
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGermanium- and tin-bridged diazulenylmethyl cations: effects of group 14 elements on the structure and properties of π-extended cations†
Masahito
Murai
 *a,
Mako
Ito
a,
Satoshi
Takahashi
a and
Shigehiro
Yamaguchi
*ab
*a,
Mako
Ito
a,
Satoshi
Takahashi
a and
Shigehiro
Yamaguchi
*ab
aDepartment of Chemistry, Graduate School of Science and Integrated Research Consortium on Chemical Science (IRCCS), Nagoya University, Furo, Chikusa, Nagoya 464-8602, Japan. E-mail: masahito.murai@chem.nagoya-u.ac.jp; yamaguchi@chem.nagoya-u.ac.jp
bInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University, Furo, Chikusa, Nagoya 464-8601, Japan
First published on 22nd May 2023
Abstract
Diazulenylmethyl cations bridged with a germanium and tin moiety were synthesized. In these cations, the nature of these elements has impacts on the chemical stability and photophysical properties. Upon aggregation, these cations exhibit absorption bands in the near-infrared region, which are slightly blue-shifted compared to those of silicon-bridged congeners.
Cationic π-conjugated compounds have attracted considerable attention due to their wide range of applications1 including conducting materials,1c,d dyes for photothermal conversion and bioimaging,1g and sensitizers for photoreactions.1e While various types of cationic π-systems have been developed to date, their properties largely depend on the nature of the π-skeleton employed and fine-tuning of suitable counter anions. A key issue in the design of novel classes of cationic π-skeletons is the need to attain effective charge delocalization that can furnish high chemical stability and unusual electronic structures.2 Another important matter for imparting π-skeletons with valuable functionality is controlling the molecular arrangement in the aggregated and solid state.3
In this context, we have recently disclosed silicon-bridged diazulenylmethyl cations 1 as a new class of stable carbocations (Fig. 1),4 where the positive charge is effectively delocalized over the π-skeleton, reflecting the highly polarizable character of azulenyl moieties.5,6 Specifically, the connection of two azulenyl groups at the 1-position to the carbocation center, while reinforcing the skeleton in a planar fashion with a Si-bridge, is crucial for the high chemical stability of the system. This stabilization mode is different from that found in conventional cationic π-systems, where nitrogen- and oxygen-based electron-donating groups are usually used.2 The Si-bridge also affects the molecular arrangement in the condensed state.7 The observed offset π-stacked arrangement is due to the steric congestion of the substituents on the silicon bridge and the positioning of the counter anions. Consequently, J-type aggregates3b with sharp absorption and emission bands in the near-infrared (NIR) region were formed.8 Such NIR chromophores have a promising variety of prospective applications in fields, such as aggregation-induced staining for bioimaging.1g,8b,d Hence, with this prominent π-skeleton in hand, further diversification of its properties became our next subject. A promising approach to achieving this objective would be via the introduction of other main group elements at the bridging position.9 Herein, we have introduced the heavier group 14 elements, Ge and Sn,10,11 into diazulenylmethyl cations. Relative to silicon,10 these heavier elements are characterized by larger atomic radii, lower electronegativity, less effective orbital interactions with the π-conjugated skeleton, and heavy-atom effects. We synthesized Ge- and Sn-bridged diazulenylmethyl cations 2 and 3 (Fig. 1), and investigated the effects of these heavier elements on the stability, structures, and properties in solution as well as in the aggregated and solid states.
 | ||
| Fig. 1 Design and structures of diazulenylmethyl cations 1–3 with group 14 elements as bridging moieties. | ||
Following the synthetic method reported for the Si-bridged diazulenylmethyl cation 1,4 Ge- or Sn-bridged 2PF6 and 3PF6 were synthesized in three steps from 2-iodoazulene (Scheme 1). The key step is the cyclization of diazulenyl-substituted precursors 4 and 5 under Vilsmeier formylation conditions,4 to produce 2Cl and 3Cl, respectively. While the reaction of 4 at 60 °C selectively afforded 2Cl, similar to the synthesis of the silicon congener 1Cl, a lower reaction temperature was required for the cyclization of 5 in order to prevent the formation of unidentified products that are likely formed via cleavage of the C–Sn bond. Subsequently, 2Cl and 3Cl were treated with KPF6 to afford 2PF6 and 3PF6, which were isolated as deep blue solids.
 | ||
| Scheme 1 Synthesis of 2 and 3. | ||
Whereas 2 and 3 are formally classified as secondary carbocations, they are stable toward air and moisture and they can be stored in air without any precautions irrespective of the counter anions. The chemical stability of 2PF6 and 3PF6 was assessed by determining their pKR+ values, which represent their affinity toward a hydroxide ion.12 The pKR+ values were estimated to be 10.5 and 11.5 for 2PF6 and 3PF6, respectively (Fig. S1†). These values are higher than those of Si-bridged 1PF6 (pKR+ = 9.7)4 and trioxatriangulenium hexafluorophosphate ([TOTA][PF6]: pKR+ = 9.3),12 the latter of which is a representative stable trityl cation (the structure is shown in Fig. S1†). This comparison demonstrates the importance of the nature of heavier elements for chemical stability.
Single crystals of 2PF6 suitable for X-ray diffraction analysis were obtained by the slow evaporation of a CH2Cl2/hexane solution at room temperature (Fig. 2a). In the crystalline state, 2PF6 exhibits a curved π-conjugated skeleton, in which the PF6− anion sits in the bay area with hydrogen bonds between the fluorine and the hydrogen atoms of the two azulene and methene moieties. Similar to the structure of 1PF6,4 the bond length alternation in the seven-membered ring of 2PF6 is small (1.377(8)–1.403(8) Å), whereby the C1–C2 bond (1.372(9) Å) exhibits a pronounced double bond character (Fig. S2†). This structure stands in contrast to the structure of the parent azulene, which exhibits a more pronounced bond length alternation in its seven-membered ring (1.3679(8)–1.4155(8) Å).13 The relatively small bond length alternation in 2PF6 reflects a large contribution of an aromatic tropylium ion, which is responsible for the effective delocalization of the positive charge and thereby its high chemical stability. The most significant difference is the geometry of the central six-membered ring, wherein the Ge atom sits slightly above/below the plane formed by the remaining five carbon atoms, while 1PF6 exhibits a more planar geometry. This difference likely results from the longer endocyclic Ge–C bond lengths (Ge–C2, 1.946(6) Å) and the smaller C2–Ge–C2′ angle in 2PF6 (98.5°) compared to those in 1PF6 (Si–C2, 1.862(3) Å; C2–Si–C2′, 99.8°).4
 | ||
| Fig. 2 (a) The molecular structure of 2PF6 with thermal ellipsoids at 50% probability and (b) molecular arrangement of 2PF6 in the crystal state. Blue: Ge, orange: P, yellow: F, white: H, gray: C. While the unit cell of 2PF6 contains two crystallographically independent molecules, only one of them is shown in (a). The relevant distances for the F⋯H–C hydrogen bonds and the representative C⋯C distance between the two adjacent seven-membered rings are shown. | ||
In the crystal state, molecules of 2PF6 adopt an offset π-stacked arrangement, in which the outer seven-membered rings overlap with the closest interfacial distances of 3.39 and 3.42 Å (Fig. 2b). This packing mode is similar to that observed for 1PF6,4 which implies that this is a common feature of the structurally constrained diazulenomethene skeleton that is probably a result of the location of the PF6− anion and the steric repulsion of the vertically oriented substituents on the Ge or Si bridge (Fig. 2 and S6†).7
The electronic effects resulting from the Ge- and Sn-bridges were studied by measuring the photophysical and electrochemical properties of 2PF6 and 3PF6. In the UV-visible absorption spectra of 2PF6 and 3PF6 in CH2Cl2, intense absorption bands were observed with maximum wavelengths (λabs) at 639 and 637 nm, respectively, which are slightly blue-shifted compared to that of 1PF6 (λabs = 646 nm) (Fig. 3a). The λabs of 2PF6 and 3PF6 did not significantly change upon changing the solvent from CH2Cl2 to THF to DMSO (Table S2†). It should be noted that 2PF6 has a larger molar absorption coefficient (ε) than 1PF6 and 3PF6. The cyclic voltammogram of 2PF6 in CH2Cl2 showed reversible redox waves for a one-electron reduction, similar to that of 1PF6, while that of 3PF6 only showed an irreversible reduction wave (Fig. S8†). The cathodic peak potentials (Epc) follow the order 1PF6 (−0.73 V, E1/2 = −0.69 V) < 2PF6 (−0.79 V, E1/2 = −0.74 V) < 3PF6 (−0.84 V) relative to the ferrocene/ferrocenium couple (Fc/Fc+). These results suggest that the energy levels of the LUMOs of cationic diazulenylmethene skeletons increase upon introducing progressively heavier bridging elements.
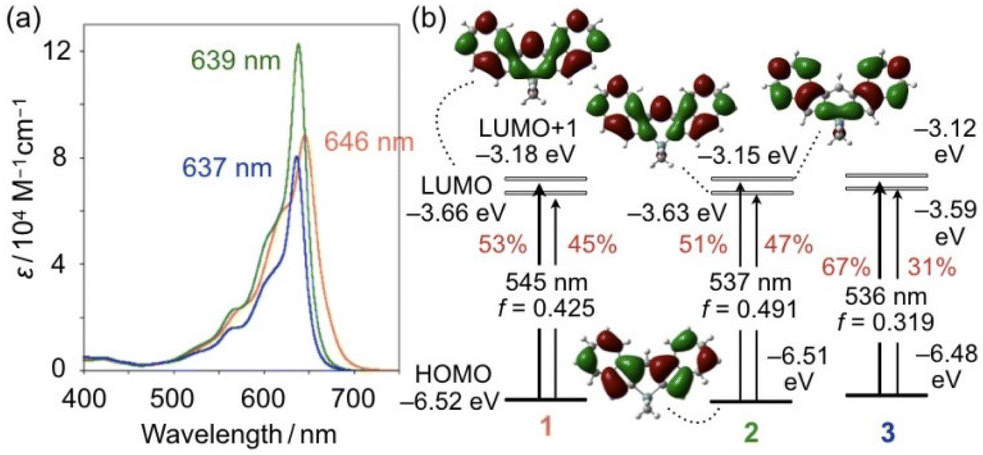 | ||
| Fig. 3 (a) UV–vis absorption spectra of 1PF6 (orange), 2PF6 (green), and 3PF6 (blue) in CH2Cl2. (b) Kohn–Sham molecular orbitals and electronic transitions of 1–3 calculated at the M06/6-311+G(d,p) (for Si, Ge, C, and H) and the LANL2DZ (for Sn) levels of theory with CH2Cl2 using the polarizable continuum model. The red values in (b) represent the percentage probability for each S0–S1 electronic transition. | ||
To gain insights into the effects of bridging elements on the structures and electronic properties, density-functional theory (DFT) calculations were conducted for 1, 2, and 3 without including the counter anions (Fig. S6†) at the M06/6-311+G(d,p) (for Si, Ge, C, and H) and LANL2DZ (for Sn) levels of theory in CH2Cl2 using the polarizable continuum model (PCM). The optimized structure for 2 exhibits an endocyclic Ge–C bond length of 1.953 Å and a C–Ge–C angle of 98.3°, both of which are comparable to those obtained from the X-ray diffraction analysis (Fig. S2†). Although a satisfactory level of crystal data could not be obtained for 3, the structural optimization revealed an endocyclic Sn–C bond length of 2.143 Å and a C–Sn–C angle of 93.5°, which are much longer and smaller than the corresponding values of 1 and 2 (Fig. S3†). This is an important structural effect caused by the bridging elements.
The HOMO and LUMO energy levels increase in the order 1 < 2 < 3, albeit the differences between 1–3 are subtle (Fig. 3b). This tendency itself is consistent with the Epc values obtained from cyclic voltammetry. Notably, while a σ*–π* orbital interaction is observed between the exocyclic SiMe2 moiety and the diazulenylmethene π-skeleton in the LUMO of 1,14 no such orbital interaction is observed in the LUMOs of 2 and 3 when the MOs are represented with a threshold value of 0.017 (Fig. 3b and S4†).
Electrostatic potential (ESP) maps for 2 and 3 obtained at the same level of theory showed that a positive charge is effectively delocalized over the concave area of the molecules (Fig. S5†), which is similar to that for 1. The natural charge at the methene carbon atom for each compound was obtained by natural population analysis (NPA). The obtained values for 1 (−0.026), 2 (−0.026), and 3 (−0.028) are comparable, which suggests that the impact of the heavier bridging elements on the delocalization of the positive charge is limited. On the other hand, slight differences between the LUMO levels of 1, 2, and 3 might be due to the differences in their stabilities as demonstrated by the assessment of their pKR+ values (Fig. S1†), albeit a detailed rationale for these differences remains unclear at this stage (vide supra).
Time-dependent (TD) DFT calculations at the same level furnished the energies for the electronic S0–S1 transitions for 1–3. The energy values are consistent with the order of the experimentally observed hypsochromic shifts of λmax in the order 1 > 2 > 3 (Fig. 3b). These calculations also show that 2 exhibits the highest oscillator strength (f), which is consistent with the fact that 2PF6 has the largest ε value among the three compounds. It should also be noted here that S0–S1 transitions for 1–3 consist of HOMO–LUMO and HOMO–LUMO+1 transitions. Given that the contribution of the HOMO–LUMO+1 transition decreases, the oscillator strength value, and thereby the molar absorption coefficient, tend to increase. The HOMO and LUMO+1 of 1–3 consist of a combination of the HOMO and LUMO of the two parent azulene units. Thus, the S0–S1 transition in 2 has the least azulene character, which is most likely responsible for it having the largest molar absorption coefficient among the three compounds.
Even though the long E–C bonds in 2PF6 and 3PF6 result in distorted structures (Fig. S3†), these compounds retain the J-type aggregation behavior in solution, which is accompanied by absorption bands in the NIR region as observed for 1PF6.3b,4 When the methylcyclohexane (MCH) content of a 1,2-dichloroethane (DCE)/MCH solution of 2PF6 was increased, the intensity of the original absorption band at 646 nm gradually decreased and a new broad band emerged at around 800 nm. The new absorption band became more prominent in a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 (v/v) DCE/MCH solution (λmax = 810 nm) and further blue-shifted to 773 nm in a 10:90 DCE/MCH solution (Fig. 4b). The powder X-ray diffraction pattern of the resulting aggregates of 2PF6, which were collected by filtration from the 10:90 DCE/MCH solution, showed good agreement with a pattern simulated from its crystal packing arrangement (Fig. S9†). This result implies that the offset molecular arrangement observed in the crystalline state is also formed, at least partly, in the aggregated state in solution. Compound 3PF6 also exhibited a bathochromically shifted absorption band (λmax = 751 nm) when the DCE/MCH ratio was increased to 10:90 (Fig. 4c). The λmax values of the aggregates exhibited hypsochromic shifts that follow the order 1PF6 > 2PF6 > 3PF6. Although there is no direct evidence due to the lack of crystal data for 3PF6, we infer from the crystal structure observed for 2PF6 that the heavier elements distort the central six-membered ring, which reduces the degree of overlap of the adjacent molecules in the aggregated state, thus resulting in a blue-shifted absorption band in the NIR region. Notably, the difference in the λmax in the aggregated state between 3PF6 and 1PF6 is 42 nm, while the difference between their monodispersed states is only 9 nm (Fig. 3a).
:80 (v/v) DCE/MCH solution (λmax = 810 nm) and further blue-shifted to 773 nm in a 10:90 DCE/MCH solution (Fig. 4b). The powder X-ray diffraction pattern of the resulting aggregates of 2PF6, which were collected by filtration from the 10:90 DCE/MCH solution, showed good agreement with a pattern simulated from its crystal packing arrangement (Fig. S9†). This result implies that the offset molecular arrangement observed in the crystalline state is also formed, at least partly, in the aggregated state in solution. Compound 3PF6 also exhibited a bathochromically shifted absorption band (λmax = 751 nm) when the DCE/MCH ratio was increased to 10:90 (Fig. 4c). The λmax values of the aggregates exhibited hypsochromic shifts that follow the order 1PF6 > 2PF6 > 3PF6. Although there is no direct evidence due to the lack of crystal data for 3PF6, we infer from the crystal structure observed for 2PF6 that the heavier elements distort the central six-membered ring, which reduces the degree of overlap of the adjacent molecules in the aggregated state, thus resulting in a blue-shifted absorption band in the NIR region. Notably, the difference in the λmax in the aggregated state between 3PF6 and 1PF6 is 42 nm, while the difference between their monodispersed states is only 9 nm (Fig. 3a).
 | ||
| Fig. 4 UV-vis-NIR absorption spectra of (a) 1PF6, (b) 2PF6, and (c) 3PF6 in various ratios of 1,2-dichloroethane (DCE)/methylcyclohexane (MCH) (v/v). Fluorescence spectra recorded in a 10:90 (v/v) DCE/MCH mixed solvent are shown as blue dashed lines. (d) Comparison of the S0–S1 electronic transition for the dimeric motifs of 1 and 2 together with their Kohn–Sham HOMOs and LUMOs calculated at the M06/6-311+G(d,p) level of theory. The geometries for the calculations were taken from their crystal packing structures and the counter ions have not been included. | ||
To gain insight into the bathochromic shift of the absorption bands upon aggregation, DFT calculations were conducted at the M06/6-311+G(d,p) level for two adjacent molecules of 2, the coordinates of which were taken from the crystal packing structure of 2PF6 (Fig. 4d). Compared to the monomer (Fig. 3b), the dimeric motif of 2 is characterized by HOMO and LUMO energy levels that are increased by 0.42 eV and 0.24 eV, respectively, and thereby the corresponding transition energy is by 0.198 eV lower, which is in agreement with a bathochromic shift of 44 nm. This calculation also showed a significant increase in the oscillator strength (f) for the dimeric motif. The offset arrangement of the electronic transition dipole of 2, which is oriented along the long axis of the molecule (Fig. S4†), should be relevant to the increased f value, despite the limited overlap of the molecules only in the outer seven-membered ring (Fig. 2b). The difference between the absorption wavelengths of the aggregated states of 1PF6 and 2PF6 can also be reproduced by comparing the calculation results for the dimeric motifs of 1 and 2 (Fig. 4d). In the LUMO of the dimer of 2, in-phase orbital interactions across the two molecules were observed (see the dotted area in Fig. 4d), the degree of which seems smaller than that of the dimer of 1.
Although monodispersed 2PF6 and 3PF6 in solution are essentially non-emissive, fluorescence was observed upon the formation of J-type aggregates in their 10:90 DCE/MCH solutions (Fig. 4a–c). The emission maximum wavelengths of 2PF6 and 3PF6 were 828 and 820 nm, respectively, which were slightly blue-shifted compared to that of 1PF6 (845 nm). The Stokes shifts for 2PF6 (860 cm−1) and 3PF6 (1121 cm−1) were larger than that for 1PF6 (776 cm−1), which might be related to the loosely packed aggregated structure in 3PF6 that occurs due to the distorted geometry of its central six-membered ring (Fig. S3†). The quantum yields of the aggregates of 2PF6 and 3PF6 in 10:90 DCE/MCH (ΦF < 0.01) are lower than the previously reported value of the Si-bridged congeners (ΦF ≈ 0.04).4
In conclusion, Ge- and Sn-bridged diazulenylmethyl cations were synthesized and the effects of these main group bridges on the stability and photophysical properties were studied. The incorporation of these heavier elements increases the stability of the resulting cationic π-systems. Among the derivatives, Ge-bridged 2PF6 showed the largest molar absorption coefficient in its UV-visible absorption spectrum. The use of heavy group 14 elements as bridging moieties also gave rise to a central six-membered ring with a distorted geometry. This geometry, which deviates from planarity, may determine how closely the π-stacked structures align and thus govern the magnitude of the hypsochromic shift of their absorption bands in the aggregated state. Despite the incorporation of heavier elements, aggregates of these cationic π-systems are capable of emitting fluorescence, albeit their emissions are generally faint.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by CREST (JPMJCR21O5) from the Japan Society and Technology Agency (JST). The synchrotron X-ray crystallography experiment for 2PF6 was performed at the BL02B1 beamline of SPring-8. The authors are grateful to Mr. Tatsuo Hikage (Nagoya Univ.) for X-ray powder diffraction measurements.References
- (a) D. F. Duxbury, Chem. Rev., 1993, 93, 381 CrossRef CAS; (b) J. Bosson, J. Gouin and J. Lacour, Chem. Soc. Rev., 2014, 43, 2824 RSC; (c) R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357 CrossRef CAS PubMed; (d) K. Goossens, K. Lava, C. W. Bielawski and K. Binnemans, Chem. Rev., 2016, 116, 4643 CrossRef CAS PubMed; (e) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (f) Y. Haketa and H. Maeda, Chem. Commun., 2017, 53, 2894 RSC; (g) L. Feng, W. Chen, X. Ma, S. H. Liu and J. Yin, Org. Biomol. Chem., 2020, 18, 9385 RSC.
- B. W. Laursen, F. C. Krebs, M. F. Nielsen, K. Bechgaard, J. B. Christensen and N. Harrit, J. Am. Chem. Soc., 1998, 120, 12255 CrossRef CAS.
- (a) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491 CrossRef CAS PubMed; (b) F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef PubMed.
- M. Murai, M. Abe, S. Ogi and S. Yamaguchi, J. Am. Chem. Soc., 2022, 144, 20385 CrossRef CAS PubMed.
- (a) H. Xin and X. Gao, ChemPlusChem, 2017, 82, 945 CrossRef CAS PubMed; (b) H. N. Zeng, Z. M. Png and J. Xu, Chem. – Asian J., 2020, 15, 1904 CrossRef CAS PubMed; (c) J. Huang, S. Huang, Y. Zhao, B. Feng, K. Jiang, S. Sun, C. Ke, E. Kymakis and X. Zhuang, Small Methods, 2020, 4, 2000628 CrossRef CAS; (d) A. Konishi and M. Yasuda, Chem. Lett., 2021, 50, 195 CrossRef CAS.
- (a) M. Murai, E. Amir, R. J. Amir and C. J. Hawker, Chem. Sci., 2012, 3, 2721 RSC; (b) M. Murai, S.-Y. Ku, N. D. Treat, M. J. Robb, M. L. Chabinyc and C. J. Hawker, Chem. Sci., 2014, 5, 3753 RSC; (c) M. Murai, K. Takami, H. Takeshima and K. Takai, Org. Lett., 2015, 17, 1798 CrossRef CAS PubMed; (d) M. Murai, M. Yanagawa, M. Nakamura and K. Takai, Asian J. Org. Chem., 2016, 5, 629 CrossRef CAS; (e) M. Murai, S. Iba, H. Ota and K. Takai, Org. Lett., 2017, 19, 5585 CrossRef CAS PubMed.
- K. Mouri, A. Wakamiya, H. Yamada, T. Kajiwara and S. Yamaguchi, Org. Lett., 2007, 9, 93 CrossRef CAS PubMed.
- (a) J. Fabian, H. Nakazumi and M. Matsuoka, Chem. Rev., 1992, 92, 1197 CrossRef CAS; (b) G. Qian and Z. Y. Wang, Chem. – Asian J., 2010, 5, 1006 CrossRef CAS PubMed; (c) L. Dou, Y. Liu, Z. Hong, G. Li and Y. Yang, Chem. Rev., 2015, 115, 12633 CrossRef CAS PubMed; (d) M. M. Leitão, D. de Melo-Diogo, C. G. Alves, R. Lima-Sousa and I. J. Correia, Adv. Healthcare Mater., 2020, 9, 1901665 CrossRef PubMed.
- (a) A. Fukazawa and S. Yamaguchi, Chem. – Asian J., 2009, 4, 1386 CrossRef CAS PubMed; (b) S. M. Parke, M. P. Boone and E. Rivard, Chem. Commun., 2016, 52, 9485 RSC; (c) A. M. Priegert, B. W. Rawe, S. C. Serin and D. P. Gates, Chem. Soc. Rev., 2016, 45, 922 RSC.
- For the effects of Ge- and Sn-bridges, see: (a) S. Yamaguchi, Y. Itami and K. Tamao, Organometallics, 1998, 17, 4910 CrossRef CAS; (b) M. Saito, R. Haga, M. Yoshioka, K. Ishimura and S. Nagase, Angew. Chem., Int. Ed., 2005, 44, 6553 CrossRef CAS PubMed; (c) C. M. Amb, S. Chen, K. R. Graham, J. Subbiah, C. E. Small, F. So and J. R. Reynolds, J. Am. Chem. Soc., 2011, 133, 10062 CrossRef CAS PubMed; (d) H. Zhong, Z. Li, F. Deledalle, E. C. Fregoso, M. Shahid, Z. Fei, C. B. Nielsen, N. Yaacobi-Gross, S. Rossbauer, T. D. Anthopoulos, J. R. Durrant and M. Heeney, J. Am. Chem. Soc., 2013, 135, 2040 CrossRef CAS PubMed; (e) D. Tanaka, J. Ohshita, Y. Ooyama, N. Kobayashi, H. Higashimura, T. Nakanishi and Y. Hasegawa, Organometallics, 2013, 32, 4136 CrossRef CAS; (f) M. Murai, K. Matsumoto, R. Okada and K. Takai, Org. Lett., 2014, 16, 6492 CrossRef CAS PubMed; (g) M. Shimizu, D. Ryuse and T. Kinoshita, Chem. – Eur. J., 2017, 23, 14623 CrossRef CAS PubMed; (h) S. Akahori, H. Sakai, T. Hasobe, H. Shinokubo and Y. Miyake, Org. Lett., 2018, 20, 304 CrossRef CAS PubMed; (i) S. Urrego-Riveros, I.-M. Ramirez y Medina, J. Hoffmann, A. Heitmann and A. Staubitz, Chem. – Eur. J., 2018, 24, 5680 CrossRef CAS PubMed.
- For Ge-containing positively charged conjugated compounds, see: A. N. Butkevich, V. N. Belov, K. Kolmakov, V. V. Sokolov, H. Shojaei, S. C. Sidenstein, D. Kamin, J. Matthias, R. Vlijm, J. Engelhardt and S. W. Hell, Chem. – Eur. J., 2017, 23, 12114 CrossRef CAS PubMed.
- B. W. Laursen and F. C. Krebs, Chem. – Eur. J., 2001, 7, 1773 CrossRef CAS PubMed.
- B. Dittrich, F. P. A. Fabbiani, J. Henn, M. U. Schmidt, P. Macchi, K. Meindl and M. A. Spackman, Acta Cryst. B, 2018, 74, 416 CrossRef CAS PubMed.
- (a) S. Yamaguchi and K. Tamao, Bull. Chem. Soc. Jpn., 1996, 69, 2327 CrossRef CAS; (b) K. Tamao, M. Uchida, T. Izumizawa, K. Furukawa and S. Yamaguchi, J. Am. Chem. Soc., 1996, 118, 11974 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, characterization data, photophysical and spectral data, as well as crystallographic data for 2PF6. CCDC 2252079. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01491f |
| This journal is © The Royal Society of Chemistry 2023 |