
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWhy a simple vanadate is inefficient as a catalyst in the oxidation of alkanes with H2O2 – the long-standing puzzle is solved†‡
Maxim L.
Kuznetsov
 * and
Armando J. L.
Pombeiro
* and
Armando J. L.
Pombeiro
Centro de Química Estrutural, Institute of Molecular Sciences, Departamento de Engenharia Química, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal. E-mail: max@mail.ist.utl.pt
First published on 22nd May 2023
Abstract
In contrast to V(V) complexes with various organic ligands, a simple vanadate without any additive is inactive in neutral medium toward the oxidation of alkanes with H2O2. In this work, we discovered that the insufficient activation of H2O2 upon coordination to the simple vanadate – the commonly accepted reason for the low catalytic activity of the vanadate – cannot explain this phenomenon. Two main findings are reported here on the basis of DFT calculations. First, the generally accepted Fenton-like mechanism of the generation of the active oxidizing species (HO˙) in a vanadate/H2O2(aq)/MeCN system was revisited. A new mechanism based on the tremendous activation of the OOH ligand in the intermediate [V(OO)2(OOH)(H2O)] toward the homolytic O–O bond cleavage is not only feasible but significantly more favourable than the Fenton-like pathway. The surprisingly low activation barrier calculated for the HO˙ generation (15.4 kcal mol−1) demonstrates the efficiency of this process. The presence of easily oxidizable non-innocent OO ligands in this intermediate explains such an activation. Second, it was found that the generated HO˙ radicals may be easily captured by the V atom soon after their formation followed by the elimination of the molecular oxygen. This side reaction of the H2O2 dismutation efficiently consumes the produced HO˙ radicals decreasing their concentration in the reaction mixture and preventing the following oxidation of alkanes.
Introduction
Alkanes represent a cheap and abundant carbon raw material, and their functionalization into various valuable organic products is of tremendous importance.1–13 The main challenge associated with such transformations is the high chemical inertness of alkanes that requires the application of catalysts and strong oxidants. One of the most popular types of the catalysts used for the oxidation of alkanes is based on transition metal complexes or oxides. Vanadium is an element exhibiting several non-zero oxidation states, which permits its easy participation in various redox processes and enables broad applications of this element and its compounds in a number of catalytic reactions with the involvement of electron transfer (for reviews see ref. 13–33). Being the 6th most abundant transition metal on Earth, vanadium is quite cheap, and its usage as a catalyst is attractive from the economic point of view.Several vanadium complexes with organic ligands (e.g. quinolin-8-olate, pyridine-2-carboxylate, triethanolaminate, bipyridines, phenanthrolines, terephthalohydrazides, NO2-donor Schiff bases, etc.) demonstrated a high catalytic activity in the oxidation of alkanes with hydrogen peroxide.34–38 Moreover, the simple vanadate VO3− was found to be active in these processes but only in the presence of either an appropriate additive (e.g. pyrazine-2-carboxylic acid, PCAH)36,39 or a strong acid.40 In the first case, the in situ formation of V-complexes with the additive occurs in solution, and these complexes serve as active catalytic forms. In the second case, the acidic medium provokes oligomerization of the vanadate, and the active catalytic form is divanadate.
The generally accepted Fenton-like mechanism includes (i) the formation of the H2O2 adduct with a catalyst molecule, (ii) proton transfer leading to a hydroperoxo intermediate, (iii) elimination of the HOO˙ radical with the reduction of the vanadium atom to V(IV), (iv) formation of the second H2O2 adduct, (v) second H-transfer and (vi) elimination of the HO˙ radical with the oxidation of the V(IV) atom39 (Scheme 1). The formed HO˙ radical directly oxidizes an alkane molecule via a hydrogen atom abstraction. The key feature of this mechanism is the direct participation of the metal centre in the electron transfer processes. At the HOO˙ generation step, the V(V) atom serves as an oxidant, while at the HO˙ formation step, the V(IV) centre is a reducing agent.
 | ||
| Scheme 1 Generally accepted mechanism of HO˙ formation from H2O2 catalysed by V(V) complexes. | ||
Meanwhile, the simple monomeric vanadate without any additive in the neutral medium – reaction conditions most attractive from the economic and environmental viewpoints – is not active toward the oxidation of alkanes with H2O2.39,41 The commonly accepted reason is the insufficient activation of H2O2 by the vanadate toward HO˙ production that leads to a high activation barrier for this process. It is expected that the introduction of an appropriate organic ligand or a second vanadate unit (in the case of divanadate) into the vanadate molecule increases the potential of the catalyst to activate H2O2 and decreases the activation energy. However, to the best of our knowledge, this hypothesis was never proved either experimentally or theoretically.
Recently, the authors paid attention to the fact that inorganic salts of some metals bearing only a single stable non-zero oxidation state (SSOS metals such as Al, Ga, Zn, etc.) are quite active as catalysts for the oxidation of alkanes with H2O2.42–44 The mechanism of a principally new type was proposed for these systems. This mechanism is based on the participation of a non-innocent (redox active) ligand in the catalyst molecule (or intermediate), whereas the metal oxidation state is not altered (the “non-innocent ligand mechanism”). It includes (i) the formation of a H2O2 adduct with the catalyst molecule, (ii) deprotonation of H2O2, (iii) addition of a second H2O2 molecule to give a key intermediate I bearing simultaneously the H2O2 and OOH− ligands and (iv) homolytic HO–OH bond cleavage in the H2O2 molecule affording HO˙ and complex II with the hydroperoxyl radical ligand (Scheme 2). The OOH− ligand in I is non-innocent and undergoes intramolecular oxidation upon HO–OH bond rupture. This oxidation stabilizes one of the products of the coordinated H2O2 decomposition and enables tremendous activation of H2O2 toward homolysis.
 | ||
| Scheme 2 Non-innocent ligand mechanism of HO˙ formation from H2O2 catalysed by salts of SSOS metals. | ||
The non-innocent nature of the carbonate ligand and the hydrogen peroxide molecule in the Fenton like process between [Co(H2O)6]2+ and H2O2 as well as the non-innocent character of the Cp ligand in the radical generation from the complexes [Cp2Ti(η1-OOtBu)L] (L = Cl−, OTf−, Br−, OEt2, Et3P) were also previously reported.45–48
The initial goal of this study was two-fold, i.e. (i) to confirm (or disprove) that the HO˙ generation from H2O2 catalysed by the simple vanadate has a high activation barrier and, for this reason, this catalyst is not active and (ii) to verify if the non-innocent ligand mechanism may effectively operate also for catalysts with a metal exhibiting various oxidation states (such as vanadium) or if it is feasible only for the SSOS metals.
The results obtained demonstrated that the non-innocent ligand mechanism is not only feasible but more favourable than the conventionally accepted pathway, and the latter should be revisited. Another unexpected finding indicated that the low activity of the simple vanadate in this reaction is associated not with a high activation barrier of the HO˙ generation but with an efficient side reaction of H2O2 decomposition into O2 and H2O catalysed by vanadate. The results of this study are described and discussed below.
Computational details
The full geometry optimization of all structures and transition states (TS) has been carried out at the density functional theory (DFT) level using the PBE0 functional49 with the atom-pairwise dispersion correction and the Becke–Johnson damping scheme D3BJ50 with the help of the Gaussian-09 program package.51 Recently, this functional demonstrated the excellent performance in the theoretical treatment of the activation of various main group bonds with (Ni,Pd)-based transition metal catalysts.52 The geometry optimization was performed by using the aug-cc-pVDZ basis set for all atoms with consideration of the solvent effects applying the polarizable continuum model in the “solvent model of density” (SMD) version53 with acetonitrile as the solvent typically used in the experimental studies of the alkane oxidation with H2O2 catalysed by V species.34,36–40 Cartesian d and f basis functions (6d, 10f) were used in all calculations. No symmetry operations have been applied for any of the structures calculated. Singlet biradical structures were calculated using the broken symmetry approach54–56 applied with the Guess = Mix Gaussian keyword.The Hessian matrix was calculated analytically for the optimized structures in order to prove the location of correct minima (no imaginary frequencies) or saddle points (only one imaginary frequency), and to estimate the thermodynamic parameters, the latter being calculated at a 298.15 K temperature and 1 atm pressure. The nature of all transition states was investigated by the analysis of vectors associated with the imaginary frequency and by the calculations of the intrinsic reaction coordinates (IRC) using the Gonzalez–Schlegel method.57–59
All possible geometrical isomers were calculated for all V complexes, and the most stable isomers are discussed (if not stated otherwise).
Results
In this section, first, equilibria in the vanadate/H2O2(aq)/MeCN catalytic system and the nature of the active catalytic species are discussed; second, the conventional mechanism of HO˙ formation from H2O2 catalysed by the simple vanadate is analysed; third, the feasibility of the non-innocent ligand mechanisms is discussed; and fourth, the reasons for the low activity of the simple vanadate in the generation of the HO˙ radicals are uncovered.Equilibria in the vanadate/H2O2(aq)/MeCN system
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)2(OH)(H2O)] (1·H2O) with a tetrahedral arrangement of the ligands which contains one water molecule, while the octahedral complexes [V(O)2(OH)(H2O)3] (1·3H2O) do not exist. In contrast, the most stable form of the H2O2 adduct is the trigonal bipyramid complex [V(O)2(OH)(H2O2)(H2O)] (1·H2O2·H2O) and this complex is the first reactive species toward the formation of the peroxo and diperoxo complexes of vanadium.
O)2(OH)(H2O)] (1·H2O) with a tetrahedral arrangement of the ligands which contains one water molecule, while the octahedral complexes [V(O)2(OH)(H2O)3] (1·3H2O) do not exist. In contrast, the most stable form of the H2O2 adduct is the trigonal bipyramid complex [V(O)2(OH)(H2O2)(H2O)] (1·H2O2·H2O) and this complex is the first reactive species toward the formation of the peroxo and diperoxo complexes of vanadium.
 | ||
| Scheme 3 Equilibria between HVO3 and water or hydrogen peroxide adducts (Gibbs free energies are indicated in parentheses in kcal mol−1 relative to that of HVO3, and the most stable H2O2 adduct is boxed; here and further, only the most stable geometrical isomers are shown). | ||
O)(OH)2(OOH)(H2O)] (2·H2O) or to the OH ligand affording [V(O)2(OOH)(H2O)2] (3·2H2O) (Scheme 4). The latter pathway is slightly more kinetically favourable. The H-transfer occurs with the assistance of a water molecule which stabilizes the 6-membered transition states TS1 and TS2 (Fig. 1). The effect of water on the H-transfer in V-species has previously been discussed in detail.36,40 For both channels, the following water liberation results in more stable tetrahedral complexes 2 and 3·H2O. Among all the mentioned hydroperoxo complexes, 2 is the most stable one and it is formed through the 1·H2O2·H2O → 3·2H2O → 3·H2O → 2 pathway. The second H-transfer in 2 from the OOH ligand either to the hydroxo or to the oxo ligand leads to the formation of the monoperoxo complex [V(O)(OH)(OO)(H2O)] (4·H2O) or [V(OH)3(OO)] (5), respectively. The first process is both kinetically and thermodynamically more feasible.
 | ||
| Scheme 4 Mechanisms of the formation of monoperoxo complexes (Gibbs free energies are indicated in parentheses in kcal mol−1 relative to that of HVO3; the initial and final species of this step and the most plausible pathway are indicated). | ||
 | ||
| Fig. 1 Equilibrium structures of TS1 and TS2. | ||
O, OH or OOH ligands in 4·H2O2 (Scheme 5). The calculations indicate that the most favourable route is the 4·H2O → 4·H2O2 → 7 → 6·H2O one. The second H-transfer affords the diperoxo species [V(OH)(OO)2(H2O) (9·H2O). Finally, one more substitution of H2O for H2O2 in 9·H2O and the proton transfer yield the hydroperoxo diperoxo complex [V(OOH)(OO)2(H2O)] (10·H2O). The last two species are the most thermodynamically stable ones in the vanadate/H2O2(aq)/MeCN system. The discussed equilibria are easily established, and the activation barriers for the formation of the most stable intermediates do not exceed 12.4 kcal mol−1.
 | ||
| Scheme 5 Mechanisms of the formation of diperoxo complexes (Gibbs free energies are indicated in parentheses in kcal mol−1 relative to that of HVO3; the initial species of this step and the most stable complexes in the vanadate/H2O2(aq)/MeCN system are boxed). | ||
Conventional Fenton-like mechanism of HO˙ generation
The general scheme of the currently accepted Fenton-like mechanism of HO˙ radical formation from H2O2 catalysed by a V(V) species was discussed in Introduction and it is shown in Scheme 1. The active catalytic forms in the vanadate/H2O2(aq)/MeCN system, which initiate this mechanism, are the V(V) hydroperoxo complexes 2, 6·H2O and 10·H2O.O)(OH)2(H2O)2] (11·2H2O), [V(O)(OO)(H2O)2] (12·2H2O) and [V(OO)2(H2O)] (13·H2O) (Scheme 6). In the case of complexes 2 and 6·H2O, this process occurs via an associative mode including initial addition of two or one H2O molecules, respectively, and the following HOO˙ elimination (Scheme S1 in the ESI‡). In the case of complex 10·H2O, the HOO˙ generation is realized dissociatively. The V–OOH bond cleavage is accompanied by the monotonous increase of the total energy of the system. The potential energy surface (PES) scan in 6·H2O indicates that until a V–OOH distance of ca. 2.41 Å is achieved, the singlet closed shell configuration is the most stable one (Fig. 2, left). At a longer distance, the singlet biradical configuration with unpaired electrons localized at the V atom and the leaving OOH group becomes more stable (Fig. 2, right).
 | ||
| Scheme 6 Conventional mechanisms of HO˙ generation (Gibbs free energies are indicated in parentheses in kcal mol−1 relative to that of HVO3, and the most favourable pathway is boxed). | ||
 | ||
| Fig. 2 PES scans for the elongation of the V–OOH bond in the complex 6·H2O (left, scans of two electronic triplet configurations are indicated) and a plot of spin density for the singlet biradical structure at a V–OOH distance of 3.6 Å (right). | ||
O)(OH)2(H2O2)(H2O)] (11·H2O2·H2O), [V(O)(OO)(H2O2)(H2O)] (12·H2O2·H2O) and [V(OO)2(H2O2)(H2O) (13·H2O2·H2O) are formed upon water substitution for H2O2 or addition of H2O2 (Scheme 6). The subsequent proton transfer from the coordinated H2O2 molecule can occur to the oxo, hydroxo or peroxo ligand furnishing the hydroperoxo V(IV) complexes [V(O)(OH)(OOH)(H2O)2] (14·2H2O), [V(OH)(OOH)(OO)(H2O)] (16·H2O) and [V(OOH)2(OO)(H2O) (17·H2O). The formation of 16·H2O occurs in two steps via complex 15·H2O. Finally, the O–O bond cleavage in the OOH ligand yields the HO˙ radical and restores the V(V) active catalytic forms.
The non-innocent ligand mechanisms of HO˙ generation
In this mechanism, the HO˙ radical is formed either from the OOH ligand in a V(V) complex or from the coordinated H2O2 molecule which are activated toward the O–O homolysis due to the presence of a non-innocent ligand in the complex.O)(OH)2(OOH)] (2), [V(O)(OOH)(OO)(H2O)] (6·H2O) and [V(OOH)(OO)2(H2O)] (10·H2O).
Thermodynamic consideration. The dissociation of the OOH ligand in the first complex produces HO˙ and [V(O)2(OH)2] (18) but it is endergonic by 35.9 kcal mol−1 (Scheme 7). Such a high value is not surprising given that there is no non-innocent ligand in 2. This value is only by 2.0 kcal mol−1 lower than that for the homolytic dissociation of free H2O2 (37.9 kcal mol−1).
 | ||
| Scheme 7 Formation of the HO˙ radical from the hydroperoxo V(V) complexes through the non-innocent ligand mechanism (Gibbs free energies are indicated in parentheses in kcal mol−1 relative to that of HVO3, and the most favourable pathway is boxed). | ||
A qualitatively different situation was found for the OOH homolyses in 6·H2O and 10·H2O. In the former species, the ΔG° value of HO˙ formation is only +5.8 kcal mol−1, whereas in the latter complex, this process is exergonic by −8.2 kcal mol−1 indicating that the generation of HO˙ from 10·H2O is thermodynamically spontaneous. The reasons of such huge thermodynamic activation of OOH in these complexes become clear from the analysis of the products of this step, [V(O)2(OO)(H2O)] (19·H2O) and [V(O)(OO)2(H2O)] (20·H2O). The unpaired electron in these doublet species is localized at the OO ligand rather than at the V atom (Fig. 3). Thus, the HO˙ elimination does not result in a change of the V oxidation state which remains at +5. The homolytic VO–OH bond cleavage produces a free HO˙ radical and an oxyl anion-radical O˙− which is coordinated to V in 19·H2O or 20·H2O. During the O–O bond rupture, the coordinated O˙− anion-radical is reduced intramolecularly, oxidizing the OO ligand and not the V centre as in the V(IV) complexes 11·2H2O, 12·2H2O and 13·H2O of the conventional mechanism. As a result, O˙− is transformed into the oxo ligand (O)2−, while the peroxo (OO)2− ligand is oxidized into the peroxyl species (OO)˙−. Therefore, the (OO)2− ligand exhibits non-innocent behaviour which permits further oxidation of the V(V) complexes 6·H2O and 10·H2O despite them bearing the metal centre in its highest oxidation state. The ability of the (OO)2− ligand to be easily oxidized is a driving force for the tremendous thermodynamic activation of OOH in the hydroperoxo V(V) complexes 6·H2O and 10·H2O.
 | ||
| Fig. 3 O–OH bond cleavage in 10·H2O, intramolecular electron transfer and spin density distribution in 20·H2O. | ||
Kinetic consideration. The low ΔG° values of HO˙ formation from 6·H2O and 10·H2O allow the assumption that these processes are controlled by kinetic factors rather than by thermodynamic ones. The analysis of the kinetic features is not trivial in this case since the initial complexes are singlet close shell structures while both reaction products are doublets. Therefore, the PES scans for the VO–OH bond cleavage in 6·H2O and 10·H2O at different spin states were firstly analysed. At the singlet close shell PES, an increase of the VO–OH distance corresponding to the heterolytic VO–OH bond cleavage is accompanied by the monotonic enhancement of the system's energy (Fig. 4). This process is unfavourable, and the elongation of the VO–OH bond until 2 Å requires 30–33 kcal mol−1.
 | ||
| Fig. 4 PES scans for the elongation of the VO–OH bond in complexes 6·H2O (A) and 10·H2O (B) (positions of the equilibrium structures of TS20 and TS21 are shown by cross and plots of spin density for 1,16·H2O and 310·H2O are provided). | ||
The adiabatic excitation of 6·H2O with the unrelaxed VO–OH bond distance to the singlet biradical state 1,16·H2O requires only 12.2 kcal mol−1. One of the unpaired electrons in 1,16·H2O is localized at the V atom while another one is mostly distributed among the peroxo ligand (Fig. 4A). The OOH ligand being bidentate in the ground state of 6·H2O becomes monodentate in 1,16·H2O. The V–O(OO) bonds elongate upon excitation from 1.82–1.83 Å to 1.96–1.99 Å which corresponds to the formation of the peroxyl ligand (OO)˙−. Thus, this excitation results in the formation of a V(IV) species due to an intramolecular redox process between the metal centre and the OO ligand. Upon increase of the VO–OH distance, the total energy on the singlet biradical PES reaches the maximum at 1.74 Å and then decreases.
The PES character of the complex 10·H2O is similar (Fig. 4B). In this case, the singlet biradical structures could be calculated only for VO–OH distances longer than 1.63 Å. The initial excitation energy to the triplet state 310·H2O is 10.4 kcal mol−1. The excitation affects only one peroxo ligand.
With the help of PES scan results, the singlet biradical transition states TS20 and TS21 which correspond to the homolytic VO–OH bond cleavage in 6·H2O and 10·H2O were located (Fig. 5). The spin density in these TSs is mostly localized at the V atom (ρs,α = 0.81 and 0.72e), one OO ligand (ρs,β = 1.05e) and the oxygen atom of the leaving HO˙ radical (ρs,α = 0.54 and 0.53e). Thus, the VO–OH bond rupture in the excited complexes 1,16·H2O and 1,110·H2O leads to the intramolecular reduction of the oxygen atom coordinated to the V atom to form the oxo ligand O2−.
 | ||
| Fig. 5 Equilibrium structures and plots of spin densities for TS20 and TS21. | ||
O)2(OH)(H2O2)(H2O)] (1·H2O2·H2O), [V(O)(OH)(OO)(H2O2)] (4·H2O2) and [V(OH)(OO)2(H2O2)] (9·H2O2). The decomposition of 1·H2O2·H2O into HO˙ and [V(O)2(OH)2(H2O)] (21·H2O) is not favourable due to the absence of any non-innocent ligands in this complex (ΔG° = 25.3 kcal mol−1). In contrast, the homolysis of H2O2 in the other two adducts is exergonic by −9.7 and −2.1 kcal mol−1 because of the presence of the redox active peroxo ligand. As in the case of the V(V) hydroperoxo species, the homolytic HO–OH bond cleavage in 4·H2O2 and 9·H2O2 results in the intramolecular oxidation of the redox active OO ligand to the OO˙− anion-radical and reduction of the formed coordinated OH group, while the oxidation state of vanadium remains intact.
 | ||
| Scheme 8 Formation of the HO˙ radical from the hydrogen peroxide V(V) complexes through the non-innocent ligand mechanism (Gibbs free energies are indicated in parentheses in kcal mol−1 relative to that of HVO3, and the most favourable pathway is boxed). | ||
The PES scan analysis allowed the localization of the singlet biradical transition states TS22 and TS23 corresponding to the homolytic HO–OH bond cleavage in 4·H2O2 and 9·H2O2 (Fig. S1 in ESI‡). The spin density in TS22 and TS23 is mostly localized at the V atom, one peroxo ligand and the leaving HO˙ radical.
Activation energies and comparison of reaction mechanisms
The inspection of the calculated mechanisms indicates the following. First, in the solution of vanadate with aqueous hydrogen peroxide, several diperoxo, monoperoxo and hydroperoxo complexes are easily formed. The diperoxo complexes 9·H2O and 10·H2O are the most thermodynamically stable in such a solution, and they are in equilibrium with other slightly less stable monoperoxo and hydroperoxo complexes 6·H2O, 7, 4·H2O and 2 (Scheme 5). The diperoxo and monoperoxo V(V) species were detected experimentally in the aqueous solution in the presence of H2O2 using the NMR technique.60–63Second, the most favourable pathway of the conventional mechanism is based on the hydroperoxo diperoxo complex 10·H2O and includes the sequence of steps 10·H2O → 13·H2O (+HOO˙) → 13·H2O2·H2O → 17·H2O → 6·H2O (+HO˙) (Scheme 6). The overall Gibbs free energy of activation for HO˙ formation in this pathway is 28.8 kcal mol−1 relative to 10·H2O. The rate determining step is the formation of the V(IV) hydrogen peroxide adduct 13·H2O2·H2O.
Third, the non-innocent ligand mechanism based on the simple homolysis of the OOH ligand in [V(OOH)(OO)2(H2O)] 10·H2O (Scheme 7) is significantly more favourable than the conventional mechanism. This pathway has an activation barrier of 15.4 kcal mol−1 relative to 10·H2O, and the rate limiting step is the VO–OH bond cleavage in 10·H2O.
Fourth, the non-innocent ligand mechanism based on the homolysis of H2O2 coordinated in [V(OH)(OO)2(H2O2)] 9·H2O2 has an activation energy of 28.1 kcal mol−1 relative to 10·H2O which is comparable to that of the conventional mechanism (Scheme 8).
Fifth, the presence of two peroxo ligands in the catalytic complexes is crucial for the highest activation of the H2O2 and OOH ligands toward homolysis. All pathways based on the monoperoxo species 2 and 6·H2O require a higher activation barrier than the pathways of the same mechanism based on the diperoxo complex 10·H2O.
Thus, one of the most important results of this work is that the mechanism involving the non-innocent peroxo ligand is not only feasible but also more favourable than the conventional Fenton-like mechanism.
Why a simple vanadate is not efficient as a catalyst for the oxidation of alkanes with H2O2?
The calculated overall activation barrier for the most plausible mechanism of HO˙ generation is 15.4 and 17.4 kcal mol−1 in terms of the Gibbs free energy and enthalpy of activation, respectively. Such low values make the generation of the HO˙ radicals in the vanadate/H2O2(aq)/MeCN system quite efficient. The latter value correlates well with the experimental activation energy of HO˙ generation in the n-Bu4NVO3/PCAH/H2O2(aq)/MeCN system which is active for the oxidation of alkanes (17 ± 2 kcal mol−1 (ref. 39)). Thus, the low activity of the simple vanadate (without any additive) as a catalyst in the oxidation of alkanes with H2O2cannot be justified by the high activation energy for this process.During the search for PES for the VO–OH bond cleavage in 10·H2O it was found that the liberating HO˙ radical can easily be trapped by the V atom soon after the formation of TS21. Indeed, the PES scan for the shortening of the V⋯OHleaving distance starting from a point on the energy curve after TS21 revealed a barrier of HO˙ capture of only 2.0 kcal mol−1 (Fig. 6, see the ESI‡ for details). As a result, the singlet biradical complex [V(O)(OH)(OO)2] (1,124) is formed that is accompanied by an extrusion of the water molecule initially coordinated in 10·H2O. The spin density in 1,124 is localized at two peroxo OO ligands (1.07e for each ligand, Fig. 7) which corresponds to the structure with two anion-radical peroxyl ligands (OO)˙−. The spin conversion from the singlet biradical to the triplet state 324 then occurs, the latter state being more stable by 0.6 kcal mol−1. Both states have similar spin distribution with some involvement of the V atom in the case of the triplet structure.
 | ||
| Fig. 6 Singlet biradical PES scans for the O–OH bond cleavage in 10·H2O (A) and for the shortening of the V⋯OHleaving distance from one of the points on the reaction path after TS21 (B). | ||
 | ||
| Fig. 7 Plots of spin density in 1,124 and 324. | ||
The formation of complex 24 from 10·H2O is highly exergonic by (−43.6)–(−44.2) kcal mol−1 (Scheme 9). Both singlet biradical and triplet complexes 24 can easily lose the molecular oxygen affording the monoperoxo species [V(O)(OH)(OO)] (4) which is stabilized by water addition to give 4·H2O. The singlet biradical channel leads to the formation of the singlet oxygen which was detected experimentally in the reactions of the V(V) species with H2O2.64 Upon these processes, one of the (OO)˙− ligands experiences further oxidation to O2, whereas another (OO)˙− ligand is reduced to the peroxo ligand (OO)2− (Fig. 8). The O2 liberation has quite a low activation barrier of 15.7 kcal mol−1 and it is exergonic by −12.5 kcal mol−1. Thus, the HO˙ radical trapping by the V centre is very efficient. Overall, this reaction channel corresponds to the catalytic dismutation of H2O2 into O2 and H2O (Scheme 10). The consumption of HO˙ in this side reaction significantly decreases the concentration of these radicals in the reaction mixture and makes their interaction with an alkane molecule inefficient.
 | ||
| Scheme 9 HO˙ radical trap upon O–OH bond cleavage in 10·H2O and the following liberation of O2. | ||
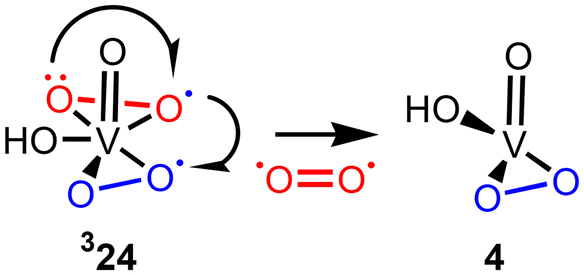 | ||
| Fig. 8 Liberation of O2 in complex 324. | ||
 | ||
| Scheme 10 Dismutation of H2O2 catalysed by the complex 4·H2O. | ||
Discussion and final remarks
The low efficiency of the simple vanadate as a catalyst in the homogeneous oxidation of alkanes with H2O2 is a long-standing puzzle. Insufficient activation of H2O2 toward the production of HO˙ radicals upon its coordination to the V centre in the simple vanadate has been postulated as the reason for such low activity. However, to the best of our knowledge, no systematic studies aimed at supporting this assumption have been undertaken. In this article, the results of the detailed mechanistic study of HO˙ formation in the vanadate/H2O2(aq)/MeCN system are reported.This work resulted in two unexpected discoveries which finally shed light on this issue. First, the commonly accepted mechanism of the vanadium(V) catalysed oxidation of alkanes with H2O2 represented in the most general form by eqn (1) and (2) was revisited. Another mechanism of HO˙ generation (eqn (3)) based on the participation of a non-innocent ligand – which was not previously considered for the V activation of H2O2toward homolysis – was found to be not only feasible but also more favourable than the conventional mechanism.
| HO–[VV]–OOH + H2O2 → HO–[VIV]–O2H2 + HOO˙ | (1) |
| HO–[VIV]–O2H2 → H2O–[VV]O + HO˙ | (2) |
| [VV]–OOH → [VV]O + HO˙ | (3) |
This new mechanism starts with the formation of the hydroperoxo diperoxo complex [V(OOH)(OO)2(H2O)] 10·H2O upon several H2O-for-H2O2 substitution and H+-transfer steps – 10·H2O being the most stable catalytic form in the vanadate/H2O2(aq)/MeCN system – and includes simple monomolecular homolytic VO–OH bond cleavage in 10·H2O affording [V(O)(OO)2(H2O)] 20·H2O and HO˙ (Scheme 11). Despite the V atom being in its highest oxidation state in 10·H2O, the further oxidation of the catalyst may occur upon VO–OH bond cleavage due to the presence of the non-innocent peroxo ligand which is oxidized instead of the metal centre. The reaction product 20·H2O, thus, represents the V(V) species bearing one peroxyl anion-radical ligand (OO)·−, and the oxidation state of the vanadium is not altered. The VO–OH bond rupture in 10·H2O yields one HO˙ radical and one peroxyl anion-radical (OO)˙− which, being coordinated to the V centre, is quite stable. Such a stabilization of one of the products explains the tremendous thermodynamic activation of the OOH ligand in 10·H2O. Thus, the role of the non-innocent ligand in the generation of the HO˙ radicals from H2O2 is not restricted to the catalysts with the metals bearing the only one stable oxidation state, reported previously.42–44 A similar mechanism is also applicable to catalysts based on the metals with variable oxidation states.
 | ||
| Scheme 11 Overall non-innocent ligand mechanism of HO˙ generation in the vanadate/H2O2(aq)/MeCN system and of the side reaction of H2O2 dismutation via complex 24 (reaction and activation (in parentheses) energies are indicated in terms of Gibbs free energies in kcal mol−1). | ||
The calculations demonstrate that the overall activation barrier of HO˙ generation in the vanadate/H2O2(aq)/MeCN system (15.4 kcal mol−1) is sufficiently low to permit efficient radical formation. The second principal result of this work indicates that the low efficiency of the simple vanadate in the oxidation of alkanes is not related to insufficient activation of H2O2by the V catalyst. Instead, an easy capture of the generated HO˙ by the V atom decreases the effective concentration of HO˙ in the reaction mixture and, therefore, hampers the following oxidation of alkanes. This side reaction corresponds to the catalytic dismutation of H2O2 into O2 and H2O (Scheme 11).
The V atom in the complex 10·H2O is an efficient HO˙ radical trap because it has an unsaturated coordination sphere. Saturation of the coordination sphere in 10·H2O by a solvent molecule (e.g. H2O) is not thermodynamically feasible (ΔG° = 5.2 kcal mol−1). In contrast, the formation of 24 from 10·H2O is highly exergonic (ΔG° = −44.2 kcal mol−1) and, additionally, the following O2 elimination also has a negative ΔG° value (−12.5 kcal mol−1).
Application of additives or V(V) complexes with various organic ligands often significantly improves the performance of the catalyst in comparison with the simple vanadate.34,36,39 Taking into account the results of this work, it is possible to speculate that such an improvement is associated not with a higher activation of H2O2 by such catalysts but with the saturation of the V coordination sphere by organic ligands. For instance, the NMR spectroscopic studies showed that complexes 26a and 26b (Chart 1) are the predominant forms of the catalyst in solution of vanadate in the presence of H2O2 and PCAH.36,39,65 These complexes have the saturated coordination sphere of the metal and, therefore, the V atom here cannot serve as an efficient HO˙ radical trap. This can explain the pronounced effect of the PCAH additive on the catalytic activity of vanadate in the alkane oxidation reaction. The verification of this hypothesis should be the subject of further investigations.
 | ||
| Chart 1 | ||
The role of the hydroperoxo V complexes as active catalytic species was revealed in the oxidation of halide anions with H2O2 catalysed by vanadium haloperoxidase enzymes or mimicking model complexes.66–69 In this reaction, the OOH ligand undergoes two-electron reduction by the Hal− anion while the latter is oxidized to the hypohalite anion OHal− which may be coordinated to the V atom (eqn (4)). The formed hypohalite oxidizes hydrogen peroxide to the molecular oxygen and water (eqn (5)), and the overall process corresponds to the dismutation of H2O2 (eqn. (4)–(7)).67 In the case of the simple vanadate with an unsaturated coordination sphere, the generated HO˙ radicals serve as such oxidizing species.

Data availability
Additional data may be found in the ESI‡ or could be requested from the authors.Author contributions
MLK – conceptualization of the work, computational studies, writing and preparation of the manuscript. AJLP – conceptualization of the work, discussion of results, proof reading, and funding.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was partially supported by the Fundação para a Ciência e a Tecnologia (FCT), Portugal, projects UIDB/00100/2020 and UIDP/00100/2020 of Centro de Química Estrutural and LA/P/0056/2020 of Institute of Molecular Sciences.References
- A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879 CrossRef CAS PubMed.
- G. B. Shul'pin, Mini-Rev. Org. Chem., 2009, 6, 95 CrossRef.
- R. H. Crabtree, Chem. Rev., 1995, 95, 987 CrossRef CAS.
- C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS PubMed.
- B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457 CrossRef CAS PubMed.
- G. Grigoropoulou, J. H. Clark and J. A. Elings, Green Chem., 2003, 5, 1 RSC.
- Alkane Functionalization, ed. A. J. L. Pombeiro and M. F. C. Guedes da Silva, J. Wiley & Sons, Hoboken, NJ, USA, 2019 Search PubMed.
- J. A. Labinger, J. Mol. Catal. A: Chem., 2004, 220, 27 CrossRef CAS.
- B. L. Conley, W. J. Tenn III, K. J. H. Young, S. K. Ganesh, S. K. Meier, V. R. Ziatdinov, O. Mironov, J. Oxgaard, J. Gonzales, W. A. Goddard III and R. A. Periana, J. Mol. Catal. A: Chem., 2006, 251, 8 CrossRef CAS.
- J. Muzart, J. Mol. Catal. A: Chem., 2007, 276, 62 CrossRef CAS.
- M. M. Díaz-Requejo and P. J. Pérez, Chem. Rev., 2008, 108, 3379 CrossRef PubMed.
- R. H. Crabtree, Chem. Rev., 2010, 110, 575 CrossRef CAS PubMed.
- J. A. L. da Silva, J. J. R. Fraústo da Silva and A. J. L. Pombeiro, Coord. Chem. Rev., 2011, 255, 2232 CrossRef CAS.
- Vanadium catalysis, ed. M. Sutradhar, J. A. L. da Silva and A. J. L. Pombeiro, Royal Society of Chemistry, Cambridge, UK, 2021 Search PubMed.
- R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Chem. Rev., 2019, 119, 2128 CrossRef CAS PubMed.
- K. Nomura and S. Zhang, Chem. Rev., 2011, 111, 2342 CrossRef CAS PubMed.
- P. Schwendt, J. Tatiersky, L. Krivosudský and M. Šimuneková, Coord. Chem. Rev., 2016, 318, 135 CrossRef CAS.
- V. Conte, A. Coletti, B. Floris, G. Licini and C. Zonta, Coord. Chem. Rev., 2011, 255, 2165 CrossRef CAS.
- A. M. F. Phillips, H. Suo, M. F. C. Guedes da Silva, A. J. L. Pombeiro and W.-H. Sun, Coord. Chem. Rev., 2020, 416, 213332 CrossRef CAS.
- M. Kirihara, Coord. Chem. Rev., 2011, 255, 2281 CrossRef CAS.
- G. Licini, V. Conte, A. Coletti, M. Mba and C. Zonta, Coord. Chem. Rev., 2011, 255, 2345 CrossRef CAS.
- H. Hagen, J. Boersma and G. van Koten, Chem. Soc. Rev., 2002, 31, 357 RSC.
- J.-Q. Wu and Y.-S. Li, Coord. Chem. Rev., 2011, 255, 2303 CrossRef CAS.
- H. Pellissier, Coord. Chem. Rev., 2015, 284, 93 CrossRef CAS.
- B. M. Weckhuysen and D. E. Keller, Catal. Today, 2003, 78, 25 CrossRef CAS.
- N. F. Dummer, J. K. Bartley and G. J. Hutchings, Adv. Catal., 2011, 54, 189 CAS.
- M. R. Maurya, A. Kumar and J. C. Pessoa, Coord. Chem. Rev., 2011, 255, 2315 CrossRef CAS.
- A. Chieregato, J. M. L. Nieto and F. Cavani, Coord. Chem. Rev., 2015, 301–302, 3 CrossRef CAS.
- J. C. Védrine, G. J. Hutchings and C. J. Kiely, Catal. Today, 2013, 217, 57 CrossRef.
- M. O. Guerrero-Pérez, Catal. Today, 2017, 285, 226 CrossRef.
- W. Chu, J. Luo, S. Paul, Y. Liu, A. Khodakov and E. Bordes, Catal. Today, 2017, 298, 145 CrossRef CAS.
- N. Mizuno and K. Kamata, Coord. Chem. Rev., 2011, 255, 2358 CrossRef CAS.
- J. J. H. B. Sattler, J. Ruiz-Martinez, E. Santillan-Jimenez and B. M. Weckhuysen, Chem. Rev., 2014, 114, 10613 CrossRef CAS PubMed.
- I. Gryca, K. Czerwińska, B. Machura, A. Chrobok, L. S. Shul'pina, M. L. Kuznetsov, D. S. Nesterov, Y. N. Kozlov, A. J. L. Pombeiro, I. A. Varyan and G. B. Shul'pin, Inorg. Chem., 2018, 57, 1824 CrossRef CAS PubMed.
- H. Mimoun, L. Saussine, E. Daire, M. Postel, J. Fischer and R. Weiss, J. Am. Chem. Soc., 1983, 105, 3101 CrossRef CAS.
- M. V. Kirillova, M. L. Kuznetsov, V. B. Romakh, L. S. Shul'pina, J. J. R. Fraústo da Silva, A. J. L. Pombeiro and G. B. Shul'pin, J. Catal., 2009, 267, 140 CrossRef CAS.
- I. S. Fomenko, A. L. Gushchin, P. A. Abramov, M. N. Sokolov, L. S. Shul'pina, N. S. Ikonnikov, M. L. Kuznetsov, A. J. L. Pombeiro, Y. N. Kozlov and G. B. Shul'pin, Catalysts, 2019, 9, 217 CrossRef.
- M. Sutradhar, N. V. Shvydkiy, M. F. C. Guedes da Silva, M. V. Kirillova, Y. N. Kozlov, A. J. L. Pombeiro and G. B. Shul'pin, Dalton Trans., 2013, 42, 11791 RSC.
- G. B. Shul'pin, Y. N. Kozlov, G. V. Nizova, G. Süss-Fink, S. Stanislas, A. Kitaygorodskiy and V. S. Kulikova, J. Chem. Soc., Perkin Trans. 2, 2001, 1351 RSC.
- M. V. Kirillova, M. L. Kuznetsov, Y. N. Kozlov, L. S. Shul'pina, A. Kitaygorodskiy, A. J. L. Pombeiro and G. B. Shul'pin, ACS Catal., 2011, 1, 1511 CrossRef CAS.
- G. B. Shul'pin and G. Süss-Fink, J. Chem. Soc., Perkin Trans. 2, 1995, 1459 RSC.
- M. L. Kuznetsov, Y. N. Kozlov, D. Mandelli, A. J. L. Pombeiro and G. B. Shul'pin, Inorg. Chem., 2011, 50, 3996 CrossRef CAS PubMed.
- A. S. Novikov, M. L. Kuznetsov, A. J. L. Pombeiro, N. A. Bokach and G. B. Shul'pin, ACS Catal., 2013, 3, 1195 CrossRef CAS.
- M. L. Kuznetsov, F. A. Teixeira, N. A. Bokach, A. J. L. Pombeiro and G. B. Shul'pin, J. Catal., 2014, 313, 135 CrossRef CAS.
- A. Burg, D. Shamir, I. Shusterman, H. Kornweitz and D. Meyerstein, Chem. Commun., 2014, 50, 13096 RSC.
- A. Burg, I. Shusterman, H. Kornweitz and D. Meyerstein, Dalton Trans., 2014, 43, 9111 RSC.
- A. G. DiPasquale, D. A. Hrovat and J. M. Mayer, Organometallics, 2006, 25, 915 CrossRef CAS PubMed.
- A. G. DiPasquale, W. Kaminsky and J. M. Mayer, J. Am. Chem. Soc., 2002, 124, 14534 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Keith, J. Kobayashi, K. Normand, A. Raghavachari, J. C. Rendell, S. S. Burant, T. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev. D.01, Gaussian Inc., 2013 Search PubMed.
- M. Steinmetz and S. Grimme, ChemistryOpen, 2013, 2, 115 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
- L. Noodleman and D. A. Case, Adv. Inorg. Chem., 1992, 38, 423 CrossRef CAS.
- E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 1999, 20, 1391 CrossRef CAS.
- E. Ruiz, A. Rodriguez-Fortea, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 2003, 24, 982 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef CAS.
- M. Bonchio, O. Bortolini, M. Carraro, V. Conte and S. Primon, J. Inorg. Biochem., 2000, 80, 191 CrossRef CAS PubMed.
- V. Conte, F. Di Furia and S. Moro, J. Mol. Catal. A: Chem., 1997, 117, 139 CrossRef CAS.
- V. Conte, F. Di Furia and S. Moro, J. Mol. Catal. A: Chem., 1994, 94, 323 CrossRef CAS.
- H. Schmidt, I. Andersson, D. Rehder and L. Pettersson, Chem. – Eur. J., 2001, 7, 251 CrossRef CAS PubMed.
- A. E. Gekhman, G. E. Amelichkina, N. I. Moiseeva, M. N. Vargaftik and I. I. Moiseev, J. Mol. Catal. A: Chem., 2000, 162, 111 CrossRef CAS.
- Y. N. Kozlov, V. B. Romakh, A. Kitaygorodskiy, P. Buglyó, G. Süss-Fink and G. B. Shul'pin, J. Phys. Chem. A, 2007, 111, 7736 CrossRef CAS PubMed.
- B. J. Hamstra, G. J. Colpas and V. L. Pecoraro, Inorg. Chem., 1998, 37, 949 CrossRef CAS.
- G. J. Colpas, B. J. Hamstra, J. W. Kampf and V. L. Pecoraro, J. Am. Chem. Soc., 1996, 118, 3469 CrossRef CAS.
- A. Butler, Coord. Chem. Rev., 1999, 187, 17 CrossRef CAS.
- Z. Chen, Coord. Chem. Rev., 2022, 457, 214404 CrossRef CAS.
Footnotes |
| † Dedicated to the memory of Prof. Georgiy B. Shul'pin. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt00967j |
| This journal is © The Royal Society of Chemistry 2023 |