
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triplex metallohelices have enantiomer-dependent mechanisms of action in colon cancer cells†
J. P. C.
Coverdale
 ab,
H.
Kostrhunova
c,
L.
Markova
c,
H.
Song
ad,
M.
Postings
a,
H. E.
Bridgewater
ae,
V.
Brabec
*c,
N. J.
Rogers
*a and
P.
Scott
*a
ab,
H.
Kostrhunova
c,
L.
Markova
c,
H.
Song
ad,
M.
Postings
a,
H. E.
Bridgewater
ae,
V.
Brabec
*c,
N. J.
Rogers
*a and
P.
Scott
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
bSchool of Pharmacy, Institute of Clinical Sciences, University of Birmingham, Edgbaston, B15 2TT, UK
cThe Czech Academy of Sciences, Institute of Biophysics, Kralovopolska 135, CZ-61265 Brno, Czech Republic
dBeijing Area Major Laboratory of Peptide and Small Molecular Drugs, School of Pharmaceutical Sciences, Capital Medical University, Beijing, 100069, China
eCentre of Exercise, Sport and Life Science, Faculty of Health and Life Sciences, Coventry University, Coventry, CV1 5FB, UK
First published on 18th April 2023
Abstract
Self-assembled enantiomers of an asymmetric di-iron metallohelix differ in their antiproliferative activities against HCT116 colon cancer cells such that the compound with Λ-helicity at the metals becomes more potent than the Δ compound with increasing exposure time. From concentration- and temperature-dependent 57Fe isotopic labelling studies of cellular accumulation we postulate that while the more potent Λ enantiomer undergoes carrier-mediated efflux, for Δ the process is principally equilibrative. Cell fractionation studies demonstrate that both enantiomers localise in a similar fashion; compound is observed mostly within the cytoskeleton and/or genomic DNA, with significant amounts also found in the nucleus and membrane, but with negligible concentration in the cytosol. Cell cycle analyses using flow cytometry reveal that the Δ enantiomer induces mild arrest in the G1 phase, while Λ causes a very large dose-dependent increase in the G2/M population at a concentration significantly below the relevant IC50. Correspondingly, G2-M checkpoint failure as a result of Λ-metallohelix binding to DNA is shown to be feasible by linear dichroism studies, which indicate, in contrast to the Δ compound, a quite specific mode of binding, probably in the major groove. Further, spindle assembly checkpoint (SAC) failure, which could also be responsible for the observed G2/M arrest, is established as a feasible mechanism for the Λ helix via drug combination (synergy) studies and the discovery of tubulin and actin inhibition. Here, while the Λ compound stabilizes F-actin and induces a distinct change in tubulin architecture of HCT116 cells, Δ promotes depolymerization and more subtle changes in microtubule and actin networks.
Introduction
We have developed several structurally distinct ranges of Fe(II) metallohelices1–6 such as that in Fig. 1(a). In all these compounds, the absolute configuration of the arrangement of ligand strands about each metal centre, and thus the sense of helicity of the assembly, is fixed independently in highly diastereoselective processes arising from the use of amino acid chiral pool derivatives containing α-phenylimines.7 The resultant hydrophobic π-stacking motifs (indicated by dashed lines) imbue the compounds with high stability to hydrolysis and overall inertness to ligand substitution, and for example the compounds of Fig. 1 do not “unfold” even at pH 1 over 10 d. Correspondingly, ranges of water-soluble compounds are readily prepared on a substantial scale. These favourable properties have allowed us and collaborators to explore various aspects of biological chemistry of the metallohelices.8–14 There is a growing body of evidence that the compounds emulate the properties of short cationic α-helical peptides; naturally occurring anticancer and antimicrobial molecules with which they share several structural features.15 | ||
| Fig. 1 Self-assembled triplex metallohelix architectures: (a) enantiomers Δ-1 and Λ-1; (b) the relatively hydrophobic ridge of Δ-1 comprising two sets of π-stacks between pendant arenes (colorised pink) and Fe-coordinated bipyridines; and (c) the opposite relatively hydrophilic face of Δ-1 showing exposed charge-bearing units and the third π-stack. | ||
Of the metallohelices, the most α-helix-like are the “triplex” classes4,6,12–18e.g.Fig. 1, so-called because they are assembled, like triplex DNA, from three antiparallel strands. The ensuing helical fold creates asymmetric shielding of the cationic charge arising from the Fe(II) centres, and as a result the structure comprises a hydrophobic ridge Fig. 1(b) opposite a patchy hydrophilic face (c). These compounds thus have amphipathic architectures reminiscent of short cationic α-helical peptides.15
Unlike another related architecture,8 the compounds 1 have no antimicrobial activity, but they do provide potent and selective anticancer systems. We have previously studied the antiproliferative activities of Λ-1 and Δ-1 in HCT116 colon cancer cells by MTT assay (96 h dosing), and in ARPE19 non-cancerous human epithelial retinal pigment cells, and observe that Λ−1 is more potent than its Δ-analogue in both cell lines under these conditions.16,17 We also expanded our library of triplex metallohelices by functionalising with an alkynyl group, and subsequently achieved post-assembly modification of the core triplex structures with a range of aromatic16 and glycosyl17 groups. In all cases we observe that the antiproliferative activity of the Λ-metallohelices is greater than their Δ-analogues in eukaryotic cancer cells.15 This led us to investigate the enantiomeric differences in more detail.
Results and discussion
Antiproliferative activities and cell uptake studies
The water soluble, optically pure triplex metallohelices Λ-1 and Δ-1 (Fig. 1) were synthesised as previously described.457Fe isotopologues (Λ-1′ and Δ-1′) of these highly stable complexes were also prepared (see ESI†) in order to allow us to readily distinguish the ferro-helix from iron naturally present in cells.We previously noted16,17 that the complex Λ-1 is significantly more potent (IC50 = 1.4 ± 0.4 μM) than the enantiomer Δ-1 (IC50 = 21 ± 2 μM) at 96 h exposure using MTT assay in HCT116 cells. We have found however in time-dependent experiments [Fig. 2(a), solid lines] that Δ-1 is in fact slightly more potent than Λ-1 at short exposure times (<24 h). By 48 h exposure however the Λ isomer is, as expected,16 the more toxic, and this continues up 96 h.
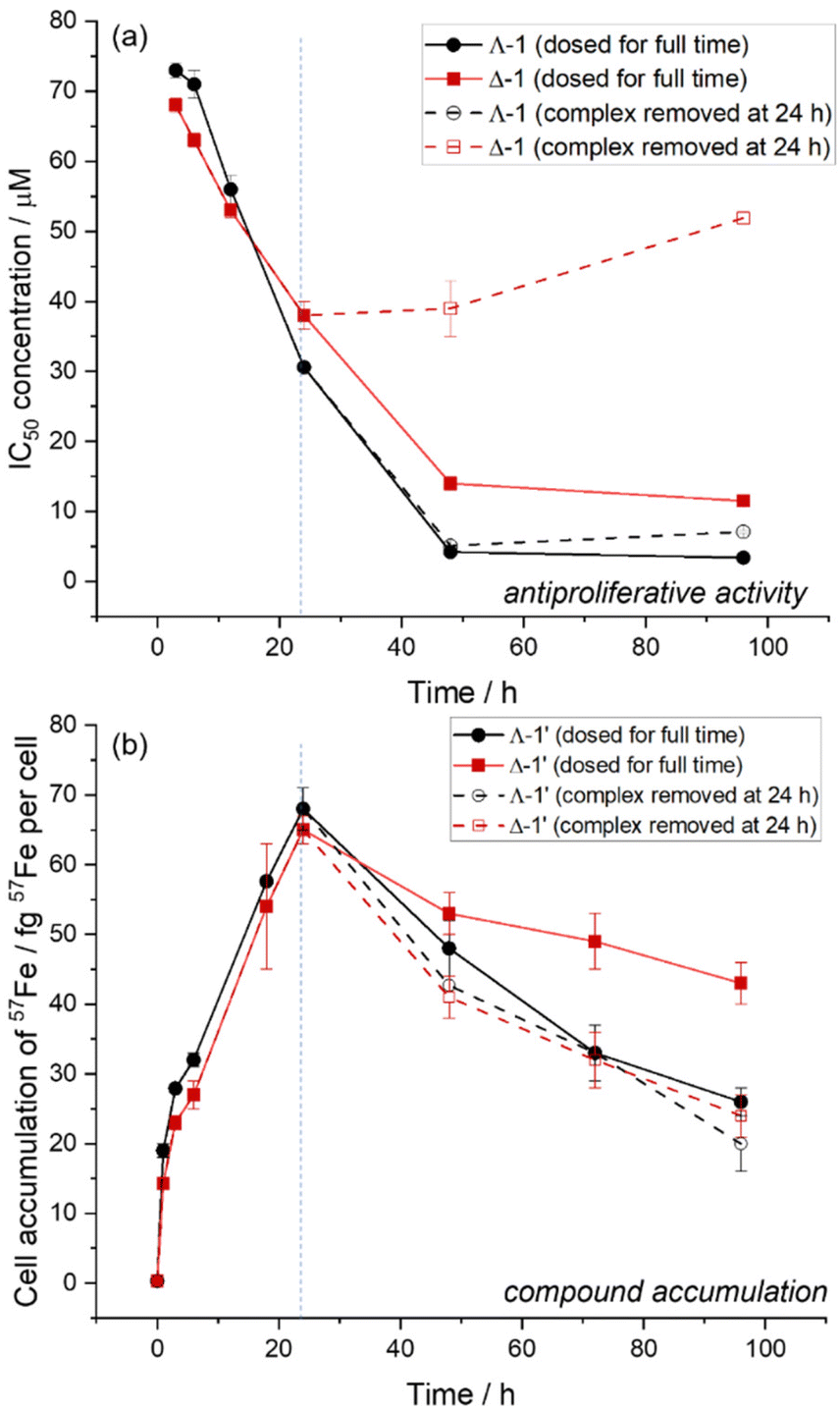 | ||
| Fig. 2 Time-dependence of antiproliferative activities and 57Fe accumulation in HCT116 cells human colorectal carcinoma (a) antiproliferative activity of Λ-1 (red) and Δ-1 (black) over 0–96 h total incubation times. IC50 concentrations (determined by SRB assay) are given for cells exposed to complex for the full incubation period (filled symbols/solid lines), or for 24 h, followed by a recovery time (open symbols/dashed lines). (b) Cellular accumulation (fg 57Fe per cell) of Λ-1′ (red) and Δ-1′ (black) in HCT116 cells, determined by ICP-MS over 0–96 h incubation times. Cells were treated with either constant exposure to 3 μM complex (filled symbols/solid lines) or 24 h exposure (3 μM) followed by recovery time in fresh medium for the remaining incubation time (open symbols/dashed lines). | ||
Time-dependent cellular accumulation of the 57Fe labelled compounds Λ-1′ and Δ-1′ was studied via ICP-MS. HCT116 cells were incubated with equimolar (3 μM) concentrations of the compounds, and the intracellular concentrations of 57Fe were determined following acid-digestion of the cell pellets. In Fig. 2(b) (solid lines) we see that over an initial 24 h period both enantiomers accumulate in the cells at a similar rate. At incubation periods longer than 24 h the 57Fe concentration per live cell decreases gradually. In this study we assume that the extracellular concentration of metallohelix remains constant over the 96 h experiment since the complexes are very stable and the extracellular volume is three orders of magnitude greater than the intracellular space. Further we know that for the Δ-1′ system at least, the cell population is not significantly declining due to cell death, since metallohelices were administered to cells at a concentration below the IC50 and cell counts were similar to untreated controls.
This behaviour is consistent with the activation of transport processes (enhanced efflux and/or reduced uptake)19 once some critical intracellular concentration of metallohelix is reached: apparently ca. 65 fg 57Fe per cell, corresponding to a micromolar intracellular concentration of metallohelix. The difference in the 24–96 h accumulations observed (Λ-1′ < Δ-1′) would be due to differences in the cellular efflux/influx responses to each enantiomer. However, it is important to note that the cells were incubated with Λ-1′ at a concentration close to the 96 h IC50, and hence we may be gradually selecting cells where the uptake is relatively slow (or efflux fast), or more generally that the remaining pool of live cells is on average more resistant to the effects of this enantiomer.
We also investigated the efflux process in isolation by replacing the metallohelix test solutions with fresh media during the incubation period at t = 24 h. Fig. 2(b) (dashed lines) shows that 57Fe concentration fell similarly for both enantiomers over the 24–96 h period indicating similar rates of efflux. However, comparison with the behaviour in the presence of 3 μM compound (solid lines) shows that while the rate of efflux is unaffected by extracellular compound concentration for Λ-1′ (black lines), for Δ-1′ (red lines) removing the compound at 24 h clearly reduces accumulation and correspondingly reduces the potency [Fig. 2(a), red lines]. This is consistent with the efflux mechanisms being different for the two enantiomers, and we postulate on the basis of the above observations that enantiomer Λ-1′ undergoes concentration-independent carrier-mediated efflux whilst for Δ-1′ the process is principally equilibrative.20,21
Turning to uptake mechanisms, we performed accumulation studies at 37 and 4 °C (Fig. 3). Cells were incubated with compound for a short period (3 or 6 h) so as to minimize cellular response (Fig. 2). In comparison with examples of drugs that are thought to enter cells via energy-requiring (i.e. ATP-dependent) carrier-mediated uptake processes (vide infra), the accumulation of 57Fe compound is affected only slightly on reducing the temperature from 37 °C to 4 °C (ca. 35% and 27% decrease in uptake for Λ-1′ and Δ-1′ at 3 h incubation time). For example, a 50-fold reduction of carboplatin accumulation is observed in liver carcinoma cells,22 and at least 10-fold reduction of imatinib in leukaemia cells23 is reported, in experiments performed at 4 °C vs. 37 °C. At 4 °C the accumulation of 57Fe per cell at both 3 h and 6 h exposure times is the same for each enantiomer (within experimental error), whereas there are observable differences at 37 °C; the accumulation of Λ-1′ is ca. 20% greater than that of Δ-1′ at this temperature. It is difficult to envisage that passive lipoidal diffusion (or endocytosis) would lead to the enantiomeric accumulation differences observed at 37 °C (although the phospholipid head groups are indeed chiral),24 and thus we conclude that the accumulation (balance of influx and efflux) of at least one enantiomer involves transporter protein mechanisms at this temperature.
 | ||
| Fig. 3 Temperature-dependent accumulation. Temperature-dependent intracellular 57Fe accumulation (fg 57Fe per cell) of complexes Λ-1′(white bars) and Δ-1′ (grey bars) in HCT116 cells, incubated with 3 μM complex for 3 h (unshaded) and 6 h (shaded). | ||
We next investigated the intracellular distribution of Λ-1′ and Δ-1′ in HCT116 cells (Fig. 4). Following incubation for 24 or 96 h, cells were separated into sub-cellular fractions using a FractionPREP™ Cell Fractionation kit (BioVision), isolating(I) a cytoskeleton/genomic DNA fraction (total cellular insoluble proteins i.e. microfilaments, microtubules, and intermediate filaments, high molecular weight DNA), (ii) a nuclear fraction (nuclear soluble proteins, including nuclear membrane proteins), (iii) a membrane fraction (organelles and organelle membrane proteins, but excluding nuclear membrane proteins), and (iv) a cytosolic fraction (total cytoplasmic soluble proteins) for subsequent ICP-MS analysis.
 | ||
| Fig. 4 Cellular distribution. Cellular distribution of 57Fe in HCT116 cells treated for either 24 h or 96 h with 3 μM complex. Cell pellets were fractioned using FractionPREP kit (Biovision), and cellular accumulation of 57Fe was determined by ICP-MS. | ||
After 24 h dosing, we observe that the enantiomers distribute similarly to one another in cells, with the majority of the 57Fe accumulation occurring in the cytoskeleton/genomic DNA fraction (Λ-1′ 94 ± 5%, Δ-1′ 93 ± 5%). Significant amounts of 57Fe were also found in the nuclear and membrane fractions, and thus while both metallohelices appear to traverse the membrane envelope and enter the nucleus, the concentration in the cytosol fraction is negligible (<0.5% of total). By 96 h the accumulation of Λ-1′ and Δ-1′ in the membrane fraction is similar to that at 24 h (consistent with saturation of membrane-binding sites) whilst 57Fe in the cytoskeleton/genomic DNA fraction has significantly decreased. A small observable reduction is also present in the nuclear fraction, particularly in the Λ-1′ enantiomer.
Flow cytometry was used to analyse the effect of increasing concentrations of both enantiomers on the HCT116 cell cycle, treating the cells for 24 h (Fig. 5). Compound Δ-1 induces a decrease in the proportion of HCT116 cells in the G2/M phase around the IC50 concentration (38 ± 2 μM at 24 h), which may indicate some arrest in the G1 phase (the G1/S checkpoint checks that the cell is ready to replicate DNA). The Λ enantiomer induces strikingly different behaviour, and there is a very large dose-dependent increase in the proportion of cells in the G2/M population of the cell cycle at concentrations significantly below the 24 h IC50 (30.6 ± 0.5 μM). Notably, the IC50 concentration for cells dosed for 24 h, and then incubated in fresh media for the remaining 72 h is only 7.1 ± 0.5 μM, which is consistent with a time-lag for the overall inhibitory cellular response from the initial effects detected in the cell cycle at 0–10 μM concentrations.
 | ||
| Fig. 5 Cell cycle analysis. Effects of Λ-1 and Δ-1 on cell-cycle profiles of HCT116 cells treated for 24 h. Cells were fixed prior to staining with propidium iodide and assessed by FACS analysis. Data for Δ-1 previously published.17 | ||
Cells in the G2 and mitotic phases both possess two copies of DNA and are detected together by flow cytometry as a G2/M population. Accumulation of cells in this G2/M population can occur a result of cell cycle arrest at: (i) the G2-M checkpoint, which occurs after the G2 phase (rapid cell growth, protein synthesis and preparation for mitosis) and ensures that cells do not proceed to mitosis until damaged or incompletely replicated DNA is sufficiently repaired; or (ii) the spindle assembly checkpoint (SAC) which occurs during mitosis and prevents progression to the anaphase (separation of the duplicated chromosomes) until each chromosome is correctly attached to the spindle. We note in this context that both enantiomers of the metallohelix are detected principally in the cytoskeletal/genomic DNA cell fraction, and we thus began to investigate the feasibility of (i) G2−M checkpoint failure as a result of the Λ-metallohelix binding to DNA, and (ii) SAC failure resulting from interference with e.g., the function of cyctoskeleton proteins.
Interactions with DNA in cell-free medium
We have mentioned that Δ-1 binds to naked calf-thymus DNA.17 The apparent binding constant of Λ-1 is very similar; log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kapp = 6.6 ± 0.3 cf. 6.5 ± 0.2 for Δ-1, as measured by displacement of the cationic DNA intercalator ethidium bromide.25 Notably however, while a plot of the reduction in dye fluorescence intensity vs. concentration of Λ-1 was essentially linear, the data for Δ-1 gave a change in slope, consistent with the presence of more than one type of event (see ESI†). Similar assays using the fluorescent minor groove binder Hoechst 33258 and the major groove binder methyl green gave the same general outcome.
Kapp = 6.6 ± 0.3 cf. 6.5 ± 0.2 for Δ-1, as measured by displacement of the cationic DNA intercalator ethidium bromide.25 Notably however, while a plot of the reduction in dye fluorescence intensity vs. concentration of Λ-1 was essentially linear, the data for Δ-1 gave a change in slope, consistent with the presence of more than one type of event (see ESI†). Similar assays using the fluorescent minor groove binder Hoechst 33258 and the major groove binder methyl green gave the same general outcome.
In linear dichroism spectroscopy (LD), long optically pure molecules such as DNA are oriented by flow in a Couette cell and the difference between the absorption of parallel and perpendicularly polarised light (LD = A‖ – A┴) is plotted against wavelength. While optically pure metallohelices such as 1 have distinctive circular dichroism spectra4 (see ESI†), they are too small to align in the Couette flow of the LD experiment, so A‖ − A┴ = 0. Nevertheless, they may give rise to induced bands in the UV/visible region if they bind in some preferred orientation to flow-oriented DNA. In particular, the presence of metal-to-ligand charge-transfer (MLCT) bands at ca. 400–650 nm in the electronic spectra of Fe(II)-based helicates/metallohelices – an otherwise spectroscopically quiet region – can give insight regarding orientation of the complex with respect to the DNA linear axis. In some cases, the angle of orientation can be calculated, and a few such studies have all led to the proposal that compounds of appropriate size bind preferentially to the major groove.1,26
Fig. 6(a) and (b) show the LD spectra of Λ- and Δ-1 respectively at various calf thymus (CT) DNA base:metallohelix ratios, at constant concentration of CT-DNA. As we increase the concentration of Λ-1, the intensity of the negative DNA band at 258 nm decreases smoothly [Fig. 6(d), black squares]. This we ascribe to reduction in linearity of the DNA molecules upon addition of the cationic complexes.26,27 For Δ-1 (red circles) there is different behaviour, including a change in slope at ca. 10 μM metallohelix. For Λ-1 there is a strongly induced LD signal at 303 nm corresponding to π–π* absorptions in the metallohelix (e) while there is almost no change for the Δ-1 system. In the MLCT region, addition of either enantiomer leads to an induced band at ca. 580 nm (f) but beyond 10:1 (DNA:Δ-1) the intensity falls, despite the growth of an adjacent broad feature (400–550 nm) that is essentially absent for the Λ-1 system.‡
 | ||
| Fig. 6 In vitro DNA interaction studies. Linear dichroism spectra of ct-DNA (600 μM) in buffer (1 mM Tris, pH 7.4 with increasing concentrations of Λ-1 (a) and Δ-1 (b) [ratios indicated are metallohelix:base pair]. (c) UV-Vis photoabsorption spectrum (6 μM, H2O) of Δ-1 for comparison. Plots of the LD signal vs. [metallohelix] are shown in (d) at 258 nm (DNA absorption region), (e) 303 nm (metallohelix π−π* absorption region), and (f) 580 nm (metallohelix MLCT absorption region). | ||
The distinct differences in behaviour that we see here for enantiomers 1 have not been reported for enantiomers of more conventional D3 symmetric metallohelices3,5,28 where the two ends of the metal–ligand assembly are chemically equivalent1 and where we would argue that the chirality is not so well expressed in the architecture. While any preferences in directional orientation Fe–Fe′ of the metallohelices 1 are undetectable since the DNA is randomly oriented 3′ → 5′ vs. 5′ → 3′ in the flow, we can see that the mode of binding of Λ-1 is quite specific in nature and given the distinct behaviour of induced π–π* bands we suggest that the hydrophobic π-stack region [Fig. 1(b)] is associated with the major groove. In further support of this we note that addition of bulky substituents on the opposite face of the metallohelix has little bearing on binding behaviour.17 In contrast for Δ-1, observations are consistent with the presence of two or more distinct binding events and the ability to access other orientations in partially-loaded DNA.
The effect of tubulin and actin inhibitors
The cytoskeleton is a complex, dynamic network of interlinked protein filaments within the cytoplasm that provides structure to eukaryotic cells, and is involved in cell signalling, cell division, and intracellular transport. To investigate the influence of Λ-1 and Δ-1 on cytoskeleton proteins (considering the intracellular compartmentalisation studies, Fig. 4) we have employed drug combination studies with known tubulin and actin inhibitors. HCT116 cells were treated with combinations of three tubulin and two actin-targeting drugs, and the cytotoxic/antiproliferative effects were determined with the MTT assay after 72 h.Nocodazole, as well as vinblastine, have been reported to act by two mechanisms.29 At low concentrations they suppress microtubule dynamics, while high concentrations result in a reduction of microtubule mass. Paclitaxel binds to β-subunits of tubulin and prevents microtubule polymer from disassembly.29 Cytochalasin D is a potent inhibitor of actin polymerization.30 Phalloidin binds F-actin and stabilizes the polymer by preventing its depolymerization.31
The HCT116 cells were simultaneously treated with Λ-1 or Δ-1 and the chosen inhibitor, in ratios reflecting their respective IC50 values. The single drug, combination doses, and corresponding induced effects were analysed using CompuSyn, Inc. (Version 1.0), to give the Combination Indices (CI) summarized in Table 1.
| Inhibitor | CI1 (Λ-1) | CI1 (Δ-1) | |||
|---|---|---|---|---|---|
| (Inhibitor: Λ/Δ-1 ratio) | Mean | Range | Mean | Range | |
| Tubulin inhibitor | Nocodazole (1:100) |
1.0406 | 0.945–1.135 | 0.9051 | 0.877–0.930 |
| Tubulin inhibitor | Vinblastine (1:100) |
0.6549*** | 0.545–0.734 | 0.7446** | 0.618–0.884 |
| Tubulin inhibitor | Paclitaxel (1:100) |
0.8259** | 0.641–0.922 | 0.8315** | 0.634–0.916 |
| Actin inhibitor | Cytochalasin D (1:10) |
0.8022** | 0.691–0.804 | 0.8590* | 0.730–0.915 |
| Actin inhibitor | Phalloidin (10:1) |
0.7472** | 0.681–0.813 | 0.6767*** | 0.597–0.756 |
The CI was used to confirm and quantify the synergism observed for co-treatment. CI of 0.9 to 1.1 indicates an additive effect, CI > 1 indicates antagonism, and CI < 1 synergism, wherein CIs of 0.85–0.9 refer to slight synergism (*), 0.7–0.85 to moderate synergism (**), 0.3–0.7 to synergism (***) and lower values to strong or very strong synergism.32
An additive effect was observed with a combination of either Λ-1 or Δ-1 with the tubulin inhibitor nocodazole, whilst combinations with the tubulin inhibitors vinblastine and paclitaxel provided moderate synergism to synergism, and combinations with the actin inhibitors Cytochalasin D and Phalloidin gave rise to slight synergism to synergism. A synergistic response indicates that the drug pair may cooperatively act on targets involved in the same process33,34 and hence further investigations into the interactions of Λ-1 or Δ-1 with tubulin and actin were carried out.
Interactions with tubulin and actin in cell-free medium
As an initial investigation, tubulin (15 μM) was incubated in a buffer (BRB80) in the presence of guanylyl-(alpha, beta)-methylene-diphosphonate (GMPCPP, 1 μM) at 37 °C for 1 h (which helps to stabilise microtubule polymers), with and without Λ-1 and Δ-1 complexes (50 μM). Samples were separated by ultracentrifugation, to separate the polymerised microtubules from the free tubulin and complex; the supernatant and re-suspended pellet were analysed by UV-Vis absorption spectroscopy and SDS-PAGE. Within error, both enantiomers were found to accumulate similarly in the pellet (with the microtubules), with 14–15% of the total complex residing in the pellet (see ESI†), equating to a 2:1 tubulin:metallohelix ratio. A tubulin polymerization assay was therefore performed using an HTS-Tubulin Polymerization Assay Biochem Kit™ (Cytoskeleton, Inc.); the time-dependent absorbance of tubulin (340 nm) was measured with or without the metallohelices at 37 °C, and following polymerization, the temperature was decreased (0.5 °C min−1) whilst the absorbance was observed for an additional 25 min, to assess the effect of the tested compounds on tubulin disassembly. Both Λ-1 and Δ-1 inhibit the polymerization of tubulin in a concentration-dependent manner (Fig. 7A and B); a ca. 20% reduction in the tubulin absorption is observed at 50 min, in the presence of either enantiomer (10 μM concentration). However, negligible effect on the tubulin depolymerization dynamics is observed in the presence Λ-1 or Δ-1 under these conditions (Fig. 7C).
 | ||
| Fig. 7 Tubulin and actin polymerization/depolymerisation dynamics in cell-free medium. (A and B) Tubulin assembly. Tubulin polymerization was performed in the absence (control, black) or presence of (A) Λ-1 (green) and (B) Δ-1 (blue) at 37 °C, and the absorbance was read at 340 nm. (C) Microtubule disassembly was induced by the decrease of temperature (0.5 °C min−1). Initial absorbance at 37 °C (t = 0) was set as 1. G-Actin polymerization (D and E) and F-Actin depolymerization (F) in the absence (control, black) or presence of (D) Λ-1 (green) and (E) Δ-1 (blue) at RT was recorded as changes in fluorescence of pyrene-conjugated actin at λEX/EM = 360/410 nm. | ||
We also investigated effects of Λ-1 and Δ-1 on the actin polymerization and depolymerisation, using an Actin Polymerization Biochem Kit™ (Cytoskeleton, Inc.); pyrene-labelled actin monomers (G-actin) was polymerized at room temperature, and the fluorescence was measured over time (λexc = 360 nm, λem = 410 nm), due to the enhanced fluorescence of pyrene-conjugated actin that occurs during polymerization (forming F-actin). The influence of Λ-1 and Δ-1 on F-actin depolymerisation was also determined using pre-formed the F-actin (1 h, 25 °C in actin polymerization buffer).
Λ-1 and Δ-1 induce almost identical inhibition of actin polymerization (at 10 μM concentration an 11% (Λ-1) and 10% (Δ-1) reduction in signal is observed at 30 min (Fig. 4C), whilst lower doses had negligible effects. However, there are clear differences between the effects Λ-1 and Δ-1 on F-actin depolymerisation; Λ-1 appears to stabilize F-actin, whilst Δ-1 promotes polymer disintegration (Fig. 7D–F), and hence it is plausible that the G2/M cell cycle arrest (Fig. 5) is associated with the stabilization of F-actin by Λ-1, whilst the G1 arrest is associated with the disruption of actin filament.35
Organization of the structural network within the cytoplasm of HCT116 cells
We next sought to gain more insight into the influence of Λ-1 and Δ-1 on the organization of the cytoskeleton within the cytoplasm; we treated the HCT116 cells with Λ-1 and Δ-1, stained with monoclonal antibodies directed against α-tubulin or with phalloidin against F-actin, and visualized by confocal fluorescence microscopy (Fig. 8 and 9). | ||
| Fig. 8 Tubulin immunofluorescence images in HCT116 cells. (1) Non-treated control; (2) cells treated with Δ-1; (3) cells treated with Λ-1. The cells were treated with 20 μM compounds for 24 h. Nuclei were stained with DAPI (blue) and tubulin with primary and secondary (Alexa Fluor® 488) antibodies (green). BF = bright field, Merge = overlay of the fluorescence channels. Samples were scanned on a confocal laser scanning microscope, and the scale bar is 25 μm. The images shown are representative of at least three independent experiments. | ||
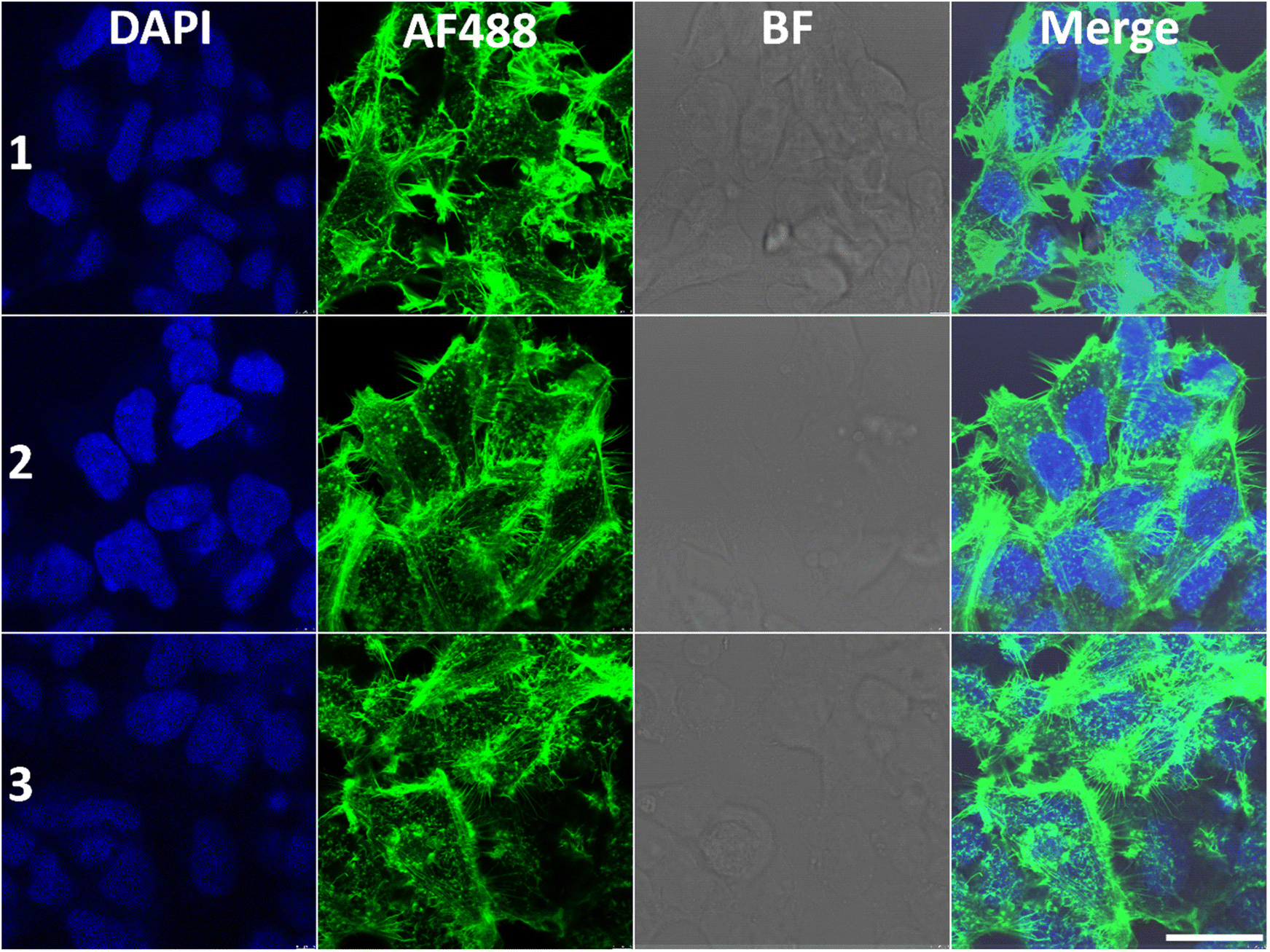 | ||
| Fig. 9 Actin immunofluorescence images in HCT116 cells. (1) Non-treated control; (2) cells treated with Δ-1; (3) cells treated with Λ-1. The cells were treated with 20 μM compounds for 24 h. Nuclei were stained with DAPI (blue) and actin with phalloidin conjugated with Alexa Fluor® 488 (green). BF = bright field, Merge = overlay of the fluorescence channels. Samples were scanned on a confocal laser scanning microscope, and the scale bar is 25 μm. The images shown are representative of at least three independent experiments. | ||
In the untreated cells, microtubules extended continuously through the cytoplasm and formed an extensive intracellular network, whilst treatment with Λ-1 induced very clear changes in the architecture of microtubule networks via retraction to the perinuclear region when compared with untreated cells (Fig. 8). The effects of Δ-1 are less obvious, but retraction to the perinuclear regions is also observable in some cells. These results are consistent with the observation that Λ-1 and Δ-1 affect microtubule dynamic in the cell-free medium (Fig. 7A–C), and have synergistic effects on the HCT116 cells with Vinblastine and Paclitaxel, but the dramatic effect on the microtubule network (Fig. 8) is only observed in cells treated with the Λ-enantiomer (Fig. 5).This specific distribution of irregular microtubule fragments in HCT116 cells treated with Λ-1 correlates with the observation that Λ-1 causes cell cycle G2/M arrest as previously demonstrated for other agents capable of disturbing tubulin assembly.36–38
We also examined whether HCT116 cells cultured in the presence of Λ-1 or Δ-1 exhibit visible changes in the organisation of actin filaments; no significant changes in the organization were observed in the presence of Δ-1 (Fig. 9), whereas the filaments appear less organised in the presence of Λ-1.
Conclusions
We have found that both enantiomers Λ-1 and Δ-1 cross the HCT116 cell membrane, and while they are subsequently found in significant quantities in all cell fractions except the cytosol, they localize principally in the cytoskeleton/genomic DNA fraction.While only subtle differences are observed between enantiomers in cellular accumulation and distribution within the first 24 h of incubation, at longer periods the transport rates and mechanisms differ. We propose, based on both enantiomer-dependent responses to concentration changes and variable temperature accumulation studies, that the cells are able to eject the substantially more potent isomer Λ-1via a carrier-mediated processes, whilst for Δ-1 the process is principally equilibrative.
Stark differences are also seen in the effects of the enantiomers on the cell cycle; Λ-1 induces a very clear dose-dependent G2/M-arrest response in the HCT116 cells, which is not observed for Δ-1. Given the observations on cellular localization and the studies on naked DNA, G2/M checkpoint failure as a result of specific DNA major groove binding is a feasible mechanism of action for Λ-1 (only).
Also noting the predominant localisation in the cytoskeleton/genomic DNA fraction, drug combination studies revealed synergism with known tubulin and actin inhibitors. Both Δ and Λ enantiomers inhibit tubulin and actin polymerisation with similar potency, but while the Λ compound stabilizes F-actin, Δ-1 promotes its depolymerization. Correspondingly, immunofluorescence imaging reveals that while Δ-1 induces subtle changes in microtubule and actin networks of HCT116 cells, Λ-1 induces a distinct change in architecture, in which the microtubules retract to the perinuclear region. There are also some changes in the architecture of the actin structures. This would point to a further feasible mechanism for Λ-1, leading to the observed G2/M arrest; spindle assembly checkpoint (SAC) failure.
Overall, we conclude that triplex metallohelices have enantiomer-dependent transport and localisation, as well as enantiomer-specific effects on processes and structure in colon cancer cells. Further, such substantial differences between enantiomers indicates that rather subtle factors are at play at the molecular level mechanism of action. We believe that this adds to the emerging body of evidence that these metallosupramolecular architectures emulate the properties of natural short cationic α-helices and represent a suitable platform for drug discovery.
Experimental
Chemical synthesis
The synthesis and full characterisation of the Fe complexes Λ-1 and Δ-1 has been previously reported.457Fe-labelled complexes Λ-1′ and Δ-1′ (see ESI†) were synthesised as follows. (S)-2-(2,2′-Bipyridin-5-ylmethoxy)-1-phenylethanamine (29 mg, 0.094 mmol, 3.0 eq.) and 2-pyridinecarboxaldehyde (10 mg, 0.094 mmol, 3.0 eq.) were dissolved in methanol (10 ml) and stirred for 24 h at ambient temperature to form a yellow solution. Anhydrous 57Fe(II) chloride (8.0 mg, 0.063 mmol, 2.0 eq.) was added and an instantaneous colour change to deep purple was observed. The solution was then heated at reflux (80 °C) for 48 h, filtered through a Celite plug, and the volatiles were evaporated under reduced pressure. The solid was dissolved in minimum methanol and pipetted into ethyl acetate (25 ml). The precipitate was collected on a fine filter paper, washed with ethyl acetate (3 × 25 ml), dissolved in methanol and the solvent was removed under reduced pressure to give the desired product as a purple solid, which was dried overnight at 50 °C in vacuo (41 mg, 0.028 mmol, 90%). The enantiomer Δ-1′ was made in same method for Λ-1′ using (R)-2-(2,2′-bipyridin-5-ylmethoxy)-1-phenyl-ethanamine. As for the natural isotopologues, the compounds are isolated as hydrates [57Fe2L3]Cl4·9H2O as determined by microanalysis and differential scanning calorimetry.Cell culture
HCT116 (human colorectal carcinoma), were purchased from the European Collection of Cell Cultures (ECACC) and grown as adherent monolayers at 310 K in a 5% CO2 humidified atmosphere in DMEM supplemented with 10% FCS and penicillin/streptomycin.Cell culture medium (DMEM) was purchased from Scientific Laboratory Supplies. Foetal calf serum (FCS) was purchased from Labtech International. Phosphate buffered saline (PBS), trypsin/EDTA and antibiotics were prepared by technical staff at the School of Life Science (University of Warwick).
In vitro growth inhibition assay
5000 cells (HCT116) were seeded per well in a 96-well plate. Stock solutions (100 μM) of Fe complexes were prepared in cell culture medium and diluted to achieve final working concentrations (100–0.01 μM). After 48 h pre-incubation, cells were exposed to the iron complexes for 0–96 h. After this time, the supernatant was removed and cells washed with PBS, before allowing 0–72 h recovery in drug-free medium. Cell viability was determined by SRB assay,39 and cell viability was measured using a ThermoFisher Multiskan FC microplate reader fitted with a 492 nm filter. IC50 values were determined in duplicate of triplicate as part of two independent experiments and standard deviations were calculated.Cell cycle analysis
HCT116 cells were seeded at a density of 5 × 105 cells per well in 6-well plates. After an overnight incubation, the cells were treated with tested compounds for 24 h. Treated and untreated cells were harvested with trypsinization, washed with PBS and fixed in 70% ethanol at 4 °C overnight. The cells were washed twice with PBS and stained with propidium iodide (50 μg ml−1) in Vindeĺs solution (10 mM Tris-Cl, pH 8.0, 10 mM NaCl, 0.1% Triton X-100, 100 μg ml−1 RNase A) for 30 min. Cell cycle profiles were taken with a FACSVerse flow cytometer (Becton Dickinson, Germany) and the data were analysed with FSC Express software. The results were obtained from three independent experiments.ICP-MS studies
Combinatorial drug tests with actin and tubulin inhibitors
The HCT116 cells were simultaneously treated with Λ-1 or Δ-1 and the particular inhibitor in ratios taking into account their respective IC50 values. The antiproliferative effect was then evaluated by the MTT method. Single drug and combination doses and corresponding induced effects were analyzed using CompuSyn, Inc. (Version 1.0). The Combination Indices (CI) values for the activity of Λ-1 or Δ-1 and actin or tubulin affecting drug combinations were calculated, and at least six entries of dose−effect relationships were employed for each drug combination.Linear dichroism studies
Flow linear dichroism (LD) spectra were measured using a Jasco J-815 spectrometer equipped with LD spectroscopy kit. The standard parameters used were: bandwidth 1 nm, response time 1 s, wavelength scan range 180–700 nm, data pitch 0.1 nm, scanning speed 100 nm min−1 and accumulation 4. Calf thymus DNA (ct-DNA) 600 μM was prepared in Trizma Buffer (pH 7.4), followed by addition of various concentration of metallohelices with the ratio to ct-DNA base pairs: 1:50, 1:25, 1:10, and 1:5. The LD spectra were obtained by subtracting the parallel absorption of the molecule from perpendicular absorption [LD = A‖ − A┴] in the presence of Laminar flow.
Tubulin binding studies
Tubulin from porcine brains was incubated at 15 μM in BRB80 buffer, in the presence of GMPCPP (1 mM) at 37 °C for 1 h with Λ-1 or Δ-1 (50 μM). The samples were spun down in an ultracentrifuge (25 psi, 10 min), the supernatant was recovered and the pellet was incubated in ice for 10 min with ice-cold BRB80 buffer (65 μL), and subsequently resuspended and stored on ice. The concentration of metallohelix in each resuspended pellet/supernatant sample was measured using UV-Vis absorption spectroscopy. All samples were measured in triplicate.Tubulin and actin polymerization in cell-free medium
Tubulin polymerization in a cell-free medium was performed using HTS-Tubulin Polymerization Assay Biochem KitTM (Cytoskeleton, Inc.) in the microplate format following the produceŕs protocol. Briefly, the absorbance of tubulin (4 mg mL−1) in general tubulin buffer with or without various concentrations of the investigated compounds at 37 °C was read at 340 nm in the kinetic mode (1 reading per minute) on SPARKTM multimode reader (Tecan). The samples were prepared in triplicate. Following the polymerization, the temperature was decreased (0.5 °C min−1), and the absorbance at 340 nm was read for additional 25 min to assess the effect of the tested compounds on tubulin disassembly.Actin Polymerization Biochem KitTM (Cytoskeleton, Inc.) was used to determine the influence of the tested compounds on actin polymerization in vitro. The method is based on the enhanced fluorescence occurring at pyrene F-actin formation from monomer pyrene G-actin. The assay was performed following the manufactureŕs protocol. Pyrene actin (0.4 mg mL−1) was polymerized in actin polymerization buffer in microplate format at room temperature on SPARKTM multimode reader (Tecan) (1 reading per minute, λex = 360 nm and λem = 410 nm). The influence of the tested compounds on F-actin depolymerization was determined following the F-actin formation (1 h, 25 °C in Actin polymerization buffer).
Microtubule and actin imaging
:500), washed and incubated with secondary antibody (Goat Anti-Rabbit-Alexa Fluor® 488, Abcam, 1:500). Following washing, the cells were mounted with ProLongTM Diamond Antifade with DAPI (Invitrogen). Imaging was performed using a Leica TCS SP8 SMD (Leica microsystems GmbH, Wetzlar, Germany). The images were sequentially scanned with first scan 405 nm (diode laser)/emission window 420 to 550 nm, second scan 488 nm (white laser)/emission window 520 to 600 (nm). Pinhole was set to 1 AU.
Author contributions
Conceptualization – V. B., N. J. R., P. S.; methodology – J. P. C. C., V. B., N. J. R.; validation – J. P. C. C., H. K., L. M., H. S., M. P., H. E. B., V. B., N. J. R., P. S.; investigation – J. P. C. C., H. K., L. M., H. S., M. P., H. E. B., N. J. R.; writing – original draft – J. P. C. C., H. K., L. M., V. B., N. J. R., P. S.; writing – review & editing J. P. C. C., V. B., N. J. R, P. S.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Warwick collegues Dr Alessio Inchingolo and Prof. Andrew McAinsh for their assistance with preliminary tubulin binding experiments, and Elizabeth Bolitho for assistance with antiproliferative and cell accumulation assays. The work of H. K., L. M., and V. B. was supported by the Czech Science Foundation [21-27514S].References
- S. E. Howson, A. Bolhuis, V. Brabec, G. J. Clarkson, J. Malina, A. Rodger and P. Scott, Nat. Chem., 2012, 4, 31–36 CrossRef CAS PubMed.
- V. Brabec, S. E. Howson, R. A. Kaner, R. M. Lord, J. Malina, R. M. Phillips, Q. M. A. Abdallah, P. C. McGowan, A. Rodger and P. Scott, Chem. Sci., 2013, 4, 4407–4416 RSC.
- S. E. Howson, G. J. Clarkson, A. D. Faulkner, R. A. Kaner, M. J. Whitmore and P. Scott, Dalton Trans., 2013, 42, 14967–14981 RSC.
- A. D. Faulkner, R. A. Kaner, Q. M. Abdallah, G. Clarkson, D. J. Fox, P. Gurnani, S. E. Howson, R. M. Phillips, D. I. Roper, D. H. Simpson and P. Scott, Nat. Chem., 2014, 6, 797 CrossRef CAS PubMed.
- R. A. Kaner, S. J. Allison, A. D. Faulkner, R. M. Phillips, D. I. Roper, S. L. Shepherd, D. H. Simpson, N. R. Waterfield and P. Scott, Chem. Sci., 2016, 7, 951–958 RSC.
- H. Song, N. J. Rogers, V. Brabec, G. J. Clarkson, J. P. C. Coverdale, H. Kostrhunova, R. M. Phillips, M. Postings, S. L. Shepherd and P. Scott, Chem. Commun., 2020, 56, 6392–6395 RSC.
- S. E. Howson, L. E. N. Allan, N. P. Chmel, G. J. Clarkson, R. J. Deeth, A. D. Faulkner, D. H. Simpson and P. Scott, Dalton Trans., 2011, 40, 10416–10433 RSC.
- D. H. Simpson, A. Hapeshi, N. J. Rogers, V. Brabec, G. J. Clarkson, D. J. Fox, O. Hrabina, G. L. Kay, A. K. King, J. Malina, A. D. Millard, J. Moat, D. I. Roper, H. Song, N. R. Waterfield and P. Scott, Chem. Sci., 2019, 10, 9708–9720 RSC.
- J. Malina, P. Scott and V. Brabec, Nucleic Acids Res., 2015, 43, 5297–5306 CrossRef CAS PubMed.
- M. Li, S. E. Howson, K. Dong, N. Gao, J. Ren, P. Scott and X. Qu, J. Am. Chem. Soc., 2014, 136, 11655–11663 CrossRef CAS PubMed.
- A. D. Zhao, S. E. Howson, C. Q. Zhao, J. S. Ren, P. Scott, C. Y. Wang and X. G. Qu, Nucleic Acids Res., 2017, 45, 5026–5035 CrossRef CAS PubMed.
- D. E. Mitchell, G. Clarkson, D. J. Fox, R. A. Vipond, P. Scott and M. I. Gibson, J. Am. Chem. Soc., 2017, 139, 9835–9838 CrossRef CAS PubMed.
- Y. Guan, Z. Du, N. Gao, Y. Cao, X. Wang, P. Scott, H. Song, J. Ren and X. Qu, Sci. Adv., 2018, 4, eaao6718 CrossRef PubMed.
- C. Zhao, H. Song, P. Scott, A. Zhao, H. Tateishi-Karimata, N. Sugimoto, J. Ren and X. Qu, Angew. Chem., Int. Ed., 2018, 57, 15723–15727 CrossRef CAS PubMed.
- H. Song, M. Postings, P. Scott and N. J. Rogers, Chem. Sci., 2021, 12, 1620–1631 RSC.
- H. Song, N. J. Rogers, S. J. Allison, V. Brabec, H. Bridgewater, H. Kostrhunova, L. Markova, R. M. Phillips, E. C. Pinder, S. L. Shepherd, L. S. Young, J. Zajac and P. Scott, Chem. Sci., 2019, 10, 8547–8557 RSC.
- H. Song, S. J. Allison, V. Brabec, H. E. Bridgewater, J. Kasparkova, H. Kostrhunova, V. Novohradsky, R. M. Phillips, J. Pracharova, N. J. Rogers, S. L. Shepherd and P. Scott, Angew. Chem., Int. Ed., 2020, 59, 14677–14685 CrossRef CAS PubMed.
- Z. Du, C. Liu, Z. Liu, H. Song, P. Scott, X. Du, J. Ren and X. Qu, Chem. Sci., 2023, 14, 506–513 RSC.
- A. Ballesta, F. Billy, J. P. C. Coverdale, J.-I. Song, C. Sanchez-Cano, I. Romero-Canelón and P. J. Sadler, Metallomics, 2019, 11, 1648–1656 CrossRef CAS PubMed.
- L. Di, P. Artursson, A. Avdeef, G. F. Ecker, B. Faller, H. Fischer, J. B. Houston, M. Kansy, E. H. Kerns, S. D. Krämer, H. Lennernäs and K. Sugano, Drug Discovery Today, 2012, 17, 905–912 CrossRef CAS PubMed.
- D. B. Kell, P. D. Dobson, E. Bilsland and S. G. Oliver, Drug Discovery Today, 2013, 18, 218–239 CrossRef CAS PubMed.
- D.-W. Shen, S. Goldenberg, I. Pastan and M. M. Gottesman, J. Cell. Physiol., 2000, 183, 108–116 CrossRef CAS PubMed.
- J. Thomas, L. Wang, R. E. Clark and M. Pirmohamed, Blood, 2004, 104, 3739–3745 CrossRef CAS PubMed.
- J. Hu, W. G. Cochrane, A. X. Jones, D. G. Blackmond and B. M. Paegel, Nat. Chem., 2021, 13, 786–791 CrossRef CAS PubMed.
- J. Malina, M. J. Hannon and V. Brabec, Nucleic Acids Res., 2008, 36, 3630–3638 CrossRef CAS PubMed.
- I. Meistermann, V. Moreno, M. J. Prieto, E. Moldrheim, E. Sletten, S. Khalid, P. M. Rodger, J. C. Peberdy, C. J. Isaac, A. Rodger and M. J. Hannon, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5069–5074 CrossRef CAS PubMed.
- J. Malina, H. Kostrhunova, V. Novohradsky, P. Scott and V. Brabec, Nucleic Acids Res., 2022, 50, 674–683 CrossRef CAS PubMed.
- S. E. Howson, A. Bolhuis, V. Brabec, G. J. Clarkson, J. Malina, A. Rodger and P. Scott, Nat. Chem., 2012, 4, 31–36 CrossRef CAS PubMed.
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS PubMed.
- J. A. May, H. Ratan, J. R. Glenn, W. Losche, P. Spangenberg and S. Heptinstall, Platelets, 1998, 9, 227–232 CrossRef CAS PubMed.
- J. A. Cooper, J. Cell Biol., 1987, 105, 1473–1478 CrossRef CAS PubMed.
- T.-C. Chou, Synergy, 2014, 1, 3–21 CrossRef.
- G. R. Zimmermann, J. Lehár and C. T. Keith, Drug Discovery Today, 2007, 12, 34–42 CrossRef CAS PubMed.
- V. Doldán-Martelli and D. G. Míguez, PLoS One, 2015, 10, e0117558 CrossRef PubMed.
- Y.-W. Heng and C.-G. Koh, Int. J. Biochem. Cell Biol., 2010, 42, 1622–1633 CrossRef CAS PubMed.
- C. H. Chang, F. Y. Yu, T. S. Wu, L. T. Wang and B. H. Liu, Toxicol. Sci., 2011, 119, 84–92 CrossRef CAS PubMed.
- A. L. Risinger, F. J. Giles and S. L. Mooberry, Cancer Treat. Rev., 2009, 35, 255–261 CrossRef CAS PubMed.
- H. Kostrhunova, J. Zajac, V. Novohradsky, J. Kasparkova, J. Malina, J. R. Aldrich-Wright, E. Petruzzella, R. Sirota, D. Gibson and V. Brabec, J. Med. Chem., 2019, 62, 5176–5190 CrossRef CAS PubMed.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation, antiproliferative activities and metallohelix accumulation and distribution in cells, DNA and tubulin biophysical experiments. See DOI: https://doi.org/10.1039/d3dt00948c |
| ‡ While it is difficult to quantitatively deconvolute the effects of metallohelix binding on DNA conformation from these trends, we note that any concentration-based increase in a metallohelix band is being significantly offset by the accompanying loss of linearity in DNA i.e. a general reduction in LD signal. In other words, we underestimate increases in any band. |
| This journal is © The Royal Society of Chemistry 2023 |