
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cancer stem cell activity of copper(II)-terpyridine complexes with aryl sulfonamide groups†
Karampal
Singh‡
,
Joshua
Northcote-Smith‡
,
Kuldip
Singh
and
Kogularamanan
Suntharalingam
 *
*
School of Chemistry, University of Leicester, Leicester LE1 7RH, UK. E-mail: k.suntharalingam@leicester.ac.uk
First published on 16th June 2023
Abstract
Copper(II)-terpyridine complexes are endowed with the ability to generate reactive oxygen species (ROS) and induce cancer cell death. Here we report the synthesis, characterisation, and anti-breast cancer stem cell (CSC) properties of a series of copper(II)-terpyridine complexes containing aryl sulfonamide groups (1–5). All of the copper(II)-terpyridine complexes adopt distorted square pyramidal geometries and are suitably stable in biologically relevant solutions (PBS and cell culture media). The p-toluene sulfonamide-bearing copper(II)-terpyridine complex 1 is 6–8-fold more potent towards breast CSCs than salinomycin (an established anti-CSC agent) and cisplatin (a metal-based anticancer drug). The copper(II)-terpyridine complex 1 also reduces the formation, size, and viability of three-dimensionally cultured mammospheres, to a similar or better extent than salinomycin and cisplatin. Mechanistic studies show that 1 successfully enters breast CSCs, generates intracellular ROS at short exposure times, partially induces endoplasmic reticulum stress, and triggers apoptosis. To the best of our knowledge, this is the first study to investigate the anti-breast CSC properties of copper(II)-terpyridine complexes.
Introduction
Despite humankind’s significant advances and breakthroughs in cancer diagnosis and treatment, cancer remains to be a global healthcare challenge.1 There are currently around 3 million people in the UK living with cancer which is expected to rise to around 5.3 million by 2040.2 On average in the UK, someone is diagnosed with cancer every 90 seconds, and the daily death toll is around 460.2 Cancer relapse and metastasis, the unfortunate return of the disease after remission and its spread throughout the body, are the leading cause of cancer associated deaths.3,4 Clinical studies have demonstrated that this phenomenon is due to, in part, the existence of cancer stem cells (CSCs).5 CSCs are a small sub-population of tumour cells with the ability to self-renew, differentiate, and initiate and maintain tumour growth.5,6 While conventional cancer therapies (chemotherapy and radiation) effectively reduce tumour mass through the passive targeting of rapidly dividing bulk cancer cells (non-CSCs), they are unable to eliminate CSCs at clinically safe doses.7–10 CSCs are intrinsically resistant and undergo controlled quiescent growth cycles.11 As a result, the CSC population not only becomes enriched after treatment but also enters a transient dormant state that may result in aggressive tumour reoccurrence and metastasis at a later date.11,12 There is therefore an urgent need to develop novel antitumour treatments that can effectively eradicate the entirety of the cancerous population (bulk cancer cells and CSCs). Although most of the chemical agents under (pre)clinical investigation as anti-CSC agents are purely organic,13–16 we and others have shown that metal complexes can exhibit clinically relevant potencies.17–19Copper is an essential trace element and the average adult human body contains 50–120 mg.20,21 Copper is vital for several biological processes including growth, cardiovascular integrity, lung elasticity, neovascularization, neuroendocrine function, and iron metabolism.22 Given the prominent role of copper in the human body, humans have evolved mechanisms to overcome copper overload.22 Copper is thus less toxic to humans than non-essential exogenous metals. This fact has prompted several investigations into copper-containing compounds as less toxic alternatives to traditional platinum-based anticancer drugs (cisplatin, carboplatin, and oxaliplatin).23–25 According to the large body work already published on the anticancer properties of copper complexes, cell toxicity and its associated mechanism of action is highly dependent on the coordinating ligands.23–25 A large percentage of the reported copper complexes kill cancer cells by inducing oxidative stress through the generation of reactive oxygen species (ROS).23,24 Other modes of action include specific organelle damage (mitochondria or endoplasmic reticulum), genomic DNA damage, anti-angiogenesis effects, cell cycle arrest, topoisomerase I,II inhibition, and proteasome disruption.23–28 The ability of copper to undergo efficient redox cycling between the copper(I) and copper(II) states under physiological conditions is central to the cytotoxic mechanism of action of most anticancer copper complexes reported.23–25
Copper(II)-terpyridine complexes are one of most widely studied family of anticancer copper agents.29–35 Copper(II)-terpyridine complexes are generally thought to act as artificial nucleases through the production of ROS via Fenton-type reactions.28 Over that last few decades, several copper(II)-terpyridine complexes (mono- and bis-terpyridine and mixed ligand systems) have been reported to effectively cleave DNA, produce ROS, and kill bulk cancer cells.23,24,28 Nevertheless, copper(II)-terpyridine complexes have never been challenged against CSCs of any tissue type. Here we report a series of new copper(II)-terpyridine complexes 1–5 (Fig. 1A) with promising anti-breast CSC activities. The terpyridine ligand within the copper(II) complexes is functionalised with various aryl sulfonamide groups to enable effective CSC uptake and enhance endoplasmic reticulum (ER) targeting. Sulfonamides are known to facilitate localisation in the ER by targeting sulfonylurea receptors on ATP-sensitive potassium cation channels in the ER membrane, similar to the mode of action of the commercial ER stain ‘ER-Tracker Red’.36–40 Further, breast CSCs are thought to be vulnerable to ER stress inducers due to their tendency to synthesise and secrete large quantities of extracellular matrix proteins, which require processing by the ER.41 Thus the presence of the sulfonamide groups within the copper(II)-terpyridine complexes 1–5 is envisaged to promote ER stress and boost breast CSC potency. The mechanism of action of the most effective copper(II)-terpyridine complex within the reported series 1, in terms of intracellular ROS generation, ER stress activation, and apoptosis induction, is also documented herein. We have previously reported a copper(II) complex containing a bulky phenanthroline ligand and a Schiff base ligand which induced oxidative ER stress, apoptosis, and immunogenic cell death of breast CSCs.42 This copper(II) complex intrinsically targeted the ER in breast CSCs due to its lipophilicity. In this work we endeavour to rationally target the ER in breast CSCs using sulfonamide groups appended to a copper(II)-containing terpyridine core.
 | ||
| Fig. 1 (A) The reaction scheme for the preparation of the copper(II)-terpyridine complexes 1–5. (B) X-ray structures of the copper(II)-terpyridine complexes 1–5 containing L1–L5. Ellipsoids are shown at 50% probability. C in grey, N in dark blue, O in red, Cl in light green, S in yellow, F in dark green, Cu in orange. H atoms and the co-crystallizing solvent molecules have been omitted for clarity. | ||
Results and discussion
Synthesis and characterisation of copper(II) complexes with aryl sulfonamide-functionalised terpyridine ligands
Five aryl sulfonamide-functionalised terpyridine ligands (L1–L5) and their corresponding copper(II) complexes (1–5) were prepared as depicted in Fig. 1A and S1.† The terpyridine ligands L1–L5 were prepared in two steps (Fig. S1†). First, 4′-chloro-2,2′:6′,2′′-terpyridine was reacted with 2-aminoethanol in the presence of potassium hydroxide in DMSO to form 2-([2,2′:6′,2′′-terpyridin]-4′-yloxy)ethan-1-amine, which was isolated as a light-brown solid in a good yield (81%). Then, 2-([2,2′:6′,2′′-terpyridin]-4′-yloxy)ethan-1-amine was reacted with various arylsulfonyl chlorides in the presence of triethylamine in dichloromethane to form L1–L5, which were isolated as white solids in reasonable to good yields (63–93%). The 4-fluorobenzenesulfonyl functionalised terpyridine ligand L4 required further purification. This was achieved by recrystallisation in ethanol. The precursor ligand (2-([2,2′:6′,2′′-terpyridin]-4′-yloxy)ethan-1-amine) and L1–L5 were characterised by 1H, 19F{1H} (for L4 and L5) and 13C NMR spectroscopy, high-resolution ESI mass spectrometry, IR spectroscopy, and elemental analysis (see ESI, Fig. S2–S22†). A downfield shift in the methylene peaks from 2.83 and 3.61 ppm in the 1H NMR spectrum for ethanolamine to 3.16 and 4.27 ppm in the 1H NMR spectrum for 2-([2,2′:6′,2′′-terpyridin]-4′-yloxy)ethan-1-amine indicated successful attachment of the ethanolamine group to the terpyridine core (Fig. S2†). Successful conversion of 2-([2,2′:6′,2′′-terpyridin]-4′-yloxy)ethan-1-amine to L1–L5 was indicated by the appearance of a sulfonamide peak (4.93–5.12 ppm) in the 1H NMR spectra of L1–L5 (Fig. S4, S6, S8, S10 and S13†). This was supported by a shift in the methylene peak closest to the sulfonamide functionality from 3.16 ppm in 2-([2,2′:6′,2′′-terpyridin]-4′-yloxy)ethan-1-amine (Fig. S2†) to 3.45–3.51 ppm in L1–L5 (Fig. S4, S6, S8, S10 and S13†). In the 1H NMR spectrum of L1, the toluene methyl group was observed at 2.36 ppm (Fig. S4†). For L4 and L5, single peaks were observed in their 19F{1H} NMR spectra, confirming the presence of fluorinated aryl groups (Fig. S12 and S15†). Distinctive molecular ion peaks corresponding to protonated (2-([2,2′:6′,2′′-terpyridin]-4′-yloxy)ethan-1-amine) or L1–L5 [M + H]+ with the appropriate isotopic pattern were observed in the positive mode of the high-resolution ESI mass spectra (Fig. S17–S22†). The purity of the aryl sulfonamide-functionalised terpyridine ligands L1–L5 was confirmed by elemental analysis.The copper(II)-terpyridine complexes 1–5 were synthesised by reacting equimolar quantities of copper(II) chloride dihydrate and L1–L5 in a methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dichloromethane mixture at room temperature for 2.5–4 h (Fig. 1A). The copper(II)-terpyridine complexes 1–5 were isolated as light blue solids in reasonable to good yields (47–83%) and characterised by high resolution ESI mass spectrometry, IR spectroscopy, elemental analysis, and single crystal X-ray diffraction (see ESI, Fig. S23–S28†). Distinctive molecular ion peaks corresponding to [M − Cl]+ with the appropriate isotopic pattern were observed in the positive mode of the high-resolution ESI mass spectra (m/z = 544.0401 a.m.u, [1–Cl]+; 530.0241 a.m.u, [2–Cl]+; 580.0396 a.m.u, [3–Cl]+; 548.0143 a.m.u, [4–Cl]+; 598.0113 a.m.u, [5–Cl]+; Fig. S24–S28†). The purity of 1–5 was established by elemental analysis.
:dichloromethane mixture at room temperature for 2.5–4 h (Fig. 1A). The copper(II)-terpyridine complexes 1–5 were isolated as light blue solids in reasonable to good yields (47–83%) and characterised by high resolution ESI mass spectrometry, IR spectroscopy, elemental analysis, and single crystal X-ray diffraction (see ESI, Fig. S23–S28†). Distinctive molecular ion peaks corresponding to [M − Cl]+ with the appropriate isotopic pattern were observed in the positive mode of the high-resolution ESI mass spectra (m/z = 544.0401 a.m.u, [1–Cl]+; 530.0241 a.m.u, [2–Cl]+; 580.0396 a.m.u, [3–Cl]+; 548.0143 a.m.u, [4–Cl]+; 598.0113 a.m.u, [5–Cl]+; Fig. S24–S28†). The purity of 1–5 was established by elemental analysis.
Light blue needle-like single crystals of 1–5 suitable for X-ray diffraction studies were obtained by slow evaporation of an acetonitrile:water (1:1) solution of 1–5 (CCDC 2258097–2258101, Fig. 1B and Tables S1 and S2†). Selected bond distances and angles associated to 1–5 are presented in Tables S3 and S4.† The copper(II) complexes 1–5 exhibit distorted square pyramidal geometries with the copper(II) centre coordinated to the corresponding terpyridine ligand L1–L5 in a tridentate manner and to two chloride ligands in a monodentate manner (Fig. 1B). The copper(II) complexes 1–5 exhibit τ5 values between 0.12–0.21 (average τ5 value = 0.17 ± 0.03), suggestive of a distorted square pyramidal geometry. The three nitrogen atoms on L1–L5 and the Cl(1) atom make up the square base while the Cl(2) atom is the apex of the square pyramidal structure. The Cu atom in 1–5 resides 0.113–0.287 Å away from the N3Cl plane. Given the distorted square pyramidal geometry of 1–5, the chloride ligands are non-equivalent and the Cu–Cl(1/2) bond lengths differ significantly. Across 1–5, the Cu–Cl(1) bond has an average length of 2.237 Å and the Cu–Cl(2) bond has an average length of 2.602 Å. Overall, the bond angles and lengths associated to 1–5 are consistent with bond parameters reported for related five-coordinate square pyramidal copper(II) complexes containing terpyridine ligands.29–31,33
Stability in biologically relevant solutions
The lipophilicity of 1–5 was determined by measuring the extent to which it partitioned between octanol and water, P. The experimentally determined logP values varied from −1.71 ± 0.08 to −2.27 ± 0.04 (Table S5†). The hydrophilic nature of 1–5 suggests that the copper(II) complexes should be readily soluble in the aqueous solutions used for cell-based studies. The experimentally determined logP value for Cu(2,2′;6′,2′′-terpyridine)Cl2 was −2.01 ± 0.08 (Table S5†). This suggests that the aryl sulfonamide moieties in 1–5 do not significantly affect lipophilicity. UV-Vis spectroscopy and ESI mass spectrometry studies were carried out to assess the stability of 1–5 in biologically relevant solutions. In PBS:DMSO (200:1) and H2O:DMSO (200:1), the absorbance bands associated to 1–5 (50 μM) remained largely unchanged over the course of 24 h at 37 °C, suggestive of stability under these conditions (Fig. S29 and S30†). In PBS:DMSO (200:1) with the added presence of 10 equivalents of ascorbic acid (a cellular reductant), the absorbance bands associated to 1–5 (50 μM) also remained unaltered, indicative of stability under biologically reducing conditions (Fig. S31†). To confirm the stability of 1–5 under reducing conditions, the ESI mass spectra of 1–5 (500 μM) in H2O:DMSO (5:1) in the presence of 10 equivalents of ascorbic acid or glutathione was recorded after incubation for 24 h at 37 °C (Fig. S32–S36†). The ESI mass spectra (positive mode) of 1–5 exhibited distinctive peaks with the appropriate isotopic pattern corresponding to the [M − Cl]+ molecular ion (Fig. S32–S36†). This further confirms that 1–5 remain intact under reducing conditions. Prior to undertaking cell-based studies, the stability of 1–5 in Mammary Epithelial Cell Growth Medium (MEGM) was assessed. In MEGM:DMSO (200:1), the UV-Vis trace associated to 1–5 (50 μM) did not markedly change over the course of 24 h at 37 °C (Fig. S37†), suggestive to sufficient stability to proceed to cell-based studies. Additional UV-Vis spectroscopy studies were conducted with concentrated solutions of 1 (taken as a representative member of the copper(II)-terpyridine series) to monitor changes in the d–d transition bands. In PBS:DMSO (10:1) and MEGM:DMSO (10:1) the wavelength of the d–d transition band associated to 1 (0.5 mM) remained largely similar over the course of 24 h at 37 °C (Fig. S38†), implying that no significant change in the copper(II) coordination environment occurred under these conditions.
Breast cancer stem cell and bulk breast cancer cell potency
The colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was used to assess the cytotoxicity of the copper(II)-terpyridine complexes 1–5 towards bulk breast cancer cells (HMLER) and breast CSC-enriched cells (HMLER-shEcad) cultured in monolayers. The IC50 values were determined from dose–response curves (Fig. S39–S43†) and are summarised in Table 1. The copper(II)-terpyridine complexes 1–5 displayed sub-micromolar potencies towards bulk breast cancer cells and breast CSCs. The copper(II)-terpyridine complexes 1–5 are approximately equipotent towards bulk breast cancer cells and breast CSCs. This suggests that 1–5 can potentially remove entire heterogeneous tumour populations (consisting of bulk cancer cells and CSCs) at a single sub-micromolar dose. The copper(II)-terpyridine complex 1 is 6-fold more potent than salinomycin (a leading anti-breast CSC agent) and 8-fold more potent than cisplatin (the most widely administered metal-based anticancer drug) towards breast CSCs.43,44 The breast CSC potency of 1 was significantly greater (p < 0.05, n = 18) than a series of clinically approved breast cancer drugs (5-fluorouracil, capecitabine, and carboplatin).45 However, gemcitabine (another clinically used anti-breast cancer agent) was more toxic than 1 toward breast CSCs (Fig. S44 and Table S6†). Furthermore 1 was significantly more potent towards breast CSCs than previously reported anti-breast CSC copper(II) complexes: 3-fold more potent than copper(II) complexes containing phenanthroline-based and indomethacin ligands,43 52-fold more toxic than copper(II) complexes containing Schiff base and naproxen ligands,46 and 18-fold more toxic than copper(II) complexes containing dithiacyclam and diclofenac ligands.47| Compound | HMLER IC50/μM | HMLER-shEcad IC50/μM | Mammosphere IC50/μM |
|---|---|---|---|
| a Reported in ref. 43, 44, 51 and 52. n.d. not determined. | |||
| 1 | 0.85 ± 0.04 | 0.69 ± 0.12 | 3.44 ± 0.03 |
| 2 | 0.85 ± 0.14 | 0.83 ± 0.01 | 2.98 ± 0.01 |
| 3 | 0.83 ± 0.06 | 0.73 ± 0.04 | 2.63 ± 0.08 |
| 4 | 0.83 ± 0.19 | 0.78 ± 0.03 | 2.99 ± 0.53 |
| 5 | 0.81 ± 0.02 | 0.79 ± 0.18 | 3.34 ± 0.36 |
| L1 | 3.32 ± 0.07 | 3.10 ± 0.25 | n.d. |
| Cu(NO3)2 | >100 | >100 | n.d. |
| Cu(terpy)Cl2 | 3.04 ± 0.07 | 2.99 ± 0.01 | 4.46 ± 0.28 |
| L 1 + CuCl2 | n.d. | 2.28 ± 0.06 | n.d. |
| Cisplatina | 2.57 ± 0.02 | 5.65 ± 0.30 | 13.50 ± 2.34 |
| Salinomycina | 11.43 ± 0.42 | 4.23 ± 0.35 | 18.50 ± 1.50 |
Control cytotoxicity studies showed that the potency of the terpyridine ligand L1 and copper nitrate towards HMLER and HMLER-shEcad cells was significantly lower (p < 0.05, n = 18) than 1 (Fig. S45, S46,† and Table 1). This suggests that the cytotoxicity of 1 towards bulk breast cancer cells and breast CSCs is likely to result from the intact copper(II) complex rather than its individual components (copper or free L1). Further, the potency of Cu(2,2′;6′,2′′-terpyridine)Cl2 towards HMLER and HMLER-shEcad cells was significantly lower (p < 0.05, n = 18) than 1 (Table 1, Fig. S47†). This suggests that the copper(II)-terpyridine unit is not solely responsible for the bulk breast cancer cell and breast CSC toxicity of 1 and that the aryl sulfonamide moiety also contributes to the potency observed for 1. The combined treatment of copper(II) chloride and free terpyridine ligand L1 showed a significant (p < 0.05) reduction in potency towards HMLER-shEcad cells compared to 1 (Fig. S48,† and Table 1). This demonstrates that the preformed copper(II)-terpyridine complex 1 is significantly (p < 0.05) better at killing breast CSCs than a mixture of its individual components.
Breast CSCs grown in anchorage-independent, serum-free cultures can generate three-dimensional structures called mammospheres.48 The ability of a given compound to disrupt mammosphere formation from single cell suspensions provides a reliable marker for in vivo and translational potential. The ability of the copper(II)-terpyridine complexes 1–5 to inhibit mammosphere formation was assessed using an inverted microscope. The addition of 1–5 (IC20 value for 5 days) to single cell suspensions of HMLER-shEcad cells markedly reduced the number (by 56–67%) and size of mammospheres formed (Fig. 2A and B). Cisplatin and salinomycin (at their respective IC20 values for 5 days) displayed similar mammosphere inhibitory properties (Fig. 2A and S49†). Specifically, cisplatin and salinomycin reduced the number of mammospheres formed by 34% and 40%, respectively (Fig. 2A). Treatment with Cu(2,2′;6′,2′′-terpyridine)Cl2 (IC20 value for 5 days) reduced the number (by 17%) and size of mammospheres formed (Fig. 2A and S50†) but to a significantly lesser extent than 1–5. This shows that the aryl sulfonamide group present in 1–5 enhances their mammosphere inhibitory effect. To establish the ability of 1–5 to reduce mammosphere viability, the colorimetric resazurin-based reagent, TOX8 was used. TOX8 is able to penetrate the three-dimensional architecture of unanchored mammospheres and access all cells that make up mammospheres (including hard-to-reach cells in the mammosphere core).49,50 The IC50 value (concentration required to reduce mammosphere viability by 50%) of 1–5 was in the micromolar range, up to 5–7-fold lower than salinomycin and cisplatin (Fig. S51† and Table 1).51,52 Cu(2,2′;6′,2′′-terpyridine)Cl2 displayed significantly lower mammosphere potency (IC50 value = 4.46 ± 0.28 μM, p < 0.05, n = 18) than 1–5, showing that the aryl sulfonamide group on 1–5 contributes to their mammosphere potency. Taken together the mammosphere studies show that 1–5 are able to effectively reduce mammosphere formation and viability, and that their capacity to do so is proportionate with the mostly widely studied anti-breast CSC agent to date, salinomycin.
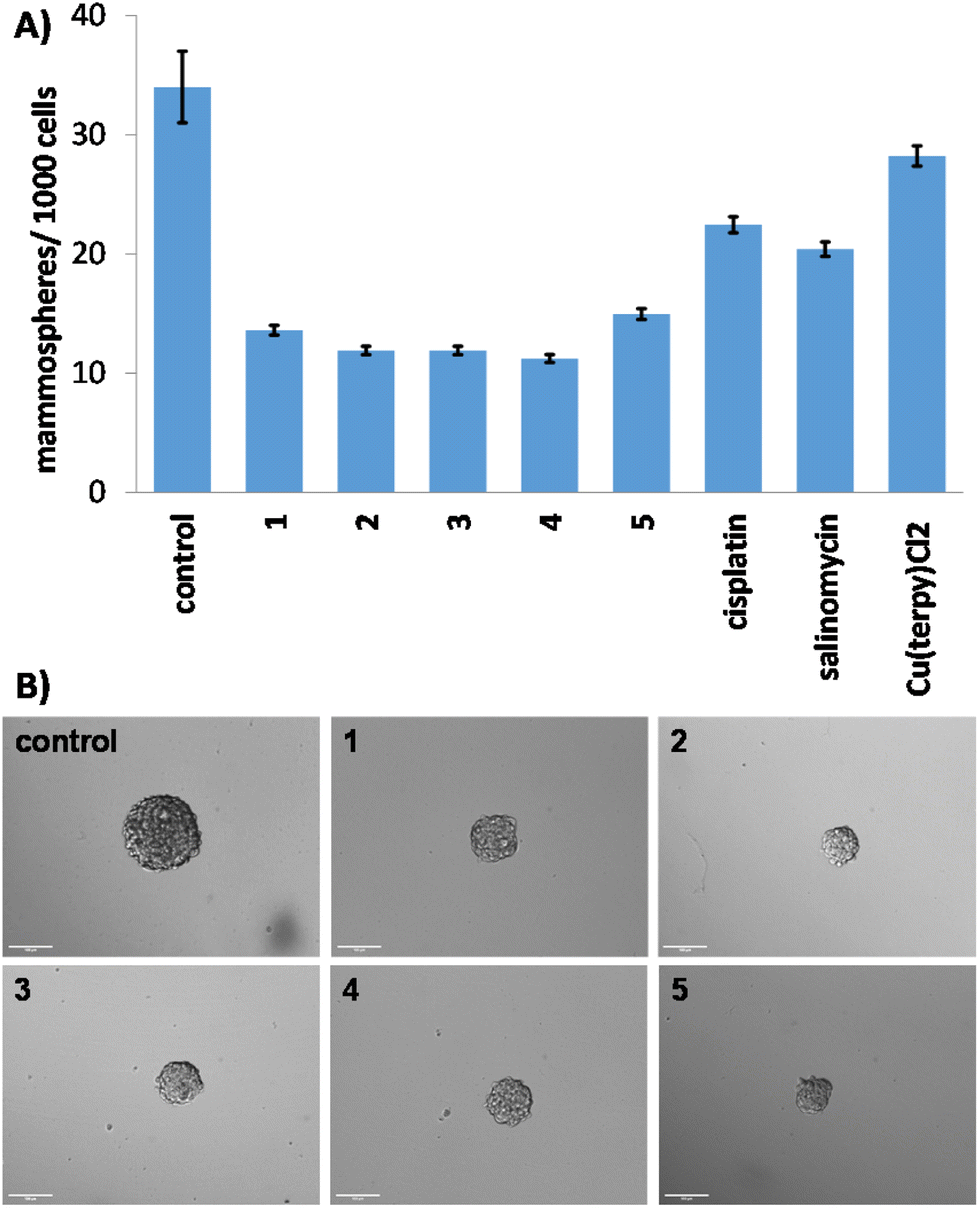 | ||
| Fig. 2 (A) Quantification of mammosphere formation with HMLER-shEcad cells untreated and treated with 1–5, cisplatin, salinomycin or Cu(2,2′;6′,2′′-terpyridine)Cl2 at their respective IC20 values for 5 days. (B) Representative bright-field images (×10) of the mammospheres in the absence and presence of 1–5 at their respective IC20 values for 5 days. | ||
Cellular uptake and reactive oxygen species generation in breast cancer stem cells
Cellular uptake studies were performed to determine the relative uptake of 1–5 by breast CSCs. HMLER-shEcad cells were dosed with 1–5 (1 μM for 24 h), harvested, digested with concentrated nitric acid, and analysed for their copper content using inductively coupled plasma mass spectrometry (ICP-MS) (Fig. 3A). All of the copper(II)-terpyridine complexes were taken up to a reasonable level (2.95 ± 0.01 to 6.78 ± 0.05 ng Cu per million cells) given the relatively low administration dose (1 μM) used. Complex 1 was taken up by breast CSCs to a significantly greater extent than 2–5. This suggests that the p-toluene sulfonamide group present in 1 facilitates greater breast CSC uptake than the other aryl sulfonamide groups present in 2–5. Control studies showed that copper(II) chloride and Cu(2,2′;6′,2′′-terpyridine)Cl2 were taken up to a lesser extent than 1 by HMLER-shEcad cells under identical conditions (Fig. 3A). This shows that the p-toluene sulfonamide-bearing ligand L1 plays an important role in the breast CSC uptake of 1. | ||
| Fig. 3 (A) Copper content (ng of Cu per 106 cells) in HMLER-shEcad cells treated with 1–5, CuCl2 or Cu(2,2′;6′,2′′-terpyridine)Cl2 (1 μM for 24 h). (B) Copper content (ng of Cu per 106 cells) in various cellular components upon treatment of HMLER-shEcad cells with 1 (1 μM for 24 h). | ||
Further cell-based studies were carried out with the p-toluene sulfonamide-bearing copper(II)-terpyridine complex 1 to provide insight into its mechanism of action. To determine where 1 localises within breast CSCs, fractionation studies were carried out. HMLER-shEcad cells were dosed with 1 (1 μM for 24 h) and the cytoplasmic, nuclear, and membrane fractions were extracted and analysed for their copper content using ICP-MS (Fig. 3B). The majority of copper was detected in the cytoplasm (80%), with a relatively small amount in the nucleus and membrane. This suggests that following entry into breast CSCs, 1 accumulates predominately in the cytoplasm. This also implies that the primary intracellular target of 1 is likely to be biomolecules located within the cytoplasm, rather than biomolecules in the nucleus such as genomic DNA or histones.
Copper(II)-terpyridine complexes have been reported to increase intracellular ROS levels in cancer cells.35 Intracellular ROS levels in breast CSCs dosed with 1 (3 μM) were measured over a 24 h period using 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA). HMLER-shEcad cells dosed with 1 showed a time-dependent increase in ROS levels compared to untreated cells over the first 3 h of exposure (30–57% increase in ROS levels from 0.5 to 3 h, Fig. 4A). At longer exposure times (6, 16 or 24 h), 1 did not statistically increase ROS levels in HMLER-shEcad cells. As the highest elevation in ROS levels was observed at short timepoints (0.5–3 h), it is likely that ROS was generated directly by 1, rather than occurring as a result of the downstream effects of cellular stress or due to cell death.
 | ||
| Fig. 4 (A) Normalised ROS activity in untreated HMLER-shEcad cells (control) and HMLER-shEcad cells treated with 1 (3 μM for 0.5, 1, 3, 6, 16, and 24 h). Error bars represent SD. (B) Cell viability (% of control) vs. concentration (μM) for 1 in HMLER-shEcad cells in the absence or presence of salubrinal (10 μM). | ||
Endoplasmic reticulum stress and mode of cell death
As 1 contains a p-toluene sulfonamide group which is theoretically able to target the ER,37–39 the ability of 1 to induce ER stress in breast CSCs was investigated. Co-treatment of 1 and salubrinal (10 μM),53 an established ER stress inhibitor, significantly reduced the cytotoxicity of 1 towards HMLER-shEcad cells (Fig. 4B). Specifically, the IC50 value of 1 towards HMLER-shEcad cells increased 3-fold (IC50 value = 2.34 ± 0.01 μM), implying that ER stress is a component of the cytotoxic mechanism of 1. To probe this further, the expression of proteins related to the unfolded protein response (UPR), an inherent cellular mechanism that attempts to correct ER stress, was determined using immunoblotting methods.54 Upon incubation of HMLER-shEcad cells with 1 (0.4–1.6 μM for 4 h) appreciably higher levels of stress-related activating transcription factor-4 (ATF-4) and C/EBP homologous protein (CHOP) were observed compared to untreated cells (Fig. 5A), indicative of ER stress.55–57 Interestingly, phosphorylated eukaryotic initiation factor 2α (phos-eIF2α), which is upstream of ATF-4 in the UPR, did not show markedly higher expression (relative to unphosphorylated eIF2α) in 1-treated HMLER-shEcad cells (Fig. 5A). Collectively, the cytotoxicity studies (in the presence of salubrinal) and the immunoblotting analysis indicate that 1 partially induces ER stress in breast CSCs. Unresolved ER stress and persistent activation of the UPR can lead to apoptosis.58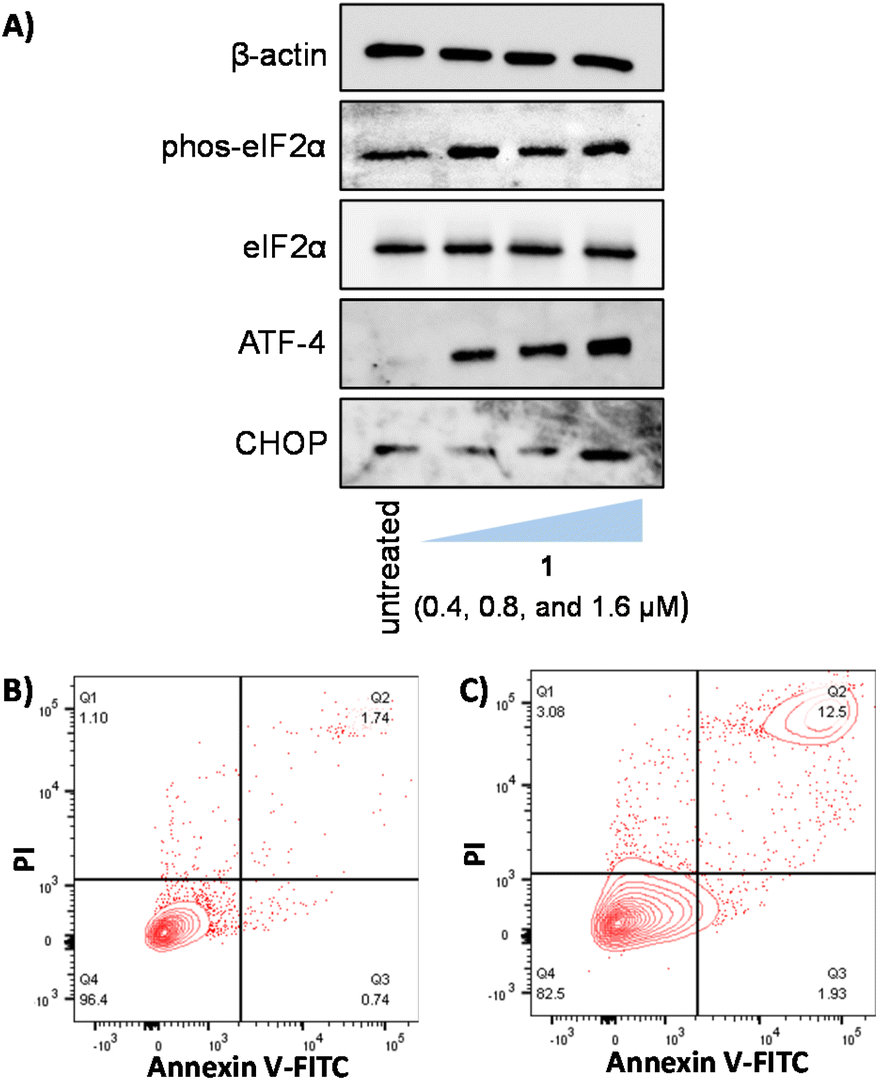 | ||
| Fig. 5 (A) Immunoblotting analysis of proteins related to ER stress. Protein expression in HMLER-shEcad cells untreated and treated with 1 (0.4, 0.8, and 1.6 μM for 4 h). FITC Annexin V-propidium iodide binding assay plots of (B) untreated HMLER-shEcad cells and (C) HMLER-shEcad cells treated with 1 (IC50 value × 2 for 48 h). | ||
Apoptosis induces morphological changes that can lead to cell membrane rearrangement. This process results in the translocation of phosphatidylserine residues to the membrane exterior which can be detected by Annexin V.59 Damaged cell membranes also facilitate propidium iodide uptake. Using a dual FITC Annexin V-propidium iodide staining flow cytometry assay, we explored the occurrence of apoptosis in HMLER-shEcad cells treated with 1. Dosage with 1 (IC50 value × 2 for 48 h) induced large populations of cells to undergo late-stage apoptosis (Fig. 5B and C). This was comparable to dosage with cisplatin (25 μM for 48 h), a well-known apoptosis inducer (Fig. S52†). Additional immunoblotting studies showed that HMLER-shEcad cells treated with 1 (0.4–1.6 μM for 24 or 48 h) exhibited a clear increase in the level of cleaved caspase 7 and cleaved PARP-1 compared to untreated control cells (Fig. S53†), indicative of caspase-dependent apoptosis.60,61 Taken together, the mechanism of action studies indicate that 1 probably induces apoptotic CSC death by elevating intracellular ROS levels and evoking ER stress.
Experimental
Instrumentation
1H, 13C{1H}, and 19F{1H} NMR were recorded at room temperature on a Bruker Avance 400 spectrometer (1H 400.0 MHz, 13C 100.6 MHz, 19F 376.5 MHz) with chemical shifts (δ, ppm) reported relative to the solvent peaks of the deuterated solvent. ICP-MS were measured using a Thermo Scientific ICAP-Qc quadrupole ICP mass spectrometer. Elemental analysis was performed commercially at the University of Cambridge.Starting materials and other reagents
4′-Chloro-2,2′:6′,2′′-terpyridine, 2-aminoethanol, potassium hydroxide, 4-methylbenzenesulfonyl chloride, benzenesulfonyl chloride, naphthalene-2-sulfonyl chloride, 4-fluorobenzenesulfonyl chloride, 4-(trifluoromethyl)benzenesulfonyl chloride, and copper(II) chloride dihydrate were purchased from Sigma-Aldrich and used without further purification. Solvents were purchased from Fisher and used without further purification. Cu(2,2′;6′,2′′-terpyridine)Cl2 was prepared according to a previously reported protocol.62Synthesis of 2-([2,2′:6′,2′′-terpyridin]-4′-yloxy)ethan-1-amine
Crushed potassium hydroxide (280 mg, 5 mmol) and 2-aminoethanol (110 μL, 1.8 mmol) were added to DMSO (5 mL). The mixture was stirred at 40 °C for 30 min, then 4′-chloro-2,2′;6′,2′′-terpyridine (286 mg, 1 mmol) was added. The mixture was stirred at 40 °C for 2 h. After allowing to cool, the mixture was diluted with DCM (30 mL) and washed with H2O (3 × 30 mL). The DCM layer was dried with Na2SO4, filtered, and the solvent was removed to yield 2-([2,2′:6′,2′′-terpyridin]-4′-yloxy)ethan-1-amine as a white solid (238 mg, 76%); 1H NMR (400 MHz, CDCl3): 8.69 (dd, 2H), 8.62 (d, 2H), 8.04 (s, 2H), 7.85 (td, 2H), 7.33 (ddd, 2H), 4.27 (t, 2H), 3.16 (t, 2H); 13C{1H} NMR (100.6 MHz, CDCl3): 167.15, 157.17, 156.07, 149.05, 136.80, 123.85, 121.34, 107.38, 70.52, 41.44; ATR-FTIR (solid, cm−1): 3242, 3097, 3051, 2980, 2951, 2874, 1580, 1557, 1468, 1440, 1405, 1361, 1349, 1339, 1203, 1155, 1087, 1028, 1009, 999, 896, 840, 813, 791, 746, 733, 696, 657, 630, 618, 573, 558, 548, 470, 404; HR ESI-MS calcd for C17H17N4O [M + H]+ 293.1402 a.m.u. found [M + H]+ 293.1400 a.m.u.; anal. calcd for C17H16N4O (%): C, 69.85; H, 5.52; N, 19.17. Found: C, 69.68; H, 5.42; N, 18.91.Synthesis of L1
2-([2,2′:6′,2′′-Terpyridin]-4′-yloxy)ethan-1-amine (136 mg, 0.47 mmol), 4-methylbenzenesulfonyl chloride (90 mg, 0.47 mmol), and triethylamine (100 μL, 0.75 mmol) were stirred in DCM (10 mL) for 16 h at ambient temperature. The mixture was diluted with DCM (20 mL), then washed with saturated NaHCO3 (5 mL) and brine (5 mL). The DCM layer was dried with anhydrous Na2SO4, filtered, and the solvent was removed to give L1 as a white solid (179 mg, 86%); 1H NMR (400 MHz, CDCl3): 8.69 (d, 2H), 8.61 (d, 2H), 7.91 (s, 2H), 7.85 (td, 2H), 7.78 (d, 2H), 7.34 (ddd, 2H), 7.28 (d, 2H), 4.93 (br t, 1H), 4.24 (t, 2H), 3.45 (m, 2H), 2.36 (s, 3H); 13C{1H} NMR (100.6 MHz, CDCl3): 166.25, 157.22, 155.72, 149.05, 143.69, 136.88, 129.86, 127.07, 124.01, 121.33, 107.12, 66.41, 42.42, 21.46; ATR-FTIR (solid, cm−1): 3305, 2979, 2968, 2921, 1600, 1582, 1563, 1454, 1446, 1433, 1467, 1392, 1365, 1353, 1322, 1308, 1250, 1199, 1157, 1110, 1089, 1065, 956, 820, 795, 766, 746, 719, 698, 671, 659, 628, 581, 548, 523, 492, 472, 406; HR ESI-MS calcd for C24H23N4O3S [M + H]+ 447.1491 a.m.u. found [M + H]+ 447.1486 a.m.u.; anal. calcd for C24H22N4O3S (%): C, 64.56; H, 4.97; N, 12.55. Found: C, 64.70; H, 5.22; N, 12.05.Synthesis of L2
2-([2,2′:6′,2′′-Terpyridin]-4′-yloxy)ethan-1-amine (151 mg, 0.52 mmol), benzenesulfonyl chloride (93 mg, 0.53 mmol), and triethylamine (110 μL, 0.83 mmol) were stirred in DCM (10 mL) for 16 h at ambient temperature. The mixture was diluted with DCM (20 mL), then washed with saturated NaHCO3 (5 mL) and brine (5 mL). The DCM layer was dried with anhydrous Na2SO4, filtered, and the solvent was removed to give L2 as a white solid (168 mg, 75%); 1H NMR (400 MHz, CDCl3): 8.68 (dd, 2H), 8.60 (d, 2H), 7.92 (m, 4H), 7.85 (td, 2H), 7.53 (m, 3H), 7.34 (ddd, 2H), 4.95 (t, 1H), 4.26 (t, 2H), 3.47 (m, 2H); 13C{1H} NMR (400 MHz, CDCl3): 166.20, 157.29, 155.76, 149.06, 139.91, 136.86, 132.85, 129.26, 127.02, 123.99, 121.33, 107.10, 66.49, 42.44; ATR-FTIR (solid, cm−1): 3239, 2980, 2971, 1581, 1561, 1468, 1444, 1407, 1363, 1338, 1330, 1256, 1190, 1164, 1126, 1091, 1073, 1048, 1040, 991, 935, 875, 855, 787, 739, 729, 690, 657, 624, 591, 566, 548, 502, 488, 470, 432, 408; HR ESI-MS calcd for C23H21N4O3S [M + H]+ 433.1334 a.m.u. found [M + H]+ 433.1324 a.m.u.; anal. calcd for C23H20N4O3S (%): C, 63.87; H, 4.66; N, 12.95. Found: C, 63.60; H, 4.72; N, 12.72.Synthesis of L3
2-([2,2′:6′,2′′-Terpyridin]-4′-yloxy)ethan-1-amine (100 mg, 0.34 mmol), naphthalene-2-sulfonyl chloride (84 mg, 0.37 mmol), and triethylamine (78 μL, 0.59 mmol) were stirred in DCM (10 mL) for 16 h at ambient temperature. The mixture was diluted with DCM (20 mL), then washed with saturated NaHCO3 (5 mL) and brine (5 mL). The DCM layer was dried with anhydrous Na2SO4, filtered, and the solvent was removed to give L3 as a white solid (148 mg, 90%); 1H NMR (400 MHz, CDCl3): 8.66 (ddd, 2H), 8.57 (m, 2H), 8.47 (d, 1H), 7.93 (d, 2H), 7.84 (m, 6H), 7.56 (m, 2H), 7.33 (ddd, 2H), 5.02 (t, 1H), 4.25 (t, 2H), 3.50 (m, 2H); 13C{1H} NMR (100.6 MHz, CDCl3): 166.12, 157.18, 155.73, 149.02, 136.80, 136.72, 134.82, 132.13, 129.72, 129.19, 128.76, 128.41, 127.87, 127.52, 123.95, 122.17, 121.32, 107.04, 66.38, 42.47; ATR-FTIR (solid, cm−1): 3241, 2981, 1582, 1561, 1468, 1442, 1405, 1363, 1339, 1320, 1203, 1155, 1131, 1089, 1073, 1038, 989, 892, 881, 867, 855, 813, 793, 741, 733, 696, 657, 636, 618, 548, 470, 406; HR ESI-MS calcd for C27H23N4O3S [M + H]+ 483.1491 a.m.u. found [M + H]+ 483.1488 a.m.u.; anal. calcd for C27H22N4O3S·H2O (%): C, 64.79; H, 4.83; N, 11.19. Found: C, 65.15; H, 4.53; N, 10.88.Synthesis of L4
2-([2,2′:6′,2′′-Terpyridin]-4′-yloxy)ethan-1-amine (100 mg, 0.34 mmol), 4-fluorobenzenesulfonyl chloride (72 mg, 0.37 mmol) and triethylamine (78 μL, 0.59 mmol) were stirred in DCM (10 mL) for 16 h at ambient temperature. The mixture was diluted with DCM (20 mL), then washed with saturated NaHCO3 (5 mL) and brine (5 mL). The DCM layer was dried with anhydrous Na2SO4, filtered, and the solvent was removed to give crude L4 as a white solid (124 mg, 80%). The isolated product required purification by recrystallisation in minimum amount of boiling ethanol to yield pure L4 as a white solid (26 mg, 17%); 1H NMR (400 MHz, CDCl3): 8.68 (dd, 2H), 8.61 (d, 2H), 7.93 (m, 4H), 7.86 (m, 2H), 7.35 (ddd, 2H), 7.18 (m, 2H), 4.96 (br t, 1H), 4.29 (t, 2H), 3.47 (m, 2H); 13C{1H} NMR (100.6 MHz, CDCl3): 166.14, 157.34, 155.72, 149.07, 136.88, 129.85, 129.76, 124.03, 121.35, 116.62, 116.39, 107.07, 66.42, 42.43; 19F{1H} NMR (376.5 MHz, CDCl3): −104.92; ATR-FTIR (solid, cm−1): 3241, 2980, 1582, 1561, 1468, 1442, 1405, 1365, 1339, 1188, 1153, 1126, 1087, 1048, 1040, 990, 939, 855, 840, 816, 795, 787, 739, 729, 708, 690, 667, 655, 624, 579, 548, 515, 482, 472, 420, 408; HR ESI-MS calcd for C23H20N4O3SF [M + H]+ 451.1240 a.m.u. found [M + H]+ 451.1241 a.m.u.; anal. calcd for C23H19N4O3SF·0.25H2O (%): C, 60.72; H, 4.32; N, 12.31. Found: C, 60.81; H, 4.27; N, 11.97.Synthesis of L5
2-([2,2′:6′,2′′-Terpyridin]-4′-yloxy)ethan-1-amine (113 mg, 0.39 mmol), 4-(trifluoromethyl)benzenesulfonyl chloride (91 mg, 0.37 mmol), and triethylamine (88 μL, 0.63 mmol) were stirred in DCM (10 mL) for 16 h at ambient temperature. The mixture was diluted with DCM (20 mL), then washed with saturated NaHCO3 (5 mL) and brine (5 mL). The DCM layer was dried with anhydrous Na2SO4, filtered, and the solvent was removed to give L5 as a white solid (162 mg, 84%); 1H NMR (400 MHz, CDCl3): 8.68 (ddd, 2H), 8.60 (dd, 2H), 8.04 (d, 2H), 7.92 (s, 2H), 7.86 (td, 2H), 7.78 (d, 2H), 7.34 (ddd, 2H), 5.12 (t, 1H), 4.29 (t, 2H), 3.51 (m, 2H); 13C{1H} NMR (100.6 MHz, DMSO-d6): 166.59, 157.09, 155.23, 149.69, 145.35, 137.83, 127.83, 126.87, 126.84, 124.99, 121.32, 107.09, 67.06, 42.37; 19F{1H} NMR (376.5 MHz, CDCl3): −63.18; ATR-FTIR (solid, cm−1): 3311, 3242, 2980, 1600, 1582, 1563, 1468, 1142, 1405, 1365, 1351, 1326, 1203, 1159, 1133, 1106, 1091, 1065, 1050, 1040, 1017, 991, 968, 960, 894, 881, 863, 846, 816, 795, 746, 733, 715, 669, 657, 630, 614, 597, 548, 470, 430, 402; HR ESI-MS calcd for C24H20N4O3SF3 [M + H]+ 501.1208 a.m.u.; found [M + H]+ 501.1206 a.m.u.; anal. calcd for C24H19N4O3SF3 (%): C, 57.60; H, 3.83; N, 11.19. Found: C, 57.60; H, 3.79; N, 11.14.Synthesis of Cu(L1)Cl2, 1
CuCl2·2H2O (34 mg, 0.20 mmol) was dissolved in MeOH (3 mL). L1 (99 mg, 0.22 mmol) in DCM (8 mL) was added to the methanolic light blue solution. Upon addition, a light blue precipitate formed. The mixture was stirred at ambient temperature for 2.5 h. The precipitate was collected and washed with cold MeOH and diethyl ether to give 1 a light blue solid (99 mg, 85%); ATR-FTIR (solid, cm−1): 3239, 2980, 2972, 1615, 1600, 1582, 1563, 1468, 1440, 1405, 1363, 1337, 1324, 1256, 1225, 1203, 1192, 1157, 1130, 1091, 1075, 1040, 1019, 991, 956, 939, 875, 867, 855, 846, 813, 797, 739, 729, 690, 657, 626, 589, 570, 548, 502, 482, 470, 410; HR ESI-MS calcd for C24H22ClCuN4O3S [M − Cl]+ 544.0397 a.m.u. found [M − Cl]+ 544.0401 a.m.u.; anal. calcd for C24H22Cl2CuN4O3S·0.5H2O (%): C, 48.86; H, 3.93; N, 9.50. Found: C, 48.90; H, 3.78; N, 9.24.Synthesis of Cu(L2)Cl2, 2
CuCl2·2H2O (23 mg, 0.13 mmol) was dissolved in MeOH (2 mL). L2 (50 mg, 0.12 mmol) in DCM (4 mL) was added to the methanolic light blue solution. Upon addition, a light blue precipitate formed. The mixture was stirred at ambient temperature for 2.5 h. The precipitate was collected and washed with cold MeOH and diethyl ether to give 2 a light blue solid (32 mg, 42%); ATR-FTIR (solid, cm−1): 3241, 3048, 1613, 1602, 1567, 1475, 1437, 1402, 1376, 1361, 1320, 1250, 1221, 1203, 1155, 1087, 1075, 1056, 1040, 1019, 964, 947, 869, 846, 801, 794, 768, 746, 729, 684, 655, 657, 630, 589, 577, 568, 548, 496, 480, 470, 418, 393; HR ESI-MS calcd for C23H20ClCuN4O3S [M − Cl]+ 530.0240 a.m.u. found [M − Cl]+ 530.0241 a.m.u.; anal. calcd for C23H20Cl2CuN4O3S·0.5H2O (%): C, 47.96; H, 3.68; N, 9.73. Found: C, 47.64; H, 3.28; N, 9.46.Synthesis of Cu(L3)Cl2, 3
CuCl2·2H2O (23 mg, 0.13 mmol) was dissolved in MeOH (2 mL). L3 (75 mg, 0.16 mmol) in DCM (4 mL) was added to the methanolic light blue solution. Upon addition, a light blue precipitate formed. The mixture was stirred at ambient temperature for 2.5 h. The precipitate was collected and washed with cold MeOH and diethyl ether to give 3 a light blue solid (32 mg, 38%); ATR-FTIR (solid, cm−1): 3240, 2980, 1613, 1602, 1571, 1472, 1437, 1402, 1376, 1361, 1318, 1221, 1153, 1133, 1075, 1056, 1040, 1019, 964, 892, 867, 855, 803, 793, 741, 727, 697, 657, 645, 630, 616, 566, 548, 472, 418, 411, 397; HR ESI-MS calcd for C27H22ClCuN4O3S [M − Cl]+ 580.0397 a.m.u. found [M − Cl]+ 580.0396 a.m.u.; anal. calcd For C27H22Cl2CuN4O3S·0.5H2O (%): C, 51.80; H, 3.70; N, 8.95. Found: C, 51.54; H, 3.31; N, 8.81.Synthesis of Cu(L4)Cl2, 4
CuCl2·2H2O (30 mg, 0.18 mmol) was dissolved in MeOH (2 mL). L4 (75 mg, 0.17 mmol) in DCM (2 mL) was added to the methanolic light blue solution. Upon addition, a light blue precipitate formed. The mixture was stirred at ambient temperature for 3 h. The precipitate was collected and washed with cold MeOH and diethyl ether to give 4 a light blue solid (66 mg, 64%); ATR-FTIR (solid, cm−1): 3376, 3241, 3091, 3065, 2980, 2866, 1615, 1602, 1571, 1475, 1468, 1437, 1402, 1376, 1361, 1339, 1320, 1227, 1164, 1141, 1102, 1091, 1061, 1040, 1019, 960, 884, 801, 715, 655, 610, 560, 548, 480, 428, 416, 391; HR ESI-MS calcd for C23H19ClCuN4O3SF [M − Cl]+ 548.0146 a.m.u. found [M − Cl]+ 548.0143 a.m.u.; anal. calcd for C23H19Cl2CuN4O3SF (%): C, 47.23; H, 3.27; N, 9.58. Found: C, 46.93; H, 3.26; N, 9.18.Synthesis of Cu(L5)Cl2, 5
CuCl2·2H2O (27 mg, 0.16 mmol) was dissolved in MeOH (2 mL). L5 (80 mg, 0.16 mmol) in DCM (5 mL) was added to the methanolic light blue solution. Upon addition, a light blue precipitate formed. The mixture was stirred at ambient temperature for 4 h. The precipitate was collected and washed with cold MeOH and diethyl ether to give 5 a light blue solid (84 mg, 83%); ATR-FTIR (solid, cm−1): 3363, 3241, 3104, 2982, 1600, 1571, 1496, 1475, 1454, 1437, 1402, 1376, 1359, 1332, 1295, 1227, 1157, 1091, 1054, 1038, 1019, 954, 869, 842, 801, 729, 713, 696, 654, 647, 628, 570, 648, 513, 480, 418, 393; HR ESI-MS calcd for C24H19ClCuN4O3SF3 [M − Cl]+ 598.0115 a.m.u. found [M − Cl]+ 598.0113 a.m.u.; anal. calcd for C24H19Cl2CuN4O3SF3 (%): C, 45.40; H, 3.02; N, 8.82. Found: C, 45.34; H, 2.88; N, 8.69.X-ray single crystal diffraction analysis
Single crystals of copper(II) complexes 1–5 were obtained by slow evaporation of an acetonitrile:water (1:1) solution of 1–5. Crystals suitable for X-ray diffraction analysis were selected and mounted on a Bruker Apex 2000 CCD area detector diffractometer using standard procedures. Data was collected using graphite-monochromated Mo-Kα radiation (λ = 0.71073) at 150(2) K. Crystal structures were solved and refined using the Bruker SHELXTL software.63–65 All hydrogen atoms were located by geometrical calculations, and all non-hydrogen atoms were refined anisotropically. The structures have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2258097–2258101†).
Measurement of water–octanol partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P)
P)
The logP value for 1–5 and Cu(2,2′;6′,2′′-terpyridine)Cl2 was determined using the shake-flask method and UV-Vis spectroscopy. The 1-octanol used in this experiment was pre-saturated with water. A DMSO solution of 1–5 or Cu(2,2′;6′,2′′-terpyridine)Cl2 (10 μL, 10 mM) was incubated with 1-octanol (495 μL) and H2O (495 μL) in a 1.5 mL tube. The tube was shaken at room temperature for 24 h. The two phases were separated by centrifugation and the 1–5 or Cu(2,2′;6′,2′′-terpyridine)Cl2 content in each phase was determined by UV-vis spectroscopy. The reported logP values are the average of three independent experiments (n = 3).
Cell lines and cell culture conditions
The human mammary epithelial HMLER and HMLER-shEcad cell lines were kindly donated by Prof. R. A. Weinberg (Whitehead Institute, MIT). HMLER and HMLER-shEcad cells were maintained in Mammary Epithelial Cell Growth Medium (MEGM) with supplements and growth factors (BPE, hydrocortisone, hEGF, insulin, and gentamicin/amphotericin-B). The cells were grown at 310 K in a humidified atmosphere containing 5% CO2.Monolayer cytotoxicity studies
Exponentially growing cells were seeded at a density of approximately 5 × 103 cells per well in 96-well flat-bottomed microplates and allowed to attach for 24 h prior to addition of compounds. Various concentrations of the test compounds (0.0004–100 μM) were added and incubated for 72 h at 37 °C (total volume 200 μL). Stock solutions of the compounds were prepared as 10 mM DMSO solutions and diluted using cell media. The final concentration of DMSO in each well was ≤1 %. After 72 h, 20 μL of MTT (4 mg mL−1 in PBS) was added to each well and the plates incubated for an additional 4 h at 37 °C. The media/MTT mixture was removed and DMSO (100 μL per well) was added to dissolve the formazan precipitates. The optical density was measured at 550 nm using a 96-well multiscanner autoreader. Absorbance values were normalised to (DMSO-containing) control wells and plotted as concentration of test compound versus % cell viability. IC50 values were interpolated from the resulting dose dependent curves. The reported IC50 values are the average of three independent experiments (n = 18).Tumoursphere formation and viability assay
HMLER-shEcad cells (5 × 103) were plated in ultralow-attachment 96-well plates (Corning) and incubated in MEGM supplemented with B27 (Invitrogen), 20 ng mL−1 EGF and 4 μg mL−1 heparin (Sigma) for 5 days. Studies were also conducted in the presence of 1–5, Cu(2,2′;6′,2′′-terpyridine)Cl2, cisplatin, and salinomycin (0–133 μM). Mammospheres treated with 1–5, Cu(2,2′;6′,2′′-terpyridine)Cl2, cisplatin, and salinomycin (IC20 values for 5 days) were counted and imaged using an inverted microscope. The viability of the mammospheres was determined by addition of a resazurin-based reagent, TOX8 (Sigma). After incubation for 16 h, the fluorescence of the solutions was read at 590 nm (λex = 560 nm). Viable mammospheres reduce the amount of the oxidised TOX8 form (blue) and concurrently increase the amount of the fluorescent TOX8 intermediate (red), indicating the degree of mammosphere cytotoxicity caused by the test compound. Fluorescence values were normalised to DMSO-containing controls and plotted as concentration of test compound versus % mammospheres viability. IC50 values were interpolated from the resulting dose dependent curves. The reported IC50 values are the average of three independent experiments, each consisting of two replicates per concentration level (n = 6).Cellular uptake
To measure the cellular uptake of 1–5, Cu(2,2′;6′,2′′-terpyridine)Cl2, and copper(II) chloride about 1 million HMLER-shEcad cells were treated with 1–5, Cu(2,2′;6′,2′′-terpyridine)Cl2 or copper(II) chloride (1 μM) at 37 °C for 24 h. After incubation, the media was removed, the cells were washed with PBS (2 mL × 3) and harvested. The number of cells was counted at this stage, using a haemocytometer. This mitigates any cell death induced by 1–5, Cu(2,2′;6′,2′′-terpyridine)Cl2 or copper(II) chloride at the administered concentration and experimental cell loss. The cellular pellets were dissolved in 65% HNO3 (250 mL) overnight. A cellular pellet of HMLER-shEcad cells treated with 1 was also used to determine the copper content in the nuclear, cytoplasmic and membrane fractions. The Thermo Scientific NE-PER Nuclear and Cytoplasmic Extraction Kit was used to extract and separate the nuclear, cytoplasmic and membrane fractions. The fractions were dissolved in 65% HNO3 (250 μL final volume) overnight. All samples were diluted 17-fold with water and analysed using inductively coupled plasma mass spectrometry (ICP-MS, Thermo Scientific ICAP-Qc quadrupole ICP mass spectrometer). Copper levels are expressed as mass of Cu (ng) per million cells. Results are presented as the mean of four determinations for each data point.Intracellular ROS assay
HMLER-shEcad cells (5 × 103) were seeded in each well of a 96-well plate. After incubating the cells overnight, they were treated with 1 (3 μM for 0.5–24 h) and incubated with 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (20 μM) for 90 min. The intracellular ROS level was determined by measuring the fluorescence of the solutions in each well at 529 nm (λex = 504 nm).Immunoblotting analysis
HMLER-shEcad cells (5 × 106) were incubated with 1 (0.4–1.6 μM for 4 h or 24 h or 48 h) at 37 °C. HMLER-shEcad cells were harvested and isolated as pellets. SDS-PAGE loading buffer (64 mM Tris-HCl (pH 6.8)/9.6% glycerol/2%SDS/5% β-mercaptoethanol/0.01% Bromophenol Blue) was added to the pellets, and this was incubated at 95 °C for 10 min. Cell lysates were resolved by 4–20% sodium dodecylsulphate polyacylamide gel electrophoresis (SDS-PAGE; 200 V for 25 min) followed by electro transfer to polyvinylidene difluoride membrane, PVDF (350 mA for 1 h). Membranes were blocked in 5% (w/v) non-fat milk in PBST (PBS/0.1% Tween 20) and incubated with the appropriate primary antibodies (Cell Signalling Technology). After incubation with horseradish peroxidase-conjugated secondary antibodies (Cell Signalling Technology), immune complexes were detected with the ECL detection reagent (BioRad) and analysed using a chemiluminescence imager (Bio-Rad ChemiDoc Imaging System).Annexin V-propidium iodide assay
HMLER-shEcad cells were incubated with and without 1 (IC50 value × 2 for 48 h) and cisplatin (25 μM for 48 h) at 37 °C. Cells were harvested from adherent cultures by trypsinisation. The FITC Annexin V/Dead Cell Apoptosis Kit was used. The manufacture's (Thermo Fisher Scientific) protocol was followed to carry out this experiment. Briefly, untreated and treated cells (1 × 106) were suspended in 1× Annexin binding buffer (100 μL) (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4), then 5 μL FITC Annexin V and 1 μL PI (100 μg mL−1) were added to each sample and incubated at room temperature for 15 min. After which more 1× Annexin binding buffer (400 μL) was added while gently mixing. The cells were analysed using a FACSCanto II flow cytometer (BD Biosciences) (10000 events per sample were acquired) at the University of Leicester FACS Facility. The FL1 channel was used to assess Annexin V binding and the FL2 channel was used to assess PI uptake. Cell populations were analysed using the FlowJo software (Tree Star).
Conclusions
In summary we report the synthesis and characterisation of a series of novel terpyridine ligands functionalised with aryl sulfonamide groups (L1–L5) and their corresponding copper(II) complexes (1–5). The anti-breast CSC properties of the copper(II) complexes 1–5 was also explored. X-ray crystallography studies showed that copper(II)-terpyridine complexes 1–5 adopt five-coordinate square pyramidal geometries (τ5 = 0.12–0.21). Biophysical studies indicated that 1–5 are remarkably soluble and stable in biologically relevant solutions. The latter was demonstrated by a series of UV-Vis spectroscopy and mass spectrometry experiments conducted in PBS and cell culture media (with and without cellular reductants) under physiological conditions. The biological stability of 1–5 is highly attractive from a translational standpoint as most anticancer copper(II) complexes reported in the literature are susceptible to degradation, structural transformation or copper leaching in biological solutions, especially in the presence of cellular reductants. The copper(II)-terpyridine complexes 1–5 exhibited sub-micromolar potencies towards bulk breast cancer cells and breast CSCs grown in monolayers. The rationale for preparing 1–5 with ER-targeting aryl sulfonamide groups was to endow selectivity for breast CSCs over bulk breast cancer cells, however, the cytotoxicity data suggests that this was not realised. The p-toluene sulfonamide-bearing copper(II)-terpyridine complex 1 displayed significantly better potency towards breast CSCs than cisplatin and salinomycin. Notably, 1 also displayed impressive mammosphere inhibitory effects with respect to the number and size of mammosphere formed and their viability. Of note, the mammosphere potency of 1 was 4–5-fold greater than cisplatin and salinomycin. Mechanism of action studies showed that 1 enters breast CSCs, accumulates in the cytoplasm, increases intracellular ROS levels, partially induces ER stress, and prompts apoptosis. Our results re-emphasise the therapeutic potential of metal-terpyridine complexes and more specifically shows that copper(II)-terpyridine complexes could make effective anti-breast CSC agents.Author contributions
Conceptualisation, K.S., J.N.S, and K.S.; methodology, K.S., J.N.S, K.S. and K.S.; formal analysis, K.S., J.N.S, K.S. and K.S.; investigation, K.S., J.N.S, K.S. and K.S.; writing—original draft preparation, K.S., J.N.S, and K.S.; writing—review and editing, K.S., J.N.S, and K.S.; supervision, K.S.; project administration, K.S.; funding acquisition, K.S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. S. is supported by an EPSRC New Investigator Award (EP/S005544/1) and the University of Leicester. XRD crystallography at the University of Leicester is supported by an EPSRC Core Equipment Award (EP/V034766/1). We also thank the Advanced Imaging Facility (RRID:SCR_020967) at the University of Leicester for support.References
- Nat. Cancer, 2020, 1, 1–2.
- Macmillan Cancer Support. Statistics fact sheet 2022. https://www.macmillan.org.uk/dfsmedia/1a6f23537f7f4519bb0cf14c45b2a629/9468-10061/2022-cancer-statistics-factsheet. (accessed April 2023).
- G. P. Gupta and J. Massagué, Cell, 2006, 127, 679–695 CrossRef CAS PubMed.
- P. Butow, L. Sharpe, B. Thewes, J. Turner, J. Gilchrist and J. Beith, Oncology, 2018, 32, 32–38 Search PubMed.
- M. F. Clarke, N. Engl. J. Med., 2019, 380, 2237–2245 CrossRef CAS PubMed.
- L. V. Nguyen, R. Vanner, P. Dirks and C. J. Eaves, Nat. Rev. Cancer, 2012, 12, 133–143 CrossRef CAS PubMed.
- L. N. Abdullah and E. K. Chow, Clin. Transl. Med., 2013, 2, 3 Search PubMed.
- V. D'Andrea, S. Guarino, F. M. Di Matteo, M. Maugeri Sacca and R. De Maria, Geka Chiryo, 2014, 35, 257–259 Search PubMed.
- M. Dean, T. Fojo and S. Bates, Nat. Rev. Cancer, 2005, 5, 275–284 CrossRef CAS PubMed.
- K. Rycaj and D. G. Tang, Int. J. Radiat. Biol., 2014, 90, 615–621 CrossRef CAS PubMed.
- W. Chen, J. Dong, J. Haiech, M. C. Kilhoffer and M. Zeniou, Stem Cells Int., 2016, 2016, 1740936 Search PubMed.
- M. Marzagalli, F. Fontana, M. Raimondi and P. Limonta, Cancers, 2021, 13(376), 1–22 Search PubMed.
- E. Batlle and H. Clevers, Nat. Med., 2017, 23, 1124–1134 CrossRef CAS PubMed.
- F. Y. Du, Q. F. Zhou, W. J. Sun and G. L. Chen, World J. Stem Cells, 2019, 11, 398–420 CrossRef PubMed.
- J. Kaiser, Science, 2015, 347, 226–229 CrossRef CAS PubMed.
- F. Marcucci, C. Rumio and F. Lefoulon, Front. Oncol., 2016, 6, 1–22 Search PubMed.
- A. Johnson, J. Northcote-Smith and K. Suntharalingam, Trends Chem., 2021, 3, 47–58 CrossRef CAS.
- K. Laws and K. Suntharalingam, ChemBioChem, 2018, 19, 2246–2253 CrossRef CAS PubMed.
- J. Northcote-Smith and K. Suntharalingam, Curr. Opin. Chem. Biol., 2023, 72, 102237 CrossRef CAS PubMed.
- J. L. Burkhead and J. F. Collins, Adv. Nutr., 2021, 13, 681–683 CrossRef PubMed.
- A. C. Ross, Modern nutrition in health and disease, Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, 11th edn, 2014 Search PubMed.
- N. R. Council, Copper in Drinking Water, The National Academies Press, Washington, DC, 2000 Search PubMed.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Anticancer Agents Med. Chem., 2009, 9, 185–211 CrossRef CAS PubMed.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- P. Ji, P. Wang, H. Chen, Y. Xu, J. Ge, Z. Tian and Z. Yan, Pharmaceuticals, 2023, 16, 234 CrossRef CAS PubMed.
- C. Molinaro, A. Martoriati, L. Pelinski and K. Cailliau, Cancers, 2020, 12(2863), 1–27 Search PubMed.
- Z. Zhang, H. Wang, M. Yan, H. Wang and C. Zhang, Mol. Med. Rep., 2017, 15, 3–11 CrossRef CAS PubMed.
- A. Erxleben, Coord. Chem. Rev., 2018, 360, 92–121 CrossRef CAS.
- J. Karges, K. Xiong, O. Blacque, H. Chao and G. Gasser, Inorg. Chim. Acta, 2021, 516, 120137 CrossRef CAS.
- K. Suntharalingam, D. J. Hunt, A. A. Duarte, A. J. P. White, D. J. Mann and R. Vilar, Chem. – Eur. J., 2012, 18, 15133–15141 CrossRef CAS PubMed.
- K. Suntharalingam, A. J. P. White and R. Vilar, Inorg. Chem., 2010, 49, 8371–8380 CrossRef CAS PubMed.
- J.-W. Liang, Y. Wang, K.-J. Du, G.-Y. Li, R.-L. Guan, L.-N. Ji and H. Chao, J. Inorg. Biochem., 2014, 141, 17–27 CrossRef CAS PubMed.
- J. Grau, A. Caubet, O. Roubeau, D. Montpeyó, J. Lorenzo and P. Gamez, ChemBioChem, 2020, 21, 2348–2355 CrossRef CAS PubMed.
- A. Kumar, J. Prakash Chinta, A. Kumar Ajay, M. Kumar Bhat and C. P. Rao, Dalton Trans., 2011, 40, 10865–10872 RSC.
- J. Rani and S. Roy, ChemMedChem, 2023, 18, e202200652 CrossRef PubMed.
- L. Fang, G. Trigiante, R. Crespo-Otero, C. S. Hawes, M. P. Philpott, C. R. Jones and M. Watkinson, Chem. Sci., 2019, 10, 10881–10887 RSC.
- W. Song, B. Dong, Y. Lu, X. Kong, A. H. Mehmood and W. Lin, Anal. Methods, 2019, 11, 4450–4455 RSC.
- H. Xiao, P. Li, X. Hu, X. Shi, W. Zhang and B. Tang, Chem. Sci., 2016, 7, 6153–6159 RSC.
- Y. Tang, A. Xu, Y. Ma, G. Xu, S. Gao and W. Lin, Sci. Rep., 2017, 7, 12944 CrossRef PubMed.
- ER-Tracker™ Red (BODIPY™ TR Glibenclamide), for live-cell imaging. https://www.thermofisher.com/order/catalog/product/E34250#/E34250. (accessed April 2023).
- Y. X. Feng, E. S. Sokol, C. A. Del Vecchio, S. Sanduja, J. H. L. Claessen, T. A. Proia, D. X. Jin, F. Reinhardt, H. L. Ploegh, Q. Wang and P. B. Gupta, Cancer Discovery, 2014, 4, 702–715 CrossRef CAS PubMed.
- P. Kaur, A. Johnson, J. Northcote-Smith, C. Lu and K. Suntharalingam, ChemBioChem, 2020, 21, 3618–3624 CrossRef CAS PubMed.
- J. N. Boodram, I. J. McGregor, P. M. Bruno, P. B. Cressey, M. T. Hemann and K. Suntharalingam, Angew. Chem., Int. Ed., 2016, 55, 2845–2850 CrossRef CAS PubMed.
- A. Eskandari and K. Suntharalingam, Chem. Sci., 2019, 10, 7792–7800 RSC.
- A. Johnson, C. Olelewe, J. H. Kim, J. Northcote-Smith, R. T. Mertens, G. Passeri, K. Singh, S. G. Awuah and K. Suntharalingam, Chem. Sci., 2023, 14, 557–565 RSC.
- C. Lu, A. Eskandari, P. B. Cressey and K. Suntharalingam, Chem. – Eur. J., 2017, 23, 11366–11374 CrossRef CAS PubMed.
- A. Johnson, L. Iffland-Mühlhaus, J. Northcote-Smith, K. Singh, F. Ortu, U.-P. Apfel and K. Suntharalingam, Dalton Trans., 2022, 51, 5904–5912 RSC.
- G. Dontu, W. M. Abdallah, J. M. Foley, K. W. Jackson, M. F. Clarke, M. J. Kawamura and M. S. Wicha, Genes Dev., 2003, 17, 1253–1270 CrossRef CAS PubMed.
- R. Mezencev, L. Wang and J. F. McDonald, J. Ovarian Res., 2012, 5, 30 CrossRef CAS PubMed.
- W. Senkowski, X. Zhang, M. H. Olofsson, R. Isacson, U. Höglund, M. Gustafsson, P. Nygren, S. Linder, R. Larsson and M. Fryknäs, Mol. Cancer Ther., 2015, 14, 1504–1516 CrossRef CAS PubMed.
- C. Lu, K. Laws, A. Eskandari and K. Suntharalingam, Dalton Trans., 2017, 46, 12785–12789 RSC.
- A. Eskandari, A. Kundu, S. Ghosh and K. Suntharalingam, Angew. Chem., Int. Ed., 2019, 58, 12059–12064 CrossRef CAS PubMed.
- M. Boyce, K. F. Bryant, C. Jousse, K. Long, H. P. Harding, D. Scheuner, R. J. Kaufman, D. Ma, D. M. Coen, D. Ron and J. Yuan, Science, 2005, 307, 935–939 CrossRef CAS PubMed.
- C. Hetz, Nat. Rev. Mol. Cell Biol., 2012, 13, 89–102 CrossRef CAS PubMed.
- T. W. Fawcett, J. L. Martindale, K. Z. Guyton, T. Hai and N. J. Holbrook, Biochem. J., 1999, 339(Pt 1), 135–141 CrossRef CAS PubMed.
- H. P. Harding, I. Novoa, Y. Zhang, H. Zeng, R. Wek, M. Schapira and D. Ron, Mol. Cell, 2000, 6, 1099–1108 CrossRef CAS PubMed.
- C. Y. Liu and R. J. Kaufman, J. Cell Sci., 2003, 116, 1861–1862 CrossRef CAS PubMed.
- J. Faitova, D. Krekac, R. Hrstka and B. Vojtesek, Cell. Mol. Biol. Lett., 2006, 11, 488–505 CAS.
- P. J. Quinn, Subcell. Biochem., 2002, 36, 39–60 CAS.
- G. M. Cohen, Biochem. J., 1997, 326(Pt 1), 1–16 CrossRef CAS PubMed.
- F. J. Oliver, G. de la Rubia, V. Rolli, M. C. Ruiz-Ruiz, G. de Murcia and J. M. Murcia, J. Biol. Chem., 1998, 273, 33533–33539 CrossRef CAS PubMed.
- W. Henke, S. Kremer and D. Reinen, Inorg. Chem., 1983, 22, 2858–2863 CrossRef CAS.
- G. Sheldrick, Program for Area Detector Absorption Correction, Institute for Inorganic Chemistry, University of Göttingen, Germany, 1996 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2258097–2258101. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01294h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |