
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntibacterial properties of phosphine gold(I) complexes with 5-fluorouracil†
Ricardo
Ferrando
ab,
Scott G.
Mitchell
 bc,
Elena
Atrián-Blasco
*b and
Elena
Cerrada
*a
bc,
Elena
Atrián-Blasco
*b and
Elena
Cerrada
*a
aDepartamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea-ISQCH, Universidad de Zaragoza-CSIC, 50009 Zaragoza, Spain. E-mail: ecerrada@unizar.es
bInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, Zaragoza 50009, Spain. E-mail: e.atrian.blasco@csic.es
cCIBER de Bioingeniería, Biomateriales y Nanomedicina, Instituto de Salud Carlos III, 28029 Madrid, Spain
First published on 30th June 2023
Abstract
New gold(I) complexes with coordination to 5-fluorouracil (5-FU), an anticancer drug with antibacterial properties, have been synthesised and characterised, and are the first reported examples of 5-FU–Au compounds. These new complexes show high solution stability, even in the presence of a cysteine derivative, and so were evaluated as antibacterial compounds against model Gram-positive and Gram-negative bacteria. All the complexes show excellent antibacterial activity against Gram-positive B. subtilis, most of them improving the activity of 5-FU alone. Furthermore, these new complexes are also active against Gram-negative E. coli, where [Au(5-FU)(PTA)], the complex with the smallest phosphane, is the most bactericidal, 32 times more active than 5-FU on its own.
Introduction
5-Fluorouracil (2,4-dihydroxy-5-fluoropyrimidine; 5-FU) is an analogue of the nucleobase uracil with a fluorine atom at the C-5 position. It is used for the treatment of a wide range of cancers: colorectal, breast and head and neck cancers, since the discovery of its activity in 1957.1 But, it is against colorectal cancer where 5-FU shows the best activity.2,3 In fact, 5-FU is administered together with leucovorin as co-adjuvants in the treatment of colon cancer with oxaliplatin.4,5 Its mechanism of action consists of the inhibition of thymidylate synthase, a key enzyme for the production of DNA, in an irreversible manner; and incorporation of its metabolites into RNA and DNA, causing DNA damage.6 5-FU is also applied to the treatment of some skin cancers, Bowen's disease and actinic keratosis.7 In addition to its anticancer properties, 5-FU has been shown to have potent antimicrobial effects against different bacterial pathogens.8,9 It seems that the effect of 5-FU would be dual by blocking the production of virulence factors and biofilm besides bacterial growth.10–12The discovery of the antimicrobial properties of K[Au(CN)2] against Mycobacterium tuberculosis by R. Koch at the end of the 19th century13 encouraged researchers to consider gold complexes as potential antibacterial agents. Since then, many examples of gold(I) complexes have shown promising activity against a broad spectrum of microorganisms.14–17 One of the most studied gold(I) complexes is auranofin (2,3,4,6-tetraacetyl-1-thio-β-D-glucopyranosato-S-triethylphosphane gold(I)), which has been widely studied displaying high efficacy mainly against Gram-positive bacteria, including multidrug-resistant strains.18–28 As in the case of its anticancer activity, the mechanism of action behind its antibacterial activity is the inhibition of thioredoxin reductase (TrxR).29
Although many examples of complexes based on metals such as platinum30–32 and silver33–37 coordinated to 5-FU and its derivatives have been studied, to the best of our knowledge, only two examples of gold(I) complexes have been described with 5-FU derivatives (5FdUrd, 5-fluoro-2′-deoxyuridine and Tegafur, 5-fluoro-1-(2-tetrahydrofuryl)-2,4(1H,3H-pyrimidinedione).38
With this background, we describe here the synthesis of new gold(I) 5-fluorouracil complexes, the first reported 5-FU–Au coordination compounds, with the phosphanes PTA (1,3,5-triaza-7-phosphaadamantane), DAPTA (3,7-diacetyl-1,3,7-triaza-5-phosphabicyclo[3.3.1]nonane), PPh3 (triphenylphosphane) and TPPTS (trisodium 3,3′,3′′-phosphanetriyltri(benzene-1-sulfonate). The antibacterial activity of these complexes has been studied against two model bacterial strains, E. coli and B. subtilis.
Experimental
General
Solvents were used as received without purification or drying. The starting materials [AuCl(PR3)],39–41 PTA,42 DAPTA43 and TPPTS44 were prepared as previously reported. All other reagents were commercially available and used without further purification. 1H, 13C{1H}, 19F, and 31P{1H} spectra were recorded on a Bruker Avance 400 or a Bruker ARX 300 spectrometer. Chemical shifts (δ, ppm) were reported relative to the solvent peaks in the 1H, 13C spectra or external 85% H3PO4 or CFCl3 in 31P or 19F spectra. IR spectra were recorded in the range of 4000–200 cm−1 on a PerkinElmer Spectrum 100 spectrophotometer on solid samples using an ATR accessory. C, H, and N analyses were carried out with a PerkinElmer 2400 Series 2 microanalyzer.Synthesis of the complexes
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). MALDI MS m/z (%): 236.367 (23) [M − TPPTS]+. Anal. calcd (%) for C22H14AuFN2Na3O11PS3 (894.445): C, 29.54; H, 1.58; N, 3.13. Found: C, 29.15; H, 1.51; N, 2.91.
O). MALDI MS m/z (%): 236.367 (23) [M − TPPTS]+. Anal. calcd (%) for C22H14AuFN2Na3O11PS3 (894.445): C, 29.54; H, 1.58; N, 3.13. Found: C, 29.15; H, 1.51; N, 2.91.
Solution chemistry
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O (80:20) containing the gold complexes (0.03 mmol) with an equimolecular amount of N-acetyl cysteine methyl ester (NAC-ME). The corresponding NMR spectra were recorded immediately after preparation, at time 0 and after 24 h.
:D2O (80:20) containing the gold complexes (0.03 mmol) with an equimolecular amount of N-acetyl cysteine methyl ester (NAC-ME). The corresponding NMR spectra were recorded immediately after preparation, at time 0 and after 24 h.
Bacterial proliferation assays
The bacterial strains used, Bacillus subtilis (Ehrenberg 1835) Cohn 1872 – CECT 35 and Escherichia coli (Migula 1895) Castellani and Chalmers 1919 – CECT 101, were acquired from the Colección Española de Cultivos Tipo (CECT) as lyophilized bacteria. The bacterial cell bank suspensions were thawed and inoculated on an agar plate and on a liquid medium (nutrient broth – NB – for B. subtilis and LB (Miller) broth – LB – for E. coli) at 37 °C for 24 h with mild agitation. A first subculture was performed to assure the viability of the strain. A dilution from these culture solutions (second subculture and so on) was used for the following tests, corresponding to an inoculum of 107 CFU mL−1. Considering that the dilution to the well will be 1:100, stock solutions were prepared – in DMSO – ×100 more concentrated than the final concentration in the well (409.6 mg mL−1, 204.8 mg mL−1, 102.4 mg mL−1, and so on).
Preparation of bacteria for scanning electron microscopy
In a 24-well plate, 500 μL of bacteria culture (107 CFU mL−1) was added per well and was treated with 5-FU, 1 and 5 at their corresponding MIC and 1/2× MIC. A positive control of bacteria without any treatment and a negative control of only culture medium were included, following the same protocol. Once the plates were prepared, bacteria were incubated at 37 °C for 24 h with mild agitation. After this time, bacterial suspensions were transferred into 1.5 mL centrifugation tubes for fixation and were centrifuged at 3000 rpm for 15 minutes. The pellet was washed twice with sterile PBS and then resuspended in 1.5 mL of a 2.5% glutaraldehyde solution in 10 mM PBS with pH 7.2. The suspensions were incubated in a tube rotator for 2 hours. Next, bacteria were washed with PBS (×1) and ultrapure H2O (×3). The fixed bacteria were then washed with PBS (×3) and dehydrated with increasing concentrations of EtOH (30%, 50%, 70%, 90%, 96%, and 100%) for 10 minutes each. Finally, 10 μL of the suspensions were transferred onto silicon wafer squares, allowed to dry and then coated with 14 nm of carbon.Results and discussion
Synthesis and characterisation of gold complexes
Deprotonation of 5-FU with KOH in methanol, followed by the addition of [AuCl(PR3)] (PR3 = PTA, DAPTA, PPh3 and TPPTS) afforded the corresponding mononuclear derivatives [Au(5-FU)(PR3)] (PR3 = PTA, 1; DAPTA, 2; PPh3, 3 and TPPTS, 4) (Scheme 1) as off-white air-stable solids. | ||
| Scheme 1 Synthesis of 5-FU gold(I) derivatives, 1–5. | ||
In the 1H NMR spectra of the new derivatives, the signal corresponding to the pyrimidine proton at position 6 is observed at around 7.50 ppm, as a doublet in the case of complex 2 with a JH–F of 5.5 Hz, which implies an up-field shift compared to that of free 5-FU (7.75 ppm). In the case of the PTA complexes (1 and 5) and the TPPTS complex (4), the signal appears as a broad signal, and in the PPh3 derivative (3), the signal appears together with the multiplet due to PPh3. The signal of an N–H proton is only clearly detected in the cases of PTA and DAPTA derivatives, as a broad singlet centred at 10.3 ppm for [Au(5-FU)(PTA)] (1) and at 7.98 ppm for the DAPTA analogue. It is not possible to distinguish between the coordination to the nitrogen atoms N1 and N3 with the available NMR data. Deprotonation of either N1 or N3 is very dependent on the solvent used in the reaction, as different solvents stabilize the different anions involved in deprotonation. Thus, deprotonation of the nitrogen at position N1 occurs in DMSO as solvent, while anions at both nitrogen positions are formed in an aqueous alkaline solution; in addition, more polar solvents stabilize the formation of the corresponding N3 anion.45,46 Since the reactions have been carried out in methanol, a solvent with an intermediate polarity between that of DMSO and water, the deprotonation and subsequent gold coordination could happen at both nitrogen positions. Besides, methylene protons of the PTA and DAPTA fragment integration with respect to the pyrimidine H(6) proton, confirming the 5-FU:PTA ratios of 1:1 for both complexes, which is also measured in the TPPTS derivative (integration of Ph and H(6) signals).
The 31P{1H} NMR spectra in DMSO-d6 at room temperature display a singlet in all the complexes. In the case of [Au(5-FU)(PTA)] (1), the spectrum in D2O shows a singlet at −51.6 ppm and a small signal at −52.3 ppm, which could indicate a fast interchange of the coordination position between N1 and N3 atoms of the pyrimidine moiety. Accordingly, NMR experiments have been performed at low temperatures in MeOD only in the case of complex [Au(5-FU)(DAPTA)] (2), which showed enough solubility in MeOD even at below 0 °C. Therefore, the 31P{1H} NMR spectrum of 2 at 213 K exhibits the formation of two signals at −23.7 and −27.8 ppm (Fig. 1), which appeared as a singlet at room temperature. The resolution of two singlets at low temperatures could be indicative of gold coordination to both nitrogen atoms after being deprotonated in methanol, as mentioned above, or to the formation of the two isomers in solution.
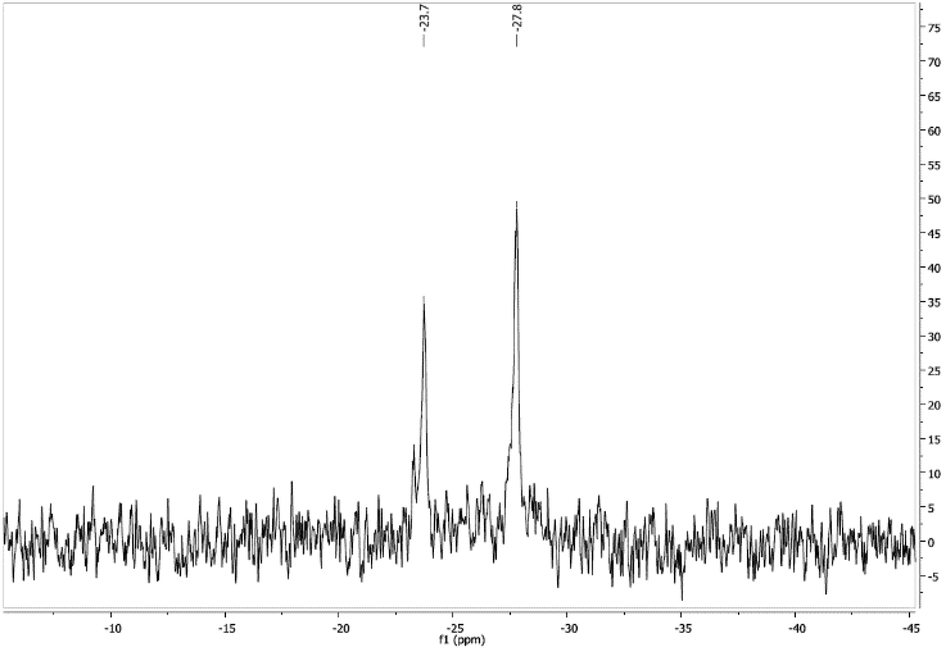 | ||
| Fig. 1 31P{1H} NMR spectrum of [Au(5-FU)(DAPTA)] (2) in MeOD at 213 K. | ||
According to Abdrakhimova et al.,46 C6 resonance is more strongly deshielded in the 13C{1H} NMR spectra of the derivatives compared to 5-FU, which is in agreement with the coordination of the metallic centre to the N1 atom, a consequence of deprotonation of the N1 atom in 5-FU.
The addition of two equivalents of KOH and the corresponding chloro-gold phosphine to 5-FU afforded the dinuclear derivative only in the case of PTA [Au2(5-FU)(PTA)2] (5), probably due to the higher spatial requirement of DAPTA, PPh3 and TPPTS, which are bulkier than PTA,47–49 resulting in stronger steric hindrance and making the coordination of two AuPR3 moieties more difficult. The signal of the N–H proton is not observed in the 1H NMR spectrum of 5, confirming the deprotonation of both nitrogen atoms and the coordination of both Au-PTA units. Methylene protons of the PTA fragment integration in a 24:1 relationship with respect to the pyrimidine H(6) proton, confirming the 5-FU:PTA ratio of 1:2. Besides, these signals are slightly more complicated than those observed in the mononuclear [Au(5-FU)(PTA)] (1), as the signals of the methylene protons of NCH2N overlap in a multiplet between 4.66 and 4.51 ppm, and methylene protons in the NCH2P moiety are seen as two singlets at 4.36 and 4.32 ppm, indicating that the two different Au-PTA fragments are inequivalent, as their chemical environment is not the same. This inequivalence can also be detected in the 31P{1H} NMR spectrum, where two singlets are observed at −51.9 and −52.7 ppm, with a ratio of 1:1, meaning that both phosphorus atoms are linked to Au in a similar manner, although in a different environment.
Experimental and calculated absorption spectra of 5-FU, and its different anions and tautomers, have been widely studied by Raman spectroscopy45,50 and IR spectroscopy.38,51,52 In the case of 5-FU, a group of complex and strong absorption bands for the carbonyl groups appear in the range from 1720 to 1650 cm−1, together with the C–H absorption band (Fig. S11†). Upon coordination of gold to the nitrogen atoms, these bands decrease in intensity and appear shifted to lower frequencies: 1636 and 1587 cm−1 for 1, 1534 and 1443 cm−1 for 2, 1651 and 1599 cm−1 for 3, 1588 and 1619 cm−1 for 4 and 1630 and 1585 cm−1 for 5. This shift indicates a reduction of the double-bond character of CO by the coordination of gold so that the pair of electrons of the carbonyl group move to favour a nitrogen–Au(I) bond, as has been stated in other examples of N–M coordination in 5-FU complexes38,53 and also in the study of the formation of 5-FU anions and dianions.52 Furthermore, the numerous N–H bands observed in the spectrum of 5-FU decrease in number and intensity in the case of the mononuclear derivatives 1–4, disappearing in the spectrum of dinuclear complex 5. Unfortunately, no single crystals were obtained for any of the complexes to confirm the structure in the solid state. The low solubility of these compounds in any solvent suitable for single crystal growth hinders the formation of crystals suitable for X-ray diffraction.
Stability of gold complexes in solution
The stability of the new complexes was analysed by UV-vis absorption spectroscopy in PBS solution (pH = 7.4). 5 × 10−5 M solutions were prepared by diluting DMSO stock solutions of the complexes in PBS buffered at pH 7.4. The resulting solutions were monitored over 24 h at 37 °C. The spectra of the complexes (Fig. S12†) show an absorption band with low intensity at around 260 nm, which could be assigned to π → π* intra-ligand transitions. These bands remain without any changes in shape and non-apparent red or blue shifts are observed in all the new derivatives, in addition to a lack of absorbance at around 500 nm over 24 h, which is characteristic of gold nanoparticle formation, implying substantial stability of the chromophore under physiological conditions. Changes in the baseline intensity for complexes 1, 3 and 5 are due to low solubility in PBS preventing a homogeneous solution being obtained at the studied concentration.Besides, the reactivity of the complexes towards model nucleophiles was studied by mixing them with N-acetyl cysteine methyl ester (NAC-ME) in equimolecular amounts.54 The resulting solutions were analysed by 1H NMR over time (Fig. S13†). NAC-ME underwent auto-oxidation, displaying new signals at around 3, 1.8 and 4.4 ppm after 24 h, as a consequence of sulphur bridge formation. All the complexes seemed to be inert with respect to ligand exchange reactions with this nucleophile, since no new signals were detected apart from those obtained from the auto-oxidation of NAC. Moreover, the solution did not change colour, indicating that there was no reduction of Au(III) to Au(0), confirming the high stability of the complexes.
Antibacterial activity of the gold(I)–5-FU complexes
As previously mentioned, 5-FU has shown antibacterial activity against different bacterial strains, both Gram-negative and Gram-positive. Its activity arises mostly from the suppression of virulence genes and actions, quenching quorum-sensing in bacteria and preventing the formation of biofilms.12,55,56 In this study, we have found 5-FU to be more active against Gram-positive B. subtilis than against Gram-negative E. coli (Table 1). In general, all the compounds presented higher antibacterial activity than 5-fluorouracil alone, confirming the fact that the incorporation of a metallic centre improves this activity, as previously observed for a silver(I) complex with 5-FU.33 Furthermore, the antibacterial activity of these gold(I) complexes against B. subtilis and E. coli is in the same range shown by auranofin and its derivatives of similar structure. It has been shown that the mechanism of action of auranofin against Gram-positive bacteria arises from the inhibition of TrxR after the coordination of the Au–phosphine unit to its cysteine residues.21,29| Compound | E. coli | B. subtilis | ||
|---|---|---|---|---|
| MIC | MBC | MIC | MBC | |
| 5-FU | 1024 | >1024 | 64 | 128 |
| 1 | 32 | 64 | 8 | 16 |
| 2 | 64 | 128 | 16 | 32 |
| 3 | >1024 | >1024 | 1 | 1 |
| 4 | 256 | 512 | 64 | 128 |
| 5 | 128 | 128 | 2 | 4 |
With the exceptions of compound 3 for E. coli and compound 4 for B. subtilis, the addition of gold–phosphane units to 5-FU increased the inhibitory activity up to ×64. All complexes possessed bactericidal activity, with MBC/MIC ratios lower than 4 in all cases. Complexes 1 and 5 show the highest activity against both strains. The improvement of the activity is even stronger when comparing the MIC and MBC values in nM instead of μg mL−1 (Table S1†).
Gram-negative bacteria have a more complex and robust cell wall and a highly developed efflux-mediated resistance mechanism, which confers them with greater resistance capacity against antibiotics. Multi-drug resistant Gram-negative bacteria are of special concern and great research effort must be put into developing new effective antibiotics. In general, 5-FU and its Au(I) complexes are more active against the Gram-positive B. subtilis than against the Gram-negative E. coli, as has been previously observed for metallic complexes and antibiotics in general.57
Nevertheless, it is interesting to observe differences in the activity of the complexes depending on the size and hydrophobicity of their phosphane ligands – which could be extrapolated to the complex. Complex 3, which has the most hydrophobic phosphane, PPh3, presents the lowest antibacterial activity against E. coli but in turn, it is the most active against B. subtilis. On the other hand, complex 1 is the most bactericidal complex against E. coli, with a non-insignificant MBC value of 64 μg mL−1. Based on these results, higher hydrophilicity and smaller size improve the bactericidal activity of the complexes against the Gram-negative model bacteria E. coli. This has been previously observed for gold complexes, improving their activity against Gram-negative bacteria with the use of smaller alkylphosphines.28,58 Given these results, it would be interesting to further study the size and hydrophilicity of gold complexes to increase their bactericidal activity against Gram-negative bacteria.
Gold(I)–phosphine complexes are in general mainly effective against Gram-positive bacteria and inactive in most of the cases against Gram-negative bacteria. In our case, all complexes enhance the activity of 5-fluorouracil against Gram-positive bacteria except for complex 4. Furthermore, all of them inhibit the growth of E. coli bacteria (Gram-negative) more effectively than 5-FU, except for the complex with PPh3, similar to the starting material [AuClPPh3].59 This fact suggests that the presence of the gold–phosphine unit is essential to induce an increase in the activity of the final complexes.
Besides, examples are known of complexes with the N–Au–phosphine core,60,61 as in our case, that inhibit the growth of Gram-negative bacteria, most likely because of their capacity to interact with the membrane or specific biomolecules present in the membrane.
Electron microscopy is a useful method for the observation of bacteria and the impact on their growth and death caused by antimicrobial agents. Here we used scanning electron microscopy (SEM) to further examine the effect of 5-FU and the complexes 1, 3 and 5 on E. coli and B. subtilis (Fig. 2). Bacteria were incubated for 24 h without any treatment or in the presence of the different compounds at concentrations corresponding to their MIC values. Since the MIC value for complex 3 against E. coli was not found at the tested concentrations, the highest concentration tested (1024 μg mL−1) was used for the incubation. In E. coli, the effect of complexes 1 and 5 can be observed in the cell since the surface of the bacteria loses its smooth appearance with the presence of dents and depressions. Incubation with 5-FU affects some of the bacteria in the samples, with changes in morphology and the membrane. Meanwhile, complex 3 does not have an observable effect compared to the control, at least observable in SEM. The limited ability to combat Gram-negative strains in the case of most of the inactive gold complexes is potentially linked to a decreased uptake of the drug caused by the permeability of the outer membrane barrier.62 In fact, modification of the ligands of auranofin can modulate the antimicrobial activity rendering effective complexes even against Gram-negative strains.28 In our case, the damage on the surface of the cell membrane observed by SEM studies could be indicative of an enhanced activity and higher interaction with the bacterial membrane.
 | ||
| Fig. 2 Scanning electron microscopy (SEM) images of E. coli (A–E) and B. subtilis (F–J) without treatment (control, A & F) and after incubation with 5-FU (B & G) or complexes 1 (C & H), 3 (D & I) and 5 (E & J), at their corresponding MIC (50k×–60k×) and at the highest tested concentration (1024 μg mL−1) in the case of complex 3 for E. coli. Scale bar = 1 μm. Orange arrows indicate cell damage, while blue arrows indicate biofilm matrix formation. | ||
Furthermore, an extracellular biofilm matrix can be seen in the control of E. coli and in the incubation with complex 3 attaching to several cells (Fig. 2, light blue arrows), while it is not observed in the presence of 5-FU or complexes 1 and 5. The biofilm matrix is produced by bacteria and is mainly composed of polysaccharides, proteins and extracellular DNA. It confers great protection to the bacteria within the biofilm, as well as adherence to the surface, and is thought to be key for the development of antibiotic resistance and virulence. It has been previously described that 5-FU reduces the formation of biofilms in different strains of E. coli in a dose-dependent manner.63 This inhibitory effect could be maintained in the case of the active Au(I) complexes. The effect of the metallic complexes is more evident in the case of B. subtilis. After treatment with 5-FU, the surface of the bacteria appears damaged, while complexes 1, 3 and 5 clearly produce not only disruption of the bacterial cell wall but also changes in morphology such as compression and accumulation of intracellular material at the extremes of the bacteria. In the sample of B. subtilis treated with complex 5, a mass of material surrounding the bacteria could be observed. Analysis of the sample by detection of backscattered electrons indicates the possible presence of gold in the mass, which is further confirmed by energy dispersive X-ray spectroscopy (Fig. S14†).
Conclusions
Here, we report the synthesis of new gold(I) phosphane complexes of 5-fluorouracil, an analogue of the nucleobase uracil used for the treatment of different types of cancer, including the first example of a dinuclear gold(I) complex. All the compounds demonstrated good stability under simulated physiological conditions in solution, even in the presence of N-acetylcysteine methyl ester (NAC-ME). Their activity as antibacterial agents was evaluated against two model bacterial strains: E. coli and B. subtilis. While 5-FU was shown to be the principal bactericidal agent, the introduction of the metallic centre improved the activity of the complexes, compared to 5-FU on its own. This is especially relevant in the case of the dinuclear complex, which demonstrated excellent activity against B. subtilis. Furthermore, the interaction of the compounds with bacteria was studied by scanning electron microscopy, and damage to the membrane could be observed after incubation with the tested compounds. This study offers new ways to complex known drugs with metallic centres offering a route to combined therapies as a method to increase their bioactivity and potentially combat the resistance generated in microorganisms and cancerous cells. Future work will focus on how fine tuning the chemistry, e.g., optimising the size and hydrophilicity of the complexes, can affect more specifically the additive or synergic activity of such gold(I)–fluorouracil complexes against a wider spectrum of bacteria.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by MCIN-Agencia Estatal de Investigación Grants PID2019-104379RB-C21/AEI/10.13039/501100011033, and Red Multimetdrugs (RED2018-102471-T/MCIN/AEI/10.13039/501100011033) and Gobierno de Aragón (E07_20R, Fondos FEDER “otra manera de hacer Europa”). E. A. B. and S. G. M. acknowledge the funding from the European Union's Horizon 2020 research and innovation program (Marie Skłodowska-Curie grant agreement no. 845427) and from the internationalization program “MSCA-IF_ERC” of CSIC. The authors acknowledge Servicio General de Apoyo a la Investigación-SAI (Universidad de Zaragoza) and the use of instrumentation as well as the technical advice provided by the National Facility ELECMI ICTS, node “Laboratorio de Microscopías Avanzadas” at the University of Zaragoza.References
- C. Heidelberger, N. K. Chaudhuri, P. Danneberg, D. Mooren, L. Griesbach, R. Duschinsky, R. J. Schnitzer, E. Pleven and J. Scheiner, Nature, 1957, 179, 663–666 CrossRef CAS PubMed.
- J. A. Meyerhardt and R. J. Mayer, N. Engl. J. Med., 2005, 352, 476–487 CrossRef CAS PubMed.
- D. Cunningham, W. Atkin, H. J. Lenz, H. T. Lynch, B. Minsky, B. Nordlinger and N. Starling, Lancet, 2010, 375, 1030–1047 CrossRef PubMed.
- C. Tournigand, T. Andre, F. Bonnetain, B. Chibaudel, G. Lledo, T. Hickish, J. Tabernero, C. Boni, J.-B. Bachet, L. Teixeira and A. de Gramont, J. Clin. Oncol., 2012, 30, 3353–3360 CrossRef CAS PubMed.
- T. Andre, C. Boni, L. Mounedji-Boudiaf, M. Navarro, J. Tabernero, T. Hickish, C. Topham, M. Zaninelli, P. Clingan, J. Bridgewater, I. Tabah-Fisch and A. de Gramont, N. Engl. J. Med., 2004, 2343–2351 CrossRef CAS PubMed.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef CAS PubMed.
- R. I. Ceilley, J. Dermatol. Treat., 2012, 23, 83–89 CrossRef CAS PubMed.
- J. M. Thomson and I. L. Lamont, Front. Microbiol., 2019, 10, 952 CrossRef PubMed.
- C. Oe, H. Hayashi, K. Hirata, K. Kawaji, F. Hashima, M. Sasano, M. Furuichi, E. Usui, M. Katsumi, Y. Suzuki, C. Nakajima, M. Kaku and E. N. Kodama, ACS Infect. Dis., 2020, 6, 1490–1500 CrossRef CAS PubMed.
- A. Ueda, C. Attila, M. Whiteley and T. K. Wood, Microb. Biotechnol., 2009, 2, 62–74 CrossRef CAS PubMed.
- C. Attila, A. Ueda and T. K. Wood, Appl. Microbiol. Biotechnol., 2009, 82, 525–533 CrossRef CAS PubMed.
- A. Rangel-Vega, L. Bernstein, E. A. Mandujano-Tinoco, S. J. García-Contreras and R. García-Contreras, Front. Microbiol., 2015, 6, 282 Search PubMed.
- E. R. T. Tieknik, Crit. Rev. Oncog., 2002, 42, 225 CrossRef PubMed.
- B. D. Glisic and M. I. Djuran, Dalton Trans., 2014, 43, 5950–5969 RSC.
- J. R. Stenger-Smith and P. K. Mascharak, ChemMedChem, 2020, 15, 2136–2145 CrossRef CAS PubMed.
- F. Novelli, M. Recine, F. Sparatore and C. Juliano, Farmaco, 1999, 54, 232–236 CrossRef CAS PubMed.
- R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- A. AbdelKhalek, N. S. Abutaleb, H. Mohammad and M. N. Seleem, Int. J. Antimicrob. Agents, 2019, 53, 54–62 CrossRef CAS PubMed.
- L. Aguinagalde, R. Diez-Martinez, J. Yuste, I. Royo, C. Gil, I. Lasa, M. Martin-Fontecha, N. I. Marin-Ramos, C. Ardanuy, J. Linares, P. Garcia, E. Garcia and J. M. Sanchez-Puelles, J. Antimicrob. Chemother., 2015, 70, 2608–2617 CrossRef CAS PubMed.
- T. Marzo, D. Cirri, S. Pollini, M. Prato, S. Fallani, M. I. Cassetta, A. Novelli, G. M. Rossolini and L. Messori, ChemMedChem, 2018, 13, 2448–2454 CrossRef CAS PubMed.
- J. P. Owings, N. N. McNair, Y. F. Mui, T. N. Gustafsson, A. Holmgren, M. Contel, J. B. Goldberg and J. R. Mead, FEMS Microbiol. Lett., 2016, 363, fnw148 CrossRef PubMed.
- N. S. Abutaleb and M. N. Seleem, Int. J. Antimicrob. Agents, 2020, 55, 105828 CrossRef CAS PubMed.
- A. N. Kuntz, E. Davioud-Charvet, A. A. Sayed, L. L. Califf, J. Dessolin, E. S. J. Arner and D. L. Williams, PLoS Med., 2007, 4, 1071–1086 CAS.
- M. Bonilla, A. Denicola, S. V. Novoselov, A. A. Turanov, A. Protasio, D. Izmendi, V. N. Gladyshev and G. Salinas, J. Biol. Chem., 2008, 283, 17898–17907 CrossRef CAS PubMed.
- A. R. Sannella, A. Casini, C. Gabbiani, L. Messori, A. R. Bilia, F. F. Vincieri, G. Majori and C. Severini, FEBS Lett., 2008, 582, 844–847 CrossRef CAS PubMed.
- A. Ilari, P. Baiocco, L. Messori, A. Fiorillo, A. Boffi, M. Gramiccia, T. Di Muccio and G. Colotti, Amino Acids, 2012, 42, 803–811 CrossRef CAS PubMed.
- N. Tejman-Yarden, Y. Miyamoto, D. Leitsch, J. Santini, A. Debnath, J. Gut, J. H. McKerrow, S. L. Reed and L. Eckmann, Antimicrob. Agents Chemother., 2013, 57, 2029–2035 CrossRef CAS PubMed.
- B. Wu, X. J. Yang and M. D. Yan, J. Med. Chem., 2019, 62, 7751–7768 CrossRef CAS PubMed.
- M. B. Harbut, C. Vilcheze, X. Z. Luo, M. E. Hensler, H. Guo, B. Y. Yang, A. K. Chatterjee, V. Nizet, W. R. Jacobs, P. G. Schultz and F. Wang, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4453–4458 CrossRef CAS PubMed.
- M. F. Mogilevkina and V. A. Shipachev, Koord. Khim., 1994, 20, 51–53 CAS.
- X. Y. Wang, J. Lin, X. M. Zhang, Q. Liu, Q. Xu, R. X. Tan and Z. J. Guo, J. Inorg. Biochem., 2003, 94, 186–192 CrossRef CAS PubMed.
- M. F. Mogilevkina, V. A. Shipachev, G. P. Troshkova, M. A. Mazurkova, M. P. Martynets and M. P. Bagryantseva, Pharm. Chem. J., 1999, 33, 68–70 CrossRef CAS.
- J. H. B. Nunes, F. R. G. Bergamini, W. R. Lustri, P. P. de Paiva, A. L. T. G. Ruiz, J. E. de Carvalho and P. P. Corbi, J. Fluor. Chem., 2017, 195, 93–101 CrossRef CAS.
- M. Barceló-Oliver, B. A. Baquero, A. Bauzá, Á. García-Raso, R. Vich, I. Mata, E. Molins, À. Terrón and A. Frontera, Dalton Trans., 2013, 42, 7631–7642 RSC.
- I. Okamoto, K. Iwamoto, Y. Watanabe, Y. Miyake and A. Ono, Angew. Chem., Int. Ed., 2009, 48, 1648–1651 CrossRef CAS PubMed.
- M. S. Wysor and R. E. Zollinhofer, Chemotherapy, 1972, 17, 188–199 CrossRef CAS PubMed.
- M. I. Gelfman and N. A. Kustova, Zh. Neorg. Khim., 1970, 15, 92–96 CAS.
- A. Takashi, M. T. Ken, I. Hikaru and S. Yukiyoshi, Bull. Chem. Soc. Jpn., 1989, 62, 1078–1080 CrossRef.
- Z. Assefa, B. G. McBurnett, R. J. Staples and J. P. Fackler, Inorg. Chem., 1995, 34, 4965–4972 CrossRef CAS.
- R. Uson, A. Laguna, M. Laguna, D. A. Briggs, H. H. Murray and J. P. Fackler, Inorg. Synth., 1989, 26, 85–91 CAS.
- E. Vergara, S. Miranda, F. Mohr, E. Cerrada, E. R. T. Tiekink, P. Romero, A. Mendia and M. Laguna, Eur. J. Inorg. Chem., 2007, 2926–2933, DOI:10.1002/ejic.200700112.
- D. J. Daigle, A. B. Pepperma and S. L. Vail, J. Heterocycl. Chem., 1974, 11, 407–408 CrossRef CAS.
- D. J. Darensbourg, C. G. Ortiz and J. W. Kamplain, Organometallics, 2004, 23, 1747–1754 CrossRef CAS.
- F. Joo, J. Kovacs, A. Katho, A. C. Benyei, T. Decuir, D. J. Darensbourg, A. Miedaner and D. L. Dubois, in Inorg. Synth, ed. M. Y. Darensbourg, 1998, vol. 32, pp. 1–8 Search PubMed.
- N. Markova, V. Enchev and G. Ivanova, J. Phys. Chem. A, 2010, 114, 13154–13162 CrossRef CAS PubMed.
- G. S. Abdrakhimova, M. Y. Ovchinnikov, A. N. Lobov, L. V. Spirikhin, S. P. Ivanov and S. L. Khursan, J. Phys. Org. Chem., 2014, 27, 876–883 CrossRef CAS.
- T. L. Brown and K. J. Lee, Coord. Chem. Rev., 1993, 128, 89–116 CrossRef CAS.
- L. R. Moore, E. C. Western, R. Craciun, J. M. Spruell, D. A. Dixon, K. P. O'Halloran and K. H. Shaughnessy, Organometallics, 2008, 27, 576–593 CrossRef CAS.
- D. J. Darensbourg, J. B. Robertson, D. L. Larkins and J. H. Reibenspies, Inorg. Chem., 1999, 38, 2473–2481 CrossRef CAS.
- I. Pavel, S. Cota, S. Cinta-Pinzaru and W. Kiefer, J. Phys. Chem. A, 2005, 109, 9945–9952 CrossRef CAS PubMed.
- K. Dobrosz-Teperek, Z. Zwierzchowska, W. Lewandowski, K. Bajdor, J. C. Dobrowolski and A. P. Mazurek, J. Mol. Struct., 1998, 471, 115–125 CrossRef CAS.
- V. K. Rastogi and M. A. Palafox, Spectrochim. Acta, Part A, 2011, 79, 970–977 CrossRef CAS PubMed.
- J. F. Villa, J. Gelber, N. Khoe and J. Cepeda, J. Am. Chem. Soc., 1978, 100, 4305–4307 CrossRef CAS.
- S. M. Meier-Menches, B. Aikman, D. Döllerer, W. T. Klooster, S. J. Coles, N. Santi, L. Luk, A. Casini and R. Bonsignore, J. Inorg. Biochem., 2020, 202, 110844 CrossRef CAS PubMed.
- V. Singh, M. Brecik, R. Mukherjee, J. C. Evans, Z. Svetlíková, J. Blaško, S. Surade, J. Blackburn, D. F. Warner, K. Mikušová and V. Mizrahi, Chem. Biol., 2015, 22, 63–75 CrossRef CAS PubMed.
- F. Sedlmayer, A. K. Woischnig, V. Unterreiner, F. Fuchs, D. Baeschlin, N. Khanna and M. Fussenegger, Nucleic Acids Res., 2021, 49, e73 CrossRef CAS PubMed.
- A. Frei, J. Zuegg, A. G. Elliott, M. Baker, S. Braese, C. Brown, F. Chen, C. G. Dowson, G. Dujardin, N. Jung, A. P. King, A. M. Mansour, M. Massi, J. Moat, H. A. Mohamed, A. K. Renfrew, P. J. Rutledge, P. J. Sadler, M. H. Todd, C. E. Willans, J. J. Wilson, M. A. Cooper and M. A. T. Blaskovich, Chem. Sci., 2020, 11, 2627–2639 RSC.
- M. Michaut, A. Steffen, J. M. Contreras, C. Morice, A. Paulen, I. J. Schalk, P. Plésiat and G. L. A. Mislin, Bioorg. Med. Chem. Lett., 2020, 30, 127098 CrossRef CAS PubMed.
- A. M. de Almeida, B. A. de Oliveira, P. P. de Castro, C. C. de Mendonça, R. A. Furtado, H. D. Nicolella, V. L. da Silva, C. G. Diniz, D. C. Tavares, H. Silva and M. V. de Almeida, BioMetals, 2017, 30, 841–857 CrossRef PubMed.
- J. Stenger-Smith, I. Chakraborty and P. K. Mascharak, J. Inorg. Biochem., 2018, 185, 80–85 CrossRef CAS PubMed.
- M. K. Tizotti, R. Hörner, A. G. O. de Freitas, C. B. Kempfer, A. Bottega, J. N. Rodrigues, V. M. Cóser, A. Locatelli, G. Paraginski, C. Giacomelli and M. Hörner, Inorg. Chim. Acta, 2016, 441, 78–85 CrossRef CAS.
- S. Thangamani, H. Mohammad, M. F. N. Abushahba, T. J. P. Sobreira, V. E. Hedrick, L. N. Paul and M. N. Seleem, Sci. Rep., 2016, 6, 22571 CrossRef CAS PubMed.
- C. Attila, A. Ueda and T. K. Wood, Appl. Microbiol. Biotechnol., 2009, 82, 525–533 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt01159c |
| This journal is © The Royal Society of Chemistry 2023 |