
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCisplatin binding to angiogenin protein: new molecular pathways and targets for the drug's anticancer activity†
Giarita
Ferraro
 a,
Vanessa
Sanfilippo
b,
Lorenzo
Chiaverini
c,
Cristina
Satriano
*b,
Tiziano
Marzo
*c,
Antonello
Merlino
*a and
Diego
La Mendola
c
a,
Vanessa
Sanfilippo
b,
Lorenzo
Chiaverini
c,
Cristina
Satriano
*b,
Tiziano
Marzo
*c,
Antonello
Merlino
*a and
Diego
La Mendola
c
aDepartment of Chemical Sciences, University of Naples Federico II, via Cintia 21, I-80126 Napoli, Italy. E-mail: antonello.merlino@unina.it
bNanoHybrid BioInterfaces Laboratory (NHBIL), Department of Chemical Sciences, University of Catania, viale Andrea Doria, 6, 95125 Catania, Italy. E-mail: cristina.satriano@unict.it
cDepartment of Pharmacy, University of Pisa, via Bonanno Pisano 6, 56126 Pisa, Italy. E-mail: tiziano.marzo@unipi.it
First published on 20th June 2023
Abstract
Cisplatin (CisPt), a platinum-based chemotherapeutic widely used in the treatment of various cancers, has multiple mechanisms of action, including nuclear DNA (nDNA) and mitochondrial DNA (mDNA) damage and cytoskeletal perturbations affecting, in turn, the membrane transporter activity. CisPt binding to proteins and enzymes may modulate its biochemical mechanism of action and is associated with cancer cell resistance to the drug. In this work, we investigate the interaction between cisplatin and angiogenin (Ang), a protein strongly expressed in many types of cancer and a potent angiogenic factor. The adduct formed upon reaction of CisPt with Ang (Ang@CisPt) was characterized by X-ray crystallography to evidence the exact platination site and by UV-visible (UV-vis) absorption and circular dichroism (CD) spectroscopies to shed light on any possible change in the protein conformation. Furthermore, high-resolution electrospray ionization (ESI) mass spectrometry was utilized to evaluate the Ang![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CisPt stoichiometry of the Ang@CisPt adduct. The effect of the Ang@CisPt adduct on a prostate cancer cell line (PC-3) was tested by colorimetric assays in terms of cell viability, at both levels of nuclear and mitochondrial damage, and reactive oxygen species (ROS) production. Cellular imaging by laser scanning confocal microscopy (LSM) was utilized to scrutinize the cytoskeleton actin reorganization and the lysosome and mitochondria organelle perturbation. These studies highlight the possibility of new molecular pathways and targets for CisPt activity.
:CisPt stoichiometry of the Ang@CisPt adduct. The effect of the Ang@CisPt adduct on a prostate cancer cell line (PC-3) was tested by colorimetric assays in terms of cell viability, at both levels of nuclear and mitochondrial damage, and reactive oxygen species (ROS) production. Cellular imaging by laser scanning confocal microscopy (LSM) was utilized to scrutinize the cytoskeleton actin reorganization and the lysosome and mitochondria organelle perturbation. These studies highlight the possibility of new molecular pathways and targets for CisPt activity.
Introduction
Prostate cancer is the most prevalent non-cutaneous type of cancer and the fifth leading cause of cancer-related male deaths worldwide.1,2 Current therapies to tackle this disease include taxanes, which can inhibit angiogenesis and induce apoptosis, as well as platinum-based therapies, via DNA damage and/or mitochondria-mediated apoptosis. Although combination chemotherapy regimens, including taxanes and a platinum agent, e.g., cisplatin (CisPt) or carboplatin, have been tested already in clinical trials,3 the whole mechanism of action of these drugs is not fully understood yet.Concerning CisPt, its main activity following the drug uptake in the nucleus of cells has been associated with its ability to form inter- and intra-strand crosslinks with DNA. Such adducts prevent the DNA repair and subsequently induce apoptosis of cancer cells, as a consequence of DNA replication and transcription blockage.4,5 Besides nuclear DNA (nDNA) damage, CisPt can trigger cell death via oxidative stress, which is another important mechanism involved in cisplatin toxicity.6,7 The mitochondrion is the primary target for CisPt-induced oxidative stress, resulting in protein oxidation and reduction of the mitochondrial membrane potential.4 It is noted that the cell death induced by CisPt depends on the number of reactive oxygen species (ROS).8 Indeed, mitochondrial ROS (mtROS) correlate with the mitochondrial content; the reduction of mitochondrial biogenesis by knock-down of transcription factors PGC1α or TFAM attenuates both mtROS induction and CisPt-induced apoptosis.8 Therefore, both nuclear and mitochondrial DNA (mDNA) damages are pivotal for CisPt sensitivity.
Actin cytoskeleton dynamics can modulate cisplatin-induced apoptosis by regulating the expression and function of membrane transporters.9 Recently, it was demonstrated that CisPt may affect cell cytoskeleton organization increasing the actin density and consequently the cell stiffness, in turn reducing prostate cancer cell migration and invasiveness.10
CisPt reactivity is essential to perform its anti-cancer activity. The drug exhibits a certain level of resistance including increased repair of damaged DNA, but one of the most prominent characteristics of cellular resistance to CisPt is the reduced cellular accumulation of the metal complex.11,12 As a consequence of reduced uptake or retention, the formation of Pt–DNA adducts is correspondingly decreased, so that drug cytotoxicity is reduced and cancer cells are more resistant to the Pt compound.13 A different mechanism for decreased accumulation of Pt in cancer cells has been proposed, including both a decreased influx and increased efflux.14,15
The interaction of CisPt with biomolecules in the plasma, especially in the cancer cell microenvironment, or the cytosol, may affect drug activity and availability.16–18
Angiogenin (Ang), a protein of the ribonuclease superfamily, promotes angiogenesis and is strongly overexpressed in almost all human cancers;19–21 its structure is made of three α-helices, seven β-strands, and a 310 helix (at the C-terminus) with the core stabilized by three disulfide bridges.22 Ang partially maintains the RNase-A fold and shares with the pancreatic enzyme the same catalytic triad (His13, Lys40, and His114 in Ang). We have recently shown that oxaliplatin, a third-generation platinum with a 1,2-diaminocyclohexane (DACH) carrier ligand, can bind angiogenin (Ang).23
Ang is particularly overexpressed in prostate cancer24 and its concentration positively correlates with the evolution of prostatic epithelial cells from benign to metastatic invasive progression.25 Hormonal refractory prostate cancer patients showed the highest Ang levels.26 The proliferation of prostate cancer cells is inhibited by the reduction of Ang expression or the inhibition of protein nuclear internalization.25,27,28
Thus, Ang has been proposed as a potential target for prostate cancer treatment. Indeed, the antitumor activity of CisPt significantly decreases the Ang and TNF-α levels in the tumor tissue of solid tumor-bearing mice.29 In previous works, we exploited, by in vitro tests in the PC-3 prostate cancer cell line, the capability to promote ROS formation by nanomaterials functionalized with peptides with angiogenic properties or just CisPt as well as the inhibition of cell migration.30–32
In the present work, we investigate the interaction between CisPt and Ang (i.e., we study the formation of the Ang@CisPt adduct) using X-ray crystallography, UV-visible (UV-vis) and circular dichroism (CD) spectroscopies, and high-resolution electrospray ionization (ESI) mass spectrometry. As per the cellular experiments, in this work we focused on the cytotoxicity and related perturbation/damage mechanisms in cancer cells. To this purpose, in vitro cellular studies with colorimetric assays and laser scanning confocal microscopy (SLM) on the PC-3 prostate cancer cell line have been carried out to unveil the effects of Ang@CisPt on the cells in terms of nuclear and/or mitochondrial DNA damage, ROS production, and cytoskeleton actin and organelle (lysosomes and mitochondria) perturbation.
Results and discussion
Interaction of CisPt and angiogenin
The CisPt interaction with Ang has been studied in solution by UV-vis absorption and circular dichroism spectroscopy.The UV-vis spectra of Ang in the absence (time zero) and in the presence of CisPt (protein to metal molar ratio 1:3) in 20 mM sodium citrate buffer at pH 5.1 as a function of time (from 1 up to 72 h after the mixture preparation) are reported in Fig. S1.† Spectra do not change over time if one excludes slight precipitation of the sample occurring after 72 h. Similar results were obtained in sodium/potassium phosphate buffer solutions, in agreement with what has been observed studying the reaction of the protein with oxaliplatin.23 CD spectra of the same samples show that the presence of CisPt does not produce a significant alteration in the protein secondary structure (Fig. S2†).
Crystals of the adduct formed when CisPt interacts with Ang (Ang@CisPt) have been then obtained using the soaking procedure: crystals of the metal-free protein grown in 10% PEG6K, 0.2 M sodium/potassium tartrate, and 20 mM sodium citrate pH 5.1, using a protein concentration of 0.9 mM, have been soaked for different times (from 2 days to 1 month) in a solution of the reservoir containing the drug in different protein-to-metal ratios (1:1, 1:2 and 1:3). The best structure of Ang@CisPt that we have obtained has been solved at 1.76 Å resolution and refined to Rfactor and Rfree values of 0.245/0.290 (Fig. 1). The structure presents residues 1–122 and consists of 1124 non-hydrogen atoms. Data collection and refinement statistics for this structure are reported in Table S1 of the ESI.† The overall structure of the adduct is superimposable to that of the metal-free protein. After superposition, the Cα atoms of Ang and Ang@CisPt do not differ significantly and present a root mean square deviation (r.m.s.d.) of 0.28 Å. Interestingly, at variance with what has been observed in the structure of the oxaliplatin/Ang adduct (Fig. S3†),23 in the Ang@CisPt adduct platination occurs at the side chain of His114, in the protein active site (Fig. 2). The location of the Pt center has been confirmed by inspection of the anomalous difference electron density map (Fig. 1). His residues have been identified as binding sites for CisPt and other Pt-based drugs in several studies.33–35 Refinement of B-factors of platinum and inspection of residual Fo − Fc electron density maps suggest that the Pt-containing fragment bound to Ang has a very low occupancy (0.20). This finding suggests that cisplatin presents a lower reactivity with Ang than oxaliplatin23 (where two Pt centers with occupancies of 0.40 and 0.25 + 0.35 have been identified close to the side chains of His8 and Gln12 and of His84 and Arg95, PDB code 7NPM; Fig. S3†) and that there is a significant amount of Ang that is not platinated in the presence of the drug. The different reactivities of the two Pt drugs towards Ang could be also related to the difference in their hydrolysis mechanisms. In fact, oxaliplatin hydrolysis occurs through a two-step reaction, being much slower than for cisplatin.36 Unfortunately, the Pt coordination sphere is not clearly defined, and just one ligand (NH3) beyond the ND1 atom of His114 has been modeled. An incomplete description of the Pt coordination sphere has been frequently observed in the CisPt adducts with proteins.37–39
 | ||
| Fig. 1 Structure of Ang@CisPt (PDB code: 8OO3). The Pt binding site is in the protein active site. Anomalous difference electron density map, which allows unambiguous determination of the Pt position, is reported in dark blue at 3.0σ level. Catalytically important residues are highlighted. | ||
 | ||
| Fig. 2 Details of the CisPt fragment binding site in the adduct formed by the drug with Ang. The platinum center binds to the ND1 atom of the side chain of His114. In the protein active site, the network of interactions that involve the other catalytically important residues (His13 and Lys40) and the position of solvent molecules are conserved. The conserved water molecule in the proximity of the Pt-containing fragment has been modeled as an alternative to the metal compound moiety. 2Fo − Fc electron density maps are contoured at the 1.0σ level (light blue). | ||
Additional data sets collected using crystals of the adduct obtained using different soaking times and CisPt concentrations provide similar results with electron density maps that are almost identical to that of the structure above described. Anomalous difference and Fourier difference electron density maps corresponding to the Pt site for one of these structures are reported in Fig. S4.†
To establish the exact CisPt fragment that binds to Ang, high-resolution ESI mass spectra of the Ang@CisPt adduct (see the Experimental section) were recorded and compared with those of the metal-free protein (Fig. S5†).18 Noteworthy, in the spectrum recorded on Ang incubated in the presence of cisplatin, small peaks attributable to the binding of the [Pt(NH3)2]2+ fragment are observed. Additional minor signals attributable to the protein plus a K+ ion were also detected. These results suggest, in good agreement with the crystallographic data, that the reactivity of CisPt with Ang is lower when compared to that of oxaliplatin and that a large amount of Ang is not platinated in the presence of the drug.23
Cytotoxicity and ROS production
In vitro experiments on PC-3 cells were carried out to scrutinize the cancer cells’ response to the Ang@CisPt adduct.First of all, cell viability assays were carried out to assess a dose range for the treatments under conditions of no toxicity for the drug. Indeed, from the literature data, it is known that the sensitivity of this particular cell line to CisPt is highly variable, depending on the oxidative conditions, the cell cycle, and the treatment time.40–42
The cytotoxicity was tested in PC-3 cells using two colorimetric tests to probe, as an indicator of cell viability, the nuclear protein/nuclear acid staining (using the ReadyProbes Cell Viability Imaging Kit, Invitrogen) and the reduction of the yellow (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) salt to purple formazan crystals by metabolically active cells (MTT assay), respectively. For the preparation of the Ang@CisPt adduct, CisPt was added at an equimolar ratio to Ang solution in 25 mM Tris-HCl (pH 7.4) buffer and incubated overnight in a thermomixer at 37 °C and 400 rpm.
Fig. 3 shows the results of cell viability experiments, probed in terms of DNA damage (nuclear staining) or perturbation of mitochondrial activity (MTT test).
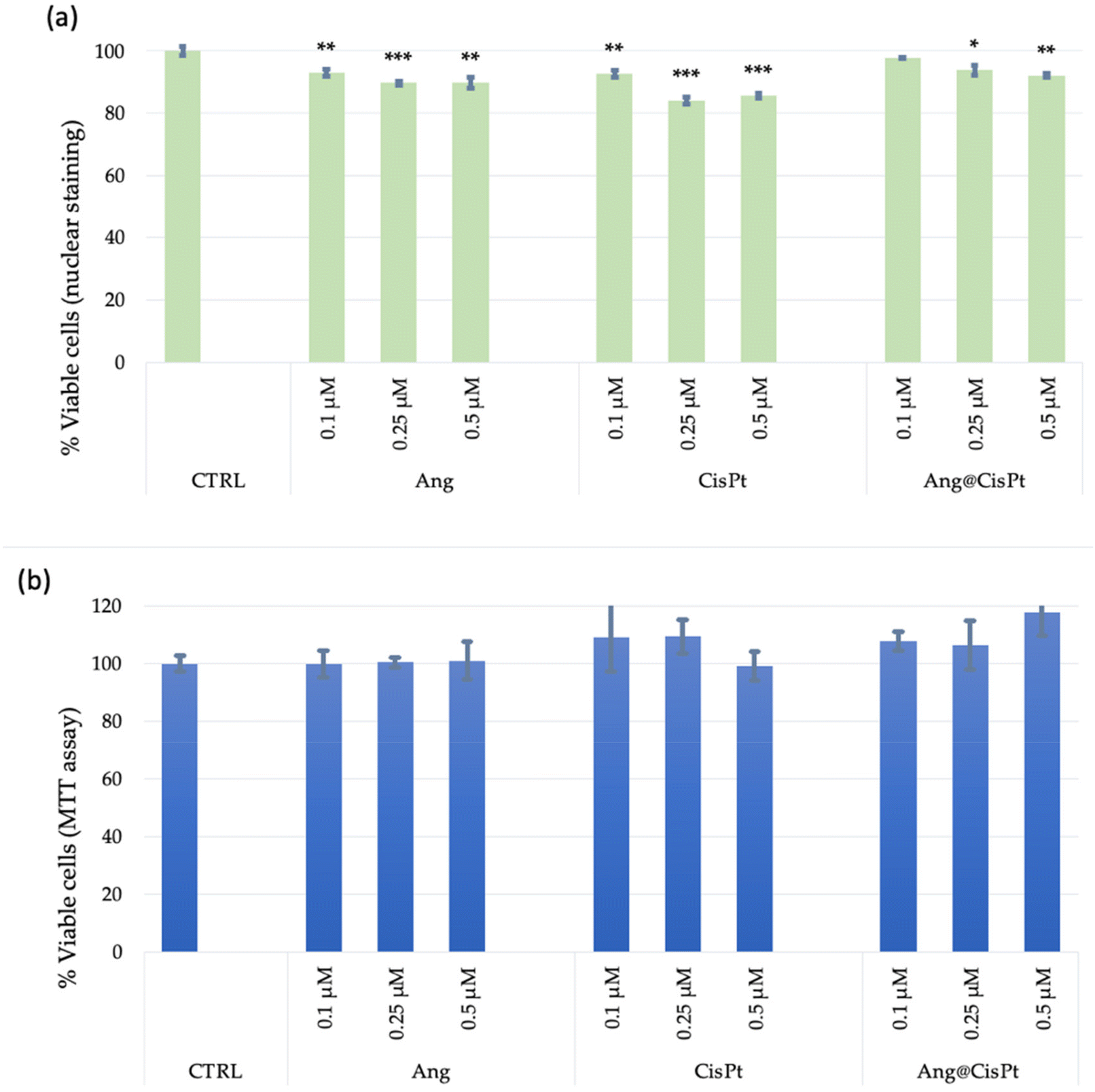 | ||
| Fig. 3 Cell viability analyses carried out using the Blue/Green Imaging kit for total/dead cells (a) or MTT assay (b) on PC-3 cells (p. 28), after 24 h of incubation with Ang, CisPt or Ang@CisPt. The bars represent the average ± standard error of the mean (SEM) values of three independent experiments performed in triplicate. Statistical analysis was performed using Student's t-test. (*) p < 0.05 and (***) p < 0.001, vs. untreated cells (CTRL). | ||
To be noted, only the viability assay detected at the level of DNA damage indicated a statistically significant effect on cell viability, in a dose-dependent manner, with a decrease in viable cells of about 20% for the treatments with CisPt and approximately 10% for the treatments either with Ang or with Ang@CisPt, respectively (Fig. 3a). In contrast, under the tested experimental conditions, no significant changes in cell viability were instead detected in terms of mitochondrial activity, i.e., by the MTT assay (Fig. 3b). Under the investigated experimental conditions, the IC50 value for CisPt was found at the concentrations of 50–100 μM (data not shown), according to the spread values ranging approximately from 20 to 130 μM found in the literature.7,43,44
These divergences can be explained by considering that prostate cancer cells, and specifically the PC-3 line, generate high levels of ROS including H2O2 and superoxide, in turn, and the ROS level increases with the aggressiveness of the cells.44 The higher values of the IC50 determined by the MTT assay should be considered in this context, where the conversion of the tetrazolium salt i.e. (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) to formazan might be overestimated owing to the presence in a high concentration of ROS.
Although the MTT assay of cell viability did not show a significant change in the cellular metabolic activity of mitochondria under all the considered conditions, the analysis of mitochondrial ROS, especially superoxide,46 by the MitoSOX-based assay (Fig. 4), showed that: (i) although the used concentrations of CisPt were not able to induce cytotoxicity, the cells treated with CisPt alone exhibited a dose-dependent effect on the production of mitochondrial ROS; (ii) the treatment with Ang alone did not induce any significant ROS production; and (iii) the Ang@CisPt adduct at the concentrations of 0.1 μM and 0.5 μM but not at 0.25 μM induced a ROS production significantly higher than those observed for the cells treated with CisPt alone at the corresponding concentration. The latter observation can be explained on the basis of what is discussed above, namely the partial binding. As evidenced by both X-ray and solution experiments on the CisPt–Ang binding, only a minor part of CisPt binds the protein. Additionally, under the applied conditions for the ROS production assay, a lack of linearity in the dose-dependent effects – in the case of the adduct – could be envisioned. The resulting effect, in fact, depends on the amount of adduct that forms that, in turn, may slightly differ. Nevertheless, the overall trend is confirmed by our findings i.e. the CisPt–Ang adduct seems more effective in inducing ROS production.
 | ||
| Fig. 4 Mitochondrial ROS levels measured by the MitoSOX assay on PC-3 cells (p. 28), after 24 h of incubation with Ang, CisPt and Ang@CisPt. The bars represent the average ± standard error of the mean (SEM) values of three independent experiments performed in triplicate. Statistical analysis was performed using Student's t-test. (**) p < 0.01, (***) p < 0.001, and (****) p < 0.0001 vs. untreated cells (CTRL); (#) p < 0.05 and (###) p < 0.001 vs. CisPt; the results are expressed as the ratio of MitoSOX intensities of the diverse samples to the emission of the CTRL sample. | ||
The increased ROS production upon treatment of PC-3 cells with CisPt alone or in combination with Ang is consistent with the already known effect of platinum drugs, but determining whether there is a correlation with apoptosis induction is not straightforward to be established because of the inherently high concentration of ROS in prostate cancer lines.45
As previously mentioned, CisPt is known to induce the production of ROS in target cells, altering the normal cellular physiological functions by interaction with various biomolecules, including carbohydrates, nucleic acids, unsaturated fatty acids, and proteins. Mitochondrial dysfunction caused by CisPt culminates in mitochondria-mediated apoptosis. According to the literature,47 our results point to the promoting effect in terms of ROS production by the Ang@CisPt adduct with respect to the drug alone, which suggests a more effective role of the CisPt/protein interaction to impair the synthesis of electron transport chain proteins encoded by mDNA instead of the nDNA damage.
Cellular internalization and organelle perturbation analyses by confocal microscopy
Fig. 5 shows the quantitative analysis of lysosome and mitochondria organelle staining, respectively with LysoTracker and MitoTracker Deep Red, for untreated and treated PC-3 cells. Especially concerning the change in the MitoTracker Deep Red emission, and in agreement with the trend observed for the MitoSOX assay described above, a higher organelle perturbation was detected upon the treatment with CisPt and, even more (and statistically significant) with the adduct. This finding suggests a synergic effect of the protein and the drug; the adduct likely favors the already preferential binding of CisPt to mDNA in the cancer cells, which, in turn, can block ATP synthesis.48 | ||
| Fig. 5 Average values of fluorescence emission for PC-3 cells (p. 28) treated for 2 h with Ang, CisPt, Ang@CisPt_t0, and Ang@CisPt_t12 (concentration 0.1 μM) and stained with LysoTracker Red (λex/em = 543/550–600 nm) and MitoTracker Deep Red (λex/em = 633/650–655 nm). Mean ± S.E.M. of at least 3 experiments is reported. Student's t-test statistical analysis: (*) = p < 0.05 vs. untreated cells (CTRL). | ||
Cytoskeleton actin analysis by confocal microscopy
Fig. 6 shows the representative micrographs of cytoskeleton actin stained by the ActinGreen probe with the quantitative analysis of fluorescence emission. | ||
| Fig. 6 Representative micrographs (scale bar = 20 μm) and quantitative analysis of the sum of all voxel values on PC-3 cells (p. 28) after 2 h of incubation with Ang, CisPt, and Ang@CisPt (concentration 0.1 μM). Nuclear staining with Hoechst, (λex/em = 405/425–450 nm) and actin staining with ActinGreen (ex/em = 488/500–530 nm). Bars represent mean ± S.E.M. of at least 3 experiments. Statistical analysis was performed using Student's t-test: (*) = p < 0.05 and (**) = p < 0.01 vs. untreated cells (CTRL); (#) = p < 0.05 vs. Ang; and (§) = p < 0.05 vs. CisPt. | ||
According to the literature, actin filaments (F-actin), formed by twisting two strands of the monomeric globular protein (G-actin), have a highly dynamic structure, with F-actin reversibly polymerizing and depolymerizing during cellular functions. CisPt increases the cell stiffness in several human prostate cells via stabilization of F-actin.10 The impaired F-actin dynamics may modulate the expression and function of membrane transporters carrying CisPt.49Fig. 6 shows for the Ang@CisPt-treated cells an intermediate change in the cytoskeletal structures, namely lower than that induced by CisPt alone (high perturbation compared to the untreated cells, with about 400% fluorescence increase by the green-stained actin and the evidence of thickened actin filaments mostly confined along the cellular membrane) but higher than the very low or null perturbation found for Ang-treated cells.
Conclusion
In this work we have studied for the first time the interaction of CisPt with human angiogenin, both the drug and the protein being the main actors in prostate cancer therapy/pathology.The crystallographic structure of the Ang@CisPt adduct indicates that selective platination occurs at the protein active site, where the Pt center is anchored to the ND1 atom of the catalytically important His114 side chain, and that the overall protein structure is not affected by the drug binding.
UV-vis absorption and CD spectroscopies and ESI-mass spectrometry provided evidence that: (i) CisPt binds Ang also in solution, although upon incubation of the protein with CisPt, there is a significant amount of metal-free protein; (ii) the [Pt(NH3)2]2+ fragment coordinates Ang in the Ang@CisPt adduct; (iii) no significant perturbation of the overall protein structure occurs in solution.
In vitro cellular experiments with a human prostate cancer cell line (PC-3), carried out at a drug concentration below the IC50, indicated that, although the mitochondria metabolism is not significantly affected by the treatment with the Ang@CisPt adduct under the analyzed experimental conditions, a similar decrease in the cell viability (about 20% less viable cells with respect to the negative control of untreated PC-3) was observed. These results from the cell viability assay are based on the nuclear staining of total/dead cells, for the cells treated either with CisPt or its adduct with the Ang. To be noted, the analysis of mitochondrial ROS production pointed out a higher efficiency of the Ang@CisPt adduct with respect to the drug alone in promoting the ROS production, as confirmed also by the quantitative analysis of the organelle perturbation performed by confocal microscopy imaging.
Moreover, the Ang@CisPt adduct showed to impair the F-actin dynamics, thus acting on the cellular stiffness, which is another hypothesized mechanism of CisPt action to target cancer cells.
Overall, it emerges as gathering insights into the mechanism of action of approved Pt-based anticancer complexes toward protein targets, and specifically Ang might potentially contribute to the optimization and development of currently available protocols.
Altogether, our results revealed that multiple pathways involving Ang can be affected by CisPt treatment. These results are relevant because Ang is an important marker in prostate cancer, positively correlating with prognosis and chemotherapy treatment effectiveness. In this frame, from the obtained results, we can affirm that – potentially – the concomitant administration of CisPt with agents capable of limiting the Ang cell uptake might be beneficial for the final CisPt pharmacological outcome. Additionally, our experiments indicate – even if preliminarily – that treating prostate cancer cells with CisPt might determine a reduced Ang concentration in the cell that, in turn, might advantageously determine reduced invasiveness and metastatization.
Experimental
Spectroscopic characterization of the interaction between CisPt and Ang
Human wild-type angiogenin (Ang) was expressed and purified as detailed in the work by Marzo et al.23 CisPt was purchased from Sigma Chemical Co.The binding of CisPt to Ang was evaluated by collecting UV-vis absorption and CD spectra of the protein in the presence of the drug. The UV-vis spectra of the adduct have been collected as a function of time (each hour for 5 hours and then after 24 h and 72 h). The spectra have been collected using a JASCO V-560 UV-vis spectrophotometer in the range of 240–500 nm and a protein concentration of 0.5 mg mL−1 (35 μM) in 20 mM sodium citrate buffer at pH 5.1 (at a protein-to-metal molar ratio of 1:3). Other experimental parameters were bandwidth 2.0 nm, scanning speed 200 nm min−1, data pitch 1.0 nm, and optical path-length quartz cell 0.1 cm.
The CD spectra of Ang in the absence and in the presence of CisPt after 24 and 72 h of incubation at 37 °C have been collected from 200 nm to 260 nm (intrinsic region; 0.1 cm path length quartz cuvette) in 20 mM sodium citrate buffer at pH 5.1. The spectra have been recorded on a Jasco J-715 spectropolarimeter equipped with a Peltier thermostatic cell holder (Model PTC-348WI) using a protein concentration of 0.2 mg mL−1 (14 μM) and with an increasing amount of the drug (protein-to-metal molar ratios = 1:1, 1:2, and 1:3). The spectra were obtained by averaging three scans and converting the signal to mean residue ellipticity in units of deg cm2 dmol−1. Other experimental settings were scanning speed 50 nm min−1, bandwidth 2.0 nm, resolution 1.0 nm, sensitivity 50 mdeg, and response 2 s.
Crystallization, X-ray diffraction data collection, structure solution and refinement of the Ang@CisPt adduct
Ang was crystallized as previously reported.23 Briefly, the protein (10 mg mL−1) was crystallized by hanging drop vapor diffusion using 10% PEG6K, 20 mM sodium citrate pH 5.1, and 0.2 M sodium/potassium tartrate as a reservoir at 20 °C. Crystals of the adduct were obtained by treating protein crystals with a solution of the reservoir containing CisPt at a protein-to-metal ratio of 1:3.
Diffraction data for the Ang@CisPt adduct were collected at the XRD2 beamline of Elettra Synchrotron, Trieste, Italy. All data sets were indexed, integrated, and scaled using Autoproc.50 Data collection statistics are reported in Table S1.† Initial phase calculations were performed by molecular replacement using the Ang structure with PDB code 1ANG51 as the starting model. Several rounds of restrained individual atomic displacement parameter refinement, energy minimization, and individual B-factor refinement were carried out using Refmac;52 model building and visualization of the model and of the electron density maps were performed using Coot.53 Refinement statistics are reported in Table S1.† Pt atom position searching has been carried out by analyzing 2Fo − Fc and anomalous difference electron density maps. The model geometry was validated using the PDB validation server. The structure was deposited in the PDB under the accession code 8OO3. All structure figures were drawn with PyMOL (DeLano Scientific LLC, San Carlos, CA, USA).
Sample preparation and characterization of the interaction between CisPt and Ang by high resolution ESI MS
The stock solution of CisPt was prepared with LC-MS grade water to a final concentration of 10−3 M and used immediately after its preparation. The stock solution of the protein was prepared at 10−3 M with LC-MS grade water. Appropriate aliquots of these stock solutions were mixed and diluted to a final protein concentration of 10−4 M and a protein-to-metal molar ratio of 1:5. The resulting mixture was incubated at 37 °C for 48 h. After that time, an aliquot of the mixture was further diluted with LC-MS water to a final protein concentration of 10−6 M. 0.1% v/v of LC-MS grade formic acid was added before infusion in the mass spectrometer. The ESI mass spectra were acquired through a direct infusion at 7 μL min−1 in an Orbitrap high-resolution mass spectrometer (Thermo, San Jose, CA, USA), equipped with a HESI source. The ESI source parameters were as follows: spray voltage (+): 3400.00; capillary temperature: 290.00 °C; sheath gas: 24.00; aux gas: 5.00; spare gas: 0.00; max spray current (+): 100.00; probe heater temp.: 50.00; and S-lens RF level: 60.00. For acquisition, Xcalibur 4.2 software (Thermo) was used. A nominal resolution (at m/z 200) of 140000 was used.54
In vitro cellular experiments
CisPt was purchased from Santa Cruz Biotechnology. Adducts of the protein and the drug (at a 1:1 molar ratio) were prepared by incubation at 37 °C in a thermomixer under stirring at 400 r.p.m overnight (Ang@CisPt).
PC-3 cells were seeded at a density of 10 × 103 cells per well in 96-well plates and maintained for 24 h in a complete medium under standard culture conditions. Afterwards, the complete medium was replaced with RPMI 1640 medium supplemented with 1% (v/v) FBS and treated with Ang, CisPt, and Ang@CisPt at the concentrations of 0.1, 0.25, and 0.5 μM. The cells were incubated for 24 h.
The nuclear staining of total cells or only dead cells was performed by incubating at 37 °C for 30 min the cells respectively with the NucBlue® Live reagent (excitation/emission: 360/460 nm) and the NucGreen® Dead reagent (excitation/emission: 504/528 nm). Afterwards, the solutions from triplicate dishes were collected by mechanical scraping of the wells and unified to record the fluorescence emission spectra on a PerkinElmer LS55 fluorimeter. For data analysis, the ratio between NucBlue and NucGreen signals was reported as the percentage of viable cells with respect to the untreated cells.
For the MTT assay, the cytotoxicity was determined by incubating the cells at 37 °C with the MTT solution (5 mg mL−1 concentration) and then detecting the enzymatic reduction of MTT to the insoluble purple formazan product by dissolving the crystals with 100 μL of DMSO and measuring the absorbance at 570 nm on a Varioscan spectrophotometer. All experiments were performed in triplicate and the results are presented as the mean ± standard deviation (SD). The statistical analysis was performed using Student's t-test.
For LSM imaging, PC3 cells were plated at a density of 20 × 103 cells in glass bottom dishes (WillCo-dish®, Willco Wells, B.V.) with 12 mm of glass diameter containing complete medium and maintained under standard culture conditions for 24 h.
After 2 h of treatment with 0.1 μM concentration of the different samples (Ang, CisPt, and Ang@CisPt), cells were stained at the nuclei, lysosome, and mitochondria by treatment for 15 min at 37 °C in the incubator with nuclear dye Hoechst33342 (1 μg mL−1), MitoTracker Deep Red (2 × 107 M) and LysoTracker Red (3 × 107 M), respectively. Then, cells were fixed with high purity 2% paraformaldehyde in PBS, pH = 7.3. For cytoskeleton actin staining, the fixed cells were first permeabilized with 0.02% w/v of Triton X-100 and 10% bovine serum albumin (BSA) and then treated with a high-affinity F-actin probe, conjugated to green-fluorescent Alexa Fluor® 488 dye.
The image analysis was carried out using Huygens Essential software (by Scientific Volume Imaging B.V., The Netherlands). The statistical analysis was performed using Student's t-test.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. F. and A. M. thank ELETTRA synchrotron staff for technical assistance during data collection. G. F. thanks the AIRC foundation for her 3-year FIRC fellowship (AIRC project code: 22587). T. M. and D. L. acknowledge the University of Pisa, “PRA – Progetti di Ricerca di Ateneo” Institutional Research Grants – Project No. PRA_2020_58 “Agenti innovative e nanosistemi per target molecolari nell'ambito dell'oncologia di precisione” and the Center for Instrument Sharing of the University of Pisa (CISUP). C. S. acknowledges the financial support from MUR under Grant PRIN (project code: 2017WBZFHL).References
- B. R. Alabi, S. Liu and T. Stoyanova, Pharmacol. Ther., 2022, 238, 108255 CrossRef CAS PubMed.
- R. J. Rebello, C. Oing, K. E. Knudsen, S. Loeb, D. C. Johnson, R. E. Reiter, S. Gillessen, T. Van der Kwast and R. G. Bristow, Nat. Rev. Dis. Primers, 2021, 7, 9 CrossRef PubMed.
- H. Beltran and F. Demichelis, Endocr.-Relat. Cancer, 2021, 28, T67–T78 CAS.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- T. Makovec, Radiol. Oncol., 2019, 53, 148–158 CrossRef CAS PubMed.
- N. M. Martins, N. A. G. Santos, C. Curti, M. L. P. Bianchi and A. C. Santos, J. Appl. Toxicol., 2008, 28, 337–344 CrossRef CAS PubMed.
- W. Yu, Y. Chen, J. Dubrulle, F. Stossi, V. Putluri, A. Sreekumar, N. Putluri, D. Baluya, S. Y. Lai and V. C. Sandulache, Sci. Rep., 2018, 8, 4306 CrossRef PubMed.
- M. Kleih, K. Böpple, M. Dong, A. Gaißler, S. Heine, M. A. Olayioye, W. E. Aulitzky and F. Essmann, Cell Death Dis., 2019, 10, 851 CrossRef PubMed.
- S. A. Abdellatef, R. Tange, T. Sato, A. Ohi, T. Nabatame and A. Taniguchi, BioMed Res. Int., 2015, 2015, 1–10 CrossRef PubMed.
- M. Raudenska, M. Kratochvilova, T. Vicar, J. Gumulec, J. Balvan, H. Polanska, J. Pribyl and M. Masarik, Sci. Rep., 2019, 9, 1660 CrossRef PubMed.
- S.-H. Chen and J.-Y. Chang, Int. J. Mol. Sci., 2019, 20, 4136 CrossRef PubMed.
- D. Cirri, F. Bartoli, A. Pratesi, E. Baglini, E. Barresi and T. Marzo, Biomedicine, 2021, 9, 504 CAS.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- X. Xue and X.-J. Liang, Chin. J. Cancer, 2012, 31, 100–109 CrossRef CAS PubMed.
- J. Zhou, Y. Kang, L. Chen, H. Wang, J. Liu, S. Zeng and L. Yu, Front. Pharmacol., 2020, 11, 343 CrossRef CAS PubMed.
- M. D. Hall, J. Biol. Inorg. Chem., 2022, 27, 691–694 CrossRef CAS PubMed.
- F.-X. Wang, I. Prokes, L. Song, H. Shi and P. J. Sadler, J. Biol. Inorg. Chem., 2022, 27, 695–704 CrossRef CAS PubMed.
- L. Messori, T. Marzo, E. Michelucci, I. Russo Krauss, C. Navarro-Ranninger, A. G. Quiroga and A. Merlino, Inorg. Chem., 2014, 53, 7806–7808 CrossRef CAS PubMed.
- H. Yang, L. Yuan, S. Ibaragi, S. Li, R. Shapiro, N. Vanli, K. A. Goncalves, W. Yu, H. Kishikawa, Y. Jiang, A. J. Hu, D. Jay, B. Cochran, E. C. Holland and G. Hu, Br. J. Cancer, 2022, 127, 422–435 CrossRef CAS PubMed.
- E. R. Garnett and R. T. Raines, Crit. Rev. Biochem. Mol. Biol., 2022, 57, 244–260 CrossRef CAS PubMed.
- D. Yu, J. Sun, Y. Weng, L. Luo, J. Sheng and Z. Xu, Anti-Cancer Drugs, 2021, 32, 703–708 CAS.
- D. D. Leonidas, R. Shapiro, S. C. Allen, G. V. Subbarao, K. Veluraja and K. R. Acharya, J. Mol. Biol., 1999, 285, 1209–1233 CrossRef CAS PubMed.
- T. Marzo, G. Ferraro, L. M. Cucci, A. Pratesi, Ö. Hansson, C. Satriano, A. Merlino and D. La Mendola, J. Inorg. Biochem., 2022, 226, 111657 CrossRef CAS PubMed.
- T. M. Katona, B. L. Neubauer, P. W. Iversen, S. Zhang, L. A. Baldridge and L. Cheng, Clin. Cancer Res., 2005, 11, 8358–8363 CrossRef CAS PubMed.
- N. Yoshioka, L. Wang, K. Kishimoto, T. Tsuji and G. Hu, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14519–14524 CrossRef CAS PubMed.
- P. K. Majumder, J. J. Yeh, D. J. George, P. G. Febbo, J. Kum, Q. Xue, R. Bikoff, H. Ma, P. W. Kantoff, T. R. Golub, M. Loda and W. R. Sellers, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 7841–7846 CrossRef CAS PubMed.
- M. L. Jones, C. M. Ewing, W. B. Isaacsa and R. H. Getzenberg, J. Cell. Mol. Med., 2012, 16, 193–201 CrossRef CAS PubMed.
- Y.-P. Liu, G.-F. Hu and Y.-X. Wu, Oncol. Lett., 2015, 10, 137–142 CrossRef CAS PubMed.
- M. El-Naa, M. Othman and S. Younes, Drug Des., Dev. Ther., 2016, 10, 3661–3672 CrossRef CAS PubMed.
- V. Verde, A. Longo, L. M. Cucci, V. Sanfilippo, A. Magrì, C. Satriano, C. D. Anfuso, G. Lupo and D. La Mendola, Int. J. Mol. Sci., 2020, 21, 5571 CrossRef CAS PubMed.
- A. Bellissima, L. M. Cucci, V. Sanfilippo, A. De Bonis, R. Fiorenza, S. Scirè, T. Marzo, M. Severi, D. La Mendola, V. Notarstefano, E. Giorgini and C. Satriano, ACS Appl. Bio Mater., 2023, 6, 483–493 CrossRef CAS PubMed.
- L. Riela, L. M. Cucci, Ö. Hansson, T. Marzo, D. La Mendola and C. Satriano, Inorganics, 2022, 10, 188 CrossRef CAS.
- L. Messori, T. Marzo, C. Gabbiani, A. A. Valdes, A. G. Quiroga and A. Merlino, Inorg. Chem., 2013, 52, 13827–13829 CrossRef CAS PubMed.
- L. Messori, T. Marzo and A. Merlino, J. Inorg. Biochem., 2015, 153, 136–142 CrossRef CAS PubMed.
- S. W. M. Tanley, A. M. M. Schreurs, L. M. J. Kroon-Batenburg and J. R. Helliwell, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2012, 68, 1300–1306 CrossRef CAS PubMed.
- D. M. Kweekel, H. Gelderblom and H.-J. Guchelaar, Cancer Treat. Rev., 2005, 31, 90–105 CrossRef CAS PubMed.
- L. Messori and A. Merlino, Coord. Chem. Rev., 2016, 315, 67–89 CrossRef CAS.
- A. Casini, G. Mastrobuoni, C. Temperini, C. Gabbiani, S. Francese, G. Moneti, C. T. Supuran, A. Scozzafava and L. Messori, Chem. Commun., 2007, 156–158 RSC.
- V. Calderone, A. Casini, S. Mangani, L. Messori and P. L. Orioli, Angew. Chem., Int. Ed., 2006, 45, 1267–1269 CrossRef CAS PubMed.
- J. Gumulec, J. Balvan, M. Sztalmachova, M. Raudenska, V. Dvorakova, L. Knopfova, H. Polanska, K. Hudcova, B. Ruttkay-Nedecky, P. Babula, V. Adam, R. Kizek, M. Stiborova and M. Masarik, Int. J. Oncol., 2014, 44, 923–933 CrossRef CAS PubMed.
- W. Zhu, Y. Li and L. Gao, Mol. Med. Rep., 2015, 11, 4561–4566 CrossRef CAS PubMed.
- Y. Liu, C. Yue, J. Li, J. Wu, S. Wang, D. Sun, Y. Guo, Z. Lin, D. Zhang and R. Wang, Oncol. Lett., 2018, 15, 2871–2880 Search PubMed.
- H. Yildirim and A. T. AydemiR, Balıkesir Üniversitesi Fen Bilimleri Enstitüsü Dergisi, 2020, 22, 698–708 CrossRef.
- X. Zhang, Y. Sun, P. Wang, C. Yang and S. Li, Exp. Ther. Med., 2019, 18, 2731–2738 CAS.
- B. Kumar, S. Koul, L. Khandrika, R. B. Meacham and H. K. Koul, Cancer Res., 2008, 68, 1777–1785 CrossRef CAS PubMed.
- M. Kauffman, M. Kauffman, K. Traore, H. Zhu, M. Trush, Z. Jia and Y. Li, React. Oxygen Species (Apex), 2016, 2, 361–370 Search PubMed.
- R. Marullo, E. Werner, N. Degtyareva, B. Moore, G. Altavilla, S. S. Ramalingam and P. W. Doetsch, PLoS One, 2013, 8, e81162 CrossRef PubMed.
- Y. Zhang and M. R. Maurizi, Biochim. Biophys. Acta, Mol. Basis Dis., 2016, 1862, 252–264 CrossRef CAS PubMed.
- T. Shimizu, T. Fujii and H. Sakai, Front. Cell Dev. Biol., 2020, 8, 597835 CrossRef PubMed.
- C. Vonrhein, C. Flensburg, P. Keller, A. Sharff, O. Smart, W. Paciorek, T. Womack and G. Bricogne, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 293–302 CrossRef CAS PubMed.
- K. R. Acharya, R. Shapiro, S. C. Allen, J. F. Riordan and B. L. Vallee, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 2915–2919 CrossRef CAS PubMed.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- I. Tolbatov, D. Cirri, L. Marchetti, A. Marrone, C. Coletti, N. Re, D. La Mendola, L. Messori, T. Marzo, C. Gabbiani and A. Pratesi, Front. Chem., 2020, 8, 812 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt01517c |
| This journal is © The Royal Society of Chemistry 2023 |