
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom Cu(I) and Cu(I)–Cu(II) mixed-valence clusters to 2D Cu(II) and Cu(I) coordination polymers supported by a flexible bis-tetrazole organosulfur ligand†
Olaya
Gómez-Paz
ab,
Rosa
Carballo
 *ab,
Ana B.
Lago
*c,
Inmaculada
Prieto
bd and
Ezequiel M.
Vázquez-López
ab
*ab,
Ana B.
Lago
*c,
Inmaculada
Prieto
bd and
Ezequiel M.
Vázquez-López
ab
aUniversidade de Vigo, Departamento de Química Inorgánica, Facultade de Química, 36310 Vigo, Spain
bMetallosupramolecular Chemistry Group, Galicia Sur Health Research Institute (IIS Galicia Sur), SERGAS-UVIGO, Galicia, Spain
cLaboratorio de Materiales para Análisis Químico (MAT4LL), Departamento de Química, Unidad Departamental de Química Inorgánica, Universidad de La Laguna (ULL), Tenerife, La Laguna, 38206, Spain. E-mail: alagobla@ull.edu.es
dUniversidade de Vigo, Departamento de Química Física, Facultade de Química, 36310 Vigo, Spain
First published on 18th July 2023
Abstract
Three coordination compounds from the reaction of copper(II) bromide with the flexible bis-tetrazole organosulfur ligand, 1,2-bis(1-methyl-1H-tetrazole-5-ylthio)ethane (bmtte) have been isolated and characterised. The identification of polymeric 2∞Cu2Br4(bmtte) (1), trinuclear [Cu3Br4(bmtte)2] (2) and tetranuclear [Cu2Br2(bmtte)]2 (3) compounds shows that the reaction conditions have a significant influence on the structure of the complexes formed. Moreover, two polymorphs of the 2D Cu(II)-coordination polymer 1 have been isolated and these crystallise in the monoclinic C2/m (1m) and the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (1t) space groups. The thermal stabilities and behaviour in aqueous media of compounds 1–3 were investigated along with the reactivity of compound 2 with CuBr2 and KI. The solid-state reaction between mixed-valence compound 2 with KI or the direct reaction of CuI and bmtte under microwave irradiation allowed the preparation of the polymeric 2∞Cu4I4(bmtte)2 (4). The redox behaviour of complexes 2 and 3 was analysed by cyclic voltammetry.
(1t) space groups. The thermal stabilities and behaviour in aqueous media of compounds 1–3 were investigated along with the reactivity of compound 2 with CuBr2 and KI. The solid-state reaction between mixed-valence compound 2 with KI or the direct reaction of CuI and bmtte under microwave irradiation allowed the preparation of the polymeric 2∞Cu4I4(bmtte)2 (4). The redox behaviour of complexes 2 and 3 was analysed by cyclic voltammetry.
Introduction
The synthesis and formation of bioinspired small molecular models that contain copper have been the subject of vigorous research in an effort to better understand the chemical behaviour of biological systems that involve copper. One of the major targets of these studies is the mixed-valence intermediates involved in enzymatic processes.1 For instance, the multicopper oxidases (MCOs) catalytic site is a trinuclear copper cluster that oxidises organic substrates and metal ions.2 The active site of this protein contains two closely spaced copper cations (dCu–Cu = 3.6 Å), which reversibly bind an oxygen molecule. The copper cations are fixed in the protein structure by six nitrogen atoms of the imidazole rings of histidine residues from two different chains.2 Based on these facts, it is reasonable to envisage that potential polydentate coordination modes and conformational flexibility of a ligand are important factors to favour the formation of these copper clusters. In this sense, we have recently focused on using bis-tetrazole organosulfur ligands to introduce more donor atoms, which could be useful to manipulate the structural conformation of the resulting copper complexes.3The self-assembly of a metal and a ligand can produce numerous compounds that have different structures and properties. In particular, the use of a flexible ligand can stabilise different ligand conformations to yield polymorphic supramolecular isomers, which occur as different crystal forms of the same chemical species but with different crystal packing, space groups and unit cell parameters.4 Therefore, despite polymorphism being a well-known phenomenon, very few cases have been described in the field of coordination polymers.5
Our group has a great deal of experience in investigating the utility of flexible dithioether ligands to form metallosupramolecular compounds with copper(II) halide salts under different synthetic conditions and we have isolated compounds of different dimensionalities.3,6–10 Among this kind of ligand, 1,2-bis(1-methyl-1H-tetrazole-5-ylthio)ethane (bmtte) is a multifunctional twisted organic ligand composed of two methyltetrazole groups separated by a flexible organosulfur bridge (–S–CH2–CH2–S–) that can rotate and adjust the direction of the coordinating nitrogen atoms. In the field of metallosupramolecular chemistry, this ligand is of interest because the tetrazole organosulfur derivatives contain multiple atoms that can facilitate simultaneously the coordination to one or more metal centres and the interaction with their surroundings through diverse weak interactions.
In the literature there is some structural information on several compounds based on copper(II/I) polyoxometallates (POMs = H2Mo8O262−, PMo12O403−, SiW12O403−, PW12O403−, SiMo12O404−, SiW12O404−, HSiMo12O403−) and the bmtte ligand.11–15 These compounds are 1D, 2D or 3D coordination polymers in which the bmtte ligand is able to coordinate two, three or four metal cations by chelating and/or bridging coordination modes.
In the work reported here, the coordinative behaviour of 1,2-bis(1-methyl-1H-tetrazole-5-ylthio)ethane (bmtte) towards copper(II) bromide was explored. The resulting trinuclear [Cu3Br4(bmtte)2] (2), tetranuclear [Cu2Br2(bmtte)]2 (3) and polymeric 2∞Cu2Br4(bmtte) (1) were obtained by different synthetic methods. In addition, in this structural study the formation of two polymorphic forms of the 2D polymer 1 was observed. The reactivity of the mixed-valence compound 2 was investigated and, as result, the 2D Cu(I) coordination polymer 2∞Cu4I4(bmtte) (4) was obtained. The redox behaviour of complexes 2 and 3, which contain Cu(I), was studied by cyclic voltammetry.
Results and discussion
Synthesis and spectroscopic characterisation
The reactivity of the CuBr2/bmtte system (Scheme 1) was studied using different stoichiometric ratios, synthetic methods, times, temperatures and solvents to obtain a broad overview of the system. In this way, four single crystals were isolated and structurally characterised: two 2D polymorphs (1m and 1t) of 2∞Cu2Br4(bmtte) and two 0D polynuclear complexes, namely the trinuclear [Cu3Br4(bmtte)2] (2) and the tetranuclear [Cu2Br2(bmtte)]2 (3). Efforts were made to achieve control over the preparation of compounds 1–3 (Scheme 1) by careful investigation of the synthetic processes and variables. However, our efforts to obtain the different compounds in separate batches were only successful for the 0D compounds and not for the polymorphs 1m and 1t, which crystallised concomitantly. | ||
| Scheme 1 Synthetic procedures used in the study of the CuBr2/bmtte system. For each reaction, only the major crystalline phases are listed. | ||
The polymorphs were obtained in good yields when an acetonitrile solution with an excess of copper salt (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 metal/ligand ratio) was subjected to microwave irradiation for 20 minutes. However, when the microwave reaction (30 minutes) was carried out with a 1:1 metal/ligand ratio, a mixture of 1m and trinuclear 2 was obtained. The Cu(I)/Cu(II) mixed-valence cluster 2 could be obtained as a pure phase when the reaction was performed with the same molar ratio and solvent but with stirring at room temperature for 13 days. Interestingly, shorter reaction times (8 days) with stirring yielded a mixture of 1m and 1t. The effect of the solvent was also studied and, on using the moderately polar aprotic solvent tetrahydrofuran, the Cu(II)/Cu(I) reduction was observed and only discrete compounds 2 and 3 were isolated. This reduction process occurred without apparent decomposition of the bmtte ligand. In contrast, pure Cu(I)-tetranuclear 3 was isolated when the reaction temperature and time were increased significantly in a hydrothermal synthesis performed with an excess of metal salt precursor. This hydrothermal reduction was previously observed in the preparation of the analogous chloride compound [Cu2Cl2(bmtte)]2,3 using a 4:1 metal/ligand ratio, and in the preparation of copper polyoxometallates of bmtte compounds11–15 – in this case the use of metal/ligand ratios greater than 10:1 was required. Moreover, our previous experience in the study of the reactivity of copper(II) halides with N-donor ligands containing –S–(CH2)n–S– linkers, such as bis(4-pyridylthio)methane,6 bis(2-pyridylthio)methane8 or bis(1-methyl-1H-tetrazole-5-ylthio)methane,3 showed that copper(II) reduction always occurs under hydrothermal or solvothermal conditions at high temperature on using water or other protic solvents. In many cases this process involves an S–C bond cleavage in the ligand yielding a thiolate that transform in the corresponding disulfide compound. However, compounds 2 and 3 are the first cases in which the Cu(II) to Cu(I) reduction has been observed under mild synthetic conditions, using equimolar reactant ratios, without a protic solvent in the medium and with no apparent decomposition of the ligand.
:1 metal/ligand ratio) was subjected to microwave irradiation for 20 minutes. However, when the microwave reaction (30 minutes) was carried out with a 1:1 metal/ligand ratio, a mixture of 1m and trinuclear 2 was obtained. The Cu(I)/Cu(II) mixed-valence cluster 2 could be obtained as a pure phase when the reaction was performed with the same molar ratio and solvent but with stirring at room temperature for 13 days. Interestingly, shorter reaction times (8 days) with stirring yielded a mixture of 1m and 1t. The effect of the solvent was also studied and, on using the moderately polar aprotic solvent tetrahydrofuran, the Cu(II)/Cu(I) reduction was observed and only discrete compounds 2 and 3 were isolated. This reduction process occurred without apparent decomposition of the bmtte ligand. In contrast, pure Cu(I)-tetranuclear 3 was isolated when the reaction temperature and time were increased significantly in a hydrothermal synthesis performed with an excess of metal salt precursor. This hydrothermal reduction was previously observed in the preparation of the analogous chloride compound [Cu2Cl2(bmtte)]2,3 using a 4:1 metal/ligand ratio, and in the preparation of copper polyoxometallates of bmtte compounds11–15 – in this case the use of metal/ligand ratios greater than 10:1 was required. Moreover, our previous experience in the study of the reactivity of copper(II) halides with N-donor ligands containing –S–(CH2)n–S– linkers, such as bis(4-pyridylthio)methane,6 bis(2-pyridylthio)methane8 or bis(1-methyl-1H-tetrazole-5-ylthio)methane,3 showed that copper(II) reduction always occurs under hydrothermal or solvothermal conditions at high temperature on using water or other protic solvents. In many cases this process involves an S–C bond cleavage in the ligand yielding a thiolate that transform in the corresponding disulfide compound. However, compounds 2 and 3 are the first cases in which the Cu(II) to Cu(I) reduction has been observed under mild synthetic conditions, using equimolar reactant ratios, without a protic solvent in the medium and with no apparent decomposition of the ligand.
Finally, it should be noted that among the synthetic methods tested, the diffusion method was also tried. However, this method was unsuccessful as the rapid crystallisation of the ligand precluded the formation of the copper complexes.
All of the compounds were studied by spectroscopic methods. Infrared analysis (Fig. S1†) showed the main bands of the free ligand without significant displacement, with bands due to the tetrazole ring vibration modes between 1480 and 1380 cm−1 and bands due to ν(CS) located at around 700 cm−1.
Solid UV-Vis spectra (diffuse reflectance) of all compounds showed absorption bands in the UV region between 290 and 340 nm due to intraligand π–π* transitions (Fig. S2†). For compounds 1 and 2 a band close to 400 nm is attributed to charge transfer (CT) transitions. A single and broad intense band was observed in the UV region in the spectrum of 3 but bands due to d–d transitions did not appear in the visible region, which is consistent with the presence of Cu(I) metal. In the visible region of the spectra of 1 and 2 the presence of broad and asymmetric bands at 565, 848 nm (1) and 522, 719 nm (2) is consistent with the d–d transitions expected for octahedral or square-based pyramidal copper(II) complexes.
Crystal structures of compounds 1m, 1t, 2 and 3
All of the compounds reported here were isolated as single crystals and their structures were elucidated by X-ray diffraction (Fig. S3–S6†). The significant structural parameters for compounds 1m and, 1t–3 are listed in Tables S2 and S3,† and crystal structure and refinement data are listed in Table S1.† The combination of the flexible polydentate bmtte ligand with different bromocuprate clusters resulted in the formation of the two polymorphs of a 2D coordination polymer (1m and 1t) and the Cu(II)/Cu(I)-trinuclear (2) or Cu(I)-tetranuclear (3) coordination compounds. A schematic view of each compound is shown in Fig. 1.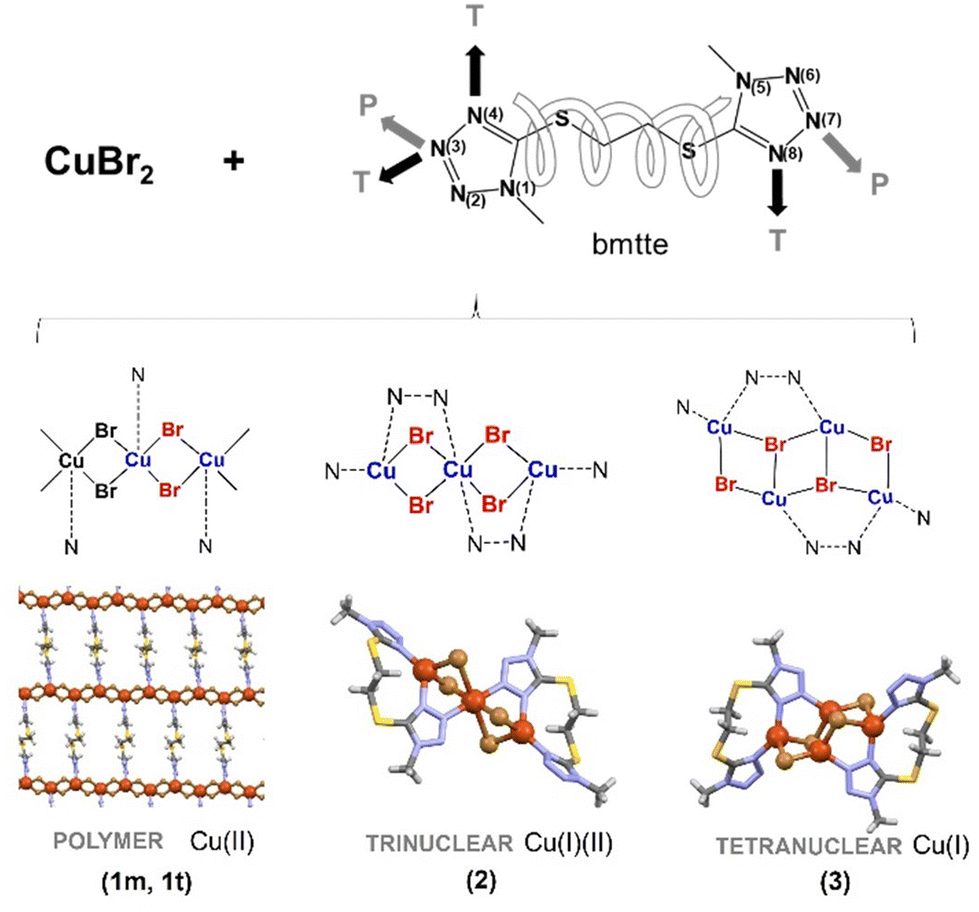 | ||
| Fig. 1 Top: The structural formula of the bmtte ligand depicting flexible fragment and binding sites in polymeric compounds (P; 1m and 1t), and trinuclear and tetranuclear compounds (T; 2, 3). Bottom: Summary of the inorganic building clusters in 1, 2 and 3 and final crystal structures. | ||
The structures can be deconstructed into two components, namely the metal cluster, which can be constituted by a dinuclear {Cu2Br4N2} unit in 1t and 1m, a trinuclear {Cu3Br4N6} in 2 and a tetranuclear unit {Cu4Br4N6} in 3, and the potentially N-hexacoordinating ligand (bmtte). In the ligand, two methyl tetrazole groups are separated by a flexible organosulfur spacer, which acts as a spring that allows adjustments in the direction of the coordination of nitrogen atoms. The nature of the ligand produces different coordination behaviour in polymeric (P in the scheme in Fig. 1) or in discrete compounds (T in the scheme in Fig. 1).
The copper bromide compounds reported here differ from the previously reported chloride analogues.3 A stable 2D coordination polymer with {Cu2Cl4N2} dinuclear units was isolated with CuCl2 and these have a different coordination mode of bmtte. Only under harsh hydrothermal conditions were tetranuclear units {Cu4X4N6} (X = Cl or Br) obtained with both halide precursors. The trinuclear mixed-valence unit {Cu3Br4N6} could only be obtained when the bromide precursor was used.
Crystal structure of the polymers 1t and 1m
The polymorphs 1t and 1m are 2D coordination polymers based on a dimeric Cu2Br4 motif bridged by the bmtte ligand. The different 2D supramolecular isomers of general formula 2∞Cu2X4(bmtte) (X = Cl3 or Br) crystallised in the space groups P (1t), C2/m (1m) and C2/c.3 It is worth noting that the polymorphism observed in these Cu–bromide coordination polymers could not be observed in the analogous 2D polymeric Cu–chloride-bmtte system.3 At first glance, the compounds 2∞Cu2Cl4(bmtte)3 and 2∞Cu2Br4(bmtte) appear to be similar but there is a fundamental difference between them since the tetrazole nitrogen atoms involved in the coordination to the cation are different in each case, with a μ-κN2 coordination mode of bmtte in 2∞Cu2Cl4(bmtte) and a μ-κN3 coordination mode in the two polymorphs of 1.
In 1t and 1m the dimeric Cu2Br4 motif with two copper(II) cations bridged by bromide atoms leads to the formation of Cu2Br4-based inorganic chains. These chains are bridged by a bis-monodentate (μ-κN3 coordination mode, Fig. S7†) bmtte ligand to yield the final 2D coordination compound. The S–C–C–S torsion angle is 169.93° in 1m and 180.0° in 1t. This observation illustrates the flexibility of the ligand, which allows the formation of the different polymorphs. In both cases, each layer incorporates molecules of the ligand with opposite axial chirality so the layers and the final structures are achiral.
The pentacoordinate Cu(II) cations in both polymorphs are in a regular square pyramidal geometry (Fig. S7†) (Addison parameter τ516 0.00 for 1m and 0.02 for 1t) with four bromide atoms in basal positions [Cu–Br distances between 2.4283(3) and 2.4500(4) Å] and a bmtte nitrogen atom at the apex [Cu–N3 distances between 2.2230(3) and 2.2441(18) Å]. In the analogous Cu–chloride compound, in the sixth coordination position and at a longer distance, there is a nitrogen atom of a bmtte molecule, but in the Cu–bromide compounds the closest (at a distance ∼3.5 Å) atom to this position is a bmtte sulfur atom. The distances between two neighbouring copper centres through the bromide atoms are 3.560 Å in 1m and 3.572 and 3.560 Å in 1t, and 13.145 Å and 12.562 and 12.425 Å through the bmtte ligand in 1m and 1t, respectively. These latter distances are clearly shorter than those observed in the Cu–chloride analogue (14.584 Å). In the layers of 1m and 1t each Cu2Br2 node is coordinated to two bmtte ligands to yield rectangular metallomacrocycles defined by six Cu(II) cations, eight bromide ligands and two bmtte ligands. In both cases, the framework can be described as a 3-c uninodal regular honeycomb net of hcb topology17 and this contains just one type of vertex, which is connected with three neighbours.
The 2D layers of 1m and 1t have a zig-zag disposition and each layer stacks with adjacent ones in a different way, i.e., parallel in 1m and slightly displaced in 1t, as shown in Fig. 2, to give the final 3D arrangement. The supramolecular arrangement is achieved by means of CHmethylene⋯N interactions (Table S5†) involving the hydrogen atoms of methylene groups and free nitrogen atoms of tetrazole fragments of neighbouring layers to form six-membered supramolecular rings with the tetrazole fragments (Fig. 2). This synthon is also reinforced by the interaction established by the bromide ions that point towards the methylene groups. Although the pattern and interactions have the same nature, stronger interactions with shorter distances are observed in 1t when compared to 1m (Table S3†). The effect of this packing on the network is also evident in the Kitaigorodskii packing indexes18 of 75% and 71% for 1t and 1m, respectively.
 | ||
| Fig. 2 (A) 2D layer disposition in 1t together with the interactions responsible for the supramolecular arrangement. (B) hbc topology in 1m and 1t together with the disposition of stacked layers in 1t. (C) Displaced and parallel final 3D arrangements in 1t and 1m, respectively. | ||
Crystal structure of the trinuclear 2
Compound 2 crystallises in the P21/n space group as a trinuclear mixed-valence copper complex with an inversion centre located at the Cu2 central atom (Fig. 3). Each trinuclear specie contains two monovalent copper ions, Cu1, and a divalent one, Cu2, located in a four- and a six-coordination sphere, respectively. Cu1 is in a distorted tetrahedral (τ4 = 0.76,19 ideal value for a tetrahedral geometry is 1) coordination environment, with two short metal–ligand bonds (Cu–N distances around 2 Å) and two long bonds with bromide atoms [Cu1–Br2 at 2.4077(5) Å and Cu1–Br1 at 2.6349(4) Å]. The divalent copper ion is in an axially elongated [4 + 2] coordination environment with two tetrazole nitrogen atoms and two bromide ions (Br1) in a trans disposition and two axial bromide ions (Br2) at a markedly longer distance [3.0865(3) Å] than the equatorial bonds. These distances are slightly shorter than the sum of the van der Waals radii of copper and bromide.20 This distorted octahedron shares edges with the two tetrahedra centred on Cu(I) atoms. The Cu⋯Cu distance through the bromide bridge (3.214 Å) is longer than the values observed previously21 and this suggests that copper–copper interactions are not present, although this distance is shorter than that in the trinuclear copper cluster of multicopper oxidases (MCOs).2 | ||
| Fig. 3 Top: Molecular structure showing the hydrophobic contour (A) and polyhedral environments in the Cu(I)/(II) trinuclear 2 (B). Bottom: Supramolecular arrangement in the crystal structure of 2 (C) showing in detail the interactions responsible for the organisation (D). | ||
The bmtte ligand acts as a bis-monodentate bridge linking Cu1 and Cu2 through the tetrazole fragment to form five-membered rings (Cu2N2Br) and as a bidentate chelating unit with Cu1 to give a nine-membered ring (CuN2S2C4). This coordinative behaviour of bmtte can be described as μ-1κ2N4,N8:2κN3 (Fig. S7†). The S–C–C–S torsion angle is 174.98° and the two ligands in the trinuclear have opposite axial chirality. The copper-tetrazole centroid-copper angle is 60.97° and the bite angle N–Cu–N is 130.34(10)°. Bromide ions, with Cu–Br–Cu angles of 70.36° and 78.75° with Br2 and Br1, respectively, bridge the two copper centres in an asymmetrical manner, with Br1 closer to Cu2 and Br2 closer to Cu1.
The nonpolar methylene spacer groups and the terminal methyl groups are oriented outwards from the molecule to define a hydrophobic contour (Fig. 3). These groups play an important role as H-donors towards the nitrogen and bromide atoms and establish several weak intermolecular C–H⋯X (X = Br, N) interactions, with C⋯X distances in the range 3.50–3.85 Å (Table S5† and Fig. 3). The resulting hydrogen-bonding synthon leads to the formation of different six-membered and eight-membered rings (Fig. 3) and each molecule interacts with six adjacent ones. In addition, an N(N6)⋯π(tetrazole C11–N4) interaction (N⋯π distance of 3.125 Å) along the b axis participates in the packing. The stabilising effect on the network of these interactions is evident in the Kitaigorodskii packing index18 of 68%.
Crystal structure of the tetranuclear 3
The Cu(I) compound 3 is isostructural with the previously reported [Cu2Cl2(bmtte)]23 and it crystallises in the P21/n space group with an inversion centre located between the Cu2 central atoms. Four Cu(I) atoms and four bromide ions form a stair-like [Cu4Br4] cluster core with each of the two bmtte ligands anchored at each end of the stair through two Cu(I) centres (Fig. 4).22,23 In the cluster, the Cu2⋯Cu2 distance of 2.638 Å indicates attractive Cu⋯Cu interactions, but the Cu1⋯Cu2 distance of 2.855 Å suggests the absence of copper–copper interactions.24 In both cases, the distances are around 0.1 Å shorter than those observed in the analogous chloride compound.
 | ||
| Fig. 4 The molecular structure of 3 (A) showing in detail the C–H⋯N interactions in the bc plane (B) and the final arrangement in the ac plane (C). | ||
The bromide ligands display μ3-Br1 and μ-Br2 bridging modes. The triply bonded bromide ligand connects Cu1 and Cu2 atoms with similar distances [2.5952(5), 2.5400(3) and 2.5264(6) Å] and Br2 is bonded to two adjacent copper atoms with Cu–Br distances of 2.5845(5) Å and 2.3881(5) Å. Each Cu(I) centre is tetrahedrally coordinated: Cu1 by two bmtte nitrogen atoms and Br1 and Br2 (τ4 = 0.7819), and Cu2 (τ4 = 0.9519) by one bmtte nitrogen atom, two μ3-Br1 and one μ-Br2, with Cu–Br distances in the range 2.3881(5)–2.5952(5) Å. The bmtte ligand uses three nitrogen atoms of the two tetrazole rings to coordinate metal centres in the same way as in 2 (μ-1κ2N4,N8:2κN3 coordination mode, Fig. S7†). A copper-tetrazole centroid-copper angle of 65.93° and an N–Cu–N bite angle of 135.96(11)° are comparable to those found in analogous chloride compounds and longer than the values observed in 2, thus suggesting a less rigid disposition in 3. Analogously, the S–C–C–S torsion angle is 169.91°and the two ligands in the tetranuclear have opposite axial chirality.
Tetrazole groups establish N⋯π interactions with neighbouring tetranuclear species in a stair-step disposition (Fig. 4), with an N6 to centroid distance of 3.125 Å. The tetrazole N1/N2/N3/N4/C11 and N5/N6/N7/N8/C21 rings, with a centroid–centroid distance of 3.843 Å, show π⋯π interactions that reinforce this disposition. The methylene spacer groups also establish C–H⋯N7 interactions with a C⋯N distance of 3.465 Å and the methyl group on the tetrazole ring participates in C–H⋯N interactions with the N2 nitrogen atom. The 3D supramolecular array is also reinforced by the contribution of different C–Hmethylene⋯Br and C–H⋯S interactions (Table S4†). As a result of these different interactions, the Kitaigorodskii packing index18 is 69%.
Supramolecular organisation for the discrete compounds 2 and 3
A Hirshfeld surface study was carried out to gain a fuller appreciation of the nature and quantitative contributions of intermolecular interactions to the supramolecular assembly of the discrete complexes 2 and 3. The Hirshfeld surfaces mapped over dnorm are shown as transparent in Fig. 5 to allow visualisation of the molecular moiety; the circular depressions (deep red) visible on the dnorm surfaces are indicative of hydrogen-bonding contacts and the tiny spots and very light-coloured regions on the surfaces designate weaker and longer contacts other than the hydrogen bonds.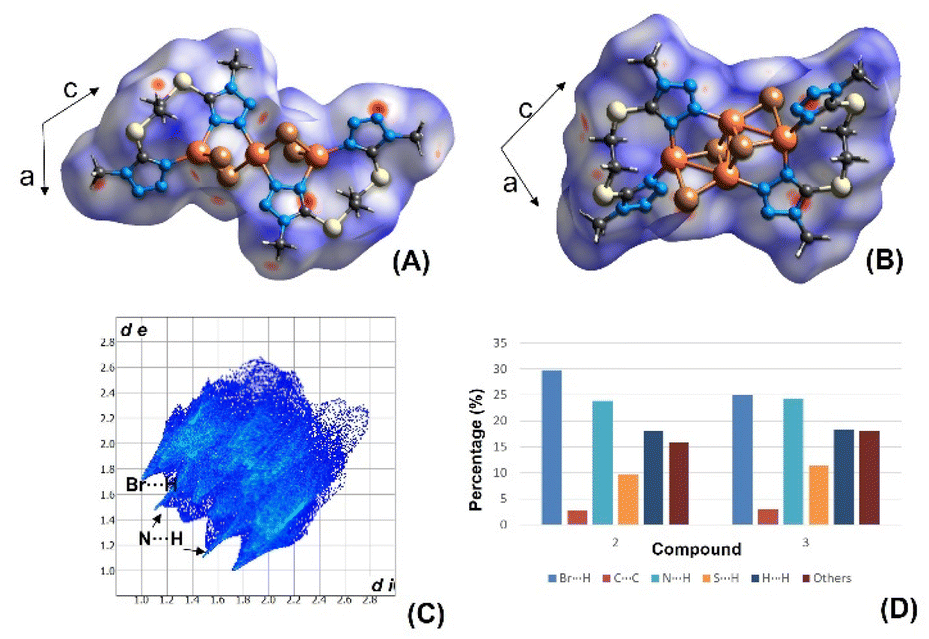 | ||
| Fig. 5 Top: Hirshfeld surfaces mapped with dnorm for 2 (A) and 3 (B) and (C) fingerprint plot for compound 2. (D) Contribution to the Hirshfeld surface area from the various close intermolecular interactions in 2 and 3. | ||
The contributions to the Hirshfeld surface area from the various close intermolecular interactions are represented in the histogram (Fig. 5). In general, the same kinds of interactions, with differences in intensities, are observed in both structures. The analysis shows the same priority (i.e., the highest contribution to the Hirshfeld surface) in both structures, with Br⋯H and N⋯H contacts being the dominant interactions. However, the relative area associated with Br⋯H contacts is significantly higher in 2 (29.8%) than in 3 (24.9%). Furthermore, and as one would expect, another common feature in the structures is the relatively low area associated with C⋯C interactions.
Fingerprint plots also show that the contacts in 2 and 3 are strikingly similar (Fig. S8 and S9, ESI†). A pair of sharp and symmetric spikes are observed in 2 and 3 and these represent N–H and Br–H contacts, which are associated mainly with C–H⋯N and C–H⋯Br hydrogen bonds. The decomposition of contributions from different interaction types, which overlap in the full fingerprint, is helpful in highlighting graphically the surface regions that are involved in the main intermolecular interactions (Fig. S8 and S9†). The H/H contacts, which correspond to the van de Waals interactions, represent less than 20% in the Hirshfeld surfaces, thus showing that H-bonds rather than dispersion forces mostly stabilise these lattices. The coordination environment of the copper metal centres gives rise to some weak interactions in 3 but these are negligible in 2 due to the more inaccessible copper coordination environments.
Thermal and aqueous stabilities
The thermal stability of compounds 1m and 1t was investigated by X-ray thermodiffraction (Fig. S11–S15†) and for all compounds by thermogravimetric analysis (Fig. S10†).The thermodiffractograms of 1m and 1t confirmed that their structures are stable up to around 175 °C. Derivate thermogravimetric (DTG) analysis studies were performed for the polymorphs 1m and 1t to compare their stability. Similar behaviour was observed in the DTG curves for the two crystalline solids but it was found that thermal decomposition begins at a slightly lower temperature in phase 1m than in 1t. The results of these studies show that the thermogravimetric behaviour and thermal stability are quite similar for both polymers.
The TGA study of complexes 2 and 3 showed good thermal stability at temperatures greater than 200 °C. The experimental mass loss for each sample is in good agreement with the organic decomposition of the compounds.
We previously observed that the presence of water as a solvent could be responsible for the occurrence of chemical reactions. For example, the in situ reduction of Cu(II) observed with the simultaneous formation of the 2D copper(I) polymorphic supramolecular isomers with the bis(4-pyridylthio)methane ligand and copper(II) chloride salt6 or in the hydrothermal synthesis of 3 and its chloride analogues.3 Given this possibility, the aqueous stabilities of the different compounds were analysed.
All of the compounds showed good stability in a humid environment, but different behaviour was observed when the compounds were dissolved in an aqueous medium (Scheme S1 and Fig. S16†). Compounds 1m, 1t and 2 showed rapid evolution in contact with water. However, the X-ray powder diffractogram and the infrared spectrum (Fig. S18 and S19†) of the solid sample recovered after treatment of compound 3 with water showed that the structure remained unaltered, but the isolation of some single crystals of bmtte ligand after the evaporation of the filtered aqueous solution indicates some degree of instability.
The dark green solids 1m, 1t and 2 were partially dissolved and the remaining sample became colourless in the presence of water. Infrared analysis of the resulting colourless solids showed the presence of the characteristic bands of the bmtte ligand. Unfortunately, the small amount of sample available did not allow XRD studies to be performed. The resulting solutions were evaporated until dark green crystalline samples were obtained and PXRD analysis showed that the resulting compounds had the same structural organisation, i.e., a mixture of polymers 1m–1t together with CuBr2 impurities (Fig. S20–S22†). It can be concluded that the compounds containing copper(II) ions react faster in contact with water than the more insoluble Cu(I) compound 3. Moreover, it can be considered that the mixture of phases 1m–1t is stable upon treatment of 1m–1t and 2 with water.
Reactivity of 2
Two different reactivity tests, namely with CuBr2 and KI, were performed in order to study the reactivity of the mixed-valence compound 2, which could evolve into species that contain only Cu(II) or Cu(I).It was previously observed that in reactions with metal:ligand ratios greater than 10:1, the reduction of copper(II) to copper(I) occurs in the presence of the bmtte ligand.11–14 As a result, it was decided to investigate the behaviour of 2 in the presence of an excess of copper(II) bromide in EtOH (method A, Scheme 2). The diffractogram of the resulting solid material closely matched the simulated patterns generated from the single crystal diffraction data of the copper(II) polymeric compounds, mainly the 1m form (Fig. S23†). This finding suggests that an excess of the metal precursor favours the formation of the Cu(II) polymeric compounds. The excess CuBr2 was also detected in the diffractogram and by elemental analysis. This result is similar to that found when 2 was treated with water – again suggesting the good stability of the polymeric forms.
 | ||
| Scheme 2 Reactivity of 2 with an excess of CuBr2 and KI. | ||
In a second test, the solid-state reaction between [Cu3Br4(bmtte)2] (2) and KI was evaluated (method B, Scheme 2). The reaction yielded a crystalline solid that gave a diffractogram that differed from all of the diffraction patterns discussed above. It was decided to carry out the direct reaction between the ligand bmtte and CuI to investigate if this solid could contain a complex that included the iodide anion. After a reaction time of a few hours under microwave radiation, single crystals were isolated from the mother liquor. Single crystal X-ray diffraction enabled the compound to be identified as 2∞Cu4I4(bmtte) (4), the structure of which is described below. The simulated diffractogram of 4 is consistent with that obtained for the solid formed in the solid-state reaction between 2 (Fig. S24†) and KI. This finding indicates that a full structural reorganisation in the solid state had occurred.
The structure of 2∞Cu4I4(bmtte) (4) is shown in Fig. 6. The main structural parameters and the crystal structure and refinement data are listed in Tables S3 and S1,† respectively.
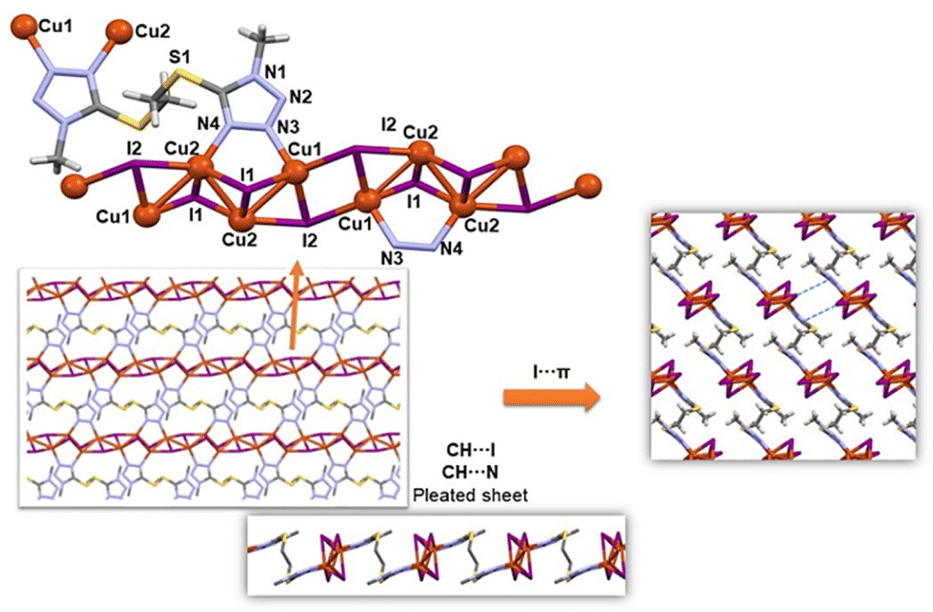 | ||
| Fig. 6 2D layer disposition in 4 together with the interactions responsible for the supramolecular arrangement. | ||
The copper(I) compound 4 is a 2D coordination polymer and, as in the Cu(II) polymorphs 1m and 1t described above, there is a chain formed by the cation and the halide. In this case the chains form rectangles that share sides, so that each copper is bound to three iodides, and each iodide is coordinated to three Cu(I) centres. The Cu–I distances are in the range 2.60–2.70 Å and the Cu1–Cu2 and Cu2–Cu2 distances are 2.7305(5) and 2.6291(7) Å, respectively. In this way, each Cu(I) cation is coordinated to three iodide ligands and one nitrogen of bmtte in a tetrahedral environment, with τ4 parameters of 0.86 for Cu1 and 0.82 for Cu2.16
The bmtte ligand shows different coordinative behaviour to that in compounds 1–3 and it acts as a bis-bidentate bridge through the N3 and N4 atoms of each heterocycle in a μ-1κN3:2κN4:3κN3′:4κN4′ coordination mode (Fig. S7†). Thus, each ligand molecule binds to four Cu(I) cations and connects the {CuI} chains to give a 2D coordination polymer. Intralayer CH⋯N and CH⋯I hydrogen bonds (Table S1†) lead to the formation of a pleated sheet (Fig. 6) that incorporates ligand molecules of opposite chirality with an S–C–C–S torsion angle of 180.0°. As in the 2D Cu(II) coordination polymers 1m and 1t, rectangular metallomacrocycles defined by six Cu(I) cations, eight iodide ligands and two bmtte ligands are formed.
The layers are packed into the 3D metallosupramolecular array by an I⋯π interaction (I1⋯centroid of C1/N2/N1/N4/N3 tetrazole ring, Fig. 6) with an I⋯centroid distance of 3.881 Å. The shortest I⋯N3 distance is 3.738 Å – a value only slightly higher than the sum of the van der Waals radii (3.7 Å). The Kitaigorodskii packing index18 is 70%.
Redox behaviour of compounds 2 and 3
The redox behaviour of complexes 2 and 3 was studied in DMSO solution by cyclic voltammetry at a glassy carbon working electrode (GCE). Representative voltammograms are shown in Fig. 7 and the measured redox potentials are summarized in Table 1. The voltammograms for the two compounds are similar and clear redox processes can be observed: two oxidations in the anodic direction and several reductions on the cathodic side. | ||
| Fig. 7 Cyclic voltammograms of 1 mM solutions of complexes 2 and 3 in DMSO, with 0.1 M tetrabutylammonium perchlorate (TBAP) as supporting electrolyte. Scan rate: 0.2 V s−1. | ||
| Compound | E pa (V) | E pc (V) |
|---|---|---|
| E pa = potential of the anodic peak; Epc = potential of the cathodic peak. In parentheses, Epc obtained when the potential is reversed after the first oxidation process. | ||
| Cu3Br4(bmtte)2 (2) | −0.07 | 0.20, −0.32/−0.67/−0.78 (−0.31) |
| 0.36 | −1.15, −1.61, −2.16, −2.90 | |
| [Cu2Br2(bmtte)]2 (3) | −0.05, 0.02 | 0.25, −0.3/−0.59/−0.78 (−0.19/−0.39) |
| 0.39 | −1.21, −1.59, −2.18, −2.91 | |
The voltammogram of [Cu3Br4(bmtte)2] (2) displays two consecutive oxidation waves, at −0.07 and 0.36 vs. Fc/Fc+, with related cathodic waves on the reverse scan. These electrochemical processes are attributed to metal-based electron transfer reactions that involve the redox couples Cu(I)/Cu(II) and Cu(II)/Cu(III), respectively, as previously observed for other copper complexes.1 A proposed scheme for the electrochemical oxidation of the complexes is shown in Scheme 3.
 | ||
| Scheme 3 Electrochemical oxidation of complexes. | ||
Several reduction waves were observed in the cathodic scan for complex 2. It should be noted that the value of the reversal potential of the anodic scan influences the electrochemical behaviour and leads to differences in the subsequent cathodic scan. Hence, voltammograms involving only the first oxidation of complex 2 show a peak at −0.31 V (Fig. S25†). The anodic to cathodic peak separation is 0.38 V, at 0.2 V s−1, and the ratio between cathodic and anodic peak currents (ipc/ipa) is lower than unity at all scan rates, thus indicating the existence of a chemical reaction after the electron transfer of the Cu(I)/Cu(II) couple.
However, when the anodic scan was reversed upon 0.6 V, new waves were observed due to the reduction of species that had been electrochemically generated in the second oxidation process. Instead of the peak mentioned above, a broad and ill-defined wave was observed in the range from −0.05 to −1 V, as can be seen in Fig. 7. This wave was assigned to successive electron transfers that occur at very similar potentials in the copper atoms (around −0.32, −0.67 and −0.78 V), which would preclude the observation of separate peaks for each cathodic process and is attributed to the multinuclear nature of the compounds.
In addition to the above, an irreversible peak was observed at −2.90 V and this can be assigned to the ligand moiety. The absence of a peak on the subsequent reverse anodic scan, even at high scan rates, results from a coupled chemical reaction that may cause instability in the reduced species (or decomposition of the reduced species).
As in complex 2, tetranuclear compound 3 showed two oxidation processes. Nevertheless, in the first oxidation two nearly overlapped waves (Ep around −0.05 and 0.02 V) were observed, rather than a single wave, and these merged together at high scan rates. As mentioned previously, the Cu(I) atoms of complex 3 are in two different chemical environments (Cu1 is coordinated by two bmtte nitrogen atoms and Br1 and Br2 atoms, and Cu2 by one bmtte nitrogen atom, two μ3-Br1 and one μ-Br2 atoms). This fact generates some interactions between Cu metal centers, which would be oxidized at different but very close potentials, leading to superimposed anodic waves. The second wave, which was observed at 0.39 V, is assigned to the redox reaction from Cu(II) to Cu(III) (see Scheme 3). It should be noted that voltammograms of complex 3 show oxidation potential values that are slightly higher than those of complex 2 and this indicates that it is more difficult to oxidise the Cu(I) and Cu(II) atoms in the tetranuclear structure.
In the cathodic scan of complex 3, reduction waves were also observed, whose peak potentials are shown in Table 1. Likewise, the electrochemical behaviour in the cathodic sweep is clearly affected by the reversal potential values of the anodic scan of the cyclic voltammograms. In contrast to complex 2, a broad wave was observed in both cases (Fig. S26†) after either of the two oxidations that complex 3 undergoes. However, when the potential was reversed after the oxidation of Cu(I) to Cu(II), the wave became narrower and shifted towards more positive potentials when compared to that observed when the second oxidation process occurred. This wave seems to be composed of two superimposed signals and these could be due to the reduction processes of Cu(II) atoms that occur at very similar potentials. This observation could be explained by the differences between the potentials depending on the chemical environment, as previously discussed for the anodic scan.
The differences observed in the cathodic scans of both complexes suggests that the compounds that generate them are different and depends on the switching potential of the anodic scan. This can be explained because the products generated in the oxidation of the complexes undergo a chemical reaction after the electron transfer reaction.
The dependence on the scan rate of the electrochemical response was investigated. The cyclic voltammograms of the complexes are similar, with a small variation in the position and appearance of the peaks (Fig. S27 and S28†). Furthermore, the anodic and cathodic peak currents show a linear dependence on the square root of the scan rate, thus indicating the diffusion-controlled nature of the processes (Fig. S29†). For the first oxidation process of complex 2, the separation between the anodic and cathodic peak (ΔEp) increased with scan speed (from 0.34 at 0.1 V s−1, to 0.41 V mV at 1 V s−1). The ΔEp value is around 0.37 mV at 0.2 V s−1, which differs from the value obtained with the Fc/Fc+ redox couple under the same experimental conditions (ΔEp = 0.15 V). This behaviour points to a relatively slow electron transfer rate.
Conclusions
The ability of the 1,2-bis(1-methyl-1H-tetrazol-5-ylthio)ethane (bmtte) ligand to support copper complexes with different oxidation states has been demonstrated by X-ray crystallography, spectroscopy and electrochemical studies. Moreover, the bmtte ligand shows flexibility in the adoption of different coordination modes that stabilise the solid state of several kinds of compounds: two polymorphs of a copper(II) 2D coordination polymer, a discrete tetranuclear copper(I) complex, a trinuclear mixed-valence solid containing tetrahedral copper(I) and tetragonally distorted octahedra copper(II) and a 2D-Cu(I) iodine coordination polymer.The synthetic methodology employed plays a key role in the formation of each compound. The complete reduction of Cu(II) to Cu(I) is favoured under hydrothermal conditions. The use of microwave radiation produced a mixed copper(II/I) valence compound on using CuBr2 and a Cu(I) polymer when the precursor was CuI. When the synthesis was carried out at room temperature there was a tendency to stabilise oxidation state II.
In most cases the crystalline packing is due to weak CH ⋯ (N, Br) hydrogen bonds. The packing of the discrete compounds is reinforced by the contribution of N⋯πtetrazole interactions and the packing of the layers of 2∞Cu4I4(bmtte) is determined by I⋯π interactions, which means that CH ⋯(N, I) hydrogen bonds are involved in the stabilisation of the layer. The evidence outlined above demonstrates that bmtte not only provides several coordination sites but is also capable of stabilising the resulting solid compound through the contribution of different weak interactions.
The study of the reactivity of the mixed-valence [Cu3Br4(bmtte)2] revealed its dynamic behaviour upon treatment with CuBr2 in solution, as it evolved to the Cu(II) 2D polymer, and upon mechanochemical treatment with KI, during which it evolved to the Cu(I) 2D 2∞Cu4I4(bmtte).
The results of electrochemical studies showed similar redox behaviour for 2 and 3, although some differences are observed, mainly due to the different chemical environments present in each compound.
Materials and methods
Materials and physical measurements
Metal salts and solvents were obtained commercially and were used as supplied. Elemental analyses (C, H, N) were carried out on a Fisons EA-1108 microanalyser. IR spectra were recorded from KBr discs (4000–400 cm−1) on a Jasco FT/IR-6100 spectrophotometer. TGA/DSC analysis profiles were obtained with a TGA-ATD/DSC SETSYS Evolution 1750 (Setaram) thermal analyser. Solid state UV-vis spectra were registered on a Jasco V670 spectrophotometer in the range 1200–200 nm. The ligand 1,2-bis(1-methyl-1H-tetrazole-5-ylthio)ethane (bmtte) was prepared as described previously.22Synthesis of the compounds
Method A: microwave irradiation. A solution of bmtte (0.228 g, 0.9 mmol) in acetonitrile (10 mL) was added to a green solution of CuBr2 (0.787 g, 3.6 mmol) in acetonitrile (10 mL). The mixture irradiated in a domestic microwave oven23 for 20 min at 700 W. A mixture of the polymorphs 1m and 1t was obtained by slow evaporation at rt (1 week) of the resulting dark green solution. Brown single crystals of 1m were manually separated. Powder X-ray diffraction of the bulk sample showed the presence of both polymorphs. Yield (1m + 1t): 46%.
Method B: stirring at room temperature. A solution of CuBr2 (0.202 g, 0.9 mmol) in acetonitrile (10 mL) and bmtte (0.234 g, 0.9 mmol) in acetonitrile (10 mL) were mixed. The mixture was stirred at room temperature for one week. A mixture of polymorphs 1m and 1t was obtained by slow evaporation at rt (1 month) of the resulting dark green solution. Brown sheeted single crystals of 1t were separated by hand. However, powder X-ray diffraction of the bulk sample also showed the presence of both polymorphs. Yield (1m + 1t): 61%.
Data for 1 (Method B) Anal. Calc. for C6H10Br4N8S2Cu2: N 15.89%, C 10.22%, H 1.43%. Found: N 14.91%, C 9.71%, H 1.25%. IR (cm−1): 1464m, 1446w, 1406m, 1389w, ν(ring); 1288w, ω(CH–CH2); 1175s, 1037m, 977m, δ(ring); γ(CH); 694s, ν(C–S). Solid state UV-Vis (nm): 323, 430, 565 (CT transition); 848 (d–d transition).
Method A: microwave irradiation. A solution of CuBr2 (0.197 g, 0.9 mmol) in acetonitrile (10 mL) was added to a solution of bmtte (0.235 g, 0.9 mmol) in acetonitrile (10 mL). The mixture was irradiated for 30 min (700 W) to give a dark green solution. A mixture of compounds 1m and 2 was isolated after slow evaporation (15 days) of the resulting solution. Brown single crystals of 2 were manually separated.
Method B: stirring at room temperature. A suspension of bmtte (0.237 g, 0.9 mmol) in acetonitrile (10 mL) was added to a green solution of CuBr2 (0.209 g, 0.9 mmol) in acetonitrile (10 mL). The mixture was stirred at room temperature for 13 days. Brown prismatic single crystals of 2 had formed after 8 days. The residual dark green solution was left to evaporate at room temperature and the resulting unidentified solid was dissolved in an iPrOH/CH3CN (1
:1) mixture. Single crystals of 2 were obtained after slow evaporation (2 months) at r.t. Yield: 80%.
Data for 2(Method B): Anal. Calc. for C12H20Br4N16S4Cu3: N 21.82%, C 14.03%, H 1.96%. Found: N 22.62%, C 14.98%, H 2.04%. IR (cm−1): 1460m, 1443w, 1411m, 1388m, ν(ring); 1280m, ω(CH–CH2); 1176s, γ(CH); 1094w, 1030m, δ(ring); 694s, ν(C–S). Solid state UV-Vis (nm): 290 (π → π* ligand transition); 325, 406, 522 (CT transition), 719 (d–d transition).
Method B: stirring at room temperature. A solution of CuBr2 (0.207 g, 0.9 mmol) in tetrahydrofuran (10 mL) was added to a suspension of bmtte (0.230 g, 0.9 mmol) in tetrahydrofuran (10 mL). The mixture was stirred for 24 h at r.t. The resulting orange solution was slowly evaporated (11 days) to give a solid material that contained some single crystals of 2 (as evidenced by single crystal X-ray diffraction). However, the powder X-ray diffraction of the bulk sample showed that 3 was the major compound.
Method C: hydrothermal synthesis. A mixture of CuBr2 (0.144 g, 0.6 mmol), bmtte (0.055 g, 0.2 mmol) and water (15 mL) was sealed in a Teflon-lined stainless steel vessel (20 mL) and heated for 72 h at 160 °C. The autoclave was cooled at a rate of 2.25 °C h−1 and colourless single crystals of 3 together with unreacted bmtte ligand were observed. Crystals of 3 were separated by hand. Yield: 12%.
Data for 3 (Method C): Anal. Calc. for C12H20Br4N16S4Cu4: N 20.55%, C 13.22%, H 1.85%. Found: N 20.97%, C 13.73%, H 1.72%. IR (cm−1): 1480m, 1440w, 1398m, 1381w, ν(ring); 1214m, ω(CH–CH2), 1160s, γ(CH), 1017w, 961w, δ(ring); 704s, ν(C–S). Solid state UV-Vis (nm): 340 (CT transition).
Method A: microwave irradiation. A mixture of CuI (0.719 g, 3.80 mmol) in acetonitrile (10 mL) and bmtte (0.260 g, 1.01 mmol) in acetonitrile (10 mL) was irradiated for 10 minutes (700 W). A small amount of unreacted reagents was filtered off and the slow evaporation of the mother liquor yielded yellow cubic crystals of 4.
Data for 4. Yield: 51%. Anal. Calc. for C6H10I4N8S2Cu4 N 11.01%, C 7.08%, H 0.99%. Found: N 11.09%, C 7.21%, H 1.01%. IR (cm−1): 1460m, 1442m, ν(ring); 1280m, ω(CH–CH2); 1173m, γ(CH); 1131m, 1102m, 1039m, δ(ring); 702s, ν(C–S). Solid state UV-Vis (nm): 300 (π → π* ligand transition); 326, 329 (CT transition).
Reactivity and stability studies
Reactivity of [Cu3Br4(bmtte)2] (2)
Crystallography
Crystallographic data for single crystals were collected on a Bruker D8 Venture diffractometer with a Photon 100 CMOS detector using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The software SMART24 was used to collect data frames, index reflections, and determine lattice parameters, SAINT for integration of intensity of reflections, and SADABS25 for scaling and empirical absorption correction. The structure was solved by dual-space algorithm using the programme SHELXT.26 All non-hydrogen atoms were refined with anisotropic thermal parameters by full-matrix least-squares calculations on F2 using the programme SHELXL26 with OLEX2.27 Hydrogen atoms were inserted at calculated positions and constrained with isotropic thermal parameters. Drawings were produced with Mercury.28 Crystal data and structure refinement parameters are reported in Table S1.† The structural data have been deposited in the Cambridge Crystallographic Data Centre (CCDC) with the reference numbers included in Table S1.†X-ray powder diffraction (PXRD) was performed using a Xpert Pro 3 (PANanlytical) diffractometer with Cu–Kα radiation (λ = 1.5406 Å) over the range 5 to 50° in steps of 0.026° (2θ) with a count time for step of 5.0 s. The programme MERCURY28 was employed to obtain theoretical powder diffractograms from single crystal data. The programme FULL PROF SUITE29 and the tool WINPLOTR30 were used to perform profile matching.
Hirshfeld surface study
Hirshfeld surfaces and their respective 2D fingerprint plots were calculated for the molecular compounds 2 and 3 with CRYSTALEXPLORER 3.1 software.31 Molecular Hirshfeld surfaces in the crystal structure are constructed based on the electron distribution calculated as the sum of spherical atom electron densities. For a given crystal structure and set of spherical atomic electron densities, the Hirshfeld surface is unique. The dnorm surface and the breakdown of two-dimensional fingerprint plots were used to analyse intermolecular interactions in the different crystal lattices. The sizes and shapes of the fingerprint illustrate the significant differences between the intermolecular interaction patterns.Cyclic voltammetry
Electrochemical measurements were carried out using an Autolab potentiostat/galvanostat (PGSTAT100). This system was equipped with a three-electrode cell, with a 2.00 mm diameter glassy carbon electrode as the working electrode (GCE), a platinum plate electrode as the counter electrode, and the Ag/AgCl reference electrode was used for electrochemical experiments.The GCE electrode was polished using alumina powder (0.05 μm) before use. Ferrocene was used as an internal reference and the redox potentials presented in this work are related to the standard ferrocene/ferrocenium redox couple (Fc/Fc+). The electrochemical data were evaluated from the first sweep.
Measurements were performed on 10−3 mol L−1 solutions of complexes in dimethylsulfoxide (DMSO) containing tetrabutylammonium perchlorate (TBAP) 0.1 mol L−1 as supporting electrolyte. Solutions were deaerated by passing a stream of nitrogen through the solution for 10 minutes prior to the measurement. The dissolution of each compound was monitored by UV-vis spectroscopy, observing no changes for 24 h.
Author contributions
Conceptualization, O. G. P., A. B. L. and R. C.; methodology, O. G. P., and I. P.; formal analysis, O. G. P., A. B. L., R. C., E. M. V. L., and I. P., writing – original draft preparation, O. G. P., R. C. and A. B. L.; writing – review and editing, A. B. L.; supervision, R. C. and E. M. V. L.; project administration, E. M. V. L.; funding acquisition, E. M. V. L. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Ministerio de Ciencia e Innovación (Spain) under research projects PID2019-110218RB-I00 and PID2020-115004RB-I00, and the project ref. 2021ECO11, funded by Fundación CajaCanarias- La Caixa. We thank the Structural Determination Service of the Universidade de Vigo-CACTI for X-ray diffraction measurements. O. G. P. thanks the” Programa de axudas á etapa predoutoral” of the Xunta de Galicia (Gain). Funding for open access charge: Universidad de La Laguna.References
- J. D. Schneider, B. A. Smith, G. A. Williams, D. R. Powell, F. Perez, G. T. Rowe and L. Yang, Inorg. Chem., 2020, 59, 5433–5446 CrossRef CAS.
- A. Sekretaryova, S. M. Jones and E. I. Solomon, J. Am. Chem. Soc., 2019, 141, 11304–11314 CrossRef CAS PubMed.
- O. Gómez-Paz, R. Carballo, A. B. Lago and E. M. Vázquez-López, Chemistry, 2020, 2, 33–49 CrossRef.
- A. Karmakar, A. Paul and A. J. L. Pombeiro, CrystEngComm, 2017, 19, 4666–4695 RSC.
- X. Sánchez-Férez, T. Solans-Monfort, M. Calvet, M. Font-Bardia and J. Pons, Inorg. Chem., 2022, 61, 4965–4979 CrossRef PubMed.
- O. Gómez-Paz, R. Carballo, A. B. Lago and E. M. Vázquez-López, CrystEngComm, 2021, 23, 4658–4666 RSC.
- A. Amoedo-Portela, R. Carballo, J. S. Casas, E. García-Martínez, A. B. Lago-Blanco;, A. Sánchez-Gónzalez, J. Sordo and E. M. Vázquez-López, Z. Anorg. Allg. Chem., 2005, 631, 2241–2246 CrossRef CAS.
- A. B. Lago, A. Amoedo, R. Carballo, E. García-Martínez and E. M. Vázquez-López, Dalton Trans., 2010, 39, 10076–10087 RSC.
- A. Amoedo, R. Carballo, E. García-Martínez, A. B. Lago and E. M. Vázquez-López, Dalton Trans., 2010, 39, 2385–2394 RSC.
- A. B. Lago, R. Carballo, E. García-Martínez and E. M. Vázquez-López, Cryst. Growth Des., 2011, 11, 59–68 CrossRef CAS.
- X. L. Wang, H. L. Hu and A. X. Tian, Cryst. Growth Des., 2010, 10, 4786–4794 CrossRef CAS.
- X. Wang, H. Hu, A. Tian, H. Lin and J. Li, Inorg. Chem., 2010, 49, 10299–10306 CrossRef CAS PubMed.
- X. Wang, Y. Wang, G. Liu, H. Hu and A. Tian, J. Cluster Sci., 2011, 22, 211–223 CrossRef CAS.
- X. Wang, Q. Gao, A. Tian, H. Hu and G. Liu, Inorg. Chim. Acta, 2012, 384, 62–68 CrossRef CAS.
- G. V. Liu and S. Z. Zhang, Zeitschrift für Kristallographie − New Crystal Structures, 2016, 231, 791–792 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedij, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- V. A. Blatov, Interpenetrating metal–organic and inorganic 3D networks: a computer-aided systematic investigation, Samara State University, Russia, 2004 Search PubMed.
- A. I. Kitaigorodskii, Molecular Crystals and Molecules, Academic Press, New York, NY, USA, 1973 Search PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- P. Majumder, J. Chakraborty, H. Adhikary, E. Kara, A. Zangrando, A. Bauza, A. Frontera and D. Das, ChemistrySelect, 2016, 1, 615 CrossRef.
- S. Argibay-Otero, O. Gómez-Paz and R. Carballo, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2017, 73, 1523–1525 CAS.
- M. Ardon, P. D. Hayes and G. Hogarth, J. Chem. Educ., 2002, 79, 1249–1251 CrossRef CAS.
- SMART and SAINT, Control and integration software, Bruker Analytical X-Ray Systems, Madison, WI, 1994 Search PubMed.
- G. M. Sheldrick, SADABS, Program for Absorption Corrections, Göttingen University, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodríguez-Monge, R. Taylor, J. Van De Streek and P. A Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- J. Rodriguez-Carvajal, Commission on Powder Diffraction (CPD), 2001, 12–19 Search PubMed.
- T. Roisnel and J. Rodríguez-Carvajal, in Proceeding of the Seventh European Powder Diffraction Conference (EPDIC 7); 2000, pp. 118–123.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman., CrystalExplorer (Version 3.1), University of Western Australia, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2264490–2264494. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01556d |
| This journal is © The Royal Society of Chemistry 2023 |