
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,2-Azolylamidino ruthenium(II) complexes with DMSO ligands: electro- and photocatalysts for CO2 reduction†
Murphy
Jennings
a,
Elena
Cuéllar
b,
Ariadna
Rojo
b,
Sergio
Ferrero
 b,
Gabriel
García-Herbosa
c,
John
Nganga
d,
Alfredo M.
Angeles-Boza
ad,
Jose M.
Martín-Alvarez
b,
Daniel
Miguel
b and
Fernando
Villafañe
*b
b,
Gabriel
García-Herbosa
c,
John
Nganga
d,
Alfredo M.
Angeles-Boza
ad,
Jose M.
Martín-Alvarez
b,
Daniel
Miguel
b and
Fernando
Villafañe
*b
aInstitute of Materials Science, University of Connecticut, 97 N. Eagleville Rd, Storrs, CT 06269, USA
bGIR MIOMeT-IU Cinquima-Química Inorgánica, Facultad de Ciencias, Campus Miguel Delibes, Universidad de Valladolid, 47011 Valladolid, Spain. E-mail: fernando.villafane@uva.es
cDepartamento de Química, Facultad de Ciencias, Universidad de Burgos, 09001 Burgos, Spain
dDepartment of Chemistry, University of Connecticut, 55 N. Eagleville Rd, Storrs, CT 06269, USA
First published on 7th November 2023
Abstract
New 1,2-azolylamidino complexes fac-[RuCl(DMSO)3(NH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(R)az*-κ2N,N)]OTf [R = Me (2), Ph (3); az* = pz (pyrazolyl, a), indz (indazolyl, b)] are synthesized via chloride abstraction from their corresponding precursors cis,fac-[RuCl2(DMSO)3(az*H)] (1) after subsequent base-catalyzed coupling of the appropriate nitrile with the 1,2-azole previously coordinated. All the compounds are characterized by 1H NMR, 13C NMR and IR spectroscopy. Those derived from MeCN are also characterized by X-ray diffraction. Electrochemical studies showed several reduction waves in the range of −1.5 to −3 V. The electrochemical behavior in CO2 media is consistent with CO2 electrocatalytic reduction. The catalytic activity expressed as [icat(CO2)/ip(Ar)] ranged from 1.7 to 3.7 for the 1,2-azolylamidino complexes at voltages of ca. −2.7 to −3 V vs. ferrocene/ferrocenium. Controlled potential electrolysis showed rapid decomposition of the Ru catalysts. Photocatalytic CO2 reduction experiments using compounds 1b, 2b and 3b carried out in a CO2-saturated MeCN/TEOA (4
C(R)az*-κ2N,N)]OTf [R = Me (2), Ph (3); az* = pz (pyrazolyl, a), indz (indazolyl, b)] are synthesized via chloride abstraction from their corresponding precursors cis,fac-[RuCl2(DMSO)3(az*H)] (1) after subsequent base-catalyzed coupling of the appropriate nitrile with the 1,2-azole previously coordinated. All the compounds are characterized by 1H NMR, 13C NMR and IR spectroscopy. Those derived from MeCN are also characterized by X-ray diffraction. Electrochemical studies showed several reduction waves in the range of −1.5 to −3 V. The electrochemical behavior in CO2 media is consistent with CO2 electrocatalytic reduction. The catalytic activity expressed as [icat(CO2)/ip(Ar)] ranged from 1.7 to 3.7 for the 1,2-azolylamidino complexes at voltages of ca. −2.7 to −3 V vs. ferrocene/ferrocenium. Controlled potential electrolysis showed rapid decomposition of the Ru catalysts. Photocatalytic CO2 reduction experiments using compounds 1b, 2b and 3b carried out in a CO2-saturated MeCN/TEOA (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) solution containing a mixture of the catalyst and [Ru(bipy)3]2+ as the photosensitizer under continuous irradiation (light intensity of 150 mW cm−2 at 25 °C, λ > 300 nm) show that compounds 1b, 2b and 3b allowed CO2 reduction catalysis, producing CO and trace amounts of formate. The combined turnover number for the production of formate and CO is ca. 100 after 8 h and follows the order 1b < 2b ≈ 3b.
:1 v/v) solution containing a mixture of the catalyst and [Ru(bipy)3]2+ as the photosensitizer under continuous irradiation (light intensity of 150 mW cm−2 at 25 °C, λ > 300 nm) show that compounds 1b, 2b and 3b allowed CO2 reduction catalysis, producing CO and trace amounts of formate. The combined turnover number for the production of formate and CO is ca. 100 after 8 h and follows the order 1b < 2b ≈ 3b.
Introduction
Research on the catalytic reduction of CO2 has been receiving intense attention since it is a key component to a sustainable future. A variety of transition metal complexes have been reported to display electrochemical CO2 reduction activities; however, novel catalytic systems are still required to cope with the social dilemma of climate change.1 Ruthenium(II) complexes are among those that are more studied, and in fact the electrochemical CO2 reductions catalyzed by cis-[Ru(bipy)2(CO)2]2+ (bipy = 2,2′-bipyridine) and cis-[Ru(bipy)2(CO)CI]+ were among the first systems reported, back in 1987.2 Since then, a plethora of Ru catalysts have been described in the context of CO2 electroreduction, most of them containing two bipy ligands coordinated cis, or other polypyridine-type ligands with similar geometries.1–3 The use of mono(bipy) complexes is limited to carbonyl complexes such as [Ru(N–N)(CO)2Cl2] (N–N = bipy or substituted bipy) or cationic derivatives derived from the substitution of the chlorido ligands by neutral ligands, and to their corresponding reduced Ru(0) species.4Reduction of carbon dioxide can also be achieved by photochemical methods. The catalysts for these processes are usually metal complexes with different accessible redox states available in both the central metal and the ligands in order to drive the multi-electron reduction process for CO2 reduction.5 In the case of Ru(II), both bis(bipy)6 and mono(bipy)7 complexes have been widely studied as photocatalysts for CO2 reduction to give CO and/or formate, depending on the reaction conditions.
The activity of the catalyst largely depends on the substituents of the ligands, which are key factors influencing both the primary coordination sphere of the transition metal center and the secondary coordination effects. The latter have been taken into consideration only very recently, when the first reports on the role of pendant groups that may lead to supramolecular arrangements have been described.8 However, synthetic methods for substituted bipy ligands are usually difficult and/or tiresome, and thus the alternative of coupling two monodentate ligands to afford a chelating diimine ligand appears as a straightforward method in order to obtain N,N-chelating ligands by accessible synthetic paths.9 1,2-Azolylamidino ligands fulfill these requirements, as they can be easily obtained in situ by the coupling reaction of 1,2-azoles and coordinated nitriles (Scheme 1). We have exploited this reaction, i.e. the metal-mediated coupling of 1,2-azoles and nitriles, so that a wide range of new 1,2-azolylamidino complexes can be thus easily obtained.10 The use of different 1,2-azoles and nitriles provides the opportunity of controlling the main features, electronic and steric, of the ligand. In particular, the electrochemical and luminescence properties of Re(I) tricarbonyl and cis-bis(bipyridyl)Ru(II) complexes containing 1,2-azolylamidino ligands, as well as the photocatalytic behaviour of the latter, have been reported.11 The same reaction using a nucleobase such as 1-methylcytosine instead of the 1,2-azole has been revealed to be a new method to incorporate biologically relevant substrates into Re(I) tricarbonyl complexes.12
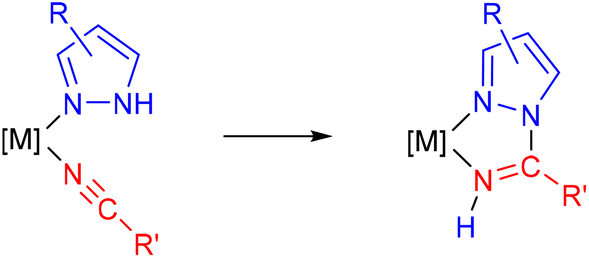 | ||
| Scheme 1 Synthesis of a 1,2-azolylamidino ligand from the (1,2-azole) precursor containing a coordinated nitrile. | ||
Another aspect of interest in the 1,2-azolylamidino ligands lies in the presence of an acidic NH group, which allows further significative reactivity, as it may be involved in noncovalent interactions. A relevant example is the ability of the fac-[Re(CO)3(Hdmpz)(HNC(Me)dmpz-κ2N,N)]+ cation to bind selectively with chloride anions by combining electrostatic attraction and hydrogen bonding of the NH groups present.13 A second exponent, this time on Mn(I), emerges from the use of a somehow uncommon nitrile, such as dicyanamide, which allowed a bimetallic Mn(I) complex containing a bridging tetradentate bis(pyrazolylamidino) ligand to be obtained, where the bromide anion plays a crucial role in its planarity, as demonstrated by DFT calculations.14
Herein, we report the synthesis and characterization of new 1,2-azolylamidino Ru(II) complexes. Their chlorido(1,2-azole) precursors are also described. The 1,2-azoles used in this work are pyrazole (pzH) and indazole (indzH), whereas the nitriles are acetonitrile (MeCN) and benzonitrile (PhCN). This allows us to study easily obtained 1,2-azolylamidino ligands with different electronic and steric properties. The behavior of the complexes synthesized as electro- and photocatalysts for the reduction of CO2 is described.
Results and discussion
Synthesis and characterization of the complexes
The synthesis of the complexes described in this work is depicted in Scheme 2. A panel of complexes with different substituents were synthesized in order to confirm the synthetic method and to study the influence of the substituents on the electrocatalytic reduction of CO2. Scheme 2 shows the mixed chlorido(1,2-azole) complexes cis,fac-[RuCl2(DMSO)3(az*H)] (1) (az* = pz, indz), which are precursors of the 1,2-azolylamidino fac-[RuCl(DMSO)3(NHC(R)az*-κ2N,N)]OTf (R = Me, Ph) complexes, as a result of coupling a coordinated pyrazole (pzH) or indazole (indzH) in 1 with acetonitrile (R = Me, 2) or benzonitrile (R = Ph, 3) (Scheme 1). Complexes 1a and 1b had been previously reported by Ferrer et al.15 and by Reisner et al.,16 respectively. Herein, we report a new synthetic procedure and its thorough characterization.
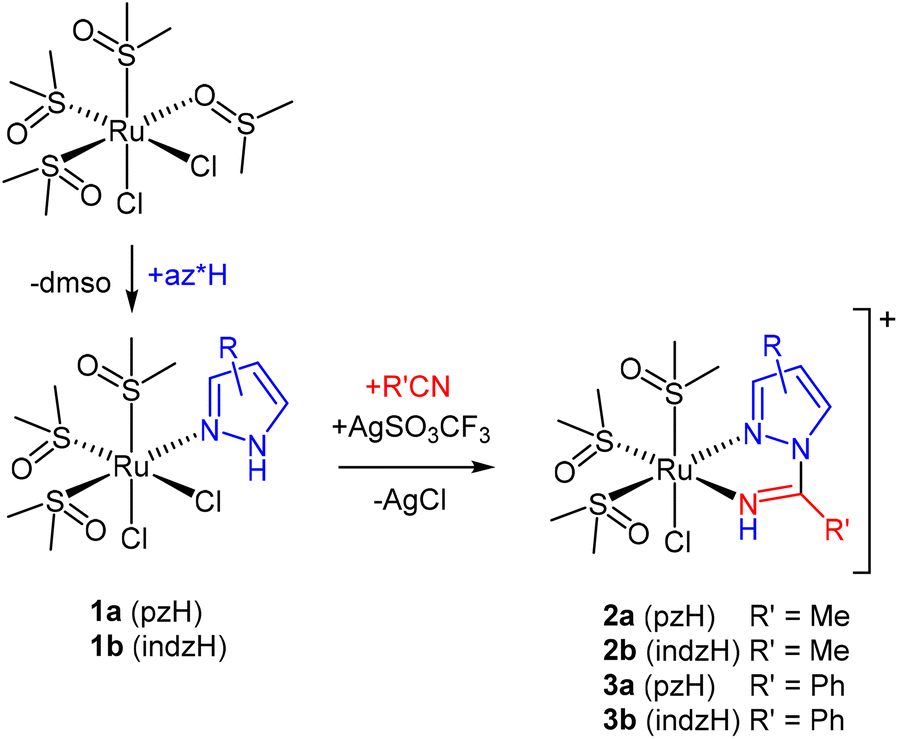 | ||
| Scheme 2 Synthesis of the new complexes. | ||
Chlorido(1,2-azole) complexes 1a and 1b were obtained by substituting the O-coordinated DMSO ligand in the parent complex cis-[RuCl2(DMSO)4] by pyrazole or indazole, respectively (Scheme 2). These synthetic methods have been improved compared to those previously described in the literature. Thus, the pyrazole complex (1a) is herein obtained at room temperature, whereas Ferrer et al.15 used refluxing methanol. Moreover, our reaction leading to the indazole complex 1b lasts for one hour at room temperature, whereas Reisner et al.16 reported an overnight reaction. The 1,2-azolylamidino complexes 2 and 3 were obtained by extracting with silver triflate one of the chlorido ligands in the presence of the nitrile, followed by the coupling of nitrile and the 1,2-azole using NaOH (aq.) as catalyst, as we have previously described.10c
The spectroscopic data are straightforward and support the proposed geometries (see the Experimental section). Complexes 2a and 2b were also characterized by single-crystal X-ray diffraction studies (Fig. 1). The distances and angles (CCDC 2221963 and 2221964†) are similar to those found in other similar Ru(II) ligands.17 Thus, the C(4)−N(3) distances (1.265(4) Å and 1.279(4) Å, respectively, for 2a and 2b) are typical of CN double bonds. In complexes 2a and 2b, the N-bound hydrogens of the 1,2-azolylamidino ligands are involved in hydrogen bonding with an oxygen atom of a triflate anion. The distances and angles detected for 2a (H(3)⋯O(4) 2.050 Å, N(3)⋯O(4) 2.885 Å, N(3)−H(3)⋯O(4) 163.4°) and 2b (H(3)⋯O(4) 2.006 Å, N(3)⋯O(4) 2.840 Å, N(3)−H(3)⋯O(4) 162.8°) may be considered as indicative of “moderate” hydrogen bonds.18
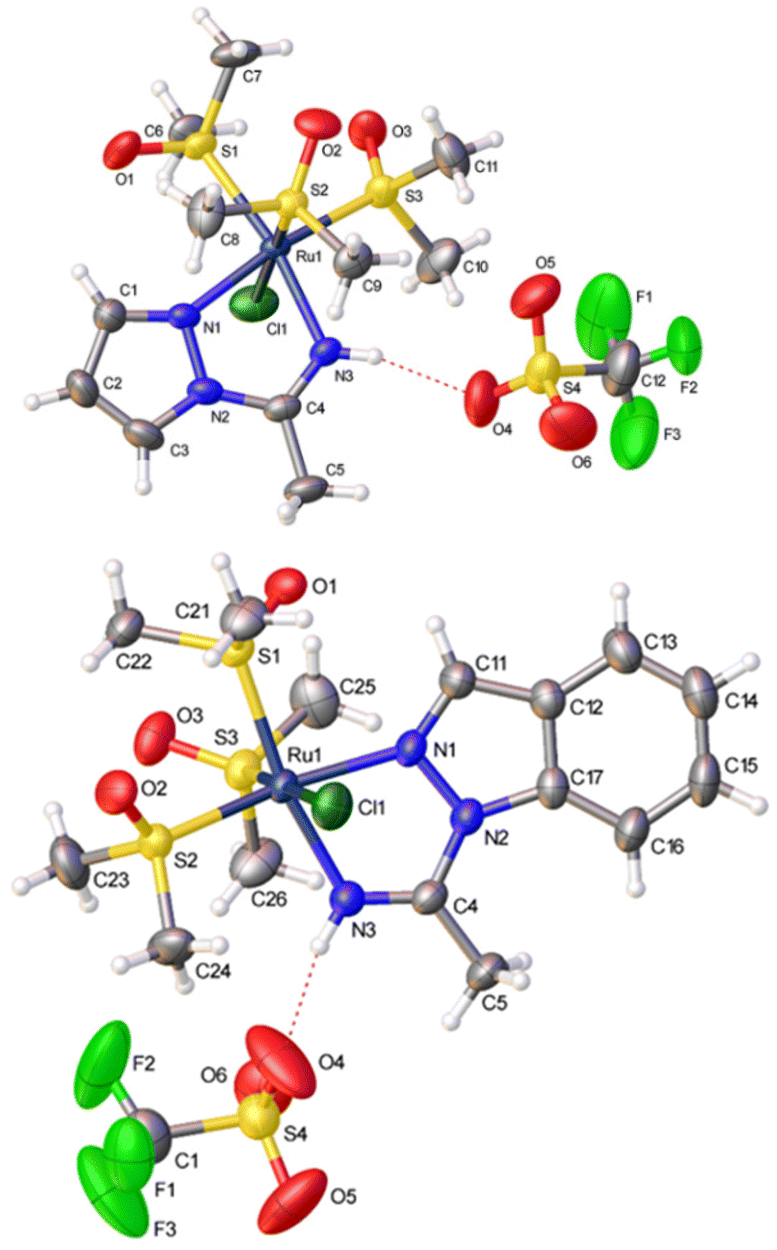 | ||
| Fig. 1 Perspective views of fac-[RuCl(DMSO)3(NHC(Me)pz-κ2N,N)](OTf), 2a (above), and fac-[RuCl(DMSO)3(NHC(Me)indz-κ2N,N)](OTf), 2b (below), showing the atom numbering. Thermal ellipsoids are drawn at 50% probability. | ||
Electrochemical studies
Table 1 gathers the observed potential values obtained from cyclic voltammetry (CV) experiments, referenced to the redox pair of ferrocenium/ferrocene, following the IUPAC recommendations.19 An AgCl/Ag (3 M NaCl) reference electrode was used, and ferrocene was added as internal calibrant always in the last experimental measurement. All the measurements were made in MeCN, once the stability of the complexes in this solvent was confirmed by NMR.| Complex | Observed Epc valuesb (cathodic scan) | i p(N2)c (at Epc) | i cat(CO2)c (at Epc) | Ratiod
|
|---|---|---|---|---|
| a The reduction potential mean value observed for ferrocenium/ferrocene (Fc+/Fc) used as an internal calibrant under the employed experimental conditions was E° = 0.443 ± 0.005 V vs. the AgCl/Ag (3 M NaCl) electrode. b Cathodic scan peaks observed under N2 unless stated otherwise. c Maximum registered cathodic current (μA) under N2, ip(N2), or under CO2, icat(CO2) taken from the peak showing greatest enhancement with CO2 addition. d Ratio between the faradaic currents observed under N2, ip(N2), or under CO2, icat(CO2). e Waves where both peaks iox and ired were observed. The value of E1/2 is given in those cases. | ||||
| 1a | −2.26, −3.00e | 45.7 (−3.00) | 79.4 (−3.10) | 1.74 |
| 1b | −2.20, −2.95 | 34.3 (−2.95) | 68.3 (−2.83) | 1.99 |
| 2a | −2.11, −2.42, −3.0 | 27.7 (−3.00) | 51.3 (−3.10) | 1.85 |
| 2b | −1.93, −2.60, −3.03 | 27.1 (−3.03) | 73.5 (−2.88) | 2.71 |
| 3a | −2.10, −2,41, −2.93e | 38.1 (−2.93) | 65.0 (−3.10) | 1.71 |
| 3b | −1.93, −2.30,− 2.80 | 53.1 (−2.80) | 195 (−2.88) | 3.67 |
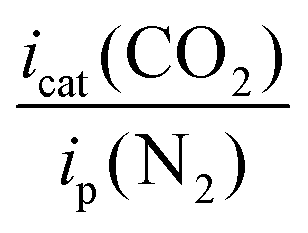
On scanning to negative potentials under N2, all the complexes display several waves, as the result of successive electron transfer reductions. This is probably the result of pyrazolylamidino ligand reduction, which occurs in RuII(bipy) systems, which display one or several waves at negative potentials that have always been assigned to bipy-based reductions.3b,6f When the same scans were carried out under a CO2 atmosphere, the indazole complexes and the pyrazole complex 2a showed a clear enhancement of the cathodic current (red lines in Fig. S1, ESI†). This electrochemical behaviour is consistent with CO2 activation, i.e. electrocatalyzed reduction. The icat(CO2)/ip(N2) ratio allows comparing the electrochemical activity of the complexes, with values ranging from 1.7 to 3.7 for the 1,2-azolylamidino complexes (Table 1). As a representative example, the results registered for the complex fac-[RuCl(DMSO)3(NHC(Ph)indz-κ2N,N)](OTf) (3b) are shown in Fig. 2. Black (N2) and red (CO2) traces overlap completely in the range of −1.25 to 0.0 V. Changing the atmosphere from N2 to CO2 generates a great enhancement of the current at potentials below −2.5 V. The rest of the experiments under the CO2 atmosphere lead to different shapes of waves associated with the electrocatalytic reduction of CO2 due to the competition at the electrode surface between CO2 consumption (related to the rate-determinining step of the catalytic cycle) and the arrival of new substrate by diffusion.1c
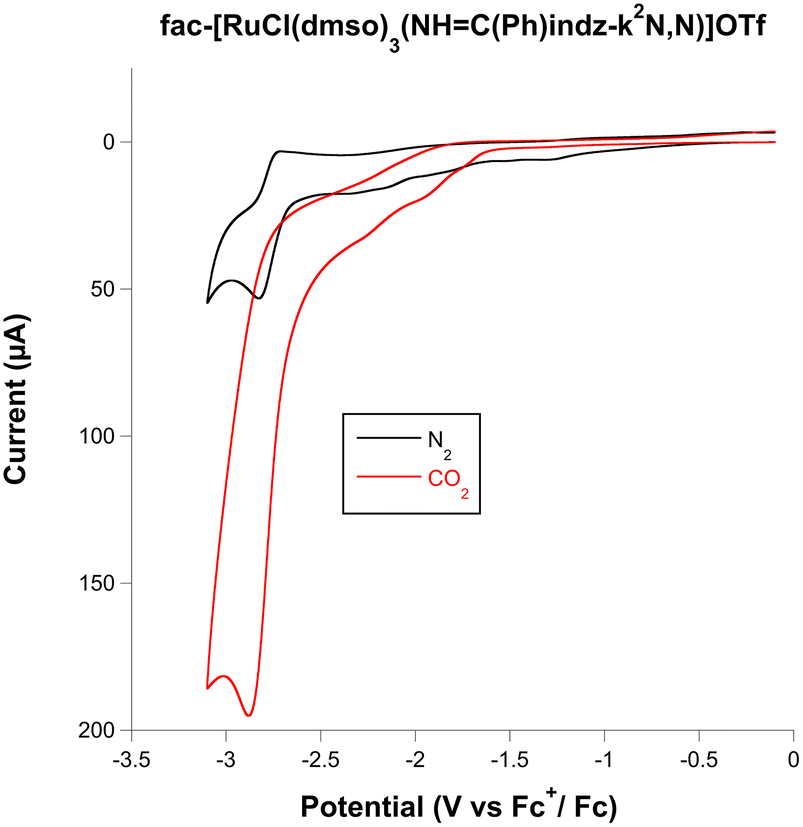 | ||
| Fig. 2 Cyclic voltammograms of 0.5 mM fac-[RuCl(DMSO)3(NHC(Ph)indz-κ2N,N)](OTf) (3b) (glassy carbon working electrode dish 3.0 mm diameter, dry MeCN, 0.1 M Bu4NPF6) under N2 (black) and after bubbling CO2 (red). | ||
The catalytic activity of these complexes is supported by the observed decrease of catalytic activity when the concentration of CO2 is progressively substituted by bubbling Ar (Fig. S13, ESI) and by the dependence of the electrocatalytic reduction of CO2 on the scan rate (Fig. S2–4, ESI) and on the concentration (Fig. S14, ESI†).
As indicated in the Introduction, there are no previous reports on the catalytic activity on CO2 reduction for complexes structurally similar to those herein described. The closest analogues might be mono(bipy)Ru(II) complexes containing two carbonyls and two chlorido ligands, which have been used both as electro- and photocatalysts for CO2 reduction.4,7 The icat(CO2)/ip(N2) ratios obtained for compounds 2 and 3 (between 1.71 and 3.67, see Table 1) are slightly below those obtained by our group for cis-Ru(bipy)2 complexes and for fac-Re(CO)3 complexes (between 2.1 and 10.8 and between 2.7 and 11.5, respectively).20,21 Although the reaction mechanism is still being discussed after more than 20 years since the first studies, there is a general consensus about the formation of reduced species that would be highly active for the reduction of CO2. These reduced species may contain RuI–RuI or Ru0–Ru0 bonds for the less hindered diimine ligands or Ru–H bonds for those containing sterically demanding substituents.4,7 In our case, all the attempts to reduce chemically either 1,2-azole (1) or 1,2-azolylamidino (2, 3) complexes led to decomposition. The possibility of Ru(0) nanoparticles being active catalysts was tested in a separate experiment and is discussed below.
In order to further evaluate the electrocatalytic activity, we performed a sequence of experiments on the indz complexes (1b, 2b, and 3b). We focused our study on these complexes since they exhibit higher icat(CO2)/ip(N2) ratios and less negative potentials for the first reduction by ca. 0.2 V, which we can attribute to the lower energy π system on the indz fragment. First, scan rate experiments were performed on each reduction step in order to determine their redox behaviour (Fig. S2–S4, ESI†). These studies showed the general instability of these complexes under catalytic conditions. Additional experiments were run either in MeCN saturated with Cl− (TBACl, Fig. S6, ESI†) or in DMSO (Table S1, ESI†) as solvent instead of MeCN. In all cases, the CVs point to the formation of species that show nonlinear responses to the scan rate experiments, showing the generation of non-freely diffusing redox species, which leads to the complexity seen in the CVs. All systems exhibited no reversibility, likely due to the instability of the species formed.
Controlled potential electrolysis (CPE) studies on indz complexes 1b, 2b and 3b were also performed following a procedure previously described.20 The experiments, carried out in MeCN using a three-electrode setup with a glassy carbon working electrode, were run at a potential 0.2 V lower than the electron addition, showing the greatest enhancement with the addition of CO2, as previously determined by CV for each indz complex. Each experiment was carried out for 6 h, while the MeCN solutions were maintained under a CO2 atmosphere and stirred. Samples of the solutions were pulled and tested with CV at 0 h, 2 h, 4 h, and 6 h (Fig. S7–S9, ESI†) under a CO2 atmosphere, bubbled with N2 for 15 min, and the scans were then repeated. The voltammograms indicate that the electrochemical activity is not lost as the reaction progresses. However, an increase in current is detected under a N2 atmosphere, indicating that a non-catalytical species that is accepting electrons is being formed (Fig. S7–S9, ESI†). After the 6 h CPE experiment, Hg was added to the reaction mixture, which was then stirred for 12 h more at r.t. Hg is known to bind to Ru nanoparticles from the solutions, removing them as active species.22 No observable changes to the activity behaviour of the complexes under CO2 were detected after the addition of Hg, suggesting that the activity of the complexes is not due to the formation of nanoparticles. HPLC experiments were also performed before and after the experiment outlined above (Fig. S11, ESI†) in order to further probe the solution stability of the indz family of complexes. The chromatograms taken at 254 nm indicate that the majority of each complex is degraded to different degradation products by the end of the experiment.
In order to test the hypothesis that the rapid formation of degradation products, including Ru(0) nanoparticles, could have been on the electrode surface, CPE experiments were run for 2 h, after which CV experiments were performed, first with the electrode “as is”, and then after cleaning the electrode with a low lint delicate task wipe saturated with dry MeCN (Fig. S10, ESI†). These experiments clearly show that a catalytically active film forms on the surface of the electrode, likely containing Ru(0) nanoparticles and possibly other Ru degradation products. This might be the cause of the less efficient catalytic abilities detected in the bulk-solution tests discussed above since rapid decomposition of the Ru complexes at the surface of the CPE electrode could be the cause behind their lack of activity. This degradation process was also examined by sonicating the working electrode with EtOH immediately after the 2 h CPE experiment in order to remove any solid species deposited on it. These EtOH mixtures were then drop-cast for TEM samples. The TEM images show a variety of electron-rich disordered clusters, thus supporting the hypothesis that Ru(0) particles are deposited on the electrode (Fig. S12, ESI†).
Photocatalytic activity
Given the electrochemical instability of the complexes, we turned our attention to the photocatalytic activity of these complexes. Although some complexes can act as photocatalysts and photosensitizers, this is not the case for 1b, 2b and 3b. Most Ru-based photocatalysts require the assistance of photosensitizers since their direct excitation leads to ligand dissociation.23 Shown in Fig. 3 are the results of the photocatalytic CO2 reduction experiments using compounds 1b, 2b and 3b carried out in a CO2-saturated acetonitrile/triethanolamine (MeCN/TEOA, 4:1 v/v) solution containing a mixture of the catalyst and [Ru(bipy)3]2+ as the photosensitizer in a glass vial with a volume of 10 mL under continuous irradiation (light intensity of 150 mW cm−2 at 25 °C, λ > 300 nm). To validate the photocatalytic data, various control experiments were carried out under different experimental conditions under irradiation with light. In the absence of [Ru(bipy)3]2+, the catalyst, or the sacrificial electron donor, TEOA, only a trace amount or no amount of product was produced, indicating that all three components are necessary for efficient CO2 activation. The formic acid produced was quantified using the protocol reported by Kubiak et al.24 The results show that compounds 1b, 2b and 3b are efficient CO2 reduction catalysts under the conditions used (Table S1, ESI†), although they also produce larger quantities of dihydrogen and trace amounts of formate.5d The source of protons is probably TEOA. The turnover number for the production of CO is ∼100 after 8 h and follows the order 1b > 2b ≈ 3b. Other complexes of the type trans(Cl)-[Ru(bipy*)(CO)2Cl2] (where bipy* is a substituted bipy) have shown TON from ca. 100 to ca. 3000.7
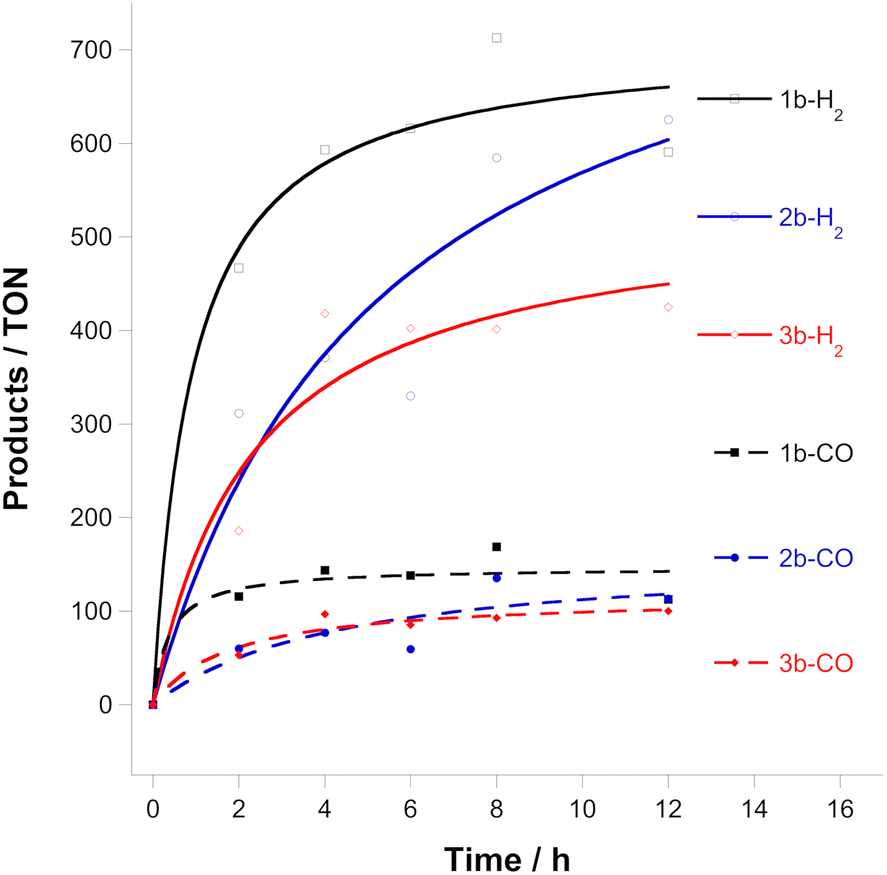 | ||
| Fig. 3 Turnover numbers for CO and H2 evolved from 0.1 mM Ru complexes, 1.6 mM [Ru(bipy)3]2+ in a CO2-saturated MeCN-TEOA solution (4:1 v/v) irradiated by >300 nm visible light. | ||
Due to the higher photochemical activity of these complexes, a series of additional experiments were conducted, trying to establish the stability of the catalysts and to further understand the process. First, the possibility of the presence of Ru(0) nanoparticles as the active catalysts in the solution was tested by adding 3 equivalents of Hg to the reaction solutions, and checking for changes in product formation. These experiments (Fig. S7–S9, ESI†) did not show significative differences with respect to those obtained with Hg-free solutions. This result led to the conclusion that Ru(0) nanoparticles are not active catalysts in this system.
The photochemical activity of 1b was also used to probe the carbon source of the complexes by analyzing the product formation of the CO2 catalysis by running the photocatalysis with a solution saturated with 13CO2 and analyzing the products by gas chromatography (GC), as well as by mass spectrometry coupled GC (Tables S4 and S5, ESI†). All the carbon monoxide detected was in the form of 13CO, showing that degradation of the complexes is not a product source; in fact, the source of CO for this reaction is the catalysis of CO2 to CO by the complexes.
Conclusions
Base-catalyzed coupling of a nitrile and a 1,2-azole previously coordinated to Ru(II) DMSO complexes allows the synthesis of new 1,2-azolylamidino complexes fac-[RuCl(DMSO)3(NHC(R)az*-κ2N,N)]OTf (R = Me, Ph; az* = pz, indz) after chloride abstraction. The 1,2-azolylamidino complexes and the parent 1,2-azole complexes are active both as electro- and photocatalysts for the reduction of CO2, as demonstrated by proof-of-concept trials that led to the production of CO and trace amounts of formate. These results open the door to new complexes displaying this interesting behaviour since, so far, the use of Ru(II) mono(bipy) complexes was limited to carbonyl complexes of the type [Ru(bipy)(CO)2X2]. The fact that 1,2-azolylamidino ligands with different electronic and steric features may be easily obtained in situ may encourage future developments in this area, given the decisive role of the presence of substituents of the diimine ligand on the electrocatalytic reduction of CO2.
Experimental section
General remarks
All manipulations were performed under an N2 atmosphere following conventional Schlenk techniques. Solvents were purified according to standard procedures. cis-[Ru(DMSO)4Cl2]25 was obtained as previously described. All other reagents were obtained from the usual commercial suppliers and used as received. Infrared spectra were recorded from solids using a Bruker Tensor 27 FTIR. Standard abbreviations are used to indicate intensity: vw = very weak, w = weak, m = medium, s = strong, and vf = very strong. NMR spectra were recorded on 500 MHz Agilent DD2 and 400 MHz Agilent MR instruments in the Laboratory of Instrumental Techniques (LTI) Research Facilities, University of Valladolid, using CDCl3 or (CD3)2CO as solvents at room temperature (r.t.). 1H and 13C NMR chemical shifts (δ) are reported in parts per million (ppm) and are referenced to tetramethylsilane (TMS), using the residual solvent peak as an internal reference. Coupling constants (J) are reported in Hz. Standard abbreviations are used to indicate multiplicity: s = singlet, d = doublet, ddd = doublet of doublet of doublets, dt = doublet of triplets, t = triplet, and m = multiplet. The full assignment of the 1H NMR spectra was supported by typical homonuclear 1H–1H correlations such as COSY, TOCSY and NOESY experiments and the assignment of 13C{1H} data was supported by HMBC and HSQC heteronuclear experiments. Numbering of pyrazole and indazole ligands for NMR assignments is shown in Fig. 4. Elemental analyses were performed on a Thermo Fisher Scientific EA Flash 2000.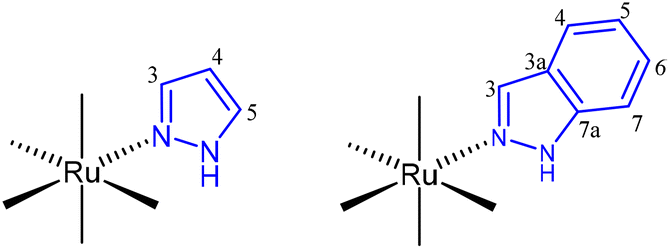 | ||
| Fig. 4 Numbering of pyrazole and indazole ligands for NMR assignment. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C(Me)pz-κ2N,N)](OTf), 2a.
AgOTf (0.051 g, 0.20 mmol) was added to a solution of 1a (0.095 g, 0.20 mmol) in MeCN (10 mL). 100 μL of aqueous 0.02 M NaOH (0.002 mmol) was then added and the mixture was stirred at r.t. for 12 h in the absence of light. The reaction mixture was filtered to remove solid AgCl and dried in vacuo. The solid was crystallized in MeCN/Et2O at −20 °C, giving a colorless microcrystalline solid, which was decanted, washed with Et2O (2 × 5 mL approximately), and dried in vacuo, yielding 0.079 g, 63%. 1H NMR (500 MHz, (CD3)2CO, r.t.) δ: 2.83 (s, HCH3 DMSO, 3 H), 3.02 (s, NC(CH3), 3 H), 3.19 (s, HCH3 DMSO, 3 H), 3.39 (s, HCH3 DMSO, 3 H), 3.55 (s, HCH3 DMSO, 3 H), 3.57 (s, HCH3 DMSO, 3 H), 3.61 (s, HCH3 DMSO, 3 H), 6.94 (t, J = 2.5 Hz, H4, 1 H), 8.78 (d, J = 2.5 Hz, H3, 1 H), 8.87 (d, J = 2.5 Hz, H5, 1 H), 11.00 (s, NH, 1 H). 13C{1H} NMR (126 MHz, (CD3)2CO, r.t.) δ: 17.8 (NC(CH3)), 44.6 (CCH3 DMSO), 45.2 (CCH3 DMSO), 45.7 (CCH3 DMSO), 46.2 (CCH3 DMSO), 47.0 (CCH3 DMSO), 47.1 (CCH3 DMSO), 111.0 (C4), 134.9 (C3), 148.7 (C5), 164.3 (NC(CH3)). IR (cm−1): 633 s, 685 m, 724 w, 759 w, 779 m, 920 w, 939 m, 964 m, 977 m, 1021 vs, 1052 w, 1086 vs, 1107 s, 1107 m, 1152 m, 1173 m, 1229 s, 1244 m, 1279 w, 1411 w, 1656 m, 2923 w, 3005 w, 3102 w, 3205 w. Calcd for C12ClF3H25N3O6RuS4: C, 22.95; H, 3.98; N, 6.69; S, 20.40. Found: C, 22.75; H, 3.91; N, 6.72; S, 20.55.
C(Me)indz-κ2N,N)](OTf), 2b.
AgOTf (0.049 g, 0.19 mmol) was added to a solution of 1b (0.100 g, 0.19 mmol) in MeCN (10 mL) and the mixture was stirred at r.t. for 12 h in the absence of light. The reaction mixture was filtered to remove solid AgCl and dried in vacuo. The solid was crystallized in MeCN/Et2O at −20 °C, giving a colorless microcrystalline solid, which was decanted, washed with Et2O (2 × 5 mL approximately), and dried in vacuo, yielding 0.801 g, 62%. 1H NMR (500 MHz, (CD3)2CO, r.t.) δ: 3.10 (s, HCH3 DMSO, 3 H), 3.24 (s, HCH3 DMSO, 3 H), 3.33 (s, NC(CH3), 3 H), 3.44 (s, HCH3 DMSO, 3 H), 3.60 (s, 2 HCH3 DMSO, 6 H), 3.65 (s, HCH3 DMSO, 3 H), 7.61 (t, J = 8.0 Hz, H6, 1 H), 7.83 (t, J = 8.0 Hz, H5, 1 H), 8.17 (d, J = 8.2 Hz, H4, 1 H), 8.19 (d, J = 8.2, H7, 1 H), 9.69 (s, H3, 1 H), 10.51 (s, NH, 1 H). 13C{1H} NMR (126 MHz, (CD3)2CO, r.t.) δ: 20.5 (NC(CH3)), 45.0–50.0 (6 C, CCH3 DMSO), 112.9 (C4), 123.3 (C7), 125.3 (C6), 125.6 (C7a), 131.3 (C5), 140 (C3a), 146.3 (C3), 165.0 (NC(CH3)). IR (cm−1): 3221 w, 3117 vw, 3022 w, 2971 vw, 1690 w, 1632 m, 1584 vw, 1511 w, 1484 m, 1468 w, 1428 m, 1409 w, 1356 w, 1316 w, 1276 s, 1249 vs, 1226 m, 1196 m, 1175 m, 1156 s, 1112 s, 1100 s, 1081 s, 1029 vs, 1019 vs, 977 m, 966 m, 943 w, 914 m, 864 w, 795 w, 756 s, 719 w, 683 m, 634 vs. Calcd for C16ClF3H27N3O6RuS4: C, 28.34; H, 3.99; N, 6.20; S, 18.89. Found: C, 28.45; H, 4.00; N, 6.39; S, 18.68.
C(Ph)pz-κ2N,N)](OTf), 3a.
AgOTf (0.051 g, 0.2 mmol) was added to a solution of 1a (0.095 g, 0.2 mmol) in (CH3)2CO (10 mL). 100 μL of PhCN and 100 μL of aqueous 0.02 M NaOH (0.002 mmol) were then added and the mixture was stirred at r.t. for 24 h in the absence of light. The reaction mixture was filtered to remove solid AgCl and dried in vacuo. The solid was crystallized in (CH3)2CO/Et2O at −20 °C, giving a yellow microcrystalline solid, which was decanted, washed with Et2O (2 × 5 mL approximately), and dried in vacuo, yielding 0.084 g, 61%. 1H NMR (500 MHz, (CD3)2CO, r.t.) δ: 3.31 (s, HCH3 DMSO, 3 H), 3.34 (s, HCH3 DMSO, 3 H), 3.47 (s, HCH3 DMSO, 3 H), 3.63 (s, HCH3 DMSO, 3 H), 3.64 (s, HCH3 DMSO, 3 H), 3.69 (s, HCH3 DMSO, 3 H), 6.97 (dd, J = 3.2, 2.1 Hz, H4 pz, 1 H), 7.59–8.04 (m, C6H5 Ph, 5 H), 8.45 (dd, J = 3.2, 0.6 Hz, H3 pz, 1 H), 9.03 (dd, J = 2.1, 0.6 Hz, H5 pz, 1 H), 11.21 (s, NH, 1 H). 13C{1H} NMR (126 MHz, (CD3)2CO, r.t.) δ: 44.6 (CCH3 DMSO), 45.2 (CCH3 DMSO), 45.7 (CCH3 DMSO), 46.2 (CCH3 DMSO), 47.0 (CCH3 DMSO), 47.1 (CCH3 DMSO), 111.6 (C4), 129.4–133.4 (5C, C Ph), 136.4 (C3), 149.7 (C5), 165.0 (NC(Ph)), 166.0 (Cipso Ph). IR (cm−1): 3508 w, 3220 w, 3138 w, 3025 w, 2925 w, 2323 w, 2287 w, 2231 w, 2188 w, 2164 w, 2140 w, 2113 w, 2083 w, 2050 w, 1982 w, 1917 w, 1626 m, 1577 w, 1527 w, 1497 w, 1456 w, 1442 m, 1415 m, 1321 w, 1274 s, 1246 vs, 1224 s, 1156 s, 1079 s, 1027 vs, 973 m, 922 m, 896 m, 760 m, 724 w, 704 m, 684 m, 636 s. Calcd for C17ClF3H27N3O6RuS4: C, 29.54; H, 3.94; N, 6.08; S, 18.56. Found: C, 29.62; H, 3.78; N, 6.01; S, 18.47.
C(Ph)indz-κ2N,N)](OTf), 3b.
AgOTf (0.051 g, 0.2 mmol) was added to a solution of 1b (0.105 g, 0.2 mmol) in (CH3)2CO (10 mL). 100 μL of PhCN was then added and the mixture was stirred at r.t. for 24 h in the absence of light. The reaction mixture was filtered to remove solid AgCl and dried in vacuo. The solid was crystallized in (CH3)2CO/Et2O at −20 °C, giving a yellow microcrystalline solid, which was decanted, washed with Et2O (2 × 5 mL approximately), and dried in vacuo, yielding 0.093 g, 63%. 1H NMR (500 MHz, (CD3)2CO, r.t.) δ: 3.31 (s, HCH3 DMSO, 3 H), 3.34 (s, HCH3 DMSO, 3 H), 3.47 (s, HCH3 DMSO, 3 H), 3.63 (s, HCH3 DMSO, 3 H), 3.64 (s, HCH3 DMSO, 3 H), 3.69 (s, HCH3 DMSO, 3 H), 6.38 (dt, J = 8.5, 0.8 Hz, H6 indz, 1 H), 7.46–7.94 (m, H4 indz, H5 indz, C6H5 Ph, 7 H), 8.17 (dd, J = 8.5, 0.8 Hz, H7 indz, 1 H), 9.80 (d, J = 0,8 Hz, H3 indz, 1 H), 10.85 (s, NH, 1 H). 13C{1H} NMR (126 MHz, (CD3)2CO, r.t.) δ: 44.5 (CCH3 DMSO), 45.2 (CCH3 DMSO), 45.8 (CCH3 DMSO), 46.3 (CCH3 DMSO), 47.0 (CCH3 DMSO), 47.4 (CCH3 DMSO), 112.2 (C6 indz), 122.7(C7a), 123.2 (C7 indz), 125.5–133.2 (7C, C4,5 indz, C Ph), 140.7 (C3a), 147.7 (C3 indz), 165.0 (NC(Ph)), 166.0 (Cipso Ph). IR (cm−1): 3264 vw, 3080 vw, 3016 vw, 2964 vw, 1310 vw, 1288 vw, 1119 m, 1094 m, 1018 m, 974 m, 918 m, 713 vw, 677 w. Calcd for C21ClF3H29N3O6RuS4: C, 34.03; H, 3.94; N, 5.68; S, 17.31. Found: C, 33.92; H, 3.75; N, 5.44; S, 17.02.
C(Me)pz-κ2N,N)](OTf), 2a.
AgOTf (0.051 g, 0.20 mmol) was added to a solution of 1a (0.095 g, 0.20 mmol) in MeCN (10 mL). 100 μL of aqueous 0.02 M NaOH (0.002 mmol) was then added and the mixture was stirred at r.t. for 12 h in the absence of light. The reaction mixture was filtered to remove solid AgCl and dried in vacuo. The solid was crystallized in MeCN/Et2O at −20 °C, giving a colorless microcrystalline solid, which was decanted, washed with Et2O (2 × 5 mL approximately), and dried in vacuo, yielding 0.079 g, 63%. 1H NMR (500 MHz, (CD3)2CO, r.t.) δ: 2.83 (s, HCH3 DMSO, 3 H), 3.02 (s, NC(CH3), 3 H), 3.19 (s, HCH3 DMSO, 3 H), 3.39 (s, HCH3 DMSO, 3 H), 3.55 (s, HCH3 DMSO, 3 H), 3.57 (s, HCH3 DMSO, 3 H), 3.61 (s, HCH3 DMSO, 3 H), 6.94 (t, J = 2.5 Hz, H4, 1 H), 8.78 (d, J = 2.5 Hz, H3, 1 H), 8.87 (d, J = 2.5 Hz, H5, 1 H), 11.00 (s, NH, 1 H). 13C{1H} NMR (126 MHz, (CD3)2CO, r.t.) δ: 17.8 (NC(CH3)), 44.6 (CCH3 DMSO), 45.2 (CCH3 DMSO), 45.7 (CCH3 DMSO), 46.2 (CCH3 DMSO), 47.0 (CCH3 DMSO), 47.1 (CCH3 DMSO), 111.0 (C4), 134.9 (C3), 148.7 (C5), 164.3 (NC(CH3)). IR (cm−1): 633 s, 685 m, 724 w, 759 w, 779 m, 920 w, 939 m, 964 m, 977 m, 1021 vs, 1052 w, 1086 vs, 1107 s, 1107 m, 1152 m, 1173 m, 1229 s, 1244 m, 1279 w, 1411 w, 1656 m, 2923 w, 3005 w, 3102 w, 3205 w. Calcd for C12ClF3H25N3O6RuS4: C, 22.95; H, 3.98; N, 6.69; S, 20.40. Found: C, 22.75; H, 3.91; N, 6.72; S, 20.55.
C(Me)indz-κ2N,N)](OTf), 2b.
AgOTf (0.049 g, 0.19 mmol) was added to a solution of 1b (0.100 g, 0.19 mmol) in MeCN (10 mL) and the mixture was stirred at r.t. for 12 h in the absence of light. The reaction mixture was filtered to remove solid AgCl and dried in vacuo. The solid was crystallized in MeCN/Et2O at −20 °C, giving a colorless microcrystalline solid, which was decanted, washed with Et2O (2 × 5 mL approximately), and dried in vacuo, yielding 0.801 g, 62%. 1H NMR (500 MHz, (CD3)2CO, r.t.) δ: 3.10 (s, HCH3 DMSO, 3 H), 3.24 (s, HCH3 DMSO, 3 H), 3.33 (s, NC(CH3), 3 H), 3.44 (s, HCH3 DMSO, 3 H), 3.60 (s, 2 HCH3 DMSO, 6 H), 3.65 (s, HCH3 DMSO, 3 H), 7.61 (t, J = 8.0 Hz, H6, 1 H), 7.83 (t, J = 8.0 Hz, H5, 1 H), 8.17 (d, J = 8.2 Hz, H4, 1 H), 8.19 (d, J = 8.2, H7, 1 H), 9.69 (s, H3, 1 H), 10.51 (s, NH, 1 H). 13C{1H} NMR (126 MHz, (CD3)2CO, r.t.) δ: 20.5 (NC(CH3)), 45.0–50.0 (6 C, CCH3 DMSO), 112.9 (C4), 123.3 (C7), 125.3 (C6), 125.6 (C7a), 131.3 (C5), 140 (C3a), 146.3 (C3), 165.0 (NC(CH3)). IR (cm−1): 3221 w, 3117 vw, 3022 w, 2971 vw, 1690 w, 1632 m, 1584 vw, 1511 w, 1484 m, 1468 w, 1428 m, 1409 w, 1356 w, 1316 w, 1276 s, 1249 vs, 1226 m, 1196 m, 1175 m, 1156 s, 1112 s, 1100 s, 1081 s, 1029 vs, 1019 vs, 977 m, 966 m, 943 w, 914 m, 864 w, 795 w, 756 s, 719 w, 683 m, 634 vs. Calcd for C16ClF3H27N3O6RuS4: C, 28.34; H, 3.99; N, 6.20; S, 18.89. Found: C, 28.45; H, 4.00; N, 6.39; S, 18.68.
C(Ph)pz-κ2N,N)](OTf), 3a.
AgOTf (0.051 g, 0.2 mmol) was added to a solution of 1a (0.095 g, 0.2 mmol) in (CH3)2CO (10 mL). 100 μL of PhCN and 100 μL of aqueous 0.02 M NaOH (0.002 mmol) were then added and the mixture was stirred at r.t. for 24 h in the absence of light. The reaction mixture was filtered to remove solid AgCl and dried in vacuo. The solid was crystallized in (CH3)2CO/Et2O at −20 °C, giving a yellow microcrystalline solid, which was decanted, washed with Et2O (2 × 5 mL approximately), and dried in vacuo, yielding 0.084 g, 61%. 1H NMR (500 MHz, (CD3)2CO, r.t.) δ: 3.31 (s, HCH3 DMSO, 3 H), 3.34 (s, HCH3 DMSO, 3 H), 3.47 (s, HCH3 DMSO, 3 H), 3.63 (s, HCH3 DMSO, 3 H), 3.64 (s, HCH3 DMSO, 3 H), 3.69 (s, HCH3 DMSO, 3 H), 6.97 (dd, J = 3.2, 2.1 Hz, H4 pz, 1 H), 7.59–8.04 (m, C6H5 Ph, 5 H), 8.45 (dd, J = 3.2, 0.6 Hz, H3 pz, 1 H), 9.03 (dd, J = 2.1, 0.6 Hz, H5 pz, 1 H), 11.21 (s, NH, 1 H). 13C{1H} NMR (126 MHz, (CD3)2CO, r.t.) δ: 44.6 (CCH3 DMSO), 45.2 (CCH3 DMSO), 45.7 (CCH3 DMSO), 46.2 (CCH3 DMSO), 47.0 (CCH3 DMSO), 47.1 (CCH3 DMSO), 111.6 (C4), 129.4–133.4 (5C, C Ph), 136.4 (C3), 149.7 (C5), 165.0 (NC(Ph)), 166.0 (Cipso Ph). IR (cm−1): 3508 w, 3220 w, 3138 w, 3025 w, 2925 w, 2323 w, 2287 w, 2231 w, 2188 w, 2164 w, 2140 w, 2113 w, 2083 w, 2050 w, 1982 w, 1917 w, 1626 m, 1577 w, 1527 w, 1497 w, 1456 w, 1442 m, 1415 m, 1321 w, 1274 s, 1246 vs, 1224 s, 1156 s, 1079 s, 1027 vs, 973 m, 922 m, 896 m, 760 m, 724 w, 704 m, 684 m, 636 s. Calcd for C17ClF3H27N3O6RuS4: C, 29.54; H, 3.94; N, 6.08; S, 18.56. Found: C, 29.62; H, 3.78; N, 6.01; S, 18.47.
C(Ph)indz-κ2N,N)](OTf), 3b.
AgOTf (0.051 g, 0.2 mmol) was added to a solution of 1b (0.105 g, 0.2 mmol) in (CH3)2CO (10 mL). 100 μL of PhCN was then added and the mixture was stirred at r.t. for 24 h in the absence of light. The reaction mixture was filtered to remove solid AgCl and dried in vacuo. The solid was crystallized in (CH3)2CO/Et2O at −20 °C, giving a yellow microcrystalline solid, which was decanted, washed with Et2O (2 × 5 mL approximately), and dried in vacuo, yielding 0.093 g, 63%. 1H NMR (500 MHz, (CD3)2CO, r.t.) δ: 3.31 (s, HCH3 DMSO, 3 H), 3.34 (s, HCH3 DMSO, 3 H), 3.47 (s, HCH3 DMSO, 3 H), 3.63 (s, HCH3 DMSO, 3 H), 3.64 (s, HCH3 DMSO, 3 H), 3.69 (s, HCH3 DMSO, 3 H), 6.38 (dt, J = 8.5, 0.8 Hz, H6 indz, 1 H), 7.46–7.94 (m, H4 indz, H5 indz, C6H5 Ph, 7 H), 8.17 (dd, J = 8.5, 0.8 Hz, H7 indz, 1 H), 9.80 (d, J = 0,8 Hz, H3 indz, 1 H), 10.85 (s, NH, 1 H). 13C{1H} NMR (126 MHz, (CD3)2CO, r.t.) δ: 44.5 (CCH3 DMSO), 45.2 (CCH3 DMSO), 45.8 (CCH3 DMSO), 46.3 (CCH3 DMSO), 47.0 (CCH3 DMSO), 47.4 (CCH3 DMSO), 112.2 (C6 indz), 122.7(C7a), 123.2 (C7 indz), 125.5–133.2 (7C, C4,5 indz, C Ph), 140.7 (C3a), 147.7 (C3 indz), 165.0 (NC(Ph)), 166.0 (Cipso Ph). IR (cm−1): 3264 vw, 3080 vw, 3016 vw, 2964 vw, 1310 vw, 1288 vw, 1119 m, 1094 m, 1018 m, 974 m, 918 m, 713 vw, 677 w. Calcd for C21ClF3H29N3O6RuS4: C, 34.03; H, 3.94; N, 5.68; S, 17.31. Found: C, 33.92; H, 3.75; N, 5.44; S, 17.02.
Electrochemical experiments
Electrochemical experiments were performed by using a Model 6012D or 604E electrochemical analyzer from CH Instruments, Inc. Cyclic voltammetry experiments were performed under either N2(g) or Ar(g) and CO2(g) in a one-compartment cell with a glassy carbon working electrode, a platinum wire counter electrode and an Ag/Ag+ (10 mM AgNO3 in acetonitrile, DMF or DMSO) reference electrode with ferrocene as an external reference. The glassy carbon working electrode was polished with 1.0 micron alumina powder, extensively rinsed with deionized water, and then polished for 60 seconds with 0.05 micron alumina powder (CH Instruments). The electrode was again rinsed with dry MeCN/DMF prior to all electrochemistry experiments. All experiments were performed by using 0.1 M TBAPF6 as the supporting electrolyte unless otherwise specified, acetonitrile as the solvent, and with Ru complexes at a concentration of 1.0 mM unless indicated otherwise.The solubility of saturated CO2 in acetonitrile has been reported to be 0.28 M at 25 °C.26 Changing the atmosphere from pure N2 (or Ar) to pure CO2 or vice versa required bubbling with the new gas for no less than five minutes. Lasting such time, the CVs obtained were the same as those obtained in the first scan under a specific atmosphere. Bubbling was maintained during the interim between scans. During the scan time, the PTFE was raised and kept above the surface of the solution to avoid agitation.
Controlled potential electrolysis
Controlled potential electrolysis (CPE) experiments were performed by using a Model 6012D electrochemical analyzer from CH Instruments, Inc. with 25 mL of an MeCN/TBAPF6 solution in a cell with a carbon-rod working electrode (∼1.9 cm2 submersed area), a silver-wire pseudoreference, and a platinum-wire counter electrode separated from the solution using a medium-porosity frit. Gas chromatography analysis of the headspace was performed on an Agilent 7820 instrument equipped with an HP-Molesieve column and a gas sampling valve, as well as a thermal conductivity detector (TCD), stainless-steel column packed with molecular sieves (60/80 mesh), and UHP He as the carrier gas (flow rate = 35 mL min−1). The operating temperatures of the injection port, the oven/column, and the detector were 100 °C, 80 °C, and 100 °C, respectively. Calibration curves for carbon monoxide and hydrogen were created by injecting known quantities of CO or H2 into the electrochemical cell and then sampling the headspace.Photocatalysis procedure
Photochemical reactions were performed in a 20 mL reaction vessel containing 10 mL of CO2-saturated MeCN–TEOA solution (4:1 v/v); the Ru catalyst was present in a concentration of 0.1 mM, whereas the initial concentration of the photosensitizer, [Ru(bipy)3]2+, was 1.6 mM. The solution was irradiated by >300 nm visible light. Analysis of the resulting compounds was performed as described in the CPE section.
Crystal structure determination for compounds 2a and 2b
Crystals were grown by slow diffusion of Et2O into concentrated solutions of the complexes in MeCN (for 2a and 2b) at −20 °C. Relevant crystallographic details can be found in the CIF. A crystal was attached to a glass fiber and transferred to an Agilent SuperNova diffractometer fitted with an Atlas CCD detector. The crystals were kept at 293(2) K during data collection. Using Olex2,27 the structures were solved with the ShelXT28 structure solution program and then the structures were refined with the ShelXL29 refinement package using least squares minimization. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were set in calculated positions and refined as riding atoms, with a common thermal parameter. All graphics were made with Olex2, and distances and angles of hydrogen bonds were calculated with PARST30 (normalized values).31Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors in Valladolid gratefully acknowledge financial support from the Spanish MINECO, Spain (PGC2018-099470-B-I00), the Junta de Castilla y León (VA130618), and the Spanish Ministerio de Ciencia e Innovación (MCIN, PID2021-124691NB-I00, funded by MCIN/AEI/10.13039/501100011033/FEDER, UE). E. C. thanks the UVa for their grant. G. G.-H. gratefully acknowledges financial support from the Junta de Castilla y León, the Spanish Ministerio de Ciencia e Innovación MICIN, and the European Union Next Generation EU/PRTR (MR6NP3). A. M. A.-B. is grateful for support from the National Science Foundation CAREER grant (CHE-1652606).References
- (a) J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC; (b) N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC; (c) R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed; (d) C. Jiang, A. W. Nichols and C. W. Machan, Dalton Trans., 2019, 48, 9454–9468 RSC; (e) S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS PubMed; (f) Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC; (g) F. Franco, C. Rettenmaier, H. S. Jeon and B. R. Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC; (h) R.-Z. Zhang, B.-Y. Wu, Q. Li, L.-L. Lu, W. Shi and P. Cheng, Coord. Chem. Rev., 2020, 422, 213436 CrossRef CAS; (i) N. W. Kinzel, C. Werlé and W. Leitner, Angew. Chem., Int. Ed., 2021, 60, 11628–11686 CrossRef CAS PubMed; (j) P. Saha, S. Amanullah and A. Dey, Acc. Chem. Res., 2022, 55, 134–144 CrossRef CAS PubMed; (k) E. Fujita, D. C. Grills, G. F. Manbeck and D. E. Polyansky, Acc. Chem. Res., 2022, 55, 616–628 CrossRef CAS PubMed; (l) G. Marcandalli, M. C. O. Monteiro, A. Goyal and M. T. M. Koper, Acc. Chem. Res., 2022, 55, 1900–1911 CrossRef CAS PubMed; (m) H.-Q. Liang, T. Beweries, R. Francke and M. Beller, Angew. Chem., Int. Ed., 2022, 61, e202200723 CrossRef CAS PubMed; (n) X. She, Y. Wang, H. Xu, S. C. E. Tsang and S. P. Lau, Angew. Chem., Int. Ed., 2022, 61, 202211396 CrossRef PubMed; (o) K. Lei and B. Y. Xia, Chem. – Eur. J., 2022, 28, e202200141 CrossRef CAS PubMed; (p) W. Nie and C. C. L. McCrory, Dalton Trans., 2022, 51, 6993–7010 RSC; (q) R. Ayyappan, I. Abdalghani, R. C. Da Costa and G. R. Owen, Dalton Trans., 2022, 51, 11582–11611 RSC; (r) Y. Yamazaki, M. Miyaji and O. Ishitani, J. Am. Chem. Soc., 2022, 144, 6640–6660 CrossRef CAS PubMed.
- (a) H. Ishida, H. Tanaka, K. Tanaka and T. Tanaka, J. Chem. Soc., Chem. Commun., 1987, 131–132 RSC; (b) H. Ishida, K. Tanaka and T. Tanaka, Organometallics, 1987, 6, 181–186 CrossRef CAS.
- (a) H. Ishida, K. Fujiki, T. Ohba, K. Ohkubo, K. Tanaka, T. Terada and T. Tanaka, J. Chem. Soc., Dalton Trans., 1990, 2, 2155–2160 RSC; (b) J. R. Pugh, M. R. M. Bruce, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1991, 30, 86–91 CrossRef CAS; (c) H. Nagao, T. Mizukawa and K. Tanaka, Inorg. Chem., 1994, 33, 3415–3420 CrossRef CAS; (d) K. Toyohara, H. Nagao, T. Mizukawa and K. Tanaka, Inorg. Chem., 1995, 34, 5399–5400 CrossRef CAS; (e) M. M. Ali, H. Sato, T. Mizukawa, K. Tsuge, M. Hagab and K. Tanaka, Chem. Commun., 1998, 249–250 RSC; (f) K. Tanaka and D. Ooyama, Coord. Chem. Rev., 2002, 226, 211–218 CrossRef CAS; (g) F. H. Haghighi, H. Hadadzadeh, H. Farrokhpour, N. Serri, K. Abdia and H. A. Rudbarib, Dalton Trans., 2014, 43, 11317–11332 RSC; (h) D. J. Boston, Y. M. F. Pachón, R. O. Lezna, N. R. de Tacconi and F. M. MacDonnell, Inorg. Chem., 2014, 53, 6544–6553 CrossRef CAS PubMed; (i) B. A. Johnson, S. Maji, H. Agarwala, T. A. White, E. Mijangos and S. Ott, Angew. Chem., Int. Ed., 2016, 55, 1825–1829 CrossRef CAS PubMed; (j) B. A. Johnson, H. Agarwala, T. A. White, E. Mijangos, S. Maji and S. Ott, Chem. – Eur. J., 2016, 22, 14870–14880 CrossRef CAS PubMed.
- (a) M.-N. Collomb-Dunand-Sauthier, A. Deronzier and R. Ziessel, J. Chem. Soc., Chem. Commun., 1994, 189–191 RSC; (b) M.-N. Collomb-Dunand-Sauthier, A. Deronzier and R. Ziessel, Inorg. Chem., 1994, 33, 2961–2967 CrossRef CAS; (c) S. Chardon-Noblat, M.-N. Collomb-Dunand-Sauthier, A. Deronzier, R. Ziessel and D. Zsoldos, Inorg. Chem., 1994, 33, 4410–4412 CrossRef CAS; (d) S. Chardon-Noblat, A. Deronzier, R. Ziessel and D. Zsoldos, Inorg. Chem., 1997, 36, 5384–5389 CrossRef CAS; (e) S. Chardon-Noblat, A. Deronzier, R. Ziessel and D. Zsoldos, J. Electroanal. Chem., 1998, 444, 253–260 CrossRef CAS; (f) C. W. Machan, M. D. Sampson and C. P. Kubiak, J. Am. Chem. Soc., 2015, 137, 8564–8571 CrossRef CAS PubMed.
- (a) A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed; (b) H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346–354 CrossRef CAS; (c) Y. Yamazaki, H. Takeda and O. Ishitani, J. Photochem. Photobiol., C, 2015, 25, 106–137 CrossRef CAS; (d) Y. Kuramochi, O. Ishitani and H. Ishida, Coord. Chem. Rev., 2018, 373, 333–356 CrossRef CAS; (e) Y.-H. Luo, L.-Z. Dong, J. Liu, S.-L. Li and Y.-Q. Lan, Coord. Chem. Rev., 2019, 390, 86–126 CrossRef CAS; (f) H. Kumagai, Y. Tamaki and O. Ishitani, Acc. Chem. Res., 2022, 55, 978–990 CrossRef CAS PubMed; (g) N. Nandal and S. L. Jain, Coord. Chem. Rev., 2022, 451, 214271 CrossRef CAS.
- (a) H. Ishida, T. Terada, K. Tanaka and T. Tanaka, Inorg. Chem., 1990, 29, 905–911 CrossRef CAS; (b) J.-M. Lehn and R. Ziessel, J. Organomet. Chem., 1990, 382, 157–173 CrossRef CAS; (c) K. Kobayashi, T. Kikuchi, S. Kitagawa and K. Tanaka, Angew. Chem., Int. Ed., 2014, 53, 11813–11817 CrossRef CAS PubMed; (d) Y. Kuramochi, M. Kamiya and H. Ishida, Inorg. Chem., 2014, 53, 3326–3332 CrossRef CAS PubMed; (e) R. R. Rodrigues, C. M. Boudreaux, E. T. Papish and J. H. Delcamp, ACS Appl. Energy Mater., 2019, 2, 37–46 CrossRef CAS; (f) R. N. Sampaio, D. C. Grills, D. E. Polyansky, D. J. Szaldaand and E. Fujita, J. Am. Chem. Soc., 2020, 142, 2413–2428 CrossRef CAS PubMed.
- (a) R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS PubMed; (b) Y. Kuramochi, K. Fukaya, M. Yoshida and H. Ishida, Chem. – Eur. J., 2015, 21, 10049–10060 CrossRef CAS PubMed; (c) Y. Kuramochi, J. Itabashi, K. Fukaya, A. Enomoto, M. Yoshida and H. Ishida, Chem. Sci., 2015, 6, 3063–3074 RSC; (d) Y. Kuramochi, J. Itabashi, M. Toyama and H. Ishida, ChemPhotoChem, 2018, 2, 314–322 CrossRef CAS.
- (a) P. L. Cheung, S. C. Kapper, T. Zeng, M. E. Thompson and C. P. Kubiak, J. Am. Chem. Soc., 2019, 141, 14961–14965 CrossRef CAS PubMed; (b) M. R. Madsen, J. B. Jakobsen, M. H. Rønne, H. Liang, H. C. D. Hammershøj, P. Nørby, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, Organometallics, 2020, 39, 1480–1490 CrossRef CAS; (c) C. Back, Y. Seo, S. Choi, M. S. Choe, D. Lee, J.-O. Baeg, H.-J. Son and S. O. Kang, Inorg. Chem., 2021, 60, 14151–14164 CrossRef CAS PubMed; (d) Z. S. Dubrawski, C. Y. Chang, C. R. Carr, B. S. Gelfand and W. E. Piers, Dalton Trans., 2022, 51, 17381–17390 RSC.
- F. Villafañe, Coord. Chem. Rev., 2017, 339, 128–137 CrossRef.
- (a) M. Arroyo, Á. López-Sanvicente, D. Miguel and F. Villafañe, Eur. J. Inorg. Chem., 2005, 4430–4437 CrossRef CAS; (b) N. Antón, M. Arroyo, P. Gómez-Iglesias, D. Miguel and F. Villafañe, J. Organomet. Chem., 2008, 693, 3074–3080 CrossRef; (c) P. Gómez-Iglesias, M. Arroyo, S. Bajo, C. Strohmann, D. Miguel and F. Villafañe, Inorg. Chem., 2014, 53, 12437–12448 CrossRef PubMed.
- (a) P. Gómez-Iglesias, F. Guyon, A. Khatyr, G. Ulrich, M. Knorr, J. M. Martín-Alvarez, D. Miguel and F. Villafañe, Dalton Trans., 2015, 44, 17516–17528 RSC; (b) E. Cuéllar, A. Diez-Varga, T. Torroba, P. Domingo-Legarda, J. Alemán, S. Cabrera, J. M. Martín-Alvarez, D. Miguel and F. Villafañe, Inorg. Chem., 2021, 60, 7008–7022 CrossRef PubMed.
- P. Gómez-Iglesias, J. M. Martín-Alvarez, D. Miguel and F. Villafañe, Dalton Trans., 2015, 44, 17478–17481 RSC.
- M. Arroyo, D. Miguel, F. Villafañe, S. Nieto, J. Pérez and L. Riera, Inorg. Chem., 2006, 45, 7018–7026 CrossRef CAS PubMed.
- M. Arroyo, P. Gómez-Iglesias, J. M. Martín-Alvarez, C. M. Alvarez, D. Miguel and F. Villafañe, Inorg. Chem., 2012, 51, 6070–6080 CrossRef CAS PubMed.
- Í. Ferrer, J. Rich, X. Fontrodona, M. Rodríguez and I. Romero, Dalton Trans., 2013, 42, 13461–13469 RSC.
- E. Reisner, V. B. Arion, A. Rufińska, I. Chiorescu, W. F. Schmid and B. K. Keppler, Dalton Trans., 2005, 2, 2355–2364 RSC.
- (a) C. J. Jones, J. A. McCleverty and A. S. Rothin, Dalton Trans., 1986, 109–111 RSC; (b) A. Romero, A. Vegas and A. Santos, J. Organomet. Chem., 1986, 310, C8–C10 CrossRef CAS; (c) J. López, A. Santos, A. Romero and A. M. Echavarren, J. Organomet. Chem., 1993, 443, 221–228 CrossRef; (d) M. R. Kollipara, P. Sarkhel, S. Chakraborty and R. Lalrempuia, J. Coord. Chem., 2003, 56, 1085–1091 CrossRef CAS; (e) P. Govindaswamy, Y. A. Mozharivskyj and M. R. Kollipara, J. Organomet. Chem., 2004, 689, 3265–3274 CrossRef CAS.
- (a) A. G. Jeffrey, in An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed; (b) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- G. Gritzner and J. Kuta, Pure Appl. Chem., 1984, 56, 461–466 CrossRef.
- E. Cuéllar, L. Pastor, G. García-Herbosa, J. Nganga, A. M. Angeles-Boza, A. Diez-Varga, T. Torroba, J. M. Martín-Alvarez, D. Miguel and F. Villafañe, Inorg. Chem., 2021, 60, 692–704 CrossRef PubMed.
- B. Merillas, E. Cuéllar, A. Diez-Varga, T. Torroba, G. García-Herbosa, S. Fernández, J. Lloret-Fillol, J. M. Martín-Alvarez, D. Miguel and F. Villafañe, Inorg. Chem., 2020, 59, 11152–11165 CrossRef CAS PubMed.
- (a) P. S. Campbell, M. H. G. Prechtl, C. C. Santini and P.-H. Haumesser, Curr. Org. Chem., 2013, 17, 414–429 CrossRef CAS; (b) Y. Na, S. Park, S. B. Han, H. Han, S. Ko and S. Chang, J. Am. Chem. Soc., 2004, 126, 250–258 CrossRef CAS PubMed.
- (a) J. M. Kelly, C. M. O'Connell and J. G. Vos, J. Chem. Soc., Dalton Trans., 1986, 253–258 RSC; (b) M.-N. Collomb-Dunand-Sauthier, A. Deronzier and R. Ziessel, J. Organomet. Chem., 1993, 444, 191–198 CrossRef CAS; (c) E. Eskelinen, M. Haukka, T. Venäläinen, T. A. Pakkanen, M. Wasberg, S. Chardon-Noblat and A. Deronzier, Organometallics, 2000, 19, 163–169 CrossRef CAS; A. Gabrielsson, S. Záliš, P. Matousek, M. Towrie and A. Vlček Jr., Inorg. Chem., 2004, 43, 7380–7388 Search PubMed.
- P. L. Cheung, C. W. Machan, A. Y. S. Malkhasian, J. Agarwal and C. P. Kubiak, Inorg. Chem., 2016, 55, 3192–3198 CrossRef CAS PubMed.
- C. E. McCusker and J. K. McCusker, Inorg. Chem., 2011, 50, 1656–1669 CrossRef CAS PubMed.
- (a) A. Gennaro, A. A. Isse and E. Vianello, J. Electroanal. Chem. Interfacial Electrochem., 1990, 289, 203–215 CrossRef CAS; (b) E. Fujita, C. Creutz, N. Sutin and D. J. Szalda, J. Am. Chem. Soc., 1991, 113, 343–353 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- (a) M. Nardelli, Comput. Chem., 1983, 7, 95–98 CrossRef CAS; (b) M. Nardelli, J. Appl. Crystallogr., 1995, 28, 659–659 CrossRef CAS.
- (a) G. A. Jeffrey and L. Lewis, Carbohydr. Res., 1978, 60, 179–182 CrossRef CAS; (b) R. Taylor and O. Kennard, Acta Crystallogr., Sect. B: Struct. Sci., 1983, 39, 133–138 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Cyclic voltammograms of complexes 1–3, solution stability tests, TEM of 2b degradation products, turnover numbers for the production of H2, CO, and HCCOH at different times for compounds 1b, 2b, and 3b, isotope labeling experiments for clarification of carbon source of CO production, 1H NMR spectra of complexes 1–3. CCDC 2221963 and 2221964 crystal structures of complexes 2a and 2b. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt01122d |
| This journal is © The Royal Society of Chemistry 2023 |