
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From cyclic (alkyl)(amino)carbene (CAAC) precursors to fluorinating reagents. Experimental and theoretical study†‡
Evelin
Gruden
 a,
Griša Grigorij
Prinčič
b,
Jan
Hočevar
b,
Jernej
Iskra
b,
Jaroslav
Kvíčala
c and
Gašper
Tavčar
*a
a,
Griša Grigorij
Prinčič
b,
Jan
Hočevar
b,
Jernej
Iskra
b,
Jaroslav
Kvíčala
c and
Gašper
Tavčar
*a
aDepartment of Inorganic Chemistry and Technology, “Jožef Stefan” Institute, Jamova cesta 39, Ljubljana, Slovenia. E-mail: gasper.tavcar@ijs.si
bDepartment of Chemistry and Biochemistry, University of Ljubljana, Faculty of Chemistry and Chemical Technology, Večna pot 113, 1000 Ljubljana, Slovenia
cDepartment of Organic Chemistry, University of Chemistry and Technology, Technická 5, 166 28 Prague 6, Prague, Czech Republic
First published on 27th June 2023
Abstract
Addition of anhydrous HF to the hydrochloride [MeCAACH][Cl(HCl)0.5] resulted in the formation of salts with high HF content. By stepwise removal of HF in vacuo, we selectively prepared [MeCAACH][F(HF)2] (3) and [MeCAACH][F(HF)3] (4). We also characterised a salt with [F(HF)4]− anions within the structure of [MeCAACH][F(HF)3.5] (5). Compounds with a lower content of HF were not accessible under vacuum conditions. MeCAAC(H)F (1) was selectively prepared by abstraction of HF from 3 with CsF or KF, while [MeCAACH][F(HF)] (2) was prepared by mixing 3 and 1 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Compound 2 proved to be quite unstable as it tends to disproportionate into 1 and 3. This observation triggered our computational study, in which the structural relationships between CAAC-based fluoropyrrolidines and dihydropyrrolium fluorides were investigated using different DFT methods. The study showed that the results were very sensitive to the computational method used. For a correct description, the quality of the triple-ζ basis set was crucial. Surprisingly, the isodesmic reaction of [MeCAACH][F] + [MeCAACH][F(HF)2] → [MeCAACH][F(HF)] + [MeCAACH][F(HF)] did not confirm the low thermodynamic stability of 2. Furthermore, the use of 3 as a nucleophilic fluorinating reagent was tested on a range of organic substrates, as it is the most stable compound in this series. It was found to have the potential to fluorinate benzyl bromides, 1- and 2-alkyl bromides, silanes and sulfonyls with good to excellent yields of the target fluorides.
:1 ratio. Compound 2 proved to be quite unstable as it tends to disproportionate into 1 and 3. This observation triggered our computational study, in which the structural relationships between CAAC-based fluoropyrrolidines and dihydropyrrolium fluorides were investigated using different DFT methods. The study showed that the results were very sensitive to the computational method used. For a correct description, the quality of the triple-ζ basis set was crucial. Surprisingly, the isodesmic reaction of [MeCAACH][F] + [MeCAACH][F(HF)2] → [MeCAACH][F(HF)] + [MeCAACH][F(HF)] did not confirm the low thermodynamic stability of 2. Furthermore, the use of 3 as a nucleophilic fluorinating reagent was tested on a range of organic substrates, as it is the most stable compound in this series. It was found to have the potential to fluorinate benzyl bromides, 1- and 2-alkyl bromides, silanes and sulfonyls with good to excellent yields of the target fluorides.
Introduction
The introduction of fluorine into organic compounds significantly alters their chemical, physical and biological properties, making them desirable compounds in medical, material and agrochemical sciences.1 In recent decades, the demand for such compounds has led to rapid development.2–4 Several fluorination methods and fluorinating reagents have been tested and are already used in industry.5,6 Nevertheless, the search for reliable, selective and easy-to-use fluorinating reagents is still ongoing. The use of hydrogen fluoride as a nucleophilic fluorinating reagent would be the most cost-effective and atom-economical.7 However, the high corrosivity, low boiling point and toxicity of gaseous HF make it difficult to handle.8 Instead, many safer HF-based reagents have been used, such as the commercially available HF–pyridine (Olah's reagent)9 or Et3N·3HF.10,11 More selective HF-based reagents have been developed to participate in specific organic transformations. DMPU–HF is less basic than HF–pyridine and Et3N·3HF and therefore suitable for reactions requiring acidic conditions.12–15 KHSO4–HF, which simultaneously exhibits high acidity and high fluoride nucleophilicity, can readily participate in the hydrofluorination of alkenes.16 Imidazolium-based fluorinating reagents are also a class of promising compounds that have been extensively studied in the past.17–22 Recently, they have been successfully used for the fluorination of alkyl and benzyl halides, tosylates, mesylates, silyl ethers, epoxides, sulfonate esters, α-haloketones and nitroarenes.23–26Three imidazolium-based fluorinating reagents suitable for the fluorination of organic26 or inorganic27,28 compounds have been prepared in our laboratory. The presence of bulky cations is particularly important for the stabilisation of fluorinated discrete anions.29 The compounds [IPrH][F], [IPrH][F(HF)] and [IPrH][F(HF)2] (IPrH = 1,3-bis(2,6-diisopropylphenyl)-1H-imidazol-3-ium) were synthesised from the corresponding NHC carbene by adding the appropriate amount of HF-based reagents or by fluorination of the corresponding chloride salt.26,30 Inspired by the simple conversion from NHC carbene to fluorinating reagent, we extended our research to another class of carbenes, the cyclic (alkyl)(amino) carbenes (CAACs). However, the electronic and hence chemical properties of CAACs differ significantly from those of NHCs.31 The smaller singlet–triplet gap of CAACs and higher electrophilicity allow them to activate small molecules under mild conditions.32 They are able to react with CO to form a stable ketene CAAC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO,33 activate molecules such as H2 and NH3 to form stable cleaved products CAACH2 and CAAC(H)NH2,34 undergo intramolecular C–H activation to form a tricycle,35 and react with a variety of sp-, sp2- and sp3-hybridised C–H groups to form CAAC(H)R compounds.35 Due to their high reactivity, CAAC carbenes could not be used as the precursors for the fluorinating reagents.
CO,33 activate molecules such as H2 and NH3 to form stable cleaved products CAACH2 and CAAC(H)NH2,34 undergo intramolecular C–H activation to form a tricycle,35 and react with a variety of sp-, sp2- and sp3-hybridised C–H groups to form CAAC(H)R compounds.35 Due to their high reactivity, CAAC carbenes could not be used as the precursors for the fluorinating reagents.
High ratio poly(hydrogen fluoride) salts were obtained starting from the corresponding hydrochloride salt [MeCAACH] [Cl(HCl)0.5] and adding anhydrous HF for fluorination as described by Matsumoto.22 By stepwise removal of HF, a fluorinated compound MeCAAC(H)F (1) and several other poly(hydrogen fluoride) salts [MeCAACH][F(HF)] (2), [MeCAACH][F(HF)2] (3), [MeCAACH][F(HF)3] (4) and [MeCAACH][F(HF)3.5] (5) were isolated and characterised. Compound 3, which is the most stable in this series, was tested as a nucleophilic fluorination reagent on organic substrates.
Results and discussion
Synthesis and characterisation of salts
The synthesis of all poly(hydrogen fluoride) compounds began with the reaction of [MeCAACH][Cl(HCl)0.5] with anhydrous HF, which was used as both reagent and solvent. After removal of the excess HF overnight to constant weight under reduced pressure of 10−2–10−3 bar, [MeCAACH][F(HF)3] (4) was obtained as a solid product in quantitative yield (Scheme 1). The compound slowly releases HF at room temperature to form the more stable trifluoride salt (3). To avoid decomposition, 4 was stored in a plastic container at −20 °C under an inert atmosphere. Single crystals of 4 suitable for X-ray analysis were obtained overnight by slow evaporation of HF during product isolation. It crystallises in the monoclinic space group P21/c and its crystal structure is shown in Fig. 1. The [F(HF)3]− anion is practically planar and has the form of a branched chain. Its structure agrees well with similar anions reported elsewhere.36 | ||
| Scheme 1 Synthesis of [MeCAACH][F(HF)3] (4) and [MeCAACH][F(HF)2] (3). aHF = anhydrous HF. | ||
 | ||
| Fig. 1 Structure of the asymmetric unit of [MeCAACH][F(HF)3] (4). The ellipsoids are drawn at 50% probability. For clarity, all H atoms on the cation are omitted, except for the atom at C2 position. Selected bond lengths (Å) and angles (°): F(1)⋯F(2) 2.345(2), F(2)⋯F(3) 2.346(3), F(2)⋯F(4) 2.379(3), C(2)–H(2) 0.950(2), N(1)–C(2) 1.273(2), F(1)–F(2)–F(3) 114.8(1), F(1)–F(2)–F(4) 122.0(1), F(3)–F(2)–F(4) 122.4(1), N(1)–C(2)–H(2) 122.5(2). | ||
Removal of 1 equiv. of HF from 4 resulted in the formation of [MeCAACH][F(HF)2] (3). Although 4 tends to release HF, the process is very slow. To ensure complete conversion to 3, removal of the volatiles had to be carried out for 2 days at room temperature under a high vacuum of 10−5–10−7 mbar (Scheme 1). Compound 3 is the most stable of all the hydrogen fluoride compounds prepared. It can be stored in plastic containers on air for long periods without decomposition and does not react with glass in the absence of moisture. Because of its stability, 3 was selected as a potential nucleophilic fluorinating reagent and its reactivity was tested on a number of organic substrates (see section 3). Single crystals of 3 suitable for X-ray analysis were obtained from a concentrated MeCN solution at −20 °C. It crystallises in the monoclinic space group P21/n and its crystal structure is shown in Fig. 2. The [F(HF)2]− anion adopts a non-linear geometry. It is bent, with the angle F(1)–F(2)–F(3) being 108.54(8)°. The bond lengths F(1)⋯F(2) and F(2)⋯F(3) of 2.263(2) Å and 2.320(2) Å, respectively, agree with those of [F(HF)2]− anions determined previously.30,37
 | ||
| Fig. 2 Structure of the asymmetric unit of [MeCAACH][F(HF)2] (3). The ellipsoids are drawn at 50% probability. For clarity, all H atoms on the cation are omitted, except for the atom at C2 position. Selected bond lengths (Å) and angles (°): F(1)⋯F(2) 2.263(2), F(2)⋯F(3) 2.320(2), C(2)–H(2) 0.950(2), N(1)–C(2) 1.275(2), F(1)–F(2)–F(3) 108.54(8), N(1)–C(2)–H(2) 122.8(2). | ||
We have observed that the reaction of [MeCAACH] [Cl(HCl)0.5] with an excess of aHF leads primarily to the formation of higher poly(hydrogen fluoride) salts. They can be made accessible by slowly removing the excess HF at lower temperatures. However, these fractions are liquid at room temperature and tend to release HF even at −20 °C. Due to the instability of the higher poly(hydrogen fluorides), we had problems with storage and characterisation. Fortunately, we were able to recover a fraction containing 3.5 equivalents of HF. This fraction contains the highest content of HF observed in this system and is stable at −20 °C. During storage of various poly(hydrogen fluoride) mixtures at −20 °C, single crystals of [MeCAACH][F(HF)3.5] (5) were repeatedly formed. Compound 5 is actually a co-crystal of [MeCAACH][F(HF)3] and [MeCAACH] [F(HF)4] in the ratio 1:1. 5 crystallises in the monoclinic space group P21/n. For reasons of clarity, only the crystal structure of [MeCAACH][F(HF)4] is shown in Fig. 3. The crystal structure of 5 is shown in the ESI in Fig. S19.‡ The [F(HF)3]− anion adopts the same branched chain form as that in compound 4. The [F(HF)4]− anion has the form of a branched-chain and represents a rare example of such an isomer. There are three possible isomers for [F(HF)4]− anions – the tetrahedral, the branched-chain and the chain type. To our knowledge, the latter has not yet been discovered,38 while the tetrahedral type is the most commonly observed.39 The branched-chain isomer has only been found in the structure of Me3N·5HF.36 In 5, the branched-chain anion is virtually centred with a particularly short inner hydrogen bond F(22)⋯F(23) of 2.275(3) Å. This is consistent with a specific [F(HF)]− subunit in the core surrounded by 3 HF molecules. Therefore, the formula can be written as [(FH)(FHF)(HF)2]−. The structure of the [F(HF)4]− anion agrees well with similar anions reported elsewhere.36
 | ||
| Fig. 3 Crystal structure of [MeCAACH][F(HF)4]. The ellipsoids are drawn at 50% probability. For clarity, all H atoms on the cation are omitted, except for the atom at C2 position. Selected bond lengths (Å) and angles (°): F(21)⋯F(22) 2.417(3), F(22)⋯F(23) 2.275(3), F(23)⋯F(24) 2.408(3), F(23)⋯F(25) 2.381(3), C(22)–H(22) 0.950(2), N(21)–C(22) 1.271(3), F(21)–F(22)–F(23) 121.8(1), F(22)–F(23)–F(24) 128.6(1), F(22)–F(23)–F(25) 112.8(1), N(21)–C(22)–H(22) 122.5(2). | ||
Since compound 3 is the most stable among the poly(hydrogen fluorides) in this series, compounds with lower HF content could not be prepared from 3 by removing HF under low pressure. Removal of another HF was not possible under high vacuum even at higher temperature. Therefore, the synthesis of MeCAAC(H)F (1) required a different approach. Compound 3 was mixed with excess CsF and suspended in toluene. The mixture was stirred for 3 days to give 1 and CsHF2 (Scheme 2). Compound 1 is very soluble in toluene and was easily separated from the salt by-products by filtration. Alternatively, KF can be used instead of CsF for the synthesis of 1. The reaction with excess KF proceeds quantitatively and yields KHF2 as a by-product. Single crystals of 1 suitable for X-ray analysis were obtained from concentrated MeCN solution at −20 °C. It crystallises in the monoclinic space group P21/c and its crystal structure is shown in Fig. 4. Compound 1 crystallises as a racemic mixture in the form of a neutral chiral compound with H and F bonded at C2 position. In the crystal structure, two MeCAAC(H)F units join together through hydrogen bonds and form dimers. Two hydrogen bonds are formed between H and F atoms at the C2 position of one molecule and the F and H atoms at the C2 position of the adjacent molecule. Consequently, a six-membered ring is formed, as shown in Fig. 5. In addition, two even shorter hydrogen bonds between the F atom and the H atoms on isopropyl wingtips C(19)–H(18A)⋯F(1) influence the stability of the dimer.
 | ||
| Scheme 2 Synthesis of MeCAAC(H)F (1) and [MeCAACH][F(HF)] (2). | ||
 | ||
| Fig. 4 Structure of the asymmetric unit of MeCAAC(H)F (1). The ellipsoids are drawn at 50% probability. For clarity, all H atoms are omitted, except for the atom at C2 position. Selected bond lengths (Å) and angles (°): C(2)–F(2) 1.450(2), C(2)–H(1) 1.000(1), N(1)–C(2) 1.397(1), N(1)–C(2)–F(1) 110.89(9), N(1)–C(2)–H(2) 110.2(1), F(1)–C(2)–H(2) 110.24(9). | ||
 | ||
| Fig. 5 Packing of two asymmetric units of MeCAAC(H)F (1). The ellipsoids are drawn at 50% probability. For clarity, all hydrogen atoms are omitted except for the one at C2 position. Selected intermolecular hydrogen bond lengths (Å) and bond angles (°): F(1)⋯C(2) 3.646(1), F(1)⋯H(2) 2.7992(7), C(2)–H(2) 1.000(1), F(1)–H(2)–C(2) 142.80(7); F(1)⋯C(18) 3.468(2), F(1)⋯H(18A) 2.506(1), C(18)–H(18A) 0.980(1), F(1)–H(18A)–C(18) 166.88(8). | ||
Initially, we expected 1 to have an ionic rather than a neutral form, just like the structurally related imidazolium-based fluoride [IPrH][F].30 It turned out that 1 prefers to form a saturated heterocyclic ring with sp3 hybridisation at the C2 position. Also, in solution, compound 1 adopts a neutral chiral form. 1H and 19F NMR measurements performed at low temperature (−30 °C) confirm this, as the peaks of the CHF group split to form a doublet corresponding to the 2JH–F geminal coupling. In the 1H NMR spectrum it appears at 5.13 ppm (2JH–F = 82.9 Hz), while in the 19F NMR spectrum it is at −105.30 ppm (2JH–F = 82.9 Hz). This agrees well with the 84 Hz value calculated at the M06-2X/def2-TZVPP level. However, if the measurement temperature is increased to room temperature, the proton–fluorine coupling disappears, which is probably due to the dissociation of 1 and a rapid exchange between the neutral and ionic forms (see Fig. S4 and S5 in the ESI‡). This exchange, although strongly endergonic, should be quite rapid at room temperature as the calculated activation Gibbs free energy does not exceed 50 kJ mol−1 (see section 2 – Computational study).
The missing [MeCAACH][F(HF)] (2) salt was prepared by dissolving 1 and 3 in MeCN and stirring for 24 hours. After removal of all volatiles, compound 2 was obtained (Scheme 2). Unfortunately, the reaction is reversible and compound 2 tends to decompose easily back into the mixture of 1 and 3. Also, crystallisation of the mixture generally resulted in single crystals of 1 or 3. The formation of single crystals was strongly dependent on the crystallisation procedure, especially on the polarity of the solvent used. In some cases, only single crystals of 1 or 3 were observed, while sometimes both crystals formed at the same time. Finally, we succeeded in structurally characterising 2 in a co-crystal with two molecules of 1. Single crystals of [MeCAACH][F(HF)]·2MeCAAC(H)F (2a) were obtained by slow evaporation of the solvent from the solution of 2 in MeCN and t-BuOH. Our results show that 2 does indeed exist but is in equilibrium with other fluorinated species. 2a crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . Its asymmetric unit consists of a central molecule of 2 and two adjacent molecules of 1. The two molecules of 1 are aligned to form interactions with the central [F(HF)]− anion. For clarity, only the structure of 2 is shown in Fig. 6. The complete asymmetric unit of 2a is shown in the ESI, Fig. S18.‡ The [F(HF)]− anion adopts a linear geometry with an F⋯F distance of 2.235(2) Å. The F⋯F distance is slightly shorter than for typical bifluoride salts (2.26–2.28 Å), but according to the literature, even shorter distances have been reported for [F(HF)]− anions (2.213(4) Å).40
. Its asymmetric unit consists of a central molecule of 2 and two adjacent molecules of 1. The two molecules of 1 are aligned to form interactions with the central [F(HF)]− anion. For clarity, only the structure of 2 is shown in Fig. 6. The complete asymmetric unit of 2a is shown in the ESI, Fig. S18.‡ The [F(HF)]− anion adopts a linear geometry with an F⋯F distance of 2.235(2) Å. The F⋯F distance is slightly shorter than for typical bifluoride salts (2.26–2.28 Å), but according to the literature, even shorter distances have been reported for [F(HF)]− anions (2.213(4) Å).40
 | ||
| Fig. 6 Structure of the asymmetric unit of [MeCAACH][F(HF)] (2). The ellipsoids are drawn at 50% probability. For clarity, all H atoms are omitted, except for the atom at C2 position. Selected bond lengths (Å) and angles (°): F(1)⋯F(2) 2.235(2), C(2)–H(2) 0.950(2), N(1)–C(2) 1.275(3), F(1)–H(1)–F(2) 168(4), N(1)–C(2)–H(2) 122.8(2). | ||
Compound 2 has a high tendency to decompose back into the reactants. Therefore, it cannot be found in solution on its own. In solution it exists only in equilibrium with 1 and 3. NMR measurements at room temperature showed no signal in the 19F NMR spectra, which is due to transitions between species as the fluorides change from one form to another. To slow down this process, the NMR measurements were repeated at a lower temperature (−30 °C). This time, 3 distinct peaks were visible in the 19F NMR spectra, assigned to compound 1 at −105.18 ppm, 2 at −144.51 ppm and 3 at −166.42 ppm. For each species only one resonance could be detected due to the rapid exchange in solution. All measured spectra are shown in the ESI, Fig. S8 and S9.‡
Computational study of the relative stability of fluoropyrrolidines and dihydropyrrolium fluorides
The limited stability of 2 compared to the mixture of 1 and 3 was the trigger for our computational study, which addressed several issues, namely (1) the relative stability of fluoropyrrolidines compared to dihydropyrrolidinium fluorides and hydrofluorides depending on the amount of HF present; (2) in the case where dihydropyrrolium fluorides are more stable, where the anions are preferentially localised; and (3) how the equilibrium of the isodesmic reaction is oriented 1 + 3 → 2 + 2, with experimental observation indicating the backward orientation.We started by looking for the stability of the respective structures 1, 2 and 3 and investigated the energies and geometries of the following systems (Scheme 3).
 | ||
| Scheme 3 Searched structures in fluoropyrrolidine–dihydropyrrolium fluoride systems. | ||
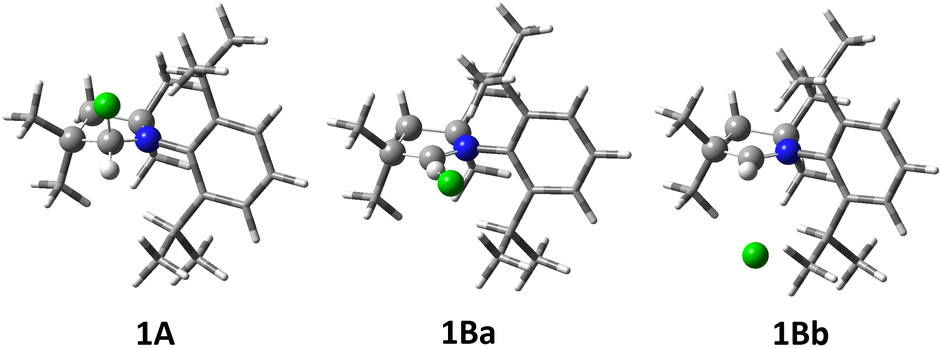 | ||
| Fig. 7 Selected computed structures of fluoropyrrolidine 1A and dihydropyrrolium fluorides 1B. For clarity, the substituents on the heterocyclic ring are drawn as tubes. | ||
 | ||
| Fig. 8 Selected computed structures of fluoropyrrolidine 2A and dihydropyrrolium dihydrogen difluorides 2B at the M06L/def2-SVP level. For clarity, the substituents on the heterocyclic ring are drawn as tubes. | ||
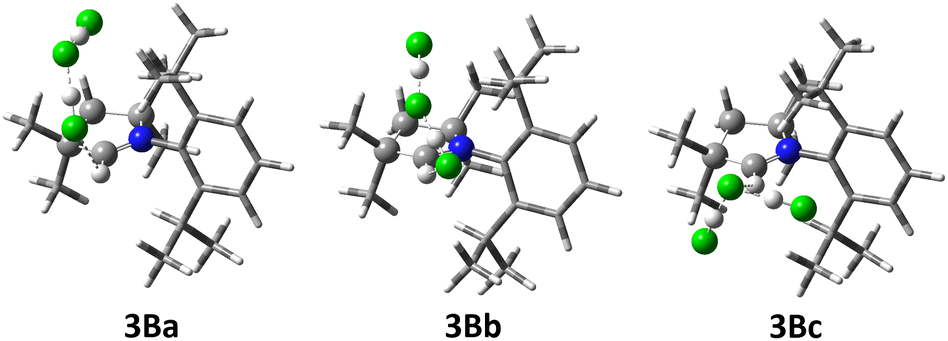 | ||
| Fig. 9 Selected computed structures of dihydropyrrolium dihydrogen trifluorides 3B at the M06L/def2-SVP level. For clarity, the substituents on the heterocyclic ring are drawn as tubes. | ||
 | ||
| Scheme 4 Isodesmic reaction of MeCAAC(H)F (1) + [MeCAACH][F(HF)2] (3) → [MeCAACH][F(HF)] (2) + [MeCAACH][F(HF)] (2). | ||
Properties of 3 as a nucleophilic fluorination reagent on organic substrates
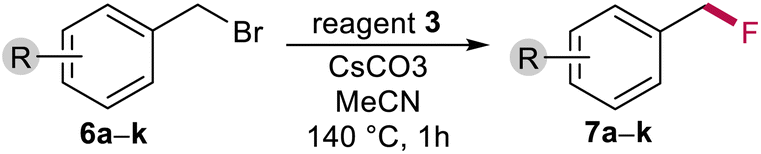
As a model reaction we chose the fluorination of benzyl bromide to its corresponding fluoride with reagent 3. As a model substrate we used 4-tert-butylbenzyl bromide 6a because of its low volatility. MeCN was used as a solvent, the base was added and the reaction was heated at 140 °C for 1 hour in a Teflon-lined high-pressure reactor. The conversion to 4-tert-butylbenzyl fluoride 7a was determined using qNMR and naphthalene as an internal standard. The results are given in Table 1 and in Table S7 in the ESI.‡|
|
|||||
|---|---|---|---|---|---|
| Entry | 3 [eq.] | Base (eq.) | 6a (%) | Conversiona | Yield 7aa (%) |
| a Conversions and yields were determined with qNMR with naphthalene as an internal standard. b 2,6-Di-tert-butylpyridine. | |||||
| 1 | 1 | — | 0 | 0 | 0 |
| 2 | 1 | DBU 2 | 0 | 100 | 22 |
| 3 | 1.1 | DIPEA 1.8 | 0 | 100 | 60 |
| 4 | 1 | t BuPyb 1 | 49 | 51 | 49 |
| 5 | 1 | NaHCO3 1 | 30 | 72 | 64 |
| 6 | 1 | Na2CO3 1 | 20 | 80 | 74 |
| 7 | 1 | K2CO3 1 | 14 | 86 | 78 |
| 8 | 1 | K2CO3 1/18-crown-6 | 0 | 99 | 98 |
| 9 | 1 | Cs2CO3 1.2 | 0 | 100 | 98 |
| 10 | 1 | Cs2CO3 0.5 | 0 | 100 | 99 |

We tested different bases in the reaction system. Amine bases such as N,N-diisopropylethylamine (DIPEA) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) gave poor yields due to the formation of an adduct with the parent benzyl bromide (Table 1, entries 1–5). Even the use of an extremely hindered base such as 2,6-di-tert-butylpyridine did not improve the yield and resulted in low conversion. Next, we tested alkali carbonates and bicarbonates (Table 1, entries 7–12). Caesium carbonate gave the best result with quantitative conversion and 98% yield of the desired benzyl fluoride 7a. We suspected that the solubility and not the type of cation plays a crucial role in the reaction, which is confirmed by the fact that when potassium carbonate is used as a base and 18-crown-6 is added to increase the solubility, the conversion and yield are quantitative (Table 1, entry 8).
Since reagent 3 contains three fluorine atoms in its structure, we reduced the loading of the reagent. The yield and conversion of 6a began to drop significantly below 0.7 equivalents of 3 (Fig. 10). For more details, see Table S8 in the ESI.‡
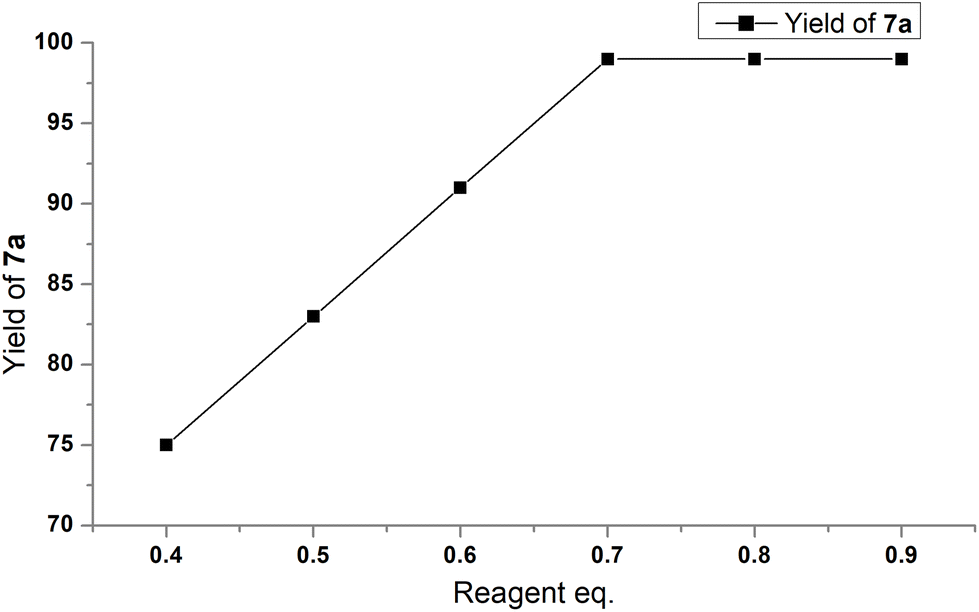 | ||
| Fig. 10 Linear increase in yield of 7avs. reagent loading. After 0.7 eq. of 3 the yield is quantitative. | ||
Reagent 3 was also tested on various substituted benzyl bromides to see how the electronic nature of the side groups affected the fluorination reaction. We were able to obtain excellent to quantitative yields regardless of the nature of the substituents. In the case of electron donating groups such as –OMe, reaction times were extended to 17 hours. The quantitative yields, even in the case of sterically hindered substates such as 2-Ph-benzyl bromide, demonstrate the excellent fluorination potential of this reagent (Table 2).
|
|
|||||
|---|---|---|---|---|---|
| 6 | Substituent | Yieldb | 6 | Substituent | Yieldb |
| a 0.1 mmol substituted benzyl alcohol, reagent 3, Cs2CO3 (1.5 eq.), naphthalene (approx. 0.1 mmol) were dissolved in MeCN (1 mL) and heated in Teflon reactor at 140 °C for 1 hour. b Conversions and yields were determined with qNMR with naphthalene as an internal standard. c Reaction time was extended to 17 hours. | |||||
| 6a | 4-tBu | 98 | 6g | 3-Cl | 94 |
| 6b | 4-NO2 | 96 | 6h | 4-Me | 98 |
| 6c | 3-OMe | 96c | 6i | H | 99 |
| 6d | 3-Br | 96 | 6j | 4-F | 97 |
| 6e | 4-Br | 100 | 6k | 2-Ph | 94 |
| 6f | 3,5-CF3 | 96 | |||
Optimised reaction conditions were then used for the fluorination of primary alkyl bromides. 1-Bromodecane was used as a model substrate. Alkyl bromides react much slower than benzyl substrates, with elimination to alkene being a competitive process. The best yield of fluorination was observed in MeCN as solvent and Cs2CO3 as base with a 91% yield of 9 and only 2% of decan-1-ene 10 after 30 hours of reaction time (Table S9 in ESI‡). Secondary bromides, however, are even less reactive and more prone to elimination. After 16 hours of reaction time at 140 °C, all the starting 2-bromooctane was consumed. The yield did not exceed 30% despite changes in temperature and reaction times. Most of the starting material was converted to 1- or 2-alkene. Due to the low reactivity of 2° bromides, we used 2° mesylates with HMPA as solvent and were able to obtain reaction yields of more than 79% of 12, which are comparable to those reported in the literature (see Table S10 in the ESI‡ for more details on optimising the reaction conditions). In addition to benzyl and alkyl substrates, we also fluorinated other substrates such as silanes, sulfonyls, acid chlorides and α-bromoketones with good to excellent yields (Scheme 5). 3 was also used as a reagent for the nucleophilic aromatic substitution of 1,4-dinitrobenzene to 4-nitrofluorobenzene in 87% yield and for fluorination of the less reactive benzyl chloride in 66% yield. After completion of the reactions, bromide salt of 3 [MeCAACH][Br] was isolated with precipitation with hexane and was regenerated as 4 in 80% yield by the use of anhydrous HF. 4 can be easily transformed to 3, which could be used for further fluorination reactions.
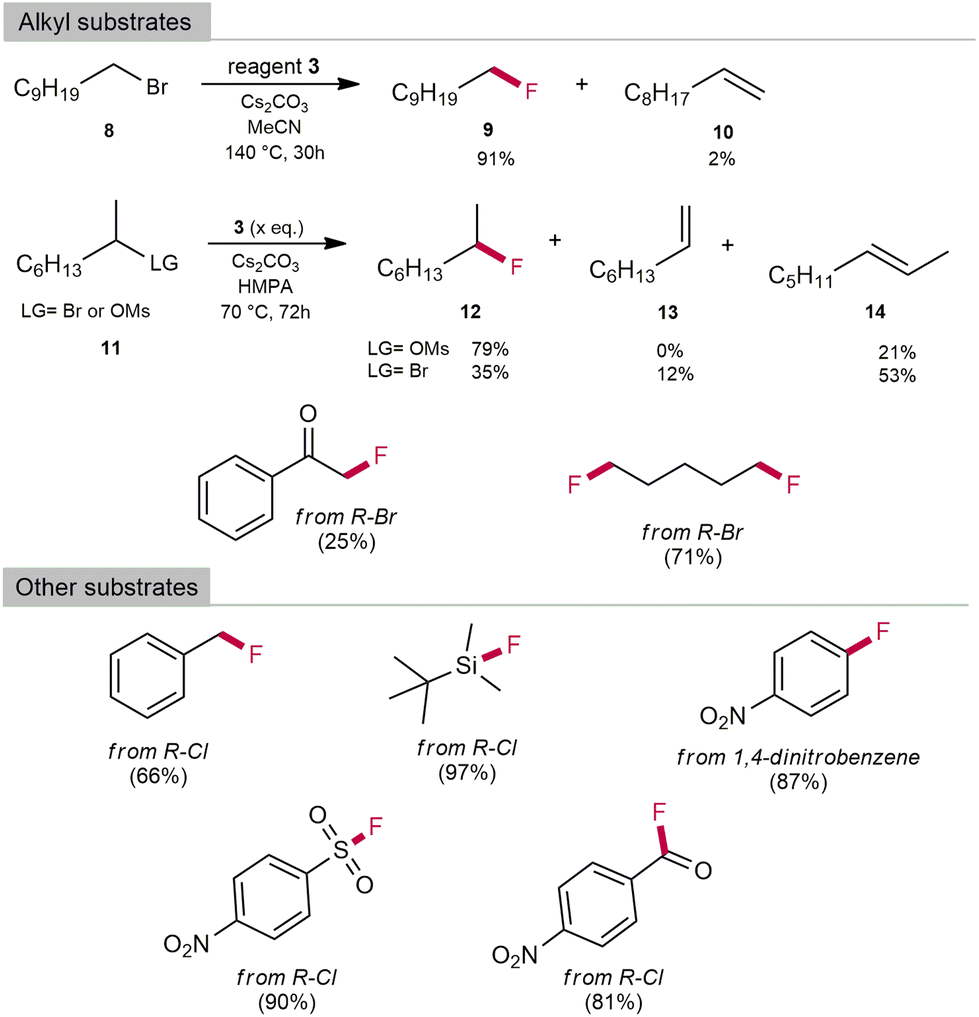 | ||
| Scheme 5 Fluorination scope of 3 (qNMR yields). 1H and 19F NMR shifts matched those previously reported (see Table 12 in the ESI‡). | ||
Experimental
Synthesis of salts
Syntheses of cyclic iminium salts were carried out under an inert atmosphere of dry argon using the standard Schlenk technique or in a glovebox (M. Braun) maintained below 0.1 ppm O2 and H2O. Reactions were carried out in FEP (tetrafluoroethylene-hexafluoropropylene) tubes, reaction vessels equipped with PTFE (polytetrafluoroethylene) valves. Anhydrous HF was handled in a vacuum line made entirely of PTFE. Samples were stored in a glovebox in PP (polypropene) plastic containers. Toluene was dried in a mixture of sodium and benzophenone, distilled under Ar atmosphere, degassed by freeze–pump–thaw cycles and stored over 3 Å molecular sieves. MeCN was degassed by freeze–thaw cycles and stored over 3 Å molecular sieves. Deuterated NMR solvents were stored over 3 Å molecular sieves in a glovebox. Cyclic iminium-based chloride salt [MeCAACH][Cl(HCl0.5)] was prepared according to the modified procedure described previously.47 Anhydrous HF (Linde, 99.995%) was dried by mixing with K2NiF6 (Advance Research Chemicals, Inc.) before use. CsF (Sigma-Aldrich, 99%) was dried overnight in vacuo at 150 °C and stored in a glovebox. NMR spectra were recorded in 5 mm glass NMR tubes with an FEP liner. Measurements were performed using a Bruker AVANCE NEO 400 MHz NMR spectrometer. Chemical shifts of 1H and 13C{1H} were referenced to the residual signals of the deuterated solvent, while 19F references were calculated according to IUPAC guidelines and are given relative to CFCl3.48 Elemental analysis was performed using the CHNS elemental analyser vario EL cube (Elementar) in CHN mode. Raman spectra were recorded using a Horiba Jobin Yvon Labram-HR spectrometer coupled with an Olympus BXFM-ILHS microscope at room temperature. The samples were excited with the 633 nm emission line of a He–Ne laser.Caution! Anhydrous HF is an extremely corrosive and highly dangerous gas. It should be handled with care in a well-ventilated hood. Always wear protective clothing, gloves and a face mask.
Crystal structure determination
Crystal data for all compounds were collected at 150 K with a Gemini A diffractometer equipped with an Atlas CCD detector using graphite-monochromated Cu Kα radiation. The data were processed using the CrysAlisPro software package.49 Analytical absorption correction was applied to all data sets.50 Structures were solved using the SHELXT programme.51 Structure refinement was performed using the SHELXL software52 implemented in the Olex2 programme package.53 Figures were made using Diamond.54Computational details
The orientation computations were accomplished by Gaussian16 program suite55 using pure M-06L functional,56 which enabled the use of the RI (resolution of identity) approach,57 together with the double-ζ def2-SVP basis set,46 which greatly accelerated the initial calculations. To better describe the anionic structures, we also used double-ζ def2-SVPD basis set58 with additional diffuse functions. No accelerating approximation for exchange integrals is available in Gaussian16. Therefore, we switched to the ORCA computational programme,41 which uses the efficient RIJCOSX42 approximation to accelerate the computations of hybrid functional computations. We used the M06-2X hybrid functional43 together with the triple-ζ def2-TZVP basis set,46 the minimally augmented ma-def2-TZVP basis set44 or the fully augmented def2-TZVPD basis set.58 Weigend's universal auxiliary basis set59 was used for the RI approximation calculations. The MeCN solvent was simulated with the SMD variant of the IEF-PCM method60 and the description of the non-covalent interactions was improved by the dispersion correction61 with the Becke–Johnson damping.62 Finally, the DSD method blending DFT and perturbation theory together with the def2-TZVPP basis set, was used for the best quality computations.45 Frequency analysis was performed for all structures to confirm them as minimae on PES and to obtain their free Gibbs energies. All calculated structures (xyz files and free Gibbs energies) are listed in the ESI.‡Details of the reactivity analysis
All chemicals, materials and solvents were purchased from commercial sources and used without further purification unless otherwise stated. Solvents (MeCN) were distilled over sodium wire, DCM was distilled over CaH2 and then stored over 3 Å molecular sieves (20% V/V) for at least 72 hours before use. 1H and 19F NMR spectra were recorded using a Bruker Ascend 600 MHz NMR spectrometer (600 MHz for 1H, 471 MHz for 19F). Chemical shifts were reported as a delta scale in ppm relative to CDCl3 (residual CHCl3δ = 7.26 ppm for 1H, 77.16 ppm for 13C). qNMR measurements were performed using a bitmap scan and T = 5T1 (relaxation time of 45 s).Conclusions
Treatment of the hydrochloride salt [MeCAACH][Cl(HCl)0.5] with anhydrous HF resulted in the formation of poly(hydrogen fluoride) salts. By removing HF under different conditions [MeCAACH][F(HF)3] (4) or [MeCAACH][F(HF)2] (3) could be obtained quantitatively. 3 is the most stable salt in this system and can be used efficiently as a nucleophilic fluorinating reagent for a range of substrates including benzyl bromides, 1- and 2-alkyl bromides, silanes and sulfonyls with good to excellent yields of desired fluorides.Reaction of 3 with a slight excess of basic CsF removed all HF from 3 and led to the formation of MeCAAC(H)F (1), a neutral chiral molecule that is highly soluble in toluene. Mixing 1 and 3 in equimolar amounts produced [MeCAACH][F(HF)] (2), which, however proved to be only partially stable as it has a tendency to decompose back into its reactants. To better understand the relationship between 1, 2 and 3, a computational study was carried out.
The computational results showed that minimally triple-ζ basis set must be used to find correct structures and energies of 1, 2 and 3. The computation of the isodesmic reaction of isolated 1 + 3 → 2 + 2 surprisingly did not confirm the experimental instability of 2, which is probably caused by the exergonic formation of 1 dimer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Slovenian Research Agency (ARRS) for financial support of the Project N1-0185 (Advanced reagents for (asymmetric) nucleophilic fluorination) and the Research Programmes P1-0045 (Inorganic Chemistry and Technology) and P1-0134 (Chemistry for sustainable development) and the Czech Science Foundation for financial support of the Project No. 21-29531K (Advanced reagents for (asymmetric) nucleophilic fluorination). The authors thank the Slovenian NMR Centre (National Institute of Chemistry) for its resources and support, Prof. Maja Ponikvar-Svet and Mira Zupančič for performing CHN elemental analysis and Asst. Prof. Evgeny Goreshnik for help with the crystal structure determination and analysis.References
- P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073–9174 CrossRef CAS PubMed.
- C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929–2942 RSC.
- C. Chatalova-Sazepin, R. Hemelaere, J. F. Paquin and G. M. Sammis, Synthesis, 2015, 2554–2569 CAS.
- R. Szpera, D. F. J. Moseley, L. B. Smith, A. J. Sterling and V. Gouverneur, Angew. Chem., Int. Ed., 2019, 58, 14824–14848 CrossRef CAS PubMed.
- S. Caron, Org. Process Res. Dev., 2020, 24, 470–480 CrossRef CAS.
- D. E. Yerien, S. Bonesi and A. Postigo, Org. Biomol. Chem., 2016, 14, 8398–8427 RSC.
- B. Baasner and E. Klauke, J. Fluorine Chem., 1982, 19, 553–564 CrossRef CAS.
- S. Liang, G. B. Hammond and B. Xu, Chem. – Eur. J., 2017, 23, 17850–17861 CrossRef CAS PubMed.
- G. Haufe, in Fluorination, ed. J. Hu and T. Umemoto, Springer Singapore, Singapore, 2020, 137–181 Search PubMed.
- G. A. Olah, T. Mathew, A. Goeppert, B. Török, I. Bucsi, X.-Y. Li, Q. Wang, E. R. Marinez, P. Batamack, R. Aniszfeld and G. K. S. Prakash, J. Am. Chem. Soc., 2005, 127, 5964–5969 CrossRef CAS PubMed.
- G. A. Olah, J. T. Welch, Y. D. Vankar, M. Nojima, I. Kerekes and J. A. Olah, J. Org. Chem., 1979, 44, 3872–3881 CrossRef CAS.
- S. Liang, F. J. Barrios, O. E. Okoromoba, Z. Hetman, B. Xu and G. B. Hammond, J. Fluorine Chem., 2017, 203, 136–139 CrossRef CAS PubMed.
- O. E. Okoromoba, G. B. Hammond and B. Xu, Org. Lett., 2015, 17, 3975–3977 CrossRef CAS PubMed.
- O. E. Okoromoba, Z. Li, N. Robertson, M. S. Mashuta, U. R. Couto, C. F. Tormena, B. Xu and G. B. Hammond, Chem. Commun., 2016, 52, 13353–13356 RSC.
- O. E. Okoromoba, J. Han, G. B. Hammond and B. Xu, J. Am. Chem. Soc., 2014, 136, 14381–14384 CrossRef CAS PubMed.
- Z. Lu, X. Zeng, G. B. Hammond and B. Xu, J. Am. Chem. Soc., 2017, 139, 18202–18205 CrossRef CAS PubMed.
- R. Hagiwara, T. Hirashige, T. Tsuda and Y. Ito, J. Fluorine Chem., 1999, 99, 1–3 CrossRef CAS.
- R. Hagiwara and Y. Ito, J. Fluorine Chem., 2000, 105, 221–227 CrossRef CAS.
- R. Hagiwara, T. Hirashige, T. Tsuda and Y. Ito, J. Electrochem. Soc., 2002, 149, D1–D6 CrossRef CAS.
- R. Hagiwara, K. Matsumoto, Y. Nakamori, T. Tsuda, Y. Ito, H. Matsumoto and K. Momota, J. Electrochem. Soc., 2003, 150, D195–D199 CrossRef CAS.
- K. Matsumoto, R. Hagiwara, R. Yoshida, Y. Ito, Z. Mazej, P. Benkič, B. Žemva, O. Tamada, H. Yoshino and S. Matsubara, Dalton Trans., 2004, 144–149 RSC.
- K. Matsumoto, T. Tsuda, R. Hagiwara, Y. Ito and O. Tamada, Solid State Sci., 2002, 4, 23–26 CrossRef CAS.
- S. Bouvet, B. Pégot, J. Marrot and E. Magnier, Tetrahedron Lett., 2014, 55, 826–829 CrossRef CAS.
- R. P. Singh and J. L. Martin, J. Fluorine Chem., 2016, 181, 7–10 CrossRef CAS.
- H. Yoshino, K. Matsumoto, R. Hagiwara, Y. Ito, K. Oshima and S. Matsubara, J. Fluorine Chem., 2006, 127, 29–35 CrossRef CAS.
- B. Alič, J. Petrovčič, J. Jelen, G. Tavčar and J. Iskra, J. Org. Chem., 2022, 87, 5987–5993 CrossRef PubMed.
- B. Alič, M. Tramšek, A. Kokalj and G. Tavčar, Inorg. Chem., 2017, 56, 10070–10077 CrossRef PubMed.
- Ž. Zupanek, M. Tramšek, A. Kokalj and G. Tavčar, Inorg. Chem., 2018, 57, 13866–13879 CrossRef PubMed.
- E. Gruden, M. Tramšek and G. Tavčar, Organometallics, 2022, 41, 41–51 CrossRef CAS.
- B. Alič and G. Tavčar, J. Fluorine Chem., 2016, 192, 141–146 CrossRef.
- M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed.
- M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed.
- V. Lavallo, Y. Canac, B. Donnadieu, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed., 2006, 45, 3488–3491 CrossRef CAS PubMed.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–441 CrossRef CAS PubMed.
- Z. R. Turner, Chem. – Eur. J., 2016, 22, 11461–11468 CrossRef CAS PubMed.
- D. Wiechert, D. Mootz, R. Franz and G. Siegemund, Chem. – Eur. J., 1998, 4, 1043–1047 CrossRef CAS.
- T. Enomoto, Y. Nakamori, K. Matsumoto and R. Hagiwara, J. Phys. Chem. C, 2011, 115, 4324–4332 CrossRef CAS.
- T. von Rosenvinge, M. Parrinello and M. L. Klein, J. Chem. Phys., 1997, 107, 8012–8019 CrossRef CAS.
- S. I. Ivlev, T. Soltner, A. J. Karttunen, M. J. Mühlbauer, A. J. Kornath and F. Kraus, Z. Anorg. Allg. Chem., 2017, 643, 1436–1443 CrossRef CAS.
- W. W. Wilson, K. O. Christe, J. Feng and R. Bau, Can. J. Chem., 1989, 67, 1898–1901 CrossRef CAS.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- S. Kozuch and J. M. L. Martin, Phys. Chem. Chem. Phys., 2011, 13, 20104–20107 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- E. Gruden and G. Tavčar, Polyhedron, 2021, 196, 115009 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. C. De Menezes, P. Granger, R. E. Hoffman and K. W. Zilm, Magn. Reson. Chem., 2008, 46, 582–598 CrossRef CAS PubMed.
- Rigaku Oxford Diffraction, CrysAlisPro, Software system, Version 1.171.41.120a (release date 26-10-2021), Rigaku Corporation, Wroclaw, Poland, 2021 Search PubMed.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887–897 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Diamond – Crystal and Molecular Structure Visualization (v4.6.8), Crystal Impact – Dr. H. Putz & Dr. K. Brandenburg GbR, Kreuzherrenstr. 102, 53227 Bonn, Germany, https://www.crystalimpact.com/diamond/ (accessed 15 May 2023).
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision C.01), Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- O. Vahtras, J. Almlöf and M. W. Feyereisen, Chem. Phys. Lett., 1993, 213, 514–518 CrossRef CAS.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
Footnotes |
| † This work is partially based on the doctoral dissertation defended by E. Gruden (2022) at Jožef Stefan International Postgraduate School. |
| ‡ Electronic supplementary information (ESI) available: NMR and Raman spectra, crystal structure data, additional computational results and reactivity analysis data. CCDC 2263384–2263388. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01476b |
| This journal is © The Royal Society of Chemistry 2023 |