
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivation of the N2 molecule by means of low-valence complexes of calcium and magnesium†
Anna
Rovaletti
 *a,
Luca
De Gioia
b,
Claudio
Greco
a and
Federica
Arrigoni
*b
*a,
Luca
De Gioia
b,
Claudio
Greco
a and
Federica
Arrigoni
*b
aDepartment of Earth and Environmental Sciences, Milano-Bicocca University, Piazza della Scienza 1, Milano, Italy. E-mail: anna.rovaletti@unimib.it
bDepartment of Biotechnology and Biosciences, Milano-Bicocca University, Piazza della Scienza 2, Milano, Italy. E-mail: federica.arrigoni@unimib.it
First published on 12th May 2023
Abstract
Nitrogen gas is a highly inert molecule and its activation under mild conditions represents a crucial goal in current research. In a recent study, the discovery of low-valence Ca(I) compounds capable of coordinating and reducing N2 was reported [B. Rösch, T. X. Gentner, J. Langer, C. Färber, J. Eyselein, L. Zhao, C. Ding, G. Frenking and S. Harder, Science, 2021, 371, 1125]. The study of low-valence alkaline earth complexes represents a new horizon in inorganic chemistry and demonstrates examples of spectacular reactivity. For example, complexes of the [BDI]2Mg2 type are selective reducing reagents in both organic and inorganic synthesis reactions. To date, however, no activity of Mg(I) complexes in the activation of the nitrogen molecule has been reported. By computational studies, in the present work, we investigated the analogies and differences of low-valence Ca(I) and Mg(I) complexes in the coordination, activation and protonation of N2. We have shown that the possibility of alkaline earth metals to employ atomic orbitals of the d type is reflected in the differences in the N2 binding energy and its coordination mode (end-on vs. side-on), as well as in the spin state of the resulting adduct (singlet vs. triplet). These divergences are finally observed in the subsequent protonation reaction, which turned out to be prohibitive in the presence of Mg.
1. Introduction
Dinitrogen (N2) is one of the most abundant molecules on our planet, forming almost 78% of the Earth's atmosphere, but at the same time, it is also one of the most difficult gases to exploit in chemical processes.1 Indeed, the strong triple bond, the low proton and electron affinity, the absence of polarisation and the high HOMO–LUMO gap (10.82 eV) govern the scarce reactivity of N2 and pose serious challenges for its industrial and biological conversion. The continuous growth in ammonia demand from humanity is industrially fulfilled by the Haber–Bosch process, which requires harsh conditions (400–600 °C/200–600 bar) to overcome N2 inertness.2 Besides being energy-intensive, the Haber–Bosch reaction uses H2 as a reductant which, in turn, is mainly produced via the unsustainable steam reforming of natural gas. Biological N2 fixation into bio-available ammonia is instead performed by metalloenzymes, known as nitrogenases, that couple protons and electrons for substrate reduction and that need considerable amounts of ATP to work.3,4Although the well-established and productive Haber–Bosch process is unlikely to be replaced in the near future, the fact that both industry and nature rely on d-block metals as catalysts for the N2 reduction reaction (N2RR) has triggered the development of alternative strategies based on transition metals, starting in the mid-1960s.5–8 The main reason is that ammonia is fundamental to sustaining human life and society's progress, so discovering the requirements for efficient N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N splitting under mild conditions and with minimal energy input is a primary goal for chemists. Furthermore, besides the “conventional” use of NH3, it has recently emerged as a promising green fuel for fuel cells, pushing forward academic and industrial interest in its fully sustainable production.9,10
N splitting under mild conditions and with minimal energy input is a primary goal for chemists. Furthermore, besides the “conventional” use of NH3, it has recently emerged as a promising green fuel for fuel cells, pushing forward academic and industrial interest in its fully sustainable production.9,10
Examples of homogeneous systems based on d-block metals for the N2RR have increased over the past decades, with crucial contributions by the groups of Chatt,11–13 Hidai,14 Schrock,15–20 Peters,21,22,23–25 Nishibayashi,26–30 Ashley31 and others.5,32,33 The partially filled d orbitals of transition metals are suited in terms of both energy and symmetry to weaken the triple bond of N2, although the latter is notably a poorly coordinating molecule. Interestingly, it has also been found that transition metals are not indispensable for N2 activation, since it can also be accomplished by f-block elements (e.g. La and Ce),34,35 by the p-block half-metal B,36–38 and by the s-block alkali metal (Li)39,40 and alkaline earth (AE) metals (Be, Ca, Sr, Ba).41–46 Activation of N2 by AE metals, in particular, has experienced huge progress in recent years thanks to several investigations performed under matrix isolation conditions. Zhou, Frenking and collaborators reported different Be-dinitrogen complexes, including the first example of the N2 side-on adduct for main group elements.42 They also isolated and spectroscopically characterized octahedral complexes of AE metals of the types M(CO)8 and M(N2)8 (M = Ca, Sr, and Ba).44,47 Here, an (n − 1)d2(n)s0(n)p0 configuration has been assigned to M, which fulfills the 18 electron rule upon coordination, behaving as a d-block element. The ability of heavier alkaline earth atoms (Ca, Sr and Ba) to employ their (n − 1)d atomic orbitals in covalent interactions has already been suggested by earlier theoretical investigations which showed how these orbitals lie close in energy to the valence np orbitals, opposite to the lighter atoms Be and Mg.48,49 Later, Harder, Frenking and coworkers showed that low valent Ca(I) is also capable of reducing N2 to give N2H2 (which then disproportionates to give 0.5N2 and 0.5N2H4), although only under very peculiar conditions.45 In particular, starting from a stable binuclear Ca(I) iodide precursor (1Ca, Fig. 1), featuring one very bulky β-diketiminate ligand (BDI = HCC(Me)N[2,6-3-pentyl-phenyl]2) per Ca(I), it was possible to serendipitously bind and activate N2 at −60 °C by (i) treatment with K/KI as the reducing agent, (ii) using an aliphatic solvent and (iii) adding tetrahydropyran (THP) or tetrahydrofuran (THF) to the solution. In this way, the crystal structures of both [(BDI)Ca(THP)]2N2 (2Ca–N2) and [(BDI)Ca(THF)]2N2 (3Ca–N2) (Fig. 1) were obtained, revealing the side-on coordination of N2, which is bound in a sandwich fashion between the two Ca atoms. DFT calculations confirmed that N2 is doubly reduced, as N22−, with each Ca(I) contributing one electron to its reduction. Interestingly, heating up 3Ca–N2 (but not 2Ca–N2) to 60 °C led to the degradation of coordinated THF molecules, i.e. the transfer of two protons from one of the OCH2 units of each THF to both N atoms of the activated N22−, resulting in N2H2 formation. All the above-described processes, if carried out in aromatic solvents such as benzene or p-xylene, lead to solvent reduction (instead of the N2 one) by their fitting between the two Ca(I) atoms. Interestingly, the same behaviour towards aromatic molecules has been previously observed for the highly reducing Mg(I) analogue [(BDI)Mg]2 (1Mg).50,51 The latter shows two important differences with respect to the Ca counterpart: (i) 1Mg features a stable and well-characterised Mg(I)–Mg(I) bond,52 while the same Ca(I)–Ca(I) interaction (as in 2Ca, Fig. 1) has not been detected, so it should form only transiently53,54 and (ii) 1Mg does not bind or reduce N2.45,55 This suggests that apparently, tiny differences among the electronic structures of the AE metals (such as between closely related Ca and Mg) may result in very different reactivities. Previous theoretical investigations indicated that in the cases of Ca, Sr and Ba, back-donation from their d orbitals to the empty π* ones of N2 is crucial for the coordination of the latter, while in the case of Be, back-donation to N2 has been ascribed to the metal p orbitals.46 Although all these elements can bind N2, only Ca(I) can effectively reduce it.
 | ||
| Fig. 1 Schematic representation of different monovalent Ca and Mg species. Species 1Ca and 1Mg have been experimentally characterized while 2Ca and 3Ca are only postulated. | ||
Furthermore, it is intriguing and puzzling at the same time that no Mg–N2 interaction has ever been detected or characterised, and so the nature of such interaction also remains completely unexplored from a theoretical standpoint. In this scenario, this work aims at shedding light on the differences between low-valent Ca(I) and Mg(I) BDI complexes in terms of their electronic structure and reactivity towards N2. In particular, we present a detailed characterisation of the N2 binding mode and energy for different M-BDI complexes (M = Ca(I), Mg(I)), accounting for the presence of both THF and THP. Then, we analysed the different Ca vs. Mg orbital contributions to the bond with N2 and investigated the effect of the two AE metals on the subsequent N2 protonation step by THF/THP degradation. Overall, our contribution will generate new knowledge on the role of different AEs in N2 activation and reduction, hopefully providing new hints for future advancements in the use of AE-based catalysts for the N2RR.
2. Results and discussion
2.1. N2 coordination to the low-valence Ca and Mg complexes
The [(BDI)Ca(THP)]2N2 and [(BDI)Ca(THF)]2N2 species (2Ca–N2 and 3Ca–N2, Fig. 1) had been crystallised and analysed through X-ray analysis (CCDC 2036242 and 2036243) in a previous experimental–theoretical work.45 We compared the two geometries stored in the CCDC database with those obtained through geometry optimisation calculations at the BP86-D3(BJ)/def2-SVP level of theory. The chosen level of theory turned out to be eligible for the description of the geometric parameters of the centrosymmetric alkaline earth metal structures. Concerning the atomic distances of the first coordination sphere of Ca(I), the maximum deviation observed is 0.02 Å for the Ca(I)–O(THP) distance in 2Ca–N2 and 0.03 Å for the Ca(I)–N(BDI) distance in 3Ca–N2 (see Fig. 2). A larger basis set – def2-TZVP – for the description of the calcium atoms and of their first coordination sphere was tested; however, the bond distance analysis showed that the use of a broader basis set gives similar results with respect to the use of a smaller def2-SVP one (see Table S1†). | ||
| Fig. 2 Optimized structures of 2Ca–N2 and 3Ca–N2. Most significant calculated and experimental (in brackets) geometry parameters are reported below each species. Colour pattern: grey, C; blue, N; red, O; green, Ca. Hydrogen atoms are omitted for clarity. | ||
In the optimised LCa(N2)CaL species (Fig. 2), the nitrogen molecule bound between the two calcium atoms shows an elongated N–N bond (1.26 Å for both 2Ca–N2 and 3Ca–N2), corresponding to the strong activation of N2.56
The natural bond orbital (NBO) analysis was carried out to study the electronic structures of the LCa(N2)CaL species. Natural atomic orbital occupancies and natural atomic charges (Table 1) indicate that the side-on bridging N2 unit is activated to an N22− anion which interacts with two Ca2+ cations. The accumulation of negative charge on the activated N2 molecule and the concomitant oxidation of Ca ions were previously observed by means of different population analysis schemes – e.g. NBO analysis and quantum theory of atoms in molecules (QTAIM) – in the Ca2N2 system.45 The resulting Ca–N(N2) distances are all equal to 2.29 Å in the presence of THF while each Ca ion lies at a distance of 2.32 and 2.30 Å from each nitrogen atom of the N2 molecule in the presence of THP, respectively (see Fig. 2).
| 2 Ca–N2 | 3 Ca–N2 | e32Mg–N2 | e33Mg–N2 | ||
|---|---|---|---|---|---|
| Ca1 | 1.74 (18) | 1.74 (18) | Mg1 | 1.80 (10) | 1.80 (10) |
| Ca2 | 1.74 (18) | 1.74 (18) | Mg1 | 1.80 (10) | 1.80 (10) |
| N1 | −0.82 (8) | −0.82 (8) | N1 | −0.83 (8) | −0.83 (8) |
| N2 | −0.82 (8) | −0.83 (8) | N1 | −0.83 (8) | −0.83 (8) |
Despite the observed structural similarity of the complexes containing THP or THF, the two compounds have experimentally shown different stability over time. In fact, while at room temperature the THF complex decomposes overnight, the THP analogue remains stable for 24 hours before, also decomposing through the evolution of H2 and N2. The difference in stability observed experimentally can be correlated to the bond strength established between the calcium ions and the dinitrogen molecule in the two complexes. The binding energy of the N2 molecule to the neutral fragment [(BDI)Ca(THP)]2 or [(BDI)Ca(THF)]2 (2Ca and 3Ca in Fig. 1) was calculated at the B3LYP-D3(BJ)/def2-TZVPP level of theory considering the species solvated in a continuum solvent. We will refer exclusively to relative binding energies (ΔΔE) for two main reasons: (i) DFT generally performs poorly in calculating absolute energies for ligand association to large metal complexes57 and (ii) the focus of the present work is to highlight eventual differences among the considered compounds in their reactivity towards N2. In the case of Ca–N2 adducts (2Ca–N2 and 3Ca–N2), we observed that N2 binds more favorably (by 6.7 kcal mol−1) to 2Ca than to 3Ca, indicating that the reaction can be favored in the presence of THP. Triplet states were investigated for the Ca–Ca reagents (2Ca and 3Ca) and the Ca–N2 products ((2Ca–N2 and 3Ca–N2) by running optimisation calculations. The results indicate that the singlet state geometries are retained in the triplet states. In any case, single states were found to be the most energetically favourable ones (energy differences between triplet and single states are 10.3 kcal mol−1 and 7.4 kcal mol−1 for 2Ca–N2 and 2Ca species and 9.7 kcal mol−1 and 13.1 kcal mol−1 for 3Ca–N2 and 3Ca species, respectively).
The BDI ligand was reported to be able to stabilise the Mg(I)–Mg(I) bond and it was proposed that Mg(BDI) systems are less reducing than their Ca(BDI) counterparts. Furthermore, Mg(BDI) complexes have not shown (so far) any reactivity towards N2. In order to verify and rationalise this Mg vs. Ca difference, we replaced the Ca(I) with Mg(I) atoms in our models. First, we verified the plausible existence of compounds of the [(BDI)Mg(THP)]2N2 or [(BDI)Mg(THF)]2N2 type (2Mg–N2 and 3Mg–N2 shown in Fig. 3). For each species, we obtained a minimum with a geometry resembling the one obtained for the Ca-containing analogues. The N2 molecule is coordinated between two Mg ions in a side-on manner, with resulting Mg–N(N2) distances of 2.15 Å and 2.18 Å in 2Mg–N2 and 2.12, 2.16 Å and 2.13, 2.15 Å in 3Mg–N2 (see Fig. 4, S1† and Table 2). Analogously to what was observed for the Ca-containing species, the two Mg atoms were oxidised to Mg(II) while the nitrogen molecule was 2-electron reduced (Table S2†). Exploration of alternative spin states led to the localisation of geometries that are significantly more stable in energy. These complexes exhibit a paramagnetic character and the nitrogen molecule is inserted between the two Mg ions in an end-on manner (e32Mg–N2 and e33Mg–N2, Fig. 4 and Fig. S1†). In more detail, these high spin (triplet) complexes with a coordinated end-on N2 molecule are much more stable with respect to the low spin (singlet) ones with a side-on N2 molecule, irrespective of the solvent. Indeed, e32Mg–N2 turned out to be 16.3 and 19.1 kcal mol−1 more stable than 2Mg–N2 and e12Mg–N2, respectively, while e33Mg–N2 is 11.8 and 17.4 kcal mol−1 more stable than 3Mg–N2 and e12Mg–N2, respectively. The end-on bridging mode of N2 shows a lower elongation of the N–N bond than the side-on coordination mode (1.22 Å and 1.23 Å for e32Mg–N2 and e33Mg–N2, respectively). Interestingly, in both e32Mg–N2 and e33Mg–N2 a contraction of the bonds between the magnesium ions and the coordinating atoms from BDI and solvent (N and O atoms) is observed, as evidenced in Table 2. Similarly to the side-on species, the end-on ones exhibit an interaction between two Mg2+ cations and a N22− anion (Table 1).
 | ||
| Fig. 3 Scheme of the N2 coordination compounds formed after insertion between the two AE(I) ions. | ||
 | ||
| Fig. 4 Optimized structures of the characterized N2 coordination modes to 2Mg. Colour pattern: grey, C; blue, N; red, O; magenta, Mg. Hydrogen atoms are omitted for clarity. | ||
| 2 Mg–N2 | 3 Mg–N2 | e32Mg–N2 | e33Mg–N2 | |
|---|---|---|---|---|
| N1–N2 | 1.29 | 1.29 | 1.22 | 1.23 |
| Mg1–N1 | 2.15 | 2.12 | 1.96 | 1.94 |
| Mg1–N2 | 2.18 | 2.16 | — | — |
| Mg2–N1 | 2.18 | 2.15 | — | — |
| Mg2–N2 | 2.15 | 2.13 | 1.96 | 1.94 |
| Mg1–N3 | 2.20 | 2.13 | 2.08 | 2.07 |
| Mg1–N4 | 2.16 | 2.16 | 2.10 | 2.07 |
| Mg2–N5 | 2.20 | 2.13 | 2.08 | 2.06 |
| Mg2–N6 | 2.16 | 2.15 | 2.10 | 2.07 |
| Mg1–O | 2.23 | 2.16 | 2.12 | 2.09 |
| Mg2–O | 2.23 | 2.18 | 2.12 | 2.10 |
In order to evaluate the effect of Ca substitution with Mg on N2 activation, we calculated N2 binding energies for LMg(I)–Mg(I)L systems (2Mg and 3Mg), as previously done for the Ca-complexes. The singlet states of 2Mg and 3Mg turned out to be much more stable with respect to their triplet state counterparts (−17.7 kcal mol−1 and −27.2 kcal mol−1, respectively), therefore they were considered as the reagents in the N2 coordination reaction. Interestingly, also for Mg(I) systems it is observed that N2 binding is favored in THP rather than in THF (by −7.7 kcal mol−1), as evidenced for the Ca(I) complexes.
Although the compounds reported by Rösch et al.45 did not show any paramagnetic character, our results for the Mg compounds motivated us to investigate the formation of a possible end-on coordination mode of N2 to both 2Ca and 3Ca. We found that only 2Ca can coordinate with N2 in an end-on fashion, forming e32Mg–N2, while in the presence of THF (i.e. for 3Ca) this binding mode is not accessible. As observed for Mg systems, e32Ca–N2 is characterised by a triplet ground state, but in this case, it is much more unstable with respect to the low spin side-on complex 2Ca–N2 (by 17.4 kcal mol−1).
Finally, considering the formation of the most stable Ae(I)–N2 adducts for both Mg (end-on coordination, triplet) and Ca (side-on coordination, singlet), it has been found that N2 binding energies are more energetically favoured in the case of Ca(I) than those in Mg(I) complexes. Indeed, considering THP solvent, the formation of 2Ca–N2 is 6.6 kcal mol−1 more favored than that of e32Mg–N2, while in THF, the formation of 3Ca–N2 is 7.6 kcal mol−1 more favored than that of 3Mg–N2.
2.2. Molecular orbital analysis
Given the unexpected differences observed between Mg and Ca compounds in terms of the coordination mode of the nitrogen molecule and the related electronic configuration, we analysed in more detail both AE(I)–AE(I) and AE(I)–N2–AE(I) interactions. As reported above, the AE(I)–AE(I) type compounds proved to be more stable in the singlet ground state for both the metal-alkaline earth elements considered in the present study.As for the Ca compounds, the computed Ca(I)–Ca(I) distance was found to be 3.93 and 3.80 Å for 2Ca and 3Ca, respectively. The molecular orbital analysis of 2Ca and 3Ca indicated that their HOMO orbitals are characterised by a σ-like bond among the two Ca ions (see Fig. 5) that contribute equally to the formation of the Ca–Ca bond by means of their s and p orbitals, as well as with a little contribution from the d orbitals. As reported in Table 3, the percentage contribution to the HOMO of the d orbitals of each Ca is around 10% for both 2Ca and 3Ca. Also in the case of the LMg(I)–Mg(I)L species the dominant interaction is characterised by a sigma bond between the two Mg atoms, in which both metal ions contribute equally almost by means of their s and p orbitals (see Fig. 5) and with a negligible (4%) participation of d orbitals.
 | ||
| Fig. 5 Graphical representation of HOMOs (isosurface cutoff = 0.05 a.u.) of the LCa(I)–Ca(I)L and LMg(I)–Mg(I)L species as well as for their energetically most stable N2 bearing complexes. | ||
| % s | % p | % d | % f | |
|---|---|---|---|---|
| 2 Ca | ||||
| Ca1 | 58.7 | 31.7 | 9.6 | |
| Ca2 | 58.4 | 31.8 | 9.8 | |
| 3 Ca | ||||
| Ca1 | 62.0 | 29.7 | 8.3 | |
| Ca2 | 59.9 | 30.5 | 9.6 | |
| 2 Mg | ||||
| Mg1 | 56.1 | 39.5 | 4.4 | |
| Mg2 | 56.1 | 39.5 | 4.4 | |
| 3 Mg | ||||
| Mg1 | 55.6 | 40.1 | 4.3 | |
| Mg2 | 54.0 | 41.7 | 4.3 | |
| 2 Ca–N2 | ||||
| Ca1 | 6.7 | 3.4 | 89.9 | |
| Ca2 | 5.3 | 4.0 | 90.7 | |
| N1 | 100 | |||
| N2 | 100 | |||
| 3 Ca–N2 | ||||
| Ca1 | 0.9 | 2.9 | 96.2 | |
| Ca2 | 1.7 | 3.3 | 95.0 | |
| N1 | 100 | |||
| N2 | 100 | |||
| e32Mg–N2 | ||||
| Mg1 | ||||
| Mg2 | ||||
| N1 | 96.7 | 3.3 | ||
| N2 | 96.7 | 3.3 | ||
| e33Mg–N2 | ||||
| Mg1 | ||||
| Mg2 | ||||
| N1 | 0.1 | 96.7 | 3.1 | 0.1 |
| N2 | 0.2 | 96.6 | 3.1 | 0.1 |
The HOMO–LUMO gap of 2Ca is 24.2 kcal mol−1 and 6.2 kcal mol−1 smaller than that of 3Ca. Interestingly, the calculated HOMO–LUMO gaps for the LMg(I)–Mg(I)L species are significantly larger (53.3 kcal mol−1 and 58.3 kcal mol−1 for 2Mg and 3Mg, respectively), and this may justify the higher stability of the systems featuring an Mg(I)–Mg(I) bond, with respect to the ones with a (hypothetical) Ca(I)–Ca(I) interaction, as suggested by previous experimental and theoretical investigations.45
We subsequently analysed the shape of the bonding orbitals in the most stable AE(I)–N2–AE(I) compounds as well as the contribution of the AE atomic orbitals to the bonding molecular orbitals. The Ca species containing a μ2-N2 molecule evidence a molecular orbital (HOMO) involving the monovalent ions and the nitrogen atoms of N2, where the latter contribute 85% to the formation of the bond thanks to the sole participation of their p orbitals (see Fig. 5). On the other hand, the two Ca ions intervene symmetrically in the formation of the bond mostly by means of their d orbitals. This contribution exceeds 95% in the case of compound 2Ca–N2 and it is around 90% in compound 3Ca–N2 (see Table 3). Our results are in accordance with what was previously reported for [(BDI)Ca(THF)]2N2 through energy decomposition with natural orbitals for chemical valence (EDA-NOCV) analysis, which showed that the interaction between the calcium complex and N2 essentially relies on Ca(I) d orbitals and N2 p orbitals.45 Interestingly, N2 binding to 2Ca and 3Ca causes an increase in HOMO–LUMO gaps by 17.8 kcal mol−1 in the presence of THP (2Ca–N2) and by 15.8 kcal mol−1 in the presence of THF (3Ca–N2), plausibly explaining why the LCa(N2)CaL species, but not the LCa–CaL ones, have been experimentally isolated and characterized.
The situation is completely different in the case of Mg complexes characterised by an N2 end-on interaction and a triplet ground state. In this case, in fact, there is no molecular orbital formed by the atomic orbitals of both Mg and N. In compounds e32Mg–N2 and e33Mg–N2, HOMO and HOMO−1 are constituted by the p orbitals of the two atoms of the N2 molecule as shown in Fig. 5. This result therefore suggests that the covalent contribution to the AE–N2 bond is larger for Ca(I) than for Mg(I), since the former can participate with its d orbitals in N2 activation (see Table 3).
2.3. N2 protonation reaction
As aforementioned, protonation of the activated N2 molecule has been experimentally observed, and it occurs by proton transfer from one of the OCH2 units of the THF ligand.45 The reaction leads to the formation of diazene (N2H2) and elimination products such as ethylene and enolate. Subsequently, diazene disproportionates at temperatures lower than −150 °C in N2 and hydrazine (N2H4). In 3Ca–N2, the protonation of N22− by one THF ligand begins with the formation of a hydrogen bond between one nitrogen of the N2 molecule and one of the two alpha protons of the spatially closest THF OCH2 unit. A similar mechanism can be envisioned for the THP counterpart which, although reported to be more stable than 3Ca–N2, decomposes unselectively at elevated temperatures. In order to highlight the effect of the Ca to Mg substitution on N22− protonation properties, we calculated the associated reaction mechanism in both cases.We started analysing the reactivity of compound 3Ca–N2, which was previously investigated through theoretical investigations using a simpler model and the pure functional BP86.45 In 3Ca–N2 the N(N2)–H(OCH2) distance is 2.67 Å while the C–H distance is 1.11 Å. As the reaction occurs, the N(N2)–H(OCH2) distance shortens up to 1.10 Å in the final product 3PCa, while the C–H distance elongates to 2.01 Å. The reaction energy was found to be 32.4 kcal mol−1, with an associated energy barrier of 36.5 kcal mol−1. The transition state structure is characterised by a geometry that is very similar to 3PCa (N(N2)–H(OCH2) = 1.25 Å, C–H = 1.57 Å, see Fig. S2†). These results are consistent with what is reported in the literature – i.e. a reaction energy of 29.0 kcal mol−1 with an activation energy of 32.1 kcal mol−1. As expected, considering 2Ca–N2, we individuated a similar reaction path. In this case, the reaction turned out to be almost isoenergetic to that of 3Ca–N2 (reaction energy equal to 30.7 kcal mol−1) but with a higher transition state energy (activation energy equal to 38.8 kcal mol−1). The starting N(N2)–H(OCH2) distance in 2Ca–N2 is 2.76 Å, which shortens to 1.08 Å in the product 2PCa, while the initial C–H distance of 1.11 Å becomes 2.35 Å in 2PCa. As for the THF case, the transition state 2TS geometry is close to the final product with an N(N2)–H(OCH2) distance of 1.28 Å and a C–H distance of 1.62 Å. In both cases, as the solvent ligand becomes deprotonated, its interaction mode with the Ca ion changes. As previously described, in the initial coordination, the oxygen atom of the solvent molecule is pointed towards the metal ions, with a corresponding distance of 2.39 Å and 2.38 Å for 2Ca–N2 and 3Ca–N2, respectively. However, once the molecule has been deprotonated by the N22− unit, the resulting electron-rich C atom approaches (Ca–C distance equals 3.42 Å and 3.39 Å in 2Ca–N2 and 3Ca–N2, respectively; 2.54 Å and 2.56 Å in 2PCa and 3PCa, respectively).
Then, we investigated the reactivity of the Mg systems, namely e32Mg–N2 and e33Mg–N2, towards N2 protonation. As previously reported, no reactivity toward N2 reduction from Mg(I) compounds has ever been reported so far. The research for a protonation product of the N2 molecule by one THP/THF ligand led to the identification of an intermediate species in both cases that only exists in the singlet state (e12IMg/e13IMg, shown in Fig. S3†) and in which the oxygen atom of THP/THF still interacts with Mg. In more detail, in e12IMg both the oxygen and deprotonated carbon atoms coordinate to the Mg ion (Mg–C(THP) = 2.20 Å, Mg–O(THP) = 2.24 Å), in analogy to the Ca-based systems, while in e13IMg only the oxygen atom of THF binds to the metal (Mg–O(THF) = 2.02 Å). Such an intermediate structure is rather unstable for both THP and THF systems (38.9 and 41.6 kcal mol−1 less stable than the reagents e32Mg–N2 and e33Mg–N2, respectively, see Fig. 6). So both e12IMg and e13IMg can evolve to a more stable product (e12PMg and e13PMg, shown in Fig. 6) in which only the deprotonated carbon atom of THP/THF coordinates with the Mg ion (Mg–C(THP) = 2.20 Å, Mg–O(THP) = 2.83 Å; Mg–C(THF) = 2.18 Å, Mg–O(THF) = 2.82). In this product, the proton of the newly formed NNH− unit points opposite to the proton-donating solvent molecule, with a C(THP)–H(NNH) distance of 4.42 Å in e12PMg and a C(THF)–H(NNH) distance of 4.57 Å in e13PMg This species is represented by a singlet ground state, which turned out to be 12–13 kcal mol−1 more stable than its triplet state counterpart (see Fig. S4†). The overall protonation reaction to form e12PMg or e13PMg is 26.8 and 32.2 kcal mol−1 endergonic, respectively. Overall, these results indicate that the reaction coordinate associated with N2 protonation for the Mg-containing systems is more complicated and more energy-demanding than for their Ca counterparts. Indeed, in Mg systems, on scanning the triplet potential energy surface along the reaction coordinate for the solvent-to-N2 proton transfer, it can be assumed that the reaction requires an activation energy of at least 40 kcal mol−1 in the case of THP and of at least 45 kcal mol−1 in the case of THF. These barriers are substantially higher than those seen for Ca compounds. On the singlet potential energy surface, instead, along the reaction coordinate for each Mg system we find an intermediate (which does not exist in the triplet state) that is 38.9 or 41.6 kcal mol−1 less stable than the reactants for THP and THF, respectively, so for the presence of both THF and THP, the reactions are more unfavorable than for the Ca complexes.
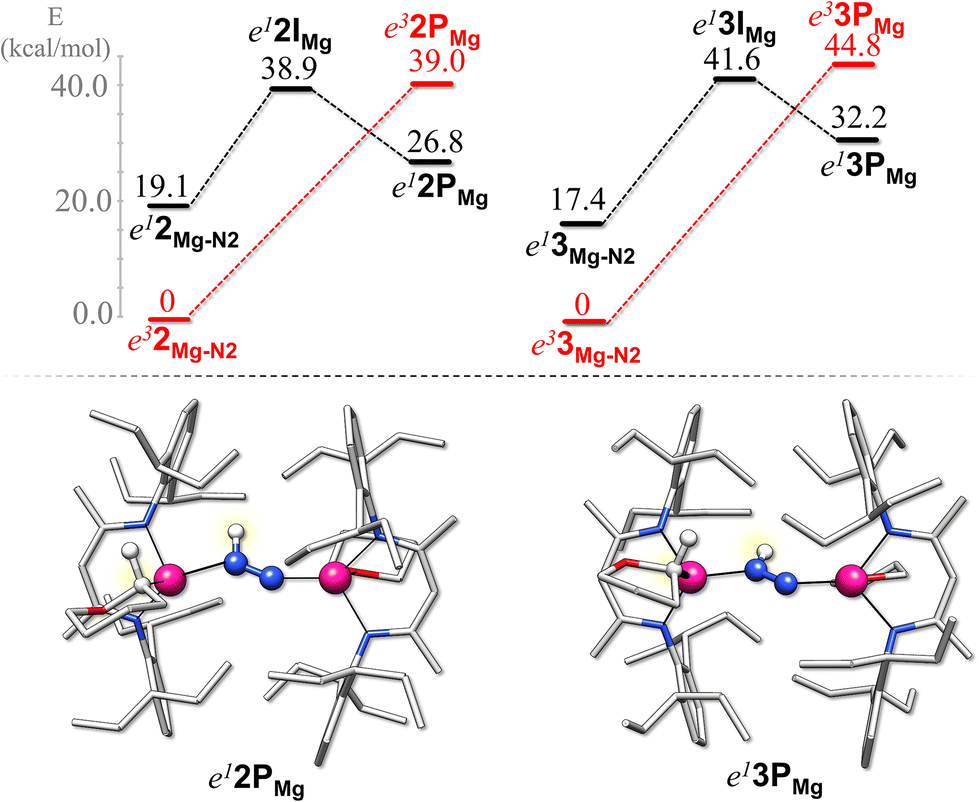 | ||
| Fig. 6 Energy profiles associated with the N2 protonation reaction for e2Mg–N2 and e3Mg–N2, considering both singlet and triplet states. Reaction energies (top) and the final product (bottom) are reported for each species. | ||
3. Conclusion
In the present work we performed a computational analysis of N2 binding and activation by low valent Mg(I) or Ca(I) BDI complexes. The main observations can be summarized as follows:- The N2 coordination mode to the Ca(I) and Mg(I) complexes is substantially different. Ca(I) systems bind N2 in a side-on manner. Here, the nitrogen molecule is equidistant from the two Ca(I) atoms, and the N–N axis is perpendicular to the Ca–Ca one. Instead, in the case of Mg(I) systems, the nitrogen molecule bridges the two metal-alkaline earth atoms with an end-on type configuration, and these species are characterised by a triplet ground state. Interestingly, coupling of the structural properties of end-on bridging nitrogen complexes to the preference of a triplet ground state has already been evidenced in the literature for Mo-containing or Sc-containing compounds.58,59
- N2 binding to Ca(I) systems is predicted to be more energetically preferred than to Mg(I). Molecular orbital analysis allowed the rationalization of this difference, which can be mainly ascribed to the participation of d orbitals in the Ca(I)–N2 interaction, but not in the Mg(I)–N2 one. This suggests a higher covalent character for the AE(I)–N2 interaction in the case of Ca than Mg.
- While in the case of Ca(I) both N2 binding and subsequent protonation occur on the singlet potential energy surface, a marked spin-crossover behavior (singlet–triplet–singlet) has been observed in the case of Mg(I) reactivity. Indeed, a first change in the spin-state (from singlet to triplet) occurs upon N2 binding, and a second one (from triplet to singlet) occurs upon N2 protonation by either THF or THP. Furthermore, we showed that N2 protonation by either THP or THF ligands is energetically prohibited for Mg systems, in contrast to what has been observed for Ca.
Overall, these results added novel insights into the current (limited) knowledge on the role of different AE metals in N2 activation/protonation, with potential implications for future innovative AE metal-based N2RR strategies. The present work is specific and limited to the Mg vs. Ca comparison, but upcoming investigations on other low valent alkaline earth metals (such as Sr and Ba) would help to further rationalize the relationship between the electronic structures of AE metals and their reactivity towards the activation and conversion of recalcitrant small molecules. Moreover, compounds of the Mg(0) type as well as low-valent magnesium–calcium complexes will be the object of future theoretical investigations since these systems with BDI seem to be able to activate inert molecules.60,61
4. Computational methods
Calculations were performed within the framework of density functional theory (DFT) with the TURBOMOLE suite of programs,62 and in particular with the 7.1 version of such software. Geometry optimisations were obtained using the BP8663,64 functional together with the def2-SVP basis set.65 Dispersion correction was taken into account by using the Grimme DFT-D3 method with Becke–Johnson damping (BJ).66 The nature of each optimized structure as the genuine minimum was confirmed by vibrational frequency calculations. As expected (in light of the large size and complexity of the molecular systems considered), some spurious imaginary vibrations, close to or below 100i cm−1, occurred. These soft modes (all corresponding to the torsional motions of the terminal methyl groups belonging to BDI ligands) are products of inaccuracies derived from numerical integration procedures employed in actual DFT methods and were thus ignored.67 Solvent effects were included by running single-point calculations on the optimised models soaked in the COSMO continuum solvent representation68 at the dielectric constant of the apolar THF solvent (ε = 7.4257). The energetics reported in the main text were refined at the B3LYP-D3(BJ)/def2-TZVPP level.69,70 For all the optimised models, atomic charges were computed by means of the natural population analysis approach.71 Orbital visualizations were produced using Chimera.72Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The computations were performed on computer resources provided by CINECA as part of the agreement with the University of Milano-Bicocca.References
- H.-P. Jia and E. A. Quadrelli, Chem. Soc. Rev., 2014, 43, 547–564 RSC.
- A. J. Martín, T. Shinagawa and J. Pérez-Ramírez, Chem, 2019, 5, 263–283 Search PubMed.
- S. F. McWilliams and P. L. Holland, Acc. Chem. Res., 2015, 48, 2059–2065 CrossRef CAS PubMed.
- L. C. Seefeldt, Z.-Y. Yang, D. A. Lukoyanov, D. F. Harris, D. R. Dean, S. Raugei and B. M. Hoffman, Chem. Rev., 2020, 120, 5082–5106 CrossRef CAS PubMed.
- Y. Roux, C. Duboc and M. Gennari, ChemPhysChem, 2017, 18, 2606–2617 CrossRef CAS PubMed.
- R. R. Schrock, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17087–17087 CrossRef CAS PubMed.
- A. Allen and C. Senoff, Chem. Commun., 1965, 621–622 RSC.
- A. D. Allen, F. Bottomley, R. O. Harris, V. Reinsalu and C. Senoff, J. Am. Chem. Soc., 1967, 89, 5595–5599 CrossRef CAS.
- G. Jeerh, M. Zhang and S. Tao, J. Mater. Chem. A, 2021, 9, 727–752 RSC.
- F. Du, W. Sun, H. Luo and C. M. Li, Mater. Rep.: Energy, 2022, 100163 CAS.
- J. Chatt, A. Pearman and R. Richards, Nature, 1975, 253, 39–40 CrossRef CAS.
- J. Chatt, A. J. Pearman and R. L. Richards, J. Chem. Soc., Dalton Trans., 1977, 1852–1860 RSC.
- J. Chatt, J. R. Dilworth and R. L. Richards, Chem. Rev., 1978, 78, 589–625 CrossRef CAS.
- M. Hidai, K. Tominari, Y. Uchida and A. Misono, J. Chem. Soc. D, 1969, 1392–1392 RSC.
- D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76–78 CrossRef CAS PubMed.
- N. C. Smythe, R. R. Schrock, P. Müller and W. W. Weare, Inorg. Chem., 2006, 45, 7111–7118 CrossRef CAS PubMed.
- W. W. Weare, R. R. Schrock, A. S. Hock and P. Müller, Inorg. Chem., 2006, 45, 9185–9196 CrossRef CAS PubMed.
- R. R. Schrock, Acc. Chem. Res., 2005, 38, 955–962 CrossRef CAS PubMed.
- R. R. Schrock, Catalysis Without Precious Metals, 2010, pp. 25–50 Search PubMed.
- L. A. Wickramasinghe, T. Ogawa, R. R. Schrock and P. Muller, J. Am. Chem. Soc., 2017, 139, 9132–9135 CrossRef CAS PubMed.
- M. J. Chalkley, M. W. Drover and J. C. Peters, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–87 CrossRef CAS PubMed.
- J. Rittle and J. C. Peters, J. Am. Chem. Soc., 2016, 138, 4243–4248 CrossRef CAS PubMed.
- C. M. Johansen, E. A. Boyd and J. C. Peters, Sci. Adv., 2022, 8, eade3510 CrossRef CAS PubMed.
- J. Fajardo Jr. and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 16105–16108 CrossRef PubMed.
- S. Kuriyama, K. Arashiba, H. Tanaka, Y. Matsuo, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Angew. Chem., Int. Ed., 2016, 55, 14291–14295 CrossRef CAS PubMed.
- Y. Nishibayashi, Nitrogen fixation, Springer, 2017, vol. 60 Search PubMed.
- Y. Nishibayashi, Transition Metal-Dinitrogen Complexes: Preparation and Reactivity, John Wiley & Sons, 2019 Search PubMed.
- H. Tanaka, K. Arashiba, S. Kuriyama, A. Sasada, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2014, 5, 1–11 Search PubMed.
- K. Arashiba, E. Kinoshita, S. Kuriyama, A. Eizawa, K. Nakajima, H. Tanaka, K. Yoshizawa and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137, 5666–5669 CrossRef CAS PubMed.
- P. J. Hill, L. R. Doyle, A. D. Crawford, W. K. Myers and A. E. Ashley, J. Am. Chem. Soc., 2016, 138, 13521–13524 CrossRef CAS PubMed.
- K. C. MacLeod and P. L. Holland, Nat. Chem., 2013, 5, 559–565 CrossRef CAS PubMed.
- C. J. Van der Ham, M. T. Koper and D. G. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- A. J. Ryan, S. G. Balasubramani, J. W. Ziller, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2020, 142, 9302–9313 CrossRef CAS PubMed.
- H. Xie, L. Shi, X. Xing and Z. Tang, Phys. Chem. Chem. Phys., 2016, 18, 4444–4450 RSC.
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed.
- M.-A. Légaré, M. Rang, G. Bélanger-Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329–1332 CrossRef PubMed.
- M.-A. Légaré, G. Bélanger-Chabot, M. Rang, R. D. Dewhurst, I. Krummenacher, R. Bertermann and H. Braunschweig, Nat. Chem., 2020, 12, 1076–1080 CrossRef PubMed.
- P. Schenk and G. Brauer, Handbook of Preparative Inorganic Chemistry, 1963, vol. 3 Search PubMed.
- R. C. Spiker Jr., L. Andrews and C. Trindle, J. Am. Chem. Soc., 1972, 94, 2401–2406 CrossRef.
- C. A. Thompson, L. Andrews and R. D. Davy, J. Phys. Chem., 1995, 99, 7913–7924 CrossRef CAS.
- G. Deng, S. Pan, G. Wang, L. Zhao, M. Zhou and G. Frenking, Angew. Chem., Int. Ed., 2020, 59, 10603–10609 CrossRef CAS PubMed.
- G. Deng, S. Pan, G. Wang, L. Zhao, M. Zhou and G. Frenking, Angew. Chem., Int. Ed., 2020, 59, 18201–18207 CrossRef CAS PubMed.
- Q. Wang, S. Pan, S. Lei, J. Jin, G. Deng, G. Wang, L. Zhao, M. Zhou and G. Frenking, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- B. Rösch, T. Gentner, J. Langer, C. Färber, J. Eyselein, L. Zhao, C. Ding, G. Frenking and S. Harder, Science, 2021, 371, 1125–1128 CrossRef PubMed.
- T.-T. Liu, D.-D. Zhai, B.-T. Guan and Z.-J. Shi, Chem. Soc. Rev., 2022, 51, 3846–3861 RSC.
- X. Wu, L. Zhao, J. Jin, S. Pan, W. Li, X. Jin, G. Wang, M. Zhou and G. Frenking, Science, 2018, 361, 912–916 CrossRef CAS PubMed.
- I. Fernández, N. Holzmann and G. Frenking, Chem. – Eur. J., 2020, 26, 14194–14210 CrossRef PubMed.
- M. Zhou and G. Frenking, Acc. Chem. Res., 2021, 54, 3071–3082 CrossRef CAS PubMed.
- C. Jones, Nat. Rev. Chem., 2017, 1, 1–9 CrossRef.
- A. Stasch and C. Jones, Dalton Trans., 2011, 40, 5659–5672 RSC.
- S. P. Green, C. Jones and A. Stasch, Science, 2007, 318, 1754–1757 CrossRef CAS PubMed.
- Y. Xie, H. F. Schaefer III and E. D. Jemmis, Chem. Phys. Lett., 2005, 402, 414–421 CrossRef CAS.
- S. Krieck, L. Yu, M. Reiher and M. Westerhausen, Eur. J. Inorg. Chem., 2010, 2010, 197–216 CrossRef.
- T. X. Gentner, B. Rösch, G. Ballmann, J. Langer, H. Elsen and S. Harder, Angew. Chem., 2019, 131, 617–621 CrossRef.
- F. Studt and F. Tuczek, J. Comput. Chem., 2006, 27, 1278–1291 CrossRef CAS PubMed.
- T. Husch, L. Freitag and M. Reiher, J. Chem. Theory Comput., 2018, 14, 2456–2468 CrossRef CAS PubMed.
- J. J. Curley, T. R. Cook, S. Y. Reece, P. Muller and C. C. Cummins, J. Am. Chem. Soc., 2008, 130, 9394–9405 CrossRef CAS PubMed.
- D. H. Woen, G. P. Chen, J. W. Ziller, T. J. Boyle, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2017, 139, 14861–14864 CrossRef CAS PubMed.
- C. Jones, Nature, 2021, 592, 687–688 CrossRef CAS.
- J. Mai, B. Rösch, N. Patel, J. Langer and S. Harder, Chem. Sci., 2023, 14, 4724–4734 RSC.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 91–100 CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- Y. Xie, H. F. Schaefer and R. B. King, J. Am. Chem. Soc., 2000, 122, 8746–8761 CrossRef CAS.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt00945a |
| This journal is © The Royal Society of Chemistry 2023 |