1, isomer A/B), 59.24 (C1, isomer A/B), 62.05 (C15, isomer A/B), 62.09 (C15, isomer A/B), 63.49 (C2, isomer A/B), 63.67 (C2, isomer A/B), 71.23 (C10, isomer A/B), 71.37 (C10, isomer A/B), 176.44 (C9, isomer A/B), 176.65 (C9, isomer A/B), 186.47 (C12, isomer A/B), 186.66 (C12, isomer A/B) ppm. 15N NMR (DMF-d7, 50.70 MHz): δ = −39.3 (N7/8, isomer A/B), −38.3 (N7/8, isomer A/B), 2.5 (N7/8, isomer A/B) ppm. 195Pt NMR (DMF-d7, 107.57 MHz): δ = −1276.2 (b, isomer A/B), −1285.0 (b, isomer A/B) ppm. EA: (C15H28N2O5Pt)(H2O): C 34.02% H 5.71% N 5.29%; found: C 33.89% H 5.63% N 5.29%. HR-MS: m/z = 534.1547 g mol−1vs. calc. 534.1540 g mol−1 [M + Na+].
Author contributions
Conceptualization, T. M. and M. S. G.; data curation, T. M., N. G., M. H., K. C., and S. M.; formal analysis, T. M., M. H., K. C., and S. M.; funding acquisition, M. A. J., M. S. G., and B. K. K.; investigation, T. M., J. W., N. G., M. H., K. C., and S. M.; methodology, T. M., M. A. J., and M. S. G.; project administration, T. M., M. A. J., M. S. G., and B. K. K.; resources, M. A. J., M. S. G., and B. K. K.; supervision, M. A. J., M. S. G., and B. K. K.; validation, T. M., J. W., N. G., M. H., K. C., and S. M.; visualization, T. M., N. G., M. H., and K. C.; writing – original draft, T. M., M. A. J., and M. S. G.; and writing – review & editing, T. M., J. W., N. G., M. H., K. C., S. M., M. A. J., M. S. G., and B. K. K.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors gratefully acknowledge the support from the University of Vienna. The authors thank the Centre for X-Ray Structure Analysis (University of Vienna) for the determination of the crystal structure. Further, the authors want to thank Martijn Dijkstra for providing oxaliplatin.References
- B. Rosenberg, L. Van Camp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS PubMed.
- B. Rosenberg, L. Vancamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed.
- B. Rosenberg and L. VanCamp, Cancer Res., 1970, 30, 1799–1802 CAS.
- B. Rosenberg, E. Renshaw, L. Vancamp, J. Hartwick and J. Drobnik, J. Bacteriol., 1967, 93, 716–721 CrossRef CAS PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653 RSC.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- P. Forgacs, Fr. Pat., 2558469A1, 1985 Search PubMed.
- L. S. Hollis, S. L. Doran, A. R. Amundsen, E. W. Stern, K. J. Ahmed and S. J. Lippard, in Inorganic Syntheses, ed. A. P. Ginsberg, 1990, pp. 283–286 Search PubMed.
- M. A. Mora, A. Raya and M. A. Mora-Ramirez, Int. J. Quantum Chem., 2002, 90, 882–887 CrossRef CAS.
- H. Yuge and T. K. Miyamoto, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 3002–3005 CrossRef.
- H. Yuge and T. K. Miyamoto, Chem. Lett., 1996, 25, 375–376 CrossRef.
- Z. X. Huang, Z. Qiu, M. Q. Cheng and X. J. Li, Chin. Chem. Lett., 1991, 2, 121–122 CAS.
- M. Hrubisko, E. Balázová, F. Kiss, J. Kovácová and V. Ujházy, Neoplasma, 1989, 36, 651–657 CAS.
- L. S. Hollis and E. W. Stern, Inorg. Chem., 1988, 27, 2826–2831 CrossRef CAS.
- L. S. Hollis, E. W. Stern, A. R. Amundsen, A. V. Miller and S. L. Doran, J. Am. Chem. Soc., 1987, 109, 3596–3602 CrossRef CAS.
- A. R. Amundsen, L. S. Hollis and E. W. Stern, EU Pat., 0174114A1, 1986 Search PubMed.
- L. S. Hollis, A. R. Amundsen and E. W. Stern, J. Am. Chem. Soc., 1985, 107, 274–276 CrossRef CAS.
- M. J. Arendse, G. K. Anderson and N. P. Rath, Inorg. Chem., 1999, 38, 5864–5869 CrossRef CAS.
- N. Farrell, J. D. Roberts, M. P. Hacker, N. Farrell, J. D. Roberts and M. P. Hacker, J. Inorg. Biochem., 1991, 42, 237–246 CrossRef CAS PubMed.
- T. W. Failes, M. D. Hall and T. W. Hambley, Dalton Trans., 2003, 1596–1600 RSC.
- V. Sicilia, L. Arnal, S. Fuertes, A. Martín and M. Baya, Inorg. Chem., 2020, 59, 12586–12594 CrossRef CAS PubMed.
- L. Arnal, S. Fuertes, A. Martín, M. Baya and V. Sicilia, Chem. – Eur. J., 2018, 24, 18743–18748 CrossRef CAS PubMed.
- H. Leopold, M. Tenne, A. Tronnier, S. Metz, I. Münster, G. Wagenblast and T. Strassner, Angew. Chem., Int. Ed., 2016, 55, 15779–15782 CrossRef CAS PubMed.
- G. Dahm, E. Borré, G. Guichard and S. Bellemin-Laponnaz, Eur. J. Inorg. Chem., 2015, 2015, 1665–1668 CrossRef CAS.
- T. Mihály, M. Bette, B. Mihály, J. Schmidt, H. Schmidt and D. Steinborn, J. Organomet. Chem., 2013, 739, 57–62 CrossRef.
- M. Chtchigrovsky, L. Eloy, H. Jullien, L. Saker, E. Ségal-Bendirdjian, J. Poupon, S. Bombard, T. Cresteil, P. Retailleau and A. Marinetti, J. Med. Chem., 2013, 56, 2074–2086 CrossRef CAS PubMed.
- E. Chardon, G. Dahm, G. Guichard and S. Bellemin-Laponnaz, Chem. – Asian J., 2013, 8, 1232–1242 CrossRef CAS PubMed.
- E. Chardon, G. Dahm, G. Guichard and S. Bellemin-Laponnaz, Organometallics, 2012, 31, 7618–7621 CrossRef CAS.
- G. J. Moxey, C. Jones, A. Stasch, P. C. Junk, G. B. Deacon, W. D. Woodul and P. R. Drago, Dalton Trans., 2009, 2630–2636 RSC.
- Y.-L. Ma, R.-J. Zhou, X.-Y. Zeng, Y.-X. An, S.-S. Qiu and L.-J. Nie, J. Mol. Struct., 2014, 1063, 226–234 CrossRef CAS.
- H. P. Varbanov, S. Göschl, P. Heffeter, S. Theiner, A. Roller, F. Jensen, M. A. Jakupec, W. Berger, M. S. Galanski and B. K. Keppler, J. Med. Chem., 2014, 57, 6751–6764 CrossRef CAS PubMed.
- S. Göschl, E. Schreiber-Brynzak, V. Pichler, K. Cseh, P. Heffeter, U. Jungwirth, M. A. Jakupec, W. Berger and B. K. Keppler, Metallomics, 2017, 9, 309–322 CrossRef PubMed.
- D. Höfer, H. P. Varbanov, A. Legin, M. A. Jakupec, A. Roller, M. Galanski and B. K. Keppler, J. Inorg. Biochem., 2015, 153, 259–271 CrossRef PubMed.
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- K. Valkó, J. Chromatogr. A, 2004, 1037, 299–310 CrossRef PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. Garcia-Ratés and F. Neese, J. Comput. Chem., 2020, 41, 922–939 CrossRef PubMed.
- B. Helmich-Paris, B. de Souza, F. Neese and R. Izsák, J. Chem. Phys., 2021, 155, 104109 CrossRef CAS PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- D. A. Pantazis and F. Neese, J. Chem. Theory Comput., 2011, 7, 677–684 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Wolfgang Weigand on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2265460. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01736b |
| This journal is © The Royal Society of Chemistry 2023 |