
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of the central atom and halido ligand on the structure, antiproliferative activity and selectivity of half-sandwich Ru(II) and Ir(III) complexes with a 1,3,4-thiadiazole-based ligand†
Radka
Křikavová
 *a,
Michaela
Romanovová
b,
Zuzana
Jendželovská
b,
Martin
Majerník
b,
Lukáš
Masaryk
a,
Pavel
Zoufalý
a,
David
Milde
c,
Jan
Moncol
d,
Radovan
Herchel
a,
Rastislav
Jendželovský
b and
Ivan
Nemec
ae
*a,
Michaela
Romanovová
b,
Zuzana
Jendželovská
b,
Martin
Majerník
b,
Lukáš
Masaryk
a,
Pavel
Zoufalý
a,
David
Milde
c,
Jan
Moncol
d,
Radovan
Herchel
a,
Rastislav
Jendželovský
b and
Ivan
Nemec
ae
aDepartment of Inorganic Chemistry, Faculty of Science, Palacký University Olomouc, 17. listopadu 12, CZ-771 46 Olomouc, Czech Republic. E-mail: radka.krikavova@upol.cz
bDepartment of Cellular Biology, Institute of Biology and Ecology, Faculty of Science, Pavol Jozef Šafárik University in Košice, Šrobárova 2, 041 54 Košice, Slovakia
cDepartment of Analytical Chemistry, Faculty of Science, Palacký University Olomouc, 17. listopadu 12, CZ-771 46 Olomouc, Czech Republic
dDepartment of Inorganic Chemistry, Faculty of Chemical and Food Technology, Slovak University of Technology in Bratislava, Bratislava SK-81237, Slovakia
eCentral European Institute of Technology, Brno University of Technology, Purkyňova 123, 61200 Brno, Czech Republic
First published on 11th August 2023
Abstract
Half-sandwich complexes [Ru(η6-pcym)(L1)X]PF6 (1, 3) and [Ir(η5-Cp*)(L1)X]PF6 (2, 4) featuring a thiadiazole-based ligand L1 (2-(furan-2-yl)-5-(pyridin-2-yl)-1,3,4-thiadiazole) were synthesized and characterized by varied analytical methods, including single-crystal X-ray diffraction (X = Cl or I, pcym = p-cymene, Cp* = pentamethylcyclopentadienyl). The structures of the molecules were analysed and interpreted using computational methods such as Density Functional Theory (DFT) and Quantum Theory of Atoms in Molecules (QT-AIM). A 1H NMR spectroscopy study showed that complexes 1–3 exhibited hydrolytic stability while 4 underwent partial iodido/chlorido ligand exchange in phosphate-buffered saline. Moreover, 1–4 demonstrated the ability to oxidize NADH (reduced nicotinamide adenine dinucleotide) to NAD+ with Ir(III) complexes 2 and 4 displaying higher catalytic activity compared to their Ru(II) analogues. None of the complexes interacted with reduced glutathione (GSH). Additionally, 1–4 exhibited greater lipophilicity than cisplatin. In vitro biological analyses were performed in healthy cell lines (CCD-18Co colon and CCD-1072Sk foreskin fibroblasts) as well as in cisplatin-sensitive (A2780) and -resistant (A2780cis) ovarian cancer cell lines. The results indicated that Ir(III) complexes 2 and 4 had no effect on human fibroblasts, demonstrating their selectivity. In contrast, complexes 1 and 4 exhibited moderate inhibitory effects on the metabolic and proliferation activities of the cancer cells tested (selectivity index SI > 3.4 for 4 and 2.6 for cisplatin; SI = IC50(A2780)/IC50(CCD-18Co)), including the cisplatin-resistant cancer cell line. Based on these findings, it is possible to emphasize that mainly complex 4 could represent a further step in the development of selective and highly effective anticancer agents, particularly against resistant tumour types.
Introduction
Conventional platinum-based anticancer drugs represent one of the most widely used groups of chemotherapeutics which have been used in clinical practice for more than 40 years.1,2 However, due to their large number of side effects and low efficacy against some types of tumours, one of the main goals of medicinal chemists is to develop new agents with higher antiproliferative activity, lower general toxicity and the ability to kill cancer cells resistant towards the biological action of conventional drugs.3–5Currently investigated promising groups of potential non-platinum drugs include ruthenium complexes (BOLD-100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6,7 and TLD14338,9), which have already entered clinical trials. Indeed, BOLD-100 is presently the most clinically advanced ruthenium-based agent, which has already received Orphan Drug Designations (ODDs) from the FDA in both gastric and pancreatic cancers.10 Based on the success of such agents, it seems rational to further investigate potentially effective anticancer complexes of not only ruthenium but also other noble metals, such as iridium which tends to form complexes with similar structural features.11–13 One of the most interesting structural types of bioactive Ru and Ir complexes is undoubtedly represented by half-sandwich complexes of the general formula [M(η6/η5-arene/arenyl)(L)(X)]0/n+, which have been widely studied for their high cytotoxicity, acceptable selectivity and different mechanisms of action compared to platinum-based anticancer drugs.14 The coordination sphere of biologically relevant half-sandwich coordination compounds is typically composed of three structural elements: (i) a η5/6-arene/yl ligand which stabilizes the oxidation state of the metal cation and can facilitate transport through a cell membrane; (ii) a monodentate ligand, often (but not necessarily) a leaving group (X, typically a halido ligand but also others)15 which readily dissociates to allow coordination of the metal atom by the target biomolecules (iii) and an auxiliary ligand L which can regulate the reactivity of the complex molecule to various biomolecules (DNA, enzymes) and even play a key role in the interactions with them through hydrogen bonds or intercalation. Furthermore, the overall charge and counterion identity are other factors which could affect solubility, cell uptake, intracellular metabolism and generally, the fate of the complexes in the biological environment.16–18
6,7 and TLD14338,9), which have already entered clinical trials. Indeed, BOLD-100 is presently the most clinically advanced ruthenium-based agent, which has already received Orphan Drug Designations (ODDs) from the FDA in both gastric and pancreatic cancers.10 Based on the success of such agents, it seems rational to further investigate potentially effective anticancer complexes of not only ruthenium but also other noble metals, such as iridium which tends to form complexes with similar structural features.11–13 One of the most interesting structural types of bioactive Ru and Ir complexes is undoubtedly represented by half-sandwich complexes of the general formula [M(η6/η5-arene/arenyl)(L)(X)]0/n+, which have been widely studied for their high cytotoxicity, acceptable selectivity and different mechanisms of action compared to platinum-based anticancer drugs.14 The coordination sphere of biologically relevant half-sandwich coordination compounds is typically composed of three structural elements: (i) a η5/6-arene/yl ligand which stabilizes the oxidation state of the metal cation and can facilitate transport through a cell membrane; (ii) a monodentate ligand, often (but not necessarily) a leaving group (X, typically a halido ligand but also others)15 which readily dissociates to allow coordination of the metal atom by the target biomolecules (iii) and an auxiliary ligand L which can regulate the reactivity of the complex molecule to various biomolecules (DNA, enzymes) and even play a key role in the interactions with them through hydrogen bonds or intercalation. Furthermore, the overall charge and counterion identity are other factors which could affect solubility, cell uptake, intracellular metabolism and generally, the fate of the complexes in the biological environment.16–18
The main aim of this study was to prepare new Ru(II) and Ir(III) half-sandwich complexes with the general formulas [Ru(η6-pcym)(L1)X]PF6 and [Ir(η5-Cp*)(L1)X]PF6, where pcym is 1-methyl-4-(propan-2-yl)benzene (para-cymene), Cp* is pentamethylcyclopentadienyl, X = Cl− or I− and L1 is a bidentate N-donor ligand derived from thiadiazole, 2-(furan-2-yl)-5-(pyridin-2-yl)-1,3,4-thiadiazole (Fig. 1). Compounds involving thiadiazole rings as scaffolds have been of great interest as core structures of antitumor agents due to their high reactivity and the presence of a toxophoric N–C–S moiety.19
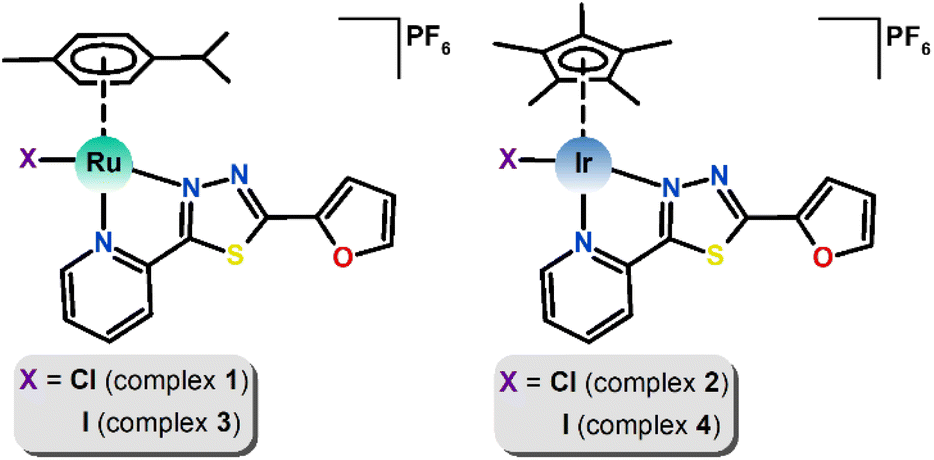 | ||
| Fig. 1 Structural formulas of [Ru(η6-pcym)(L1)X]PF6 (left) and [Ir(η5-Cp*)(L1)X]PF6 (right) complexes, where X = Cl− (1, 2) or I−(3, 4). | ||
Diverse modifications of the thiadiazole rings in various positions have led to a variety of novel compounds with a wide spectrum of pharmacological activities, such as antifungal,20 antibacterial,21 antiviral,22 anti-inflammatory,23 analgesic,24 antihelmintic25 and of particular interest is the impressive anticancer/antitumor activity.26–29 Several patents have been registered since 2008 concerning new thiadiazole ring-containing derivatives useful for the development of new anticancer drug molecules.30 To the best of our knowledge, only two works have reported on the biological investigation of Ru(II) half-sandwich complexes involving thiadiazole-based ligands. The studies focused on Ru(II) complexes with a carbonic anhydrase inhibitor acetazolamide, which were found to be inactive in vitro on all tested cell lines,31 however showed to be very potent inhibitors of tumour-associated carbonic anhydrase isoforms.32 The thiadiazole derivative in this work contains two heterocyclic substituents, i.e. the pyridine and furan rings. Pyridine is present to enable a bidentate N,N-coordination mode. This motif has been frequently used in half-sandwich ruthenium/iridium complexes as an N-donor part of bidentate ligands, such as bipyridine, azopyridine, 2-phenylpyridine, picolinate, or as a terminal monodentate ligand as a part of structure–activity studies, particularly, in the pioneering works of Sadler et al.14,33–35 The furanyl moiety brings another heterocyclic functionality and as the non-coordinating one, it could enable a varied array of non-covalent contacts due to its aromatic ring and heteroatom. Additionally, the furan ring is also an interesting moiety from the medicinal point of view, as multiple clinically approved pharmaceuticals, e.g. with antimicrobial, antiviral, anti-inflammatory, anti-ageing, and anticancer properties, contain this heterocycle in their structures.36
Furthermore, another objective of this study was to investigate changes in the biological and chemical properties (e.g. solution stability, antiproliferative activity and cytotoxicity) of the prepared complexes upon replacement of the chlorido by the iodido ligand. In previous works on various half-sandwich complexes,33,37,38 it was shown that a relevant difference in biological properties can be achieved by such structural variation. This work thus reports on half-sandwich complexes [M(η6/η5-arene/yl)(L1)X]PF6 in which the influence of two variables, i.e. M = Ru/Ir, X = Cl/I, on structural properties, solution stability and antiproliferative activity was investigated.
Results
Synthesis and basic characterization
The thiadiazole-based compound L1 (2-(furan-2-yl)-5-(pyridin-2-yl)-1,3,4-thiadiazole) used in this work was prepared by a two-step synthetic procedure (Scheme 1), which was inspired by previously published protocols.39,40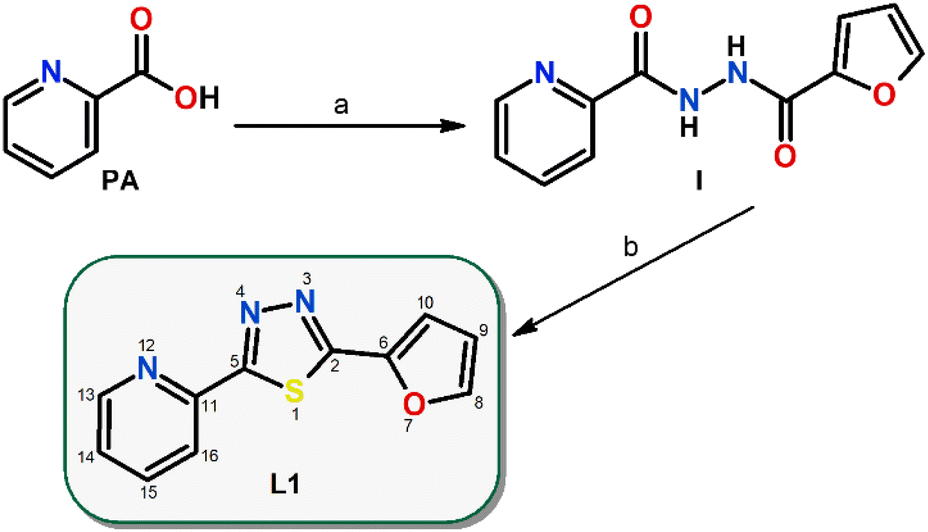 | ||
| Scheme 1 Preparation of 2-(furan-2-yl)-5-(pyridin-2-yl)-1,3,4-thiadiazole (L1) via a two-step reaction procedure: (a) 2-furoic hydrazide, dichloromethane, room temperature and (b) Lawesson's reagent, chloroform, reflux, overnight, nitrogen atmosphere, given with the atom numbering scheme. PA = pyridine-2-carboxylic acid and I = N‘-(furan-2-carbonyl)pyridine-2-carbohydrazide. | ||
First, picolinic acid and 2-furoic hydrazide were converted via a 1,1′-carbonyldiimidazole coupling reagent into N-2-furanyl-N‘-picolinoylhydrazine(I) in dichloromethane at room temperature. Second, Lawesson's reagent was employed in the cyclization reaction of I under reflux and a nitrogen atmosphere in chloroform to yield L1 (Scheme 1).
Compounds 1–4 were prepared by the reaction between the corresponding dimeric complex precursors [M(μ-Cl)(η6/η5-arene/yl)Cl]2 and L1, followed by the addition of the stabilizing PF6− counter anions (NH4PF6). During the preparation of 3 and 4, the chloride anions were precipitated by the addition of silver triflate, and then were substituted by iodide anions (KI) and again, microcrystalline products were isolated after the addition of PF6− counter anions. Compounds 1–4 were slightly soluble in water but were well soluble in organic media, such as N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), methanol, ethanol or acetone. The purity and structure of L1 and resulting complexes 1–4 were studied and confirmed by elemental analysis, mass spectrometry, FTIR, and NMR spectroscopy and crystal structures were determined by single crystal X-ray diffraction.
The ESI+ mass spectra contained peaks with m/z values and isotopic distributions attributable to the ions (e.g. {[Ru(pcym)(L1)]-H}+, {[Ir(Cp*)(L1)]-H}+, [Ru(pcym)(L1)X]+, [Ir(Cp*)(L1)X]+) agreeing well with the proposed formulas of the complex cations in 1–4 (Fig. S1–S4†).
The identity and purity of the ligand and complexes were investigated using high resolution 1H and 13C NMR spectroscopy (see the ESI, Fig. S5–S12†). The 1H signals in L1 were all significantly shifted downfield upon coordination with the metal atoms (see ESI, Fig. S10†). The most significant change was observed for the signal assigned to the C13H hydrogen, adjacent to the coordination site, i.e. pyridine nitrogen, where the coordination shift, Δδ = δcomplex − δligand, equalled 0.91 (1), 0.36 (2) 0.86 (3) and 0.38 ppm (4). Large shifts were also calculated for the hydrogen C16H with Δδ ranging between 0.39 and 0.49 ppm. In the carbon NMR spectra, which also showed significant shifting of most of the signals with respect to the spectrum of L1, the highest Δδs were observed for the signal of C16, i.e. 6.3–7.0 ppm downfield. Then the chemical shift of C13 also changed markedly, yet more so for Ru(II) complexes 1 and 3 (∼6 ppm) than for Ir(III) complexes 2 and 4 (∼3 ppm). Similarly, significant upfield shifts of ca. 4.5 ppm, and 2.5 ppm in the spectra of Ru(II) and Ir(III) complexes, respectively, were observed for the signals corresponding to the quaternary carbon C5, which lies in the vicinity of nitrogen N4, i.e. the coordination site on the 1,3,4-thiadiazole ring.
Crystal structures
Single crystals were obtained for all the herein reported coordination compounds 1–4 and their crystal structures were determined using single-crystal X-ray diffraction analysis (Table S1†). The coordination compounds consist of the complex cations and PF6− anions. All the complex cations contain the bidentate L1 ligand, η6-pcym (1 and 3) or η5-Cp* (2 and 4) ligands and monodentate halido ligands X (X = Cl− in 1 and 2, I− in 3 and 4). The overall coordination geometry can be described best as three-legged piano stool pseudooctahedral. The longest metal–ligand (M–L) bond lengths were observed for bonds with the halides (in Å, 2.394(2) in 1, 2.3857(11) in 2, 2.6986(4) in 3 and 2.6445(9) in 4), while the M–N bonds were significantly shorter (2.05–2.12 Å, Fig. 2). The distances between the centroids of the arene/yl ligands and metal atoms are shorter in the Ru complex cations (1.69 in 1 and 3vs. 1.78 Å in 2 and 4). The non-covalent interactions in the crystal structures of 1–4 are mostly of weak nature, mainly the C–H⋯π, C–H⋯S, C–H⋯F, C–H⋯Cl (1 and 2), C–H⋯I (3 and 4) hydrogen bonds. Significant non-covalent interactions are summarized in ESI (Fig. S13–S16†) and some of the selected interactions are discussed in greater detail in the Discussion paragraph (vide infra).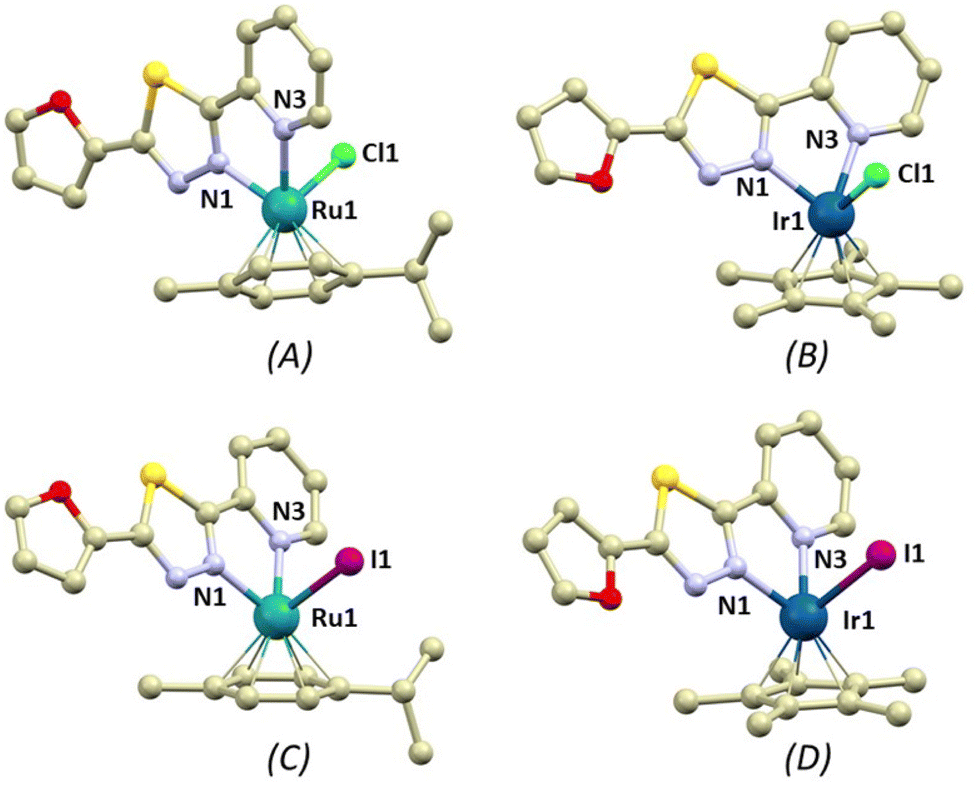 | ||
| Fig. 2 A perspective view illustrating the molecular structures of the complex cations [Ru(η6-pcym)(L1)Cl]+ (A, complex 1), [Ir(η5-Cp*)(L1)Cl]+ (B, 2), [Ru(η6-pcym)(L1)I]+ (C, 3), and [Ir(η5-Cp*)(L1)I]+ (D, 4). Hydrogen atoms have been omitted for clarity. The colour code used is as follows: light grey (carbon), green (chlorine), violet (iodine), dark blue (iridium), turquoise (ruthenium), light blue (nitrogen), red (oxygen), yellow (sulphur). Selected bond lengths (in Å): 1 (A), d(Ru1–N1) = 2.052(10), d(Ru1–N3) = 2.116(10), d(Ru1–Cl1) = 2.394(2); 2 (B), d(Ir1–N1) = 2.063(4), d(Ir1–N3) = 2.114(4), d(Ir1–Cl1) = 2.3857(11); 3 (C), d(Ru–N1) = 2.063(3), d(Ru1–N3) = 2.120(3), d(Ru1–I1) = 2.6986(4); and 4 (D), d(Ir1–N1) = 2.054(8), d(Ir1–N3) = 2.093(8), d(Ir1–I1) = 2.6445(9). | ||
Lipophilicity studies
Cytotoxicity and the ability of drugs to enter cells often correlate with their lipophilicity (hydrophobicity). Thus, one of the possible explanations for different cytotoxicity of 1–4 (see below) may be based on their different lipophilicity, which is related to their ability to enter cancer cells. Lipophilicity can be determined by the octanol/water partition coefficient (logP), which was calculated for all studied compounds: logP = −0.96 ± 0.03 (for 1), −0.25 ± 0.01 (for 2), −0.03 ± 0.02 (for 3) and −0.07 ± 0.01 (for 4). The obtained results showed that the iodido compounds (3 and 4) were more lipophilic than the chlorido ones (1 and 2). Compounds 1–4 were more lipophilic than cisplatin (CDDP, −2.21 ± 0.1).41
Solution stability
Solution stability of 1–4 was investigated by 1H NMR spectroscopy in water containing solvent mixtures of 150 μL of MeOD-d4 and 350 μL of D2O (SM1) and in 150 μL of MeOD-d4 and 350 μL of D2O with the addition of PBS (SM2, pH = 7.4, PBS stands for phosphate-buffered saline, concentration of the chloride anions in SM2 was 98 mM) (Fig. S17–S24†).In the solvent mixtures SM1 and SM2, 1–3 showed overall hydrolytic stability because their spectra did not change over time. No new signals emerged up to 48 h. To exclude immediate hydrolysis, the obtained spectra were compared to those of dehalogenated complexes 1h and 2h, which unambiguously proved no occurrence of hydrolysis. The obtained signals corresponded neither to the signals of 1h and 2h, nor to those of ligand L1 measured under the same conditions. Analogically, in SM1 compound 4 was hydrolytically stable. In contrast, though, new signals appeared in the spectrum of 4 dissolved in SM2. The chemical shifts of the new set of signals agreed perfectly with the signals of the chlorido analogue, i.e. complex 2 (Fig. 3). Many signals were overlapping, resulting in broad unresolved multiplets, however, discrete new signals appeared for C15H at 8.33 ppm (at 8.27 ppm for the iodido complex and 8.34 ppm for 2) and for C14H at 7.91 ppm (at 7.82 ppm for 4 and 7.92 ppm for 2) as well as for Cp* hydrogens at 1.77 ppm (at 1.87 ppm for 4 and 1.77 ppm for 2). Therefore, in the presence of chloride anions in solution, complex 4 undergoes gradual iodido/chlorido ligand exchange. The conversion rate of the ligand exchange for 4 in SM2 was 40% after standing at room temperature for 48 h.
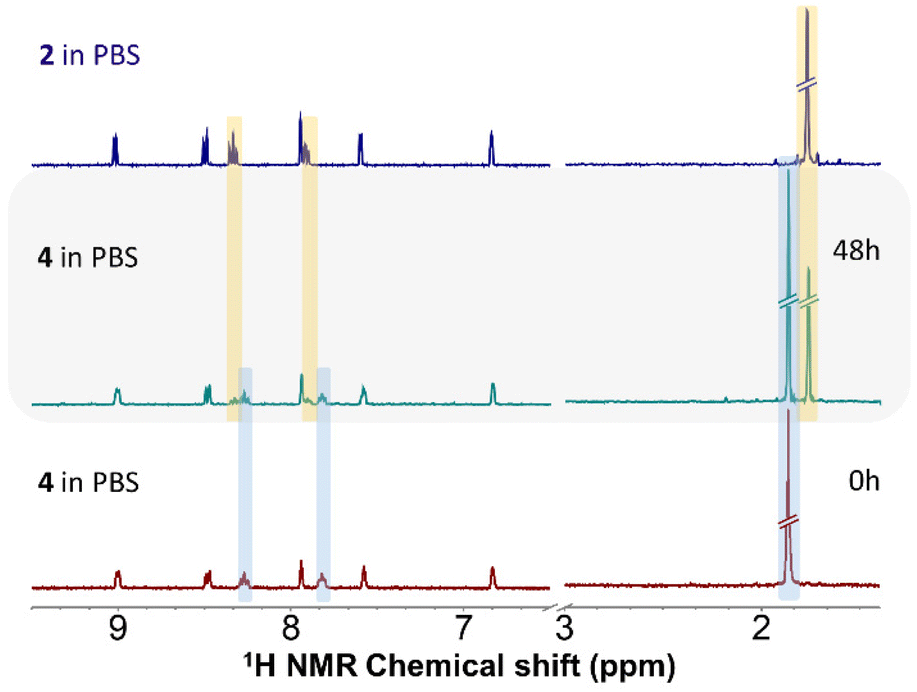 | ||
| Fig. 3 1H NMR stability study of complex 4 in SM2 (30% MeOD-d4/70% D2O with PBS), as observed at different time points (0 h or 48 h). The grey area shows the 1H NMR spectrum of 4 after 48 h confirming the occurrence of I− → Cl− ligand exchange with light blue colour denoting the signals of the original iodido complex and with yellow for the chlorido analogue. Other signals are not coloured due to overlap. For comparative purposes, the 1H spectrum of chlorido complex 2 in the same solvent mixture is shown (top). | ||
Interaction with GSH
Reduced glutathione (GSH) is a tripeptide, which often coordinates to metal centres of complex molecules and is involved in the detoxification of many anticancer metallodrugs.42,43 Importantly, it plays a vital role in the redox balance in the cell, therefore any interference with the equilibrium between GSH and its oxidized form GSSG (GSSG = glutathione disulphide) can result in pathological changes in cellular metabolism.42,44 Possible interactions with GSH of 1–4 were studied in a solvent mixture (SM3) of 150 μL of MeOD-d4 and 350 μL of D2O with PBS (pH = 7.4) with 5 molar equivalents of GSH. The structural features of complexes 1–3 remained unaltered because the positions of signals in their spectra did not change over time. In contrast, new signals appeared in the spectrum of 4, as it exhibited the same changes as described above in SM2 (i.e. the iodido/chlorido ligand exchange). The presence of GSH did not affect the conversion rate of the ligand exchange significantly, which was ca. 35% after 48 h. In addition, as far as any covalent interactions and/or GSH-related ligand exchange reactions are concerned, the results showed that none of these occurred in the interaction system, as evidenced by the unaltered aliphatic part of the 1H NMR spectra over 48 h. Similarly, complexes 1–4 were catalytically inactive in the GSH-to-GSSG oxidation reaction, since negligible (1) or no GSH transformation was evidenced in the NMR study (Fig. S25–S32†). Interestingly, with respect to the spectrum of GSH alone in the same medium, the position of two signals of GSH between 3.5 and 4.0 ppm in the spectra of all the interaction mixtures was shifted already at 0 h. These two shifted signals belong to the protons attached to carbons directly neighbouring with the two terminal COOH groups of GSH. Since no covalent interactions and/or GSH-related ligand exchange reactions were detected in the spectra, it could be suggested that the shift of the signals is related to the immediate formation of a different array of non-covalent interactions of the COOH groups of GSH with the complexes present in the mixtures. Analogical shifts of the same GSH protons were observed by Y. Q. Hao et al.45Interaction with NADH
The NADH/NAD+ system is indispensable for cellular metabolism, as it plays multiple crucial roles especially in many enzymatic events in cells. The ability of ruthenium and iridium complexes to oxidize NADH to form NAD+ has been reported in several studies.46–48 Disordering of the NADH to NAD+ ratio can lead to the disruption of various metabolic events eventually resulting in cell death.49 In a mixture (SM4) of 150 μL of MeOD-d4 and 350 μL of D2O with PBS (pH = 7.4) and 5 molar equivalents of NADH, complexes 1–3 remained intact and the positions of their signals in the spectra did not change over time up to 48 h (Fig. S33–S35†). On the other hand, similar to the above presented results, due to the presence of chloride anions in the solution, the stability of 4 was lower and again, new signals corresponding to chlorido complex 2 resulting from the ligand exchange were observed in the obtained spectra. Notably, the presence of NADH in the interaction mixture somewhat affected the iodido/chlorido exchange, whose conversion rate was 47% after 48 h (Fig. 4).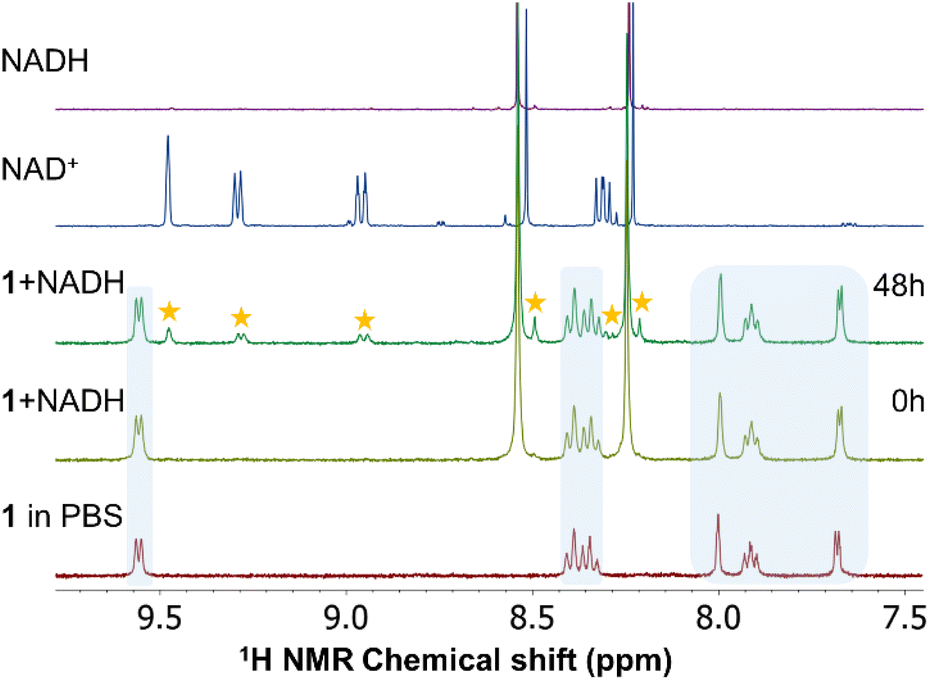 | ||
| Fig. 4 1H NMR investigation of representative compound 1 in SM4 (30% MeOD-d4/70% D2O with PBS+ 5 molar equivalents of NADH), as observed at different time points (0 h or 48 h). Asterisks denote the signals corresponding to NAD+ originating from the oxidation of NADH in the mixture with 1 (blue areas). For comparison purposes, the 1H spectra of NADH and NAD+ are shown (top). | ||
In the spectra of all compounds new signals confirming the oxidation of NADH to NAD+ were observed. Interestingly, ruthenium complexes 1 and 3 exhibited significantly lower ability to oxidize NADH (i.e. 14 and 10% NADH oxidation after 48 h, respectively) than their iridium congeners 2 and 4 (28% and 32%). In other words, the catalytic efficiency of 1–4 towards the NADH oxidation can be expressed as being ca. 0.7, 1.4, 0.5 and 1.6 molar equiv. per mol of a complex after 48 h, respectively. We did not detect the characteristic hydrido signal in the high-field region of the 1H NMR spectra. Previously reported half-sandwich Ru(II) and Ir(III) complexes (with pyridine derived ligands) were also observed as potent oxidants of NADH.47,50,51
Determination of IC50 values and effects of tested complexes on metabolic activity in healthy CCD-18Co and CCD-1072Sk human fibroblasts
To determine the IC50 (half maximal inhibitory concentration) values of tested complexes in the CCD-18Co colon and CCD-1072Sk foreskin fibroblasts we used the well-established MTT assay. The MTT assay was performed 24 and 48 h after the exposure of the cells to tested complexes. As the reference drug, we used CDDP, a well-known chemotherapeutic agent, which is generally used in the treatment of ovarian cancer. The estimated IC50 values derived from mean metabolic activity are shown in Table 1. Based on the obtained results, iridium complexes 2 and 4 which had the weakest activity against fibroblasts were chosen for subsequent experiments realized on ovarian cancer cells. Complexes 1 and 3 showed an inhibitory effect against healthy cells; nevertheless, complex 1 was also involved in further studies on the cancer cell lines for comparative purposes.| CCD-18Co | CCD-1072Sk | |||
|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | |
| 1 | 4.31 ± 1.83 | >50 | 1.92 ± 1.02 | 3.29 ± 0.88 |
| 2 | >50 | >50 | >50 | >50 |
| 3 | 3.18 ± 1.33 | >50 | 2.18 ± 1.27 | 2.63 ± 0.12 |
| 4 | >50 | >50 | >50 | >50 |
| CDDP | >50 | 11.03 ± 0.62 | >50 | 26.39 ± 8.22 |
Determination of IC50 values and effect of tested complexes on the proliferation of A2780 and A2780cis ovarian carcinoma cells
Complexes 1, 2 and 4 were chosen for a study of a potential effect on proliferation and induction of cell death in CDDP-sensitive A2780 and CDDP-resistant A2780cis ovarian carcinoma cells. At first, we determined the IC50 values of selected complexes by the MTT assay. The MTT assay was performed 24 and 48 h after exposure of the cells to tested complexes. We used CDDP as the reference drug and the estimated IC50 values derived from mean metabolic activity are shown in Table 2. The lowest IC50 values were obtained for 1 and 4. The IC50 values for 2 were not defined; therefore, this compound was excluded from subsequent analyses.| A2780 | A2780cis | |||
|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | |
| 1 | 14.90 ± 5.21 | 8.69 ± 1.75 | 16.11 ± 5.79 | 12.48 ± 4.83 |
| 2 | >25 | >25 | >25 | >25 |
| 4 | >25 | 14.70 ± 6.72 | >25 | 23.36 ± 1.03 |
| CDDP | 20.35 ± 1.39 | 4.27 ± 0.70 | >25 | 11.96 ± 2.71 |
Determination of IC50 values in healthy and cancer cell lines allowed the calculation of the selectivity index, SI = (IC50(CCD-18Co)/IC50(A2780), which was more favourable for complexes 1 and 4 with SI > 5.8 and SI > 3.4, respectively, as compared to 2.6 for CDDP. On the other hand, with the second used healthy cell line, i.e. SI2 = (IC50(CCD-1072Sk)/IC50(A2780), complex 1 was found non-selective with SI2 = 0.4, in contrast with >3.4 (4) and 6.2 (CDDP). Additionally, differences in IC50 values against sensitive and resistant cell lines enabled the calculation of the resistance factor RF, defined as RF = IC50(A2780cis)/IC50(A2780), which equals 1.4 (1), 1.6 (4) and 2.8 (CDDP).
The impact of complexes 1 and 4 on cell proliferation was assessed by evaluation of metabolic activity (Fig. S36†), cell cycle distribution (Table 3) and total cell number (Fig. 5). Tested complexes showed a time- and dose-dependent inhibitory effect on the metabolic activity of both cancer cell lines. However, a stronger effect of tested complexes was observed in CDDP-sensitive A2780 cancer cells (Table 2, Fig. S36A and C†).
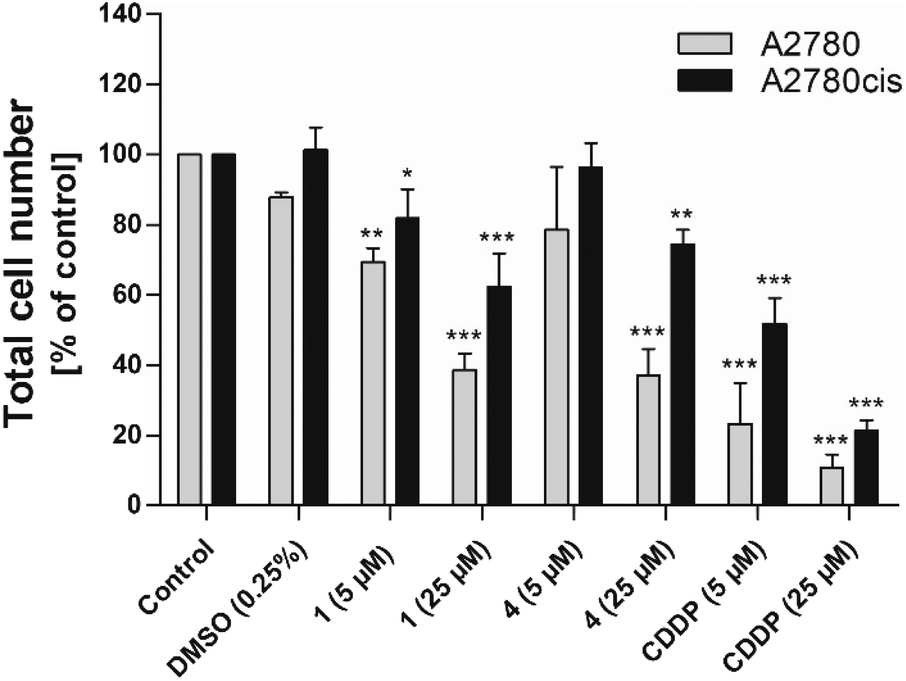 | ||
| Fig. 5 The effect of 1, 4 and CDDP on the total cell number of A2780 and A2780cis ovarian carcinoma cell lines. The total cell number was analysed 48 h after treatment of cells with selected complexes. The experimental groups were compared with the untreated control (*p < 0.05, ** p < 0.01, *** p < 0.001). | ||
| A2780 | A2780cis | |||||
|---|---|---|---|---|---|---|
| G0/G1 | S | G2/M | G0/G1 | S | G2/M | |
| Control | 64.60 ± 4.27 | 23.34 ± 2.33 | 12.06 ± 2.09 | 58.10 ± 1.07 | 28.69 ± 1.30 | 13.21 ± 0.24 |
| DMSO (0.25%) | 64.33 ± 5.36 | 24.14 ± 2.81 | 11.53 ± 2.99 | 59.01 ± 0.70 | 27.94 ± 0.49 | 13.04 ± 0.51 |
| 1 (5 μM) | 65.88 ± 6.10 | 23.50 ± 3.39 | 10.63 ± 2.77 | 58.59 ± 1.65 | 28.25 ± 1.14 | 13.16 ± 0.51 |
| 1 (25 μM) | 71.42 ± 5.56 | 18.40 ± 3.74 | 10.18 ± 2.06 | 63.66 ± 0.96** | 25.50 ± 0.84* | 10.84 ± 0.73* |
| 4 (5 μM) | 62.41 ± 6.01 | 26.53 ± 3.25 | 11.07 ± 2.96 | 58.02 ± 0.56 | 29.23 ± 0.25 | 12.76 ± 0.50 |
| 4 (25 μM) | 72.61 ± 5.51 | 19.50 ± 2.94 | 7.90 ± 2.61 | 63.51 ± 1.22** | 24.89 ± 0.47* | 11.60 ± 0.83 |
| CDDP (5 μM) | 9.49 ± 1.02*** | 85.21 ± 7.22*** | 5.30 ± 7.20 | 35.77 ± 3.22* | 30.63 ± 0.31 | 33.60 ± 3.07*** |
| CDDP (25 μM) | 66.23 ± 6.05 | 27.39 ± 1.99 | 6.38 ± 5.27 | 24.49 ± 15.82*** | 58.28 ± 17.79** | 17.23 ± 2.14 |
Analyses of the total cell number and cell cycle distribution were performed 48 h after incubation of the cells with 5 and 25 μM of complexes 1 and 4, and CDDP. Analogously to the results from the metabolic activity studies, we observed a dose-dependent decrease in the total cell number in both cancer cell lines. The effect of tested complexes was weaker in CDDP-resistant A2780cis cells in comparison with CDDP-sensitive A2780 cancer cells (Fig. 5). Flow cytometric analysis of cell cycle distribution revealed that the observed antiproliferative effect of tested complexes against CDDP-resistant cells was accompanied by increased accumulation of cells in the G1 phase of the cell cycle. These changes were attended by the reduction of cell population in the S phase of the cell cycle. In the case of complex 1, decreased percentage of cells in the G2/M phase of the cell cycle was also observed. After the exposure of CDDP-sensitive A2780 cancer cells to tested complexes we did not observe significant changes in the cell cycle distribution (Table 3).
Effect of complexes 1 and 4 on the induction of cell death in A2780 and A2780cis ovarian carcinoma cells
To determine whether the antiproliferative effect of complexes 1 and 4 was associated with the onset of cell death, we analysed cell viability and mitochondrial membrane depolarization. The analyses were performed 48 h after incubation of the cells with 5 and 25 μM of complexes 1 and 4, and CDDP. However, we did not observe any effect on the viability or mitochondrial membrane potential of used cancer cells after exposure to tested complexes. Significant changes in viability and mitochondrial membrane depolarization were achieved only after the treatment of both cancer cell lines with CDDP (Fig. S37†).Discussion
Computational evaluation of structures
It is well established that the structural and electronic properties of molecules determine their biological and pharmacological properties.52 Furthermore, any structural modification leads to the formation of a different array of non-covalent interactions. This may be of crucial importance in the biological environment, as varied non-covalent interactions of a suitable drug with its target molecule result in functional modification of the biologically relevant molecules with different consequences for cellular metabolism.53 Therefore, we took a closer look at structures and energetically available structural modifications of 1–4.The crystal structures were determined by single crystal X-ray analysis of all the reported compounds, which enabled mutual comparison between organic compound L1/complexes, Ru/Ir complexes and chlorido/iodido complexes. After analysing the crystal structures of compounds 1–4, the most striking difference was found in the conformation of ligand L1 in Ru/Ir complexes, which is evidenced by the orientation of the furanyl rings. In Ir(III) compounds 2 and 4, the furanyl rings are oriented towards the metal centre and the thiadiazole nitrogen atoms (further abbreviated as the Z conformation), whereas in Ru(II) compounds 1 and 3, they are oriented away from the metal centre (E conformation, see Fig. 2). The difference in the orientation could potentially be attributed to the different aryl ligands present in compounds 1–4 (η6-pcym in 1 and 3, η5-Cp* in 2 and 4). However, it is apparent that the aryl ligands do not induce any steric hindrance. In fact, some of the methyl groups on the aryl ligands participate in weak intramolecular C–H⋯N hydrogen bonding with the thiadiazole nitrogen atom, stabilizing the structure of the molecule by one (1 and 3) or by a bifurcated pair (2 and 4) of hydrogen bonds. The strengths of the non-covalent interactions were evaluated employing the quantum theory of atoms in molecules (QT-AIM) by means of interaction energy (Eint), and wave functions were calculated by DFT theory using the ORCA 4.2.1 program.54 The calculations were performed as single-point convergence on the appropriate fragments of the crystal structure at B3LYP and ZORA-def2-TZVP levels of theory (special basis sets: old-ZORA-TZVP for Ru and I, SARC-ZORA-TZVP for Ir).55 All QT-AIM calculations were performed using the Multiwfn package.56 It was revealed that Eint of these contacts is comparable for all the intramolecular C–H⋯N hydrogen bonds (1.5–2.3 kcal mol−1) with the weaker second interactions in the bifurcated pairs (kcal mol−1, 0.86 in 2 and 0.72 in 4).
Importantly, the presence of the PF6− anion induces significant non-covalent interactions in 1–4. Apart from the weak C–H⋯F interactions observed in all compounds, each PF6− anion forms either one (3) or two (1, 2, and 4) F⋯S interactions with the thiadiazole sulphur atom from the L1 ligand (Fig. 6A and the ESI, Fig. S13†). Although the F⋯S distances in these contacts are relatively long, they are mostly shorter than the sum of the van der Waals radii (∑Rvdw(F,S) = 3.27 Å, see ESI Fig. S13†): d(F⋯S, in Å) = 1, 3.246(7), 3.422(6); 2, 3.024(3), 3.156(4); 3, 3.212(4); and 4, 3.025(7), 3.156(8). The interaction energies for these contacts range from 0.7 to 1.9 kcal mol−1. The calculations performed using the non-covalent interaction (NCI) method57 confirm that these interactions are indeed weakly attractive (see ESI Fig. S13†). Interestingly, in the molecules adopting the Z conformation (2, and 4) the hydrogen atom in the fifth position of the furanyl ring forms a weak C–H⋯F interaction with the PF6− anion (in Å): 2, d(C19⋯F5) = 3.434(6), 4, d(C19⋯F3) = 3.459(8). Another significant distinction between the crystal packing of the E and Z compounds is the presence of the centrosymmetric R22(6)58 synthon, which is formed by hydrogen bonding between neighbouring furanyl moieties through C–H⋯O interactions (Fig. 6B and see ESI Fig. S14†). These interactions are relatively weak with long donor⋯acceptor distances (in Å, 3.406(7) in 2, 3.580(18) in 4) resulting in low Eint (1.10 kcal mol−1 in 2 and 0.50 kcal mol−1 in 4). Notably, the formation of this synthon is also supported by interactions with the halido ligands. In 2, a pair of Cl⋯O contacts are formed with d(Cl⋯O) = 3.670(4) Å, while I⋯O contacts in 4 are slightly shorter with a distance of d(I⋯O) = 3.633(8) Å. However, calculations using the electron localization function (ELF)59 indicate that these interactions are not reliant on the formation of a σ-hole on the halogen atom (see ESI Fig. S15†).60 Consequently, it is unsurprising that these interactions are only weakly attractive, as evident from the NCI plots (see ESI Fig. S15†), and exhibit relatively low calculated Eint (0.40 kcal mol−1 in 2 and 0.86 kcal mol−1 in 4).
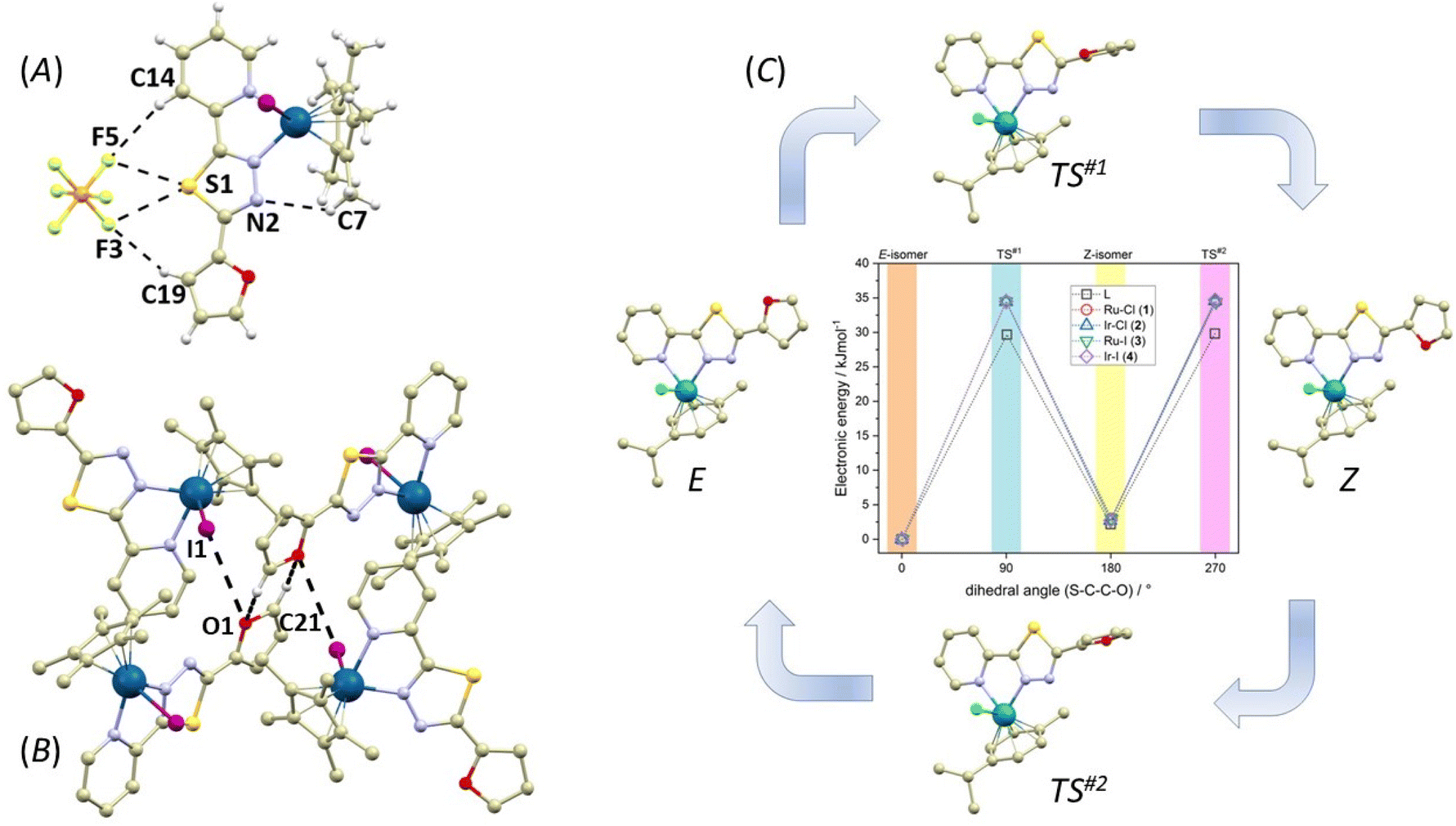 | ||
| Fig. 6 (A) Perspective view illustrating the non-covalent interactions (black dashed lines) between the PF6− anion and the complex cation in the crystal structure of 4. (B) Perspective view highlighting the interactions between the furanyl rings and iodido ligands. Hydrogen atoms, except for those involved in non-covalent interactions, have been omitted for clarity. (C) Graphical comparison displaying the relative electronic energies for the ground states of E/Z-isomers of L1 and 1–4, along with the transition states (TS#1 and TS#2) derived from DFT calculations. The colour code used is as follows: light grey (carbon), green (chlorine), violet (iodine), dark blue (iridium), turquoise (ruthenium), light blue (nitrogen), red (oxygen), yellow (sulphur). Selected lengths of non-covalent interactions (in Å): (A), d(C7⋯N2) = 3.422(8), d(C14⋯F5) = 3.660(6), d(C19⋯F3) = 3.459(8), d(F3⋯S1) = 3.156(8), d(F5⋯S1) = 3.025(7) and (B) d(C21⋯O1) = 3.580(18), d(I1⋯O1) = 3.633(8). | ||
As the orientation of the furanyl ring is different in the crystal structures of 1–4, there are E (dihedral angle S–C–C–O close to 0°) and Z (dihedral angle S–C–C–O close to 180°) isomers of L1 observed in these complexes, we decided to investigate this phenomenon also by theoretical methods employing density functional theory (DFT). Herein, ORCA 5.0 software was utilized,61 and r2SCAN functional62 was applied to all calculations together with the atom-pairwise dispersion correction (D4).63 The geometry optimization was carried out in water with the C-PCM implicit solvation model64,65 and again, Ahlrichs def2-TZVP basis set was used for all atoms, with ECP for Ru, Ir and I.66 The relative energies were investigated for both E- and Z-isomer for L1 and complexes 1–4. It is evident that the lowest electronic energy is found for the E-isomers of all compounds, and the Z-isomers have energy higher by 2–3 kJ mol−1 (Fig. 6c and see ESI Fig. S16†). The selected bond distances of optimized molecular geometries are listed in Table S2† and show nice agreement with the X-ray data, confirming good performance of the selected theoretical method. The data are also supplemented by Mayer bond orders. Generally, there is a very small variation in Ru–N and Ir–N bond distances and Mayer bond orders within 1–4. However, Mayer bond orders are significantly lower for Ir–X (X = Cl and I) bonds in 2 (0.774) and 4 (0.668) compared to Ru–X bonds in 1 (0.937) and 3 (0.934), respectively. In particular, the Ir–I bond in 4 is evidently much weaker, which is in accordance with the lower solution stability of this complex to halogen-ligand substitution (see the Solution stability section). Moreover, in all cases, the transition states (TS#1 and TS#2) were identified and confirmed by the presence of one imaginary frequency. The activation energy (energy barrier) for the E–Z isomeric reaction was found close to 34–35 kJ mol−1 for 1–4 and slightly lower for the compound L1 itself, close to 29–30 kJ mol−1, which means that the kinetics of such reaction is slowed down by the coordination of L1.
In summary, the structural and computational investigations demonstrate that the orientation of the furanyl rings in 1–4 is dominantly governed by the collective influence of weak non-covalent interactions within the crystal structure, rather than intramolecular interactions.
Biological studies
As demonstrated previously, the structure–activity modifications of this type of Ru(II) and Ir(III) with the general formula [M(η6/η5-arene/yl)(L)X]0/n+ can be achieved by the choice of both a suitable bidentate ligand L and a monodentate ligand X.18,34,67 The original idea behind such a design was that an arene/yl ligand should control lipophilicity and stabilize the oxidation state of the metal, a chelating ligand was present for ensuring additional stability and a monodentate ligand, X, was initially included as a site for activation, expectedly by aquation.33,68,69Hydrolysis of half-sandwich complexes was suggested to be an important activation step leading to the formation of covalent bonds with biomolecular targets.70 However, it has been proven that, intriguingly, it is not only the identity of X, but also the specific combination of monodentate X and chelating ligands that influences the hydrolysis rate significantly. At the same time, evidence has shown that for this type of organometallic complexes, hydrolysis does not necessarily have to be the activation step.33,71 Therefore, we thoroughly investigated the solution stability of 1–4 towards hydrolysis and interactions with selected biomolecules. It was confirmed that 1–4 were stable in water (D2O with the addition of MeOD for solubility) for a period of 48 h. This is important information because rapid hydrolysis of halido (X) half-sandwich [M(η6/η5-arene/yl)(L)(X)]+ complex cations to their aqua species [M(η6/η5-arene/yl)(L)(H2O)]2+ can result in strong binding to biomolecules, which may lead to deactivation and decrease in their cytotoxicity.33,71
Analogically, after the addition of PBS buffer (in a MeOD/D2O solution mixture) solution stability was also observed, but in this case only for 1–3. Interestingly, complex 4 underwent partial I− to Cl− ligand exchange, due to the presence of an excess of the chloride anions originating from PBS in solution. Such ligand exchange has been observed for similar half sandwich complexes previously.71,72 It is important to note that the concentration of the chloride anions in the studied solutions (98 mM) was very close to that in the extracellular environment (110 mM), but significantly higher than that in the intracellular (4 mM) environment of mammalian cells.73 It is also of importance that the extent of the ligand substitution was 40% after 48 h thus indicating slow reaction kinetics.
Similarly to previous reports on analogous half-sandwich complexes,49,511–4 catalysed the conversion of NADH to NAD+, suggesting that the mechanism of action is possibly related to changes in the redox homeostasis in the cell. These types of catalytically active complexes change the equilibrium related to the important NADH/NAD+ redox couple in cells. Such a change may have significant consequences for cellular metabolism, such as a substantial increase in ROS levels or interference with the lactate dehydrogenase-catalysed lactate–pyruvate conversions.34,35 Herein, the Ir complexes (2 and 4) were significantly more efficient oxidizers than Ru complexes (1 and 3). NADH oxidation occurred with the efficiency of ca. 0.7, 1.4, 0.5 and 1.6 molar equiv. per mol of a complex after 48 h for 1–4, respectively. Similarly, as in the solution stability study performed in the absence of NADH, complexes 1–3 remained stable and did not undergo any structural changes in the mixture containing NADH (in MeOD/D2O with PBS). On the other hand, the chloride anion presence again induced the I− to Cl− ligand exchange (47% after 48 h) for 4 also in the presence of NADH.
Furthermore, we also investigated the interactions of 1–4 with GSH and it was found that 1–4 did not react with GSH, no oxidation of GSH or formation of GSH adducts with complex molecules was observed whatsoever. Again, 1–3 were stable in this solution mixture but 4 underwent partial I− to Cl− ligand exchange to a very similar extent as in other experiments (35%).
The investigation of the effect of 1–4 on metabolic activity of healthy fibroblasts CCD-18Co and CCD-1072Sk (Table 1) revealed that after 48 h significant inhibitory activity was shown by Ru complexes only towards the CCD-1072Sk cell line and both 1 and 3 were significantly more active than CDDP. In contrast, Ir complexes 2 and 4 showed no activity (IC50 > 50 μM), which made them interesting for the following investigations. In the case of studies in cancer cell lines A2780 and A2780cis (Table 2), only 1 and 4 exhibited an inhibitory effect on metabolic activity, which was further comparable to that of CDDP in the case of 1. Unfortunately, the results showed that complex 1 was more active in healthy cells than in cancer cells. Therefore, taking into consideration the presented results, complex 4 might be understood as the most suitable candidate for further studies. Although the activity of 4 in ovarian carcinoma cells was weaker than that of 1 and CDDP, it showed the best activity profile, as it was clearly selective towards cancer over healthy cells with the selectivity index (SI = (IC50(CCD-18Co)/IC50(A2780)) > 3.4, as compared to 2.6 for CDDP. Differential selectivity of an antiproliferative agent toward cancer cells compared to healthy cells is clearly an important factor as it increases the probability of tumour-specific cytotoxicity, which is related to decreased side-effects during treatment. All in all, based on the results for complexes 1–4, it is evident that the change of the metal from Ru to Ir resulted in lower toxicity to normal cells while halide switch from Cl to I led to higher activity in cancer cells. Higher cytotoxicity of iodido with respect to chlorido Ru, Ir, Rh or Os half sandwich complexes has been previously reported in a few studies.33,37,38 However, this study reports on the situation, when the difference in the determined IC50 values between 2 and 4 is significant, and the IC50 value for 2 could not be determined up to the highest tested concentration, proving that the halide switch indeed turned on the desired activity. Additionally, although the tested complexes share partial cross-resistance with CDDP, complex 4 exhibited somewhat better ability to overcome resistance in A2780cis cells than CDDP, as the resistance factor, defined as RF = IC50(A2780cis)/IC50(A2780), dropped from 2.8 for CDDP to 1.6 for 4.
Lastly, in spite of the notably higher effect on metabolic activity of 1, both 1 and 4 seem to influence the cell cycle in a very similar manner, (Table 3) leading to the accumulation of cells in the G1 phase of the cell cycle and reduction of their number in the S and G2/M phases, which is in agreement with the previous reports.33
Because of the fact that the analysed complexes affected the metabolic activity status and cell cycle but did not influence the viability of the cells, it is possible to conclude that the effect of both 1 and 4 is cytostatic not cytotoxic. These findings agree with previously reported results. Sačková et al.74 showed that despite the decreased metabolic activity status analysed by the MTT assay, the cell viability could remain unchanged. Subsequently, cell death analyses realized by Mikešová et al.75 and Babinčák et al.76 also proved and enriched these findings. Thus, the decrease in the metabolic activity of the cells does not necessarily result from the cytotoxic action of tested compounds. In relation to our analyses, cell death induction is not the substantial cause of lower total cell number observed 48 h after application of 1 and 4. More specifically, this phenomenon seems to be preferentially associated with the antiproliferative effect of the analysed complexes. Therefore, it can be highlighted that the results of our study again confirmed that the MTT assay is a very powerful tool to assess primarily cell metabolic activity. Unfortunately, this is often overlooked in published studies that interpret the results in relation to secondary processes or states of cells, such as viability and consequently drug-induced cytotoxicity.77 Thus, in order to draw well-founded conclusions, it should be emphasized that careful and rational interpretation of the data in combination with different types of cell-based assays is necessary.
In summary, it was found that the presented complexes did not require hydrolysis as an activation step. The results of lipophilicity studies (Ir complexes are more lipophilic than Ru analogues) and NADH oxidation efficiency investigation (Ir complexes are more efficient oxidants) do not correlate with the general findings of the antiproliferative activity study, in which Ru complex 1 was labelled as the most potent one. However, looking at antiproliferative activity profile more closely, complex 1 has to be excluded from further studies, since it was shown to negatively influence metabolic activity more in healthy than in cancer cells. Therefore, overall best results were found for Ir-iodido complex 4 with possibly the most promising therapeutic index. Complex 4 was more lipophilic than its Ru analogue and was the most efficient catalyst of the NADH-to-NAD+ oxidation reaction, which could indicate that the mechanism of action is likely related to the disruption of cellular redox balance. These results highlight the importance of studies performed not only in cancer but also in normal cells to properly evaluate the pharmacological potential of anticancer drug candidates.
Nevertheless, it should be pointed out that the results for complex 4 represent a stepping stone for further studies, in which various structural modifications will be necessary in order to increase the stability of the iodido complex in the physiological environment with high chloride concentrations, as it was found for complex 4 that partial I−-to-Cl− ligand exchange occurred in such an environment. It might be speculated that this reaction could contribute to a general decrease in anticancer activity of 4, which thereby partially transforms to complex 2 (i.e. the chlorido counterpart of 4) which was evaluated as inactive up to the highest concentration level.
Experimental
Materials
Dichlorido(p-cymene)ruthenium(II) chloride dimer ([Ru(μ-Cl)(η6-pcym)Cl]2), pentamethylcyclopentadienyl iridium(III) chloride dimer ([Ir(μ-Cl)(η5-Cp*)Cl]2), ammonium hexafluoridophosphate (NH4PF6), β-nicotinamide adenine dinucleotide-reduced disodium salt hydrate (NADH), reduced glutathione (GSH), phosphate-buffered saline (PBS), silver nitrate (AgNO3), silver trifluoromethanesulfonate (AgOTf), picolinic acid, furan-2-carbohydrazide, 1,1′-carbonyldiimidazole, Lawesson's reagent, solvents (methanol (MeOH), diethyl ether (Et2O), ethanol (EtOH), N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO) and n-octanol) and deuterated solvents for NMR experiments (DMSO-d6, MeOD-d4, D2O) were supplied by VWR International (Stříbrná Skalice, Czech Republic), Sigma Aldrich (Prague, Czech Republic), Lach-Ner (Neratovice, Czech Republic) and Litolab (Chudobín, Czech Republic).Cisplatin (CDDP; cis-diamminedichloridoplatinum; CAS no.: 15663-27-1) aqueous solution (0.5 mg ml−1) was manufactured by EBEWE Pharma GmbH Nfg KG (Unterach, Austria). Stock solutions of tested compounds were prepared in dimethyl sulfoxide (DMSO; Sigma-Aldrich, St Louis, MO, USA) and further diluted to working solutions which were always freshly prepared immediately before addition to the cell culture. In all experiments, the control sample (noted as Control) and the sample with the same concentration of DMSO as the sample affected by the tested compound at the highest concentration were used to establish the effect of the solvent.
Cell cultures
The human colon fibroblasts CCD-18Co, foreskin fibroblasts CCD-1072Sk and the human ovarian carcinoma cell line A2780 were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). CDDP-resistant subline A2780cis was kindly provided by Prof. Alois Kozubík (Institute of Biophysics, Brno, Czech Republic). CCD-18Co and CCD-1072Sk cells were grown in the MEM medium (Biosera, Nuaille, France), A2780 and A2780cis cells were grown in the RPMI-1640 medium (Sigma Aldrich) at 37 °C, 95% humidity and under an atmosphere of 5% CO2. All cultivation media were supplemented with 10% fetal bovine serum (Biosera) and antibiotics (1% antibiotic-antimycotic 100× and 50 μg ml−1 gentamicin; Biosera). For maintaining the resistance of A2780cis cells, CDDP was added to the culture medium once a week at a 1 μM final concentration.For the experiments, cells were seeded in 96-well plates (MTT assay) or in 6-well plates (quantification of the cell number and viability, flow cytometry analyses) (all TPP, Trasadingen, Switzerland). Subsequently, the cells were allowed to settle for 24 h before treatment.
General methods
Electrospray ionization mass spectrometry (ESI-MS; methanol solutions) was carried out with an LCQ Fleet ion trap spectrometer (Thermo Scientific; QualBrowser software, version 2.0.7). 1H and 13C NMR spectroscopy, 1H–1H gs-COSY, 1H–13C gs-HMQC and 1H–13C gs-HMBC two-dimensional correlation experiments were performed using DMSO-d6 solutions at 298 K using a Varian spectrometer at 400 MHz (for 1H NMR) and 101 MHz (for 13C NMR); gs = gradient selected, COSY = correlated spectroscopy, HMQC = heteronuclear multiple quantum coherence, and HMBC = heteronuclear multiple bond coherence. 1H and 13C NMR spectra were calibrated against the residual DMSO signals: 1H (2.50 ppm) and 13C (39.51 ppm). The splitting of proton resonances in the reported 1H spectra is defined as s = singlet, d = doublet, dd = doublet of doublets, t = triplet, sep = septet and m = multiplet. A Jasco FT/IR-4700 spectrometer (Jasco, Easton, MD, USA) was used for the collection of the Fourier transform infrared (FTIR) spectra in the range of 400–4000 cm−1 by using the attenuated total reflection (ATR) technique on a diamond plate. Elemental analysis was performed using a Flash 2000 CHNS Elemental Analyser (Thermo Scientific).The cyclization reaction was monitored by TLC pre-coated with silica gel 60 (SIL G/UV254, 0.2 mm, Macherey-Nagel, Düren, Germany) and L1 was purified by column chromatography on Merck silica gel 60 (0.015–0.040 nm, Darmstadt, Germany).
The determination of the metal content (Ru and Ir) was performed using ICP-MS (Agilent 7700x, Agilent Japan) in the He mode to overcome potential interferences. External calibration was applied and internal standard corrections were used. Calibration solutions were prepared by diluting a multi elemental certified reference material – water calibration solution (obtained from Analytika Ltd, Czech Republic) with a concentration of 100.0 ± 0.2 mg L−1 of each metal. All samples were diluted accordingly with deionized water prior to ICP-MS analysis.
X-ray crystallography
Data collection and cell refinement of L1, 1, 2 and 4 were made using a Stoe StadiVari (Stoe & Cie GmbH, Darmstadt, Germany) diffractometer using a Pilatus3R 300K detector and microfocused X-ray source Xenocs Genix3D Cu HF (Cu Kα radiation). For compound 3, data collection was done using an XtaLAB Synergy-I diffractometer with a HyPix3000 hybrid pixel array detector and microfocused PhotonJet-I X-ray source (Cu Kα). All crystal structures were solved using a SHELXT program78 and refined by the full matrix least-squares procedure using Olex2.refine79 in OLEX2 (version 1.3).80 The multi-scan absorption corrections were applied using the program Stoe LANA software81 or CrysAlisPro 1.171.40.82a.82 The molecular structures and packing diagrams were drawn using MERCURY.83 Crystal structure refinement: all non-hydrogen atoms were refined anisotropically. The hydrogen atoms were placed at the calculated positions and they were included in the riding-model approximation with Uiso = 1.2Ueq(C) or 1.5Ueq(CH3) and d(C–H) = 0.95–0.98 Å.Synthesis of L1
The preparation of 2-(furan-2-yl)-5-(pyridin-2-yl)-1,3,4-thiadiazole (L1) proceeded in two steps. First, picolinic acid and 2-furoic hydrazide were converted via the 1,1′-carbonyldiimidazole coupling reagent into N-2-furanyl-N‘-picolinoylhydrazine(I) in dichloromethane at room temperature. Second, Lawesson's reagent was employed in the cyclization reaction of I under reflux and a nitrogen atmosphere in chloroform to yield L1 (Scheme 1).N-2-Furanyl-N‘-picolinoylhydrazine(I)
Carbonyldiimidazole (1.98 g, 12.2 mmol) and picolinic acid (1.5 g, 12.2 mmol) were stirred in 60 ml of dichloromethane for 15 min, the suspension of picolinic acid and evolution of carbon dioxide was observed. Then furoic acid hydrazide (1.54 g, 12.2 mmol) was added and the mixture was stirred for several hours, which resulted in the formation of a white precipitate. The solid obtained after evaporation of dichloromethane was washed with water and re-crystallized from acetone to give a white solid in 68% yield. EA (%) for C15H13N3O4 (MW = 231.22), calc.: C, 57.14, H, 3.92, N, 18.17. Found: C, 57.35, H, 3.94, N, 18.54. 1H NMR (400 MHz, DMSO-d6, 298 K, ppm) δ 10.60 (s, 1H), 10.42 (s, 1H), 8.70 (d, J = 4.5 Hz. 1H), 8.06–8.01 (m, 2H), 7.94–7.90 (m, 1H), 7.69–7.65 (m, 1H), 7.26 (d, J = 3.4 Hz, 1H), 6.68 (d, J = 3.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6, 298 K, ppm) 163.3, 157.0, 149.2, 148.7, 146.3, 145.7, 137.9, 127.0, 122.4, 114.5, 111.9.2-(Furan-2-yl)–5-(pyridin-2-yl)-1,3,4-thiadiazole (L1)
Compound I (900 mg, 3.89 mmol) and Lawesson's reagent (2600 mg, 6.43 mmol) in 50 ml of CHCl3 was refluxed overnight under a nitrogen atmosphere. A sticky solid which formed after evaporation of the solvent was treated with 30 ml of water, pH was adjusted to 7–8 with KHCO3 and stirred for a few hours. A light brown solid obtained by filtration was subjected to column chromatography with gradient elution (CHCl3(amylene); CHCl3 with 0.6% ethanol). Beige solid 76%. EA (%) for C11H7N3OS (Mr = 229.26), calc.: C, 57.63, H, 3.08, N, 18.33. Found: C, 57.20, H, 3.02, N, 18.02. 1H NMR (400 MHz, DMSO-d6, 298 K, ppm) δ 8.72 (d, J = 5.1 Hz, 1H, C13–H), 8.29 (d, J = 7.8 Hz, 1H, C16–H), 8.05 (m, 2H, C15–H, C8–H), 7.60 (dd, J = 7.4, 4.9 Hz, 1H, C14–H), 7.41 (d, J = 3.5 Hz, 1H, C10–H), 6.81 (dd, J = 3.5, 2.0 Hz, 1H, C9–H). 13C NMR (101 MHz, DMSO-d6, 298 K, ppm) δ 168.8 (C5), 159.3 (C2), 150.3 (C13), 147.9 (C11), 146.7 (C8), 144.7 (C6), 138.1 (C15), 126.2 (C14), 120.6 (C16), 113.1 (C9), 113.0 (C10).Syntheses of complexes 1 and 2
Complexes 1 and 2 were prepared by the reaction between [Ru(μ-Cl)(η6-pcym)Cl]2/[Ir(μ-Cl)(η5-Cp*)Cl]2 (0.05 mmol) and L1 (0.10 mmol) in MeOH (5 mL). The resulting brown suspension was stirred at ambient temperature for 24 h and this led to the preparation of an orange solution containing complex [Ru(η6-pcym)(L1)Cl]Cl (1*)/[Ir(η5-Cp*)(L1)Cl]Cl (2*). Then, an excess of NH4PF6 (0.25 mmol) was added and after stirring for 5 min at ambient temperature the solution was filtered and the solvent volume was reduced by nitrogen gas until an orange product precipitated, which was collected by filtration, washed (1 × 0.5 mL of MeOH and 3 × 1.0 mL of diethyl ether) and dried under vacuum.Syntheses of complexes 3 and 4
Nearly identical preparative procedures were used for the syntheses of 3 and 4, but before the addition of NH4PF6, a stoichiometric amount of AgOTf (0.10 mmol) was added to the orange solutions, which was further stirred at ambient temperature in the dark for 1 h. The white precipitate of AgCl was removed by filtration and KI (0.10 mmol) was added into a clear light-orange solution. The reaction mixture became darker during 2 h of stirring at ambient temperature and then, NH4PF6 (0.25 mmol) was added and after stirring for 5 min at ambient temperature, the solution was filtered and the solvent volume was reduced by nitrogen gas until a red product precipitated, which was collected by filtration, washed (1 × 0.5 mL of MeOH and 3 × 1.0 mL of diethyl ether) and dried under vacuum.Lipophilicity studies log(P)
Octanol-saturated water (OSW) and water-saturated octanol (WSO) were prepared from octanol and 0.2 M water solution of KCl by overnight stirring. The stock solutions were prepared by shaking of 1 μmol of complexes 1–4 in 11 mL of OSW for 1 h. Then the mixtures were centrifuged (5 min, 11000 rpm) and the supernatant was collected. 5 mL of this solution was studied by ICP-MS, while another 5 mL of this solution was added to 5 mL of WSO and shaken for 2 h at ambient temperature. After that, these mixtures were centrifuged and aqueous layers were carefully separated. The Ru and Ir concentrations were determined by ICP-MS (the obtained value was corrected for the adsorption effects). The LogP = log([M]WSO/[M]OSWa) equation was used for the partition coefficient calculation, [M]OSWb and [M]OSWa stands for the Ru and Ir concentration before and after partition, respectively, and [M]WSO = [M]OSWb − [M]OSWa. The experiment was conducted in triplicate and the results are presented as arithmetic mean ± SD.84
Solution stability studies
Appropriate amounts of complexes 1–4 for the preparation of a 1 mM solution were dissolved in 150 μL of MeOD-d4 and 350 μL of D2O (SM1) and in 150 μL of MeOD-d4 and 350 μL of D2O with PBS (pH = 7.4, SM2) The prepared systems were measured immediately after dissolving the solid (0 h) and then at various time intervals (up to 48 h) and incubated at 25 °C. The obtained 1H NMR spectra were calibrated against the residual signal of D2O (4.75 ppm). Note: MeOD-d4 ensured solubility of all tested complexes during 1H NMR experiments, as their solubility in water is limited.1H NMR spectra were also recorded for dehalogenated 1h and 2h species in the same medium, which were prepared from the fresh solutions of complexes 1 and 2 in 30% MeOD-d4/70% D2O by the addition of a stoichiometric amount of silver nitrate. The mixtures were shaken under aluminium foil (25 °C) for 1 h, then the formed precipitate of AgCl was centrifuged. The so obtained solutions were used for 1H NMR experiments. Compound L1 was also studied under the same conditions as the respective complexes. The stability of 1–4 was evaluated by determining the unchanged amount of complexes in solution. This was done by integrating a representative C13–H signal in the 1H NMR spectra.
Interaction with GSH
Mixture SM3 – GSH (5 mol equiv.) was added to a 1 mM solution of complexes 1–4 in 150 μL of MeOD-d4 and 350 μL of D2O with PBS (pH 7.4). The prepared solutions were measured immediately after dissolving the solid (0 h) and then at various time points (up to 48 h) and incubated at ambient temperature between the individual 1H NMR experiments. No inert atmosphere was used for the experiments, as control experiments with GSH alone in 150 μL of MeOD-d4 and 350 μL of D2O with PBS (pH 7.4) did not show any autooxidation of GSH (Fig. S38†).Interaction with NADH
The SM4–NADH (5 mol equiv.) mixture was added to a 1 mM solution of complexes 1–4 in 150 μL of MeOD-d4 and 350 μL of D2O with PBS (pH 7.4). The prepared solutions were measured immediately after dissolving the solid (0 h) and then at various time points (up to 48 h) and incubated at ambient temperature between the individual 1H NMR experiments. No inert atmosphere was used for the experiments, as control experiments with NADH alone in 150 μL of MeOD-d4 and 350 μL of D2O with PBS (pH 7.4) did not show any autooxidation of NADH (Fig. S39†).MTT assay and IC50 value evaluation
MTT assays were performed as previously reported by Kleban et al.85 to evaluate changes in the metabolic activity of CCD-18Co, CCD-1072Sk, A2780 and A2780cis cells that occurred as a consequence of treatment by complexes. MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) (Sigma Aldrich) from a stock solution (5 mg ml−1) was added to the cells in a 96-well plate (TPP) (final concentration 0.5 mg ml−1) 24 and 48 h after the treatment with tested complexes. The metabolic reaction was stopped after 4 h of incubation at 37 °C and the insoluble formazan crystals were dissolved by the addition of sodium dodecyl sulphate (SDS) at a final concentration of 3.3%. The absorbance (λ = 584 nm) was measured using a BMG FLUOstar Optima (BMG Labtechnologies GmbH, Offenburg, Germany). The results were evaluated as percentages of the absorbance of the untreated control.The IC50 values of tested complexes were extrapolated from a dose-response fit to the mean metabolic activity data using OriginPro 8.5.0 SR1 (OriginLab Corp., Northampton, MA, USA).
Based on the obtained IC50 values, relative indices were calculated, i.e. selectivity index SI = IC50(healthy cells)/IC50(A2780), and resistance factor RF = IC50(A2780cis)/IC50(A2780).
Quantification of cell number and viability
For the assessment of the total cell numbers and viability within individual experimental groups, A2780 and A2780cis cells were harvested 48 h after the treatment with selected complexes, washed with PBS and analysed using a Vi-CELL XR Cell Viability Analyzer (Beckman Coulter, Indianapolis, IN, USA). The total cell number was expressed as a percentage of the untreated control of the total cell number. Viability was expressed as a percentage of viable cells.Cell cycle analysis
Cell cycle analysis was performed as previously reported by Kleban et al.85 For flow cytometric analysis of the cell cycle, A2780 and A2780cis cells were harvested 48 h after the treatment with selected complexes, washed in cold PBS, fixed in cold 70% ethanol and kept at −20 °C overnight. Prior to analysis, cells were washed twice with PBS, resuspended in the staining solution (0.1% Triton X-100, 0.137 mg ml−1 ribonuclease A and 0.02 mg ml−1 propidium iodide – PI), incubated in the dark at room temperature (RT) for 30 min and analysed (15 × 103 cells per sample) using a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA, USA) with a 488 nm argon-ion excitation laser. Fluorescence was detected via a 585/42 nm band-pass filter (FL-2). ModFit 3.0 (Verity Software House, Topsham, ME, USA) software was used to generate DNA content frequency histograms and quantify the number of cells in the individual cell cycle phases.Detection of mitochondrial membrane depolarization
Detection of mitochondrial membrane depolarization was performed as previously reported by Mikeš et al.86 A2780 and A2780cis cells were harvested 48 h after the treatment with selected complexes, centrifuged and washed with Hank's balanced salt solution (HBSS). Subsequently, the cells were stained with 0.1 μM tetramethylrhodamine ethyl ester perchlorate (TMRE; Sigma Aldrich) in HBSS for 20 min at RT in the dark and analysed (1 × 104 cells per sample) using a flow cytometer (BD FACSCalibur). Fluorescence was detected via a 585/42 band-pass filter (FL-2). The results were analysed using FlowJo software (TreeStar Inc., Ashland, OR, USA) and are presented as percentages of the cells with normal/undissipated mitochondrial membrane potential (TMRE + cells).Statistical analysis
Data were analysed using one-way ANOVA with Tukey's post hoc test and are expressed as the mean ± standard deviation (SD) of at least three independent experiments. Significance levels are indicated in the legend for each particular figure.Conclusions
In this paper, we reported on new half-sandwich ruthenium and iridium complexes with a thiadiazole-based organic ligand 2-(furan-2-yl)-5-(pyridin-2-yl)-1,3,4-thiadiazole (L1) with the general formula [Ru(η6-pcym)(L1)X]PF6 (X = Cl for 1, I for 3) and [Ir(η5-Cp*)(L1)X]PF6 (X = Cl for 2, I for 4). The single-crystal X-ray diffraction analysis confirmed the expected composition of the complexes and revealed different orientations of the L1 ligand in Ru and Ir complex cations. This observation was explained by DFT calculations, which yielded a low relative energy barrier for reorientation of the furanyl ring. The observed orientation in the crystal structures was explained by the present non-covalent interactions in the crystal structures.Complexes were thoroughly characterized, and we proved that 1–4 were inert towards hydrolysis in water and 1–3 were also stable under pseudo-physiological conditions (in PBS, PBS with GSH and PBS with NADH). In contrast, [Ir(η5-Cp*)(L1)I]PF6 (4) was found to undergo partial I−-to-Cl− ligand exchange in such a chloride rich environment. Anyway, hydrolysis was not indispensable for activation of the complexes, as 1 and 4 exhibited antiproliferative activity not only in a CDDP-sensitive, but also in a CDDP-resistant ovarian carcinoma cell line in vitro. In addition, complexes 1–4 did not interact with GSH, but oxidized NADH to NAD+, which could be linked to their redox-mediated mechanism of biological action. Complex 4 was evaluated as the most efficient oxidant.
This study emphasised the significance of the evaluation of activity not only in cancer cells but also in normal cells to appropriately identify the pharmacological potential of anticancer drug candidates. The activity of ruthenium complex 1 was comparable to cisplatin in the tested cancer cell lines; however, it even more significantly affected the metabolic activity of normal human fibroblasts (IC50–3 μM), thus disqualifying it as a non-selective compound.
All in all, Ir-iodido complex 4 comes out of the comparison of 1–4 as the best candidate for further studies. It was shown to be more lipophilic than its Ru congener, the best NADH oxidation catalyst, exhibited activity on both cisplatin-sensitive and cisplatin-resistant ovarian carcinoma cell lines in vitro with the resistance factor, RF = IC50(A2780cis)/IC50(A2780), of 1.6 as compared to 2.8 for cisplatin. Furthermore, very good selectivity compared to healthy human fibroblasts (IC50 > 50 μM) was observed. Therefore, complex 4 may represent a solid stepping stone for further studies of structural derivatization of this type of organometallic complexes on the way to developing effective agents for the treatment of ovarian cancer, or for sensitization of resistant cancer types to anticancer agents.
In conclusion, it was unambiguously confirmed that even small changes in the structural properties of organometallic complexes can have a major effect on structure as well as biological activity. There is a complex interplay between the metal and both chelating and monodentate ligands which can modify drug behaviour in the cell leading to diverse effects on overall cellular metabolism. Herein, the desired cancer cell-specific activation was achieved by exchanging Ru for Ir and by switching the halido coligand from chloride to iodide.
Author contributions
Conceptualization: R. K., M. R., L. M., R. J., and I. N. Data curation: R. K., M. R., Z. J., M. M., L. M., D. M., R. H., R. J., and I. N. Formal analysis: R. K., M. R., Z. J., M. M., L. M., P. Z., D. M., J. M., R. H., R. J., and I. N. Funding acquisition: J. M., R. H., and R. J. Investigation: R. K., M. R., Z. J., M. M., L. M., P. Z., D. M., J. M., R. H., R. J., and I. N. Methodology: R. H., R. J., and I. N. Project administration: R. K. and R. J. Resources: D. M., J. M., R. H., R. J., and I. N. Supervision: R. J. and I. N. Validation: R. K., M. R., Z. J., M. M., R. J., and I. N. Visualization: R. K., R. H., R. J., and I. N. Writing – original draft: R. K., M. R., Z. J., M. M., L. M., R. H., R. J., and I. N. Writing – review & editing: R. K., Z. J., M. M., R. H., R. J., and I. N. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the “Open Scientific Community for Modern Interdisciplinary Research in Medicine (OPENMED)”, ITMS2014 + 313011V455 within the Operational Programme Integrated Infrastructure, funded by the ERDF, by the Slovak Research and Development Agency (APVV-19-0087) and by the Scientific Grant Agency of the Slovak Republic (Project 1/0686/23). Financial support from the institutional sources of the Department of Inorganic Chemistry, Palacký University Olomouc, Czech Republic, is also acknowledged. The authors also wish to thank Mrs Pavla Richterová for performing elemental analyses.References
- B. Rosenberg, L. Van Camp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS PubMed.
- P. Štarha, J. Hošek, Z. Trávníček and Z. Dvořák, Appl. Organomet. Chem., 2020, 34, e5785 CrossRef.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2010, 54, 3–25 CrossRef PubMed.
- J. Kralj, A. Bolje, D. S. Polančec, I. Steiner, T. Gržan, A. Tupek, N. Stojanović, S. Hohloch, D. Urankar, M. Osmak and B. Sarkar, Organometallics, 2019, 38, 4082–4092 CrossRef CAS.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- B. J. Park, P. Raha, J. Panchovich and M. Bazett, Cancers, 2023, 15, 28 CrossRef CAS PubMed.
- J. Fong, K. Kasimova, Y. Arenas, P. Kaspler, S. Lazic, A. Mandel and L. Lilge, Photochem. Photobiol. Sci., 2015, 14, 2014–2023 CrossRef CAS PubMed.
- S. Monro, K. L. Colón, H. Yin, J. Roque III, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS PubMed.
- https://www.bold-therapeutics.com/technology (accessed 08/2023).
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Chem. Soc. Rev., 2018, 47, 909–928 RSC.
- C. H. Leung, H. J. Zhong, D. S. H. Chan and D. L. Ma, Coord. Chem. Rev., 2013, 257, 1764–1776 CrossRef CAS.
- H. Hanif, M. V. Babak and C. G. Hartinger, Drug Discovery Today, 2014, 19, 1640–1648 CrossRef PubMed.
- P. Zhang and P. J. Sadler, J. Organomet. Chem., 2017, 839, 5–14 CrossRef CAS.
- P. Štarha and Z. Trávníček, Coord. Chem. Rev., 2019, 395, 130–145 CrossRef.
- A. K. Singh, D. S. Pandey, Q. Xu and P. Braunstein, Coord. Chem. Rev., 2014, 270, 31–56 CrossRef.
- C. S. Allardyce and P. J. Dyson, Platinum Met. Rev., 2001, 45, 62–69 CAS.
- H. Zhang, L. Guo, Z. Tian, M. Tian, S. Zhang, Z. Xu, P. Gong, X. Zheng, J. Zhao and Z. Liu, Chem. Commun., 2018, 54, 4421–4424 RSC.
- A. M. M. Omar and O. M. Aboulwafa, J. Heterocycl. Chem., 1986, 23, 1339–1341 CrossRef CAS.
- B. Chaturvedi, N. Tiwari and Nizamuddin, Agric. Biol. Chem., 1988, 52, 1229–1232 CAS.
- N. Demirbas, A. Demirbas, S. A. Karaoglu and E. Celik, ARKIVOC, 2005, 75–91 Search PubMed.
- F. P. Invidiata, D. Simoni, F. Scintu and N. Pinna, Farmaco, 1996, 51, 659–664 CAS.
- M. Amir, H. Kumar and S. A. Javed, Eur. J. Med. Chem., 2008, 43, 2056–2066 CrossRef CAS PubMed.
- G. Chawla, U. Kumar, S. Bawa and J. Kumar, J. Enzyme Inhib. Med. Chem., 2012, 27, 658–665 CrossRef CAS PubMed.
- S. M. El-Khawass, M. A. Khalil, A. A. Hazzaa, H. A. Bassiouny and N. F. Loutfy, Farmaco, 1986, 44, 703–709 Search PubMed.
- A. Husain, M. Rashid, M. Shaharyar, A. A. Siddiqui and R. Mishra, Eur. J. Med. Chem., 2013, 62, 785–798 CrossRef CAS PubMed.
- D. Chowrasia, C. Karthikeyan, L. Choure, M. Gupta, M. Arshad and P. Trivedi, Arabian J. Chem., 2017, 10, S2424–S2428 CrossRef CAS.
- D. A. Ibrahim, Eur. J. Med. Chem., 2009, 44, 2776–2781 CrossRef CAS PubMed.
- K. Ilango and P. Valentina, Eur. J. Med. Chem., 2010, 1, 50–53 CAS.
- R. Morigi, A. Locatelli, A. Leoni and M. Rambaldi, Recent Pat. Anticancer Drug Discov., 2015, 10, 280–297 CrossRef CAS PubMed.
- L. Biancalana, L. K. Batchelor, G. Ciancaleoni, S. Zacchini, G. Pampaloni, P. J. Dyson and F. Marchetti, Dalton Trans., 2018, 47, 9367–9384 RSC.
- S. Seršen, K. Traven, J. Kljun, I. Turel and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2019, 34, 388–393 CrossRef PubMed.
- I. Romero-Canelón, L. Salassa and P. J. Sadler, J. Med. Chem., 2013, 56, 1291–1300 CrossRef PubMed.
- A. J. Millett, A. Habtemariam, I. Romero-Canelón, G. Clarkson and P. J. Sadler, Organometallics, 2015, 34, 2683–2694 CrossRef CAS PubMed.
- Z. Liu, I. Romero-Canelón, B. Qamar, J. M. Hearn, A. Habtemariam, N. P. E. Barry, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, Angew. Chem., 2014, 126, 4022–4027 CrossRef.
- H. Saeid, H. Al-sayed and M. Bader, Alq. J. Med. Appl. Sci., 2023, 6, 44–58 Search PubMed.
- M. Kubanik, H. Holtkamp, T. Söhnel, S. M. Jamieson and C. G. Hartinger, Organometallics, 2015, 34, 5658–5668 CrossRef CAS.
- P. Štarha, Z. Dvořák and Z. Trávníček, J. Organomet. Chem., 2018, 872, 114–122 CrossRef.
- P. Zoufalý, E. Čižmár, J. Kuchár and R. Herchel, Molecules, 2020, 25, 277 CrossRef PubMed.
- A. E. Siegrist, E. Maeder, M. Duennenberger and P. Liechti, CIBA Ltd, SWXXASCH411906; Chem. Abstr., 1967, 71, 64406 Search PubMed.
- J. J. Wilson and S. J. Lippard, J. Med. Chem., 2012, 55, 5326–5336 CrossRef CAS PubMed.
- E. Hatem, N. El Banna and M.-E. Huang, Antioxid. Redox Signal., 2017, 27, 1217–1234 CrossRef CAS PubMed.
- J. Li, L. Guo, Z. Tian, S. Zhang, Z. Xu, Y. Han, R. Li, Y. Li and Z. Liu, Inorg. Chem., 2018, 57, 13552–13563 CrossRef CAS PubMed.
- G. Noctor, G. Queval, A. Mhamdi, S. Chaouch and C. H. Foyer, The Arabidopsis Book, 2011, 9, e0142 CrossRef PubMed.
- Y. Q. Hao, Y. Wu, J. Liu, G. Luo and G. Yang, J. Inclusion Phenom. Macrocyclic Chem., 2006, 54, 171–175 CrossRef CAS.
- J. J. Soldevila-Barreda, I. Romero-Canelón, A. Habtemariam and P. J. Sadler, Nat. Commun., 2015, 6, 6582 CrossRef CAS PubMed.
- S. Betanzos-Lara, Z. Liu, A. Habtemariam, A. M. Pizarro, B. Qamar and P. J. Sadler, Angew. Chem., Int. Ed., 2012, 51, 3897–3900 CrossRef CAS PubMed.
- Z. Liu, R. J. Deeth, J. S. Butler, A. Habtemariam, M. E. Newton and P. J. Sadler, Angew. Chem., Int. Ed., 2013, 52, 4194–4197 CrossRef CAS PubMed.
- P. Zhang and P. J. Sadler, Eur. J. Inorg. Chem., 2017, 1541–1548 CrossRef CAS.
- J. Li, Z. Tian, X. Ge, Z. Xu, Y. Feng and Z. Liu, Eur. J. Med. Chem., 2019, 163, 830–839 CrossRef CAS PubMed.
- J. Li, L. Guo, Z. Tian, M. Tian, S. Zhang, K. Xu, Y. Qian and Z. Liu, Dalton Trans., 2017, 46, 15520–15534 RSC.
- (a) C. Hansch and A. Leo, Exploring QSAR: Fundamentals and Applications in Chemistry and Biology, American Chemical Society, Washington, DC, 1995 Search PubMed; (b) A. V. Gubskaya, in In Quantum Biochemistry: Electronic Structure and Biological Activity, Vol. 2, ed. C. F. Matta, Wiley-VCH, Weinheim, 2010, pp. 693 Search PubMed.
- L. Masaryk, J. Orvoš, K. Słoczyńska, R. Herchel, J. Moncol, D. Milde, P. Halaš, R. Křikavová, P. Koczurkiewicz-Adamczyk, E. Pękala, R. Fischer, I. Šalitroš, I. Nemec and P. Štarha, Inorg. Chem. Front., 2022, 9, 3758–3770 RSC.
- F. Neese, Software update: The ORCA program system, version 4, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2011, 33, 580–592 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Patterns, in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals, Angew. Chem., Int. Ed. Engl., 1995, 15, 1555–1573 CrossRef.
- A. D. Becke and K. E. Edgecombe, A simple measure of electron localization in atomic and molecular systems, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- H. Wang, W. Wang and W. J. Jin, σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond, Chem. Rev., 2016, 116(9), 5072–5104, DOI:10.1021/ACS.CHEMREV.5B00527.
- F. Neese, Software Update: The ORCA Program System -Version 5.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- J. W. Furness, A. D. Kaplan, J. Ning, J. P. Perdew and J. Sun, J. Phys. Chem. Lett., 2020, 11, 8208–8215 CrossRef CAS PubMed.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- M. Garcia-Ratés and F. Neese, J. Comput. Chem., 2020, 41, 922–939 CrossRef PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- (a) D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS; (b) K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113–11123 CrossRef CAS.
- X. X. Ge, S. J. Chen, X. C. Liu, Q. H. Wang, L. J. Gao, C. F. Zhao, L. Zhang, M. X. Shao, X.-A. Yuan, L. J. Tian and Z. Liu, Inorg. Chem., 2019, 58, 14175–14184 CrossRef CAS PubMed.
- Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 4764–4776 RSC.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- N. Graf and S. J. Lippard, Adv. Drug Delivery Rev., 2012, 64, 993–1004 CrossRef CAS PubMed.
- X. Liu, M. Shao, C. Liang, J. Guo, G. Wang, X. A. Yuan, Z. Jing, L. Tian and Z. Liu, ChemBioChem, 2021, 22, 557–564 CrossRef CAS PubMed.
- W.-Y. Zhang, S. Banerjee, G. M. Hughes, H. E. Bridgewater, J.-I. Song, B. G. Breeze, G. J. Clarkson, J. P. C. Coverdale, C. Sanchez-Cano, F. Ponte, E. Sicilia and P. J. Sadler, Chem. Sci., 2020, 11, 5466–5480 RSC.
- L. Costanzo, Physiology, Elsevier, Inc, 6th edn, 2018, ISBN: 978-0-323-47881-6 Search PubMed.
- V. Sačková, P. Fedoročko, B. Szilárdiová, J. Mikeš and J. Kleban, Phytochem. Phytobiol., 2006, 82, 1285–1291 CrossRef PubMed.
- L. Mikešová, J. Mikeš, J. Kovaľ, K. Gyuraszová, L.' Čulka, J. Vargová, B. Valeková and P. Fedoročko, Photodiagn. Photodyn. Ther., 2013, 10, 470–483 CrossRef PubMed.
- M. Babinčák, R. Jendželovský, J. Košuth, M. Majerník, J. Vargová, K. Mikulášek, Z. Zdráhal and P. Fedoročko, Cancers, 2021, 13, 1646 CrossRef PubMed.
- M. Ghasemi, T. Turnbull, S. Sebastian and I. Kempson, Int. J. Mol. Sci., 2021, 22, 12827 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Bourhis, O. V. Dolomanov, R. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59–75 CrossRef CAS PubMed.
- O. Dolomanov, L. J. Bourhis, R. Gildea, J. A. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- J. Koziskova, F. Hahn, J. Richter and J. Kožíšek, Acta Chim. Slovaca, 2016, 9, 136–140 CrossRef CAS.
- CrysAlisPro 1.171.40.82a, Rigaku Oxford Diffraction, 2020 Search PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- P. Štarha, Z. Trávníček, H. Crlíková, J. Vančo, J. Kašpárková and Z. Dvořák, Organometallics, 2018, 37, 2749–2759 CrossRef.
- J. Kleban, J. Mikeš, B. Szilardiová, J. Kovaľ, V. Sačková, P. Solár, V. Horváth, J. Hofmanová, A. Kozubík and P. Fedoročko, Photochem. Photobiol., 2007, 83, 1174–1185 CrossRef CAS PubMed.
- J. Mikeš, J. Kovaľ, R. Jendželovský, V. Sačková, I. Uhrinová, M. Kello, L. Kuliková and P. Fedoročko, Photochem. Photobiol. Sci., 2009, 8, 1558–1567 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, ESI+ mass spectra, 1H NMR stability and interaction studies, crystallographic data, crystal structures and non-covalent interactions, and cellular experimental (metabolic activity, viability, MMP) results. CCDC 2266585–2266588. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01696j |
| This journal is © The Royal Society of Chemistry 2023 |