
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxidation-induced C–H bond activation in iridium pincer complexes†
Alexey V.
Polukeev
 * and
Magdalena
Tasić
* and
Magdalena
Tasić
Centre for Analysis and Synthesis, Department of Chemistry, Lund University, P.O. Box 124, SE-221 00 Lund, Sweden. E-mail: alexey.polukeev@chem.lu.se
First published on 9th May 2023
Abstract
Dehydrogenation reactions that produce molecular hydrogen are thermodynamically unfavourable. Desired is to couple them with a green driving force, such as oxidation with oxygen or an electric current. This, in turn, requires understanding of the catalyst's redox properties. Here we report oxidation of the iridium pincer complexes (POCOP)IrHCl (POCOP = 2,6-(tBu2PO)2C6H3; 1a) and (PCP)IrHCl (PCP = 2,6-(tBu2PCH2)2C6H3; 1c) that induced intramolecular C–H activation, followed by the formation of complexes with a cyclometallated tert-butyl group. Based on an electrochemical study and DFT calculations, we propose a mechanism that involves H+ loss from hydrochlorides 1a and 1c to give a highly reactive (pincer)IrCl+ compound.
Introduction
Enabling the selective, large-scale functionalization of C–H bonds has long been a tantalizing goal since it may allow for the efficient conversion of the most abundantly available carbon-based feedstocks, alkanes, into more valuable commodity chemicals and raw materials for the chemical industry.1 Much of the progress in the area is associated with iridium pincer complexes,2,3 the most efficient catalysts for alkane dehydrogenation, a reaction that converts alkanes into much more reactive olefins. One of the fundamental problems of homogeneous alkane dehydrogenation is the endothermicity of the reaction, which typically requires the presence of a sacrificial hydrogen acceptor. The same often applies to related cross-dehydrogenative coupling reactions.4 To avoid the use of a sacrificial hydrogen acceptor, attempts were made to couple dehydrogenation with secondary reactions.1,3 In this respect, there is a growing interest in using oxidation, especially with molecular oxygen, as a driving force for dehydrogenation or C–H activation in general.5 Attractive is also to use electric current as an oxidant,6 thus performing a sort of catalytic electrolysis of alkanes.While there are some successes in iridium promoted C–H bond oxidations,7 primarily utilizing a Cp*Ir fragment, redox properties and reactions of iridium pincer-ligated complexes only recently attracted the attention of researchers. Thus, it was shown that (Phebox)Ir(OAc)2(H2O) (Phebox = bis(oxazolinyl)phenyl− NCN pincer ligand) can stoichiometrically activate C–H bonds8,9via a concerted metallation deprotonation pathway;10 after reaction, this complex can be regenerated by O2.11 Conditions required for these two processes are dramatically different though, which precludes the organization of a catalytic cycle.11 With a milder oxidant, Ag2O, some conversion of mesitylene to 3,5-dimethylbenzaldehyde and 3,5-dimethylbenzoic acid by (Phebox)Ir(OAc)2(H2O) and (Phebox)Ir(OCOCF3)2(H2O) was achieved.12 Electrochemical and chemical hydride loss from (PCP)IrH2 (PCP = 2,6-(CH2PtBu2)2C6H3) were investigated;13 more recently, it was shown14,15 that, through the addition of a base, such hydride loss can generate active 14e (pincer)Ir species that are known to react with C–H bonds.3
Previously, we reported the reaction of (p-X–POCOP)IrHCl complexes 1a and 1b with several protic acids to give compounds with cyclometallated tert-butyl group (Scheme 1).16 The cleanest transformation was observed with CF3COOH. Some mechanistic studies performed17 suggested two possible mechanisms: (a) protonation of the hydride ligand and (b) redox processes at the iridium centre.
 | ||
| Scheme 1 Reactions of (p-X–POCOP)IrHCl iridium pincer complexes with CF3COOH. | ||
Results and discussion
Oxidation-induced cyclometallation
To get more insight into the latter opportunity, we investigated the reaction of complex 1a with a mild chemical oxidant, CF3COOAg. It was found that addition of one equivalent of CF3COOAg to 1a in THF (or DCM) led to an anion exchange and the formation of trifluoroacetate complex 1a-COOCF3 within seconds, while the addition of excess CF3COOAg resulted in the formation of metallic silver, cyclometallation of the tert-butyl group and the formation of compound 2a-COOCF3 within 30 min in a 90% isolated yield (Scheme 2). | ||
| Scheme 2 Reaction of complex 1a with CF3COOAg. | ||
The trifluoroacetate complex 1a-COOCF3 is characterized by resonances at 172.0 (s) and −38.98 (t, 3JPH = 12.7 Hz) ppm in the 31P{1H} and 1H NMR spectra, correspondingly, with other signals being quite featureless. For complex 2a-COOCF3, an AX system (159.1 and 113.5 ppm, d, 2JPP = 367.4 Hz) in the 31P{1H} NMR spectrum, as well as doublets from the CH3 groups at 1.42 (3JPH ≈ 15.7 Hz) and 0.95 ppm (3JPH = 12.4 Hz) and multiples from the –CH2– group at 2.79 and 2.06 ppm indicate metallation of one of the tert-butyl groups. In the 13C{1H} NMR spectrum, a signal at −3.90 ppm (dd, 2JCP1 = 25.4 Hz, 2JCP2 = 2.3 Hz) is observed, corresponding to the cyclometallated aliphatic carbon. These features closely resemble spectra of the related phosphinite complexes 2a and 2b.16
When DDQ and Me3NO were tried as alternative oxidants, mainly decomposition of 1a and dismantling of the pincer ligand were observed. However, the formation of 2a was detected when acetyl–ferrocenium hexafluorophosphate [FcAc]PF6 was used.
We have also attempted the oxidation of complex (PCP)IrHCl (1c), which is a counterpart of 1a with –CH2– groups instead of –O– in the pincer arms. The reaction with CF3COOAg was much less clean than that with 1a, but the use of [FcAc]PF6 provided the respective product with a cyclometallated tert-butyl, 2c (Scheme 3).
 | ||
| Scheme 3 Reaction of complex 1c with [FcAc]PF6. | ||
Other relevant observations include an attempt to oxidize the ruthenocene-based heterobimetallic18 complex [Ru](PCP)IrHCl (3) with CF3COOAg that led to the observation of AX-systems in the 31P{1H} NMR and a disappearance of the hydride resonance, that we interpret as the formation of structures with exo and endo cyclometallated tert-butyl groups (see ESI† for details), and the oxidation of the (PCP)IrHI (1c-I) complex with atmospheric oxygen in a C6D6 solution that provided 2c-I. Due to the instability and small amounts of available material these two examples received only a limited characterization in situ. They prove, however, that the oxidation-induced C–H bond activation may not be an uncommon reactivity pattern. Also, metallation of a tert-butyl group upon deprotection of 1d (X = O-TBDMS) under oxidative conditions was reported.19
A(na)gostic bonding in 2a-COOCF3 and related complexes
An XRD structure of complex 2a-COOCF3 is depicted in Fig. 1. Trifluoroacetate is coordinated in a monodentate mode. One of the tert-butyl C–H bonds is clearly directed towards the site trans to the four-membered metallacycle. Judging from the C–Ir = 3.16 Å and H–Ir ≈ 2.5 Å distances, this site is occupied by an agostic (3c–2e interaction) or an anagostic20,21 (Irδ−⋯Hδ+ interaction) Ir⋯H–C bond. This type of bonding was not found in the electronically very similar parent hydrido-trifluoroacetate 1a-COOCF3 or hydrido-chlorides 1a and 1c neither experimentally nor computationally, and thus is possibly a result of the four-membered metallacycle constraining the tert-butyl in a favourable conformation. It is believed that agostic bonds are characterized by an up-field shift of the bound hydrogen versus the non-coordinated one in the 1H NMR spectra, while for anagostic bonds a downfield shift should be observed.20 Even though such shifts would be averaged over the tert-butyl group, we were expecting them to be revealed in a series of related compounds 2a-COOCF3, 2a, 2c, and the p-MeO-substituted counterpart of 2c, (p-MeO-PCP)IrHCl (2e).22 However, in the experimental NMR spectra of those compounds little, if any, changes associated with the presence of a(na)gostic bonds were isolated. DFT calculations were then performed that provided some rationale. The selected data for a series of compounds studied is given in Table 1. The Ir⋯H–C bonding is well captured in the calculated structures. If the chemical shift is taken as a criterion, the bond should be classified as agostic, despite the comparatively long Ir⋯H distances.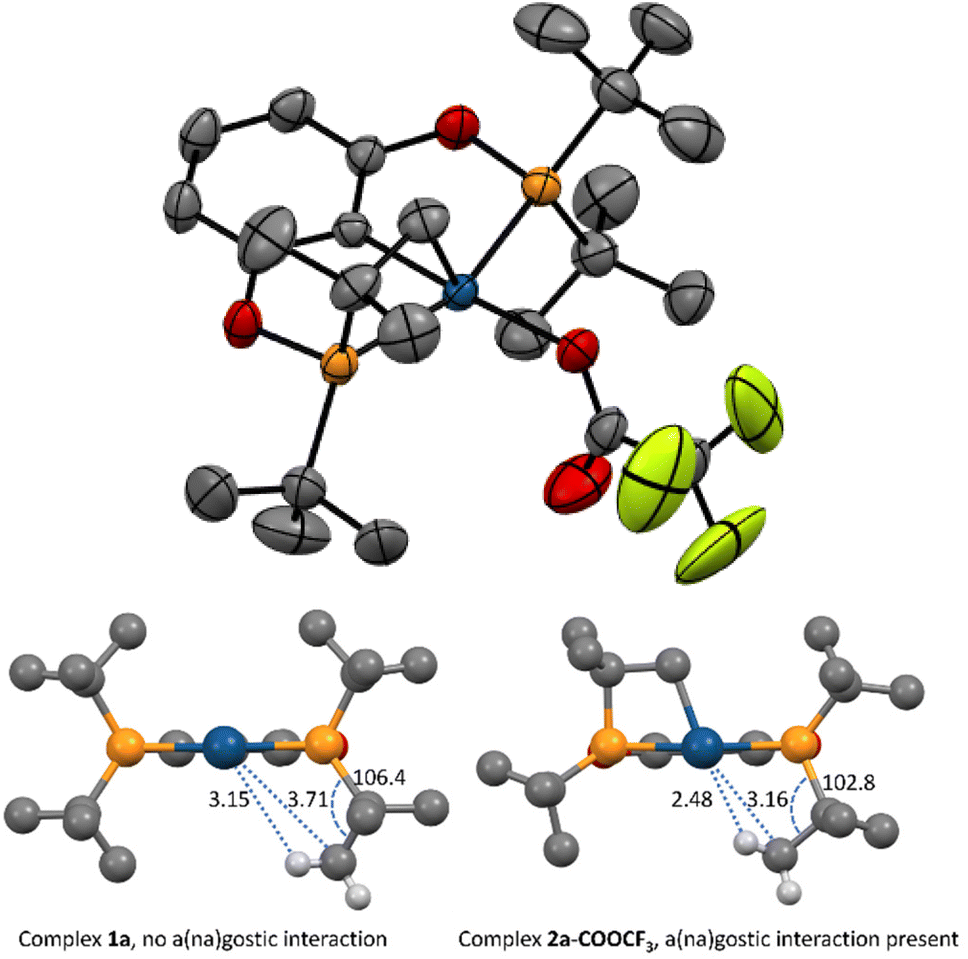 | ||
Fig. 1 Top: ORTEP plot of complex 2a-COOCF3. Ellipsoids are given at a 50% probability level. Hydrogen atoms are omitted for clarity. Bottom: a schematic comparison of XRD structures of 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 and 2a-COOCF3. Hydrogen atoms (except for one methyl group) as well as –Cl and –COOCF3 ligands are omitted for clarity. 23 and 2a-COOCF3. Hydrogen atoms (except for one methyl group) as well as –Cl and –COOCF3 ligands are omitted for clarity. | ||
| Complex | 2a-COOCF3 | 2a | 2c | 2e |
|---|---|---|---|---|
| a Experimental data for complex 2e is taken from ref. 22. | ||||
| Ir–H calc. (exp.), Å | 2.56 (2.49) | 2.48 | 2.39 | 2.40 (2.34) |
| Ir–C, calc. (exp.), Å | 3.17 (3.16) | 3.13 | 3.07 | 3.08 (3.02) |
| δ(Ir⋯H), calc. | 0.75 | −0.60 | −1.00 | −0.87 |
| δ(tBu), calc. | 1.13 | 1.15 | 1.16 | 1.18 |
| δ(tBu), exp. | 1.16 | 1.20 | 1.11 | 1.11 |
For complex 2a-COOCF3 with the weakest agostic bond, the calculated δ(Ir⋯H–C) in 1H NMR (0.75 ppm) in fact falls in the range observed for other tert-butyl type hydrogens (0.56–1.86 ppm). Presumably, this is because the bound hydrogen, in addition to the agostic bond that is expected to feature an up-field shift, is involved in a hydrogen bond with the oxygen from the –COOCF3 group (H⋯O = 2.39 Å), that is expected to feature a counter-balancing down-field shift. For complexes 2a, 2c and 2e, the agostic bond becomes stronger, and δ(Ir⋯H–C) appears as negative values. At the same time, in these compounds one of the hydrogens in the same tert-butyl group is involved in a weak hydrogen bond with the Cl ligand (H⋯Cl 2.65–2.72 Å). The latter hydrogen undergoes a down-field shift (2.83, 3.55 and 3.71 ppm in 2a, 2c and 2e, respectively) that fully counterbalances the up-field shift of the agostic hydrogen, such that the overall changes in δ(tBu) are very minor.
Electrochemical study
To gain further insight we performed an electrochemical study of 2a, 2a-COOCF3, 1a, 1a-COOCF3, as well as 1b′ (X = MeOOC–), 1c, and 1g (X = OMe) (Table 2). THF and, in a few experiments, DCM were used as solvents, glassy carbon was taken as a working electrode, and the Ag/Ag+ redox system was used as the reference electrode. Potentials are reported versus the Fc/Fc+ couple. Notably, considerable complications presumably related to the adsorption on the electrode were noted. To obtain reproducible cyclic voltammograms (CVs), the application of a negative potential at the end was required (see Fig. S5† for more details).Compound 2a revealed a partially reversible oxidation wave at Eoxp = 0.664 V (E1/2 = 0.572 V; Fig. 2). The ΔEp and ip values, when compared to those of the ferrocene couple, argued for a one-electron oxidation. Judging from the scan rate dependence of iredp/ioxp (increases upon increasing the scan rate), oxidation is possibly coupled to a chemical reaction that gives electrochemically inactive products. Also, an irreversible reduction wave was observed at Eredp = −2.556 V that corresponds to placing an electron on the LUMO. At high scan rates some unidentified electrochemically active products of the reduction were noted. A similar pattern was observed for 2a-COOCF3, with Eoxp shifted to a more positive potential.
 | ||
| Fig. 2 Cyclic voltammograms of 1a and 2a in THF, scan rate 0.05 V s−1. | ||
The oxidation of 1a revealed an irreversible wave at Eoxp = 0.721 V. If the potential range is extended, two smaller, overlapping oxidation waves are observed at ca. 1.05 and 1.15 V, as well as a reduction shoulder-like wave at ca. 0.90 V (Fig. 3). We have also briefly examined the analogues of 1a with X = MeOOC-(1b′) and MeO-(1g) (Table 2). Complex 1b′ revealed an oxidation wave shifted to a more positive potential compared to that of 1a, while for 1g two nearly coinciding oxidation waves were observed, and one reduction wave, that is close, but does not match precisely the reduction wave observed for 2g (Fig. S3†). Complex 1c revealed an irreversible oxidation wave at Eoxp = 0.476 V.
 | ||
| Fig. 3 Cyclic voltammogram of 1a in THF, scan rate 0.05 V s−1, extended range of potentials. | ||
DFT calculated one-electron oxidation potentials are in a reasonable agreement with the experimental data (Table 2). One could note a systematic shift of ca. 0.3 V between the experimental and calculated E1/2 values; presumably, this could be due to inaccuracies in the continuum solvation model.24
To sum up, most of the type 1 compounds undergo a one-electron oxidation with the formation of Ir(IV) species 1˙+; the latter undergoes subsequent transformations presumably associated with the hydride ligand, as suggested by comparison with the electrochemically more stable type 2 compounds. No unambiguous evidence that 2 is formed upon electrochemical oxidation of 1 was obtained.
Previously, Speiser et al. reported25 two reduction waves for 1c, one of which was interpreted as the formation of 2c through a so-called “square scheme” mechanism. A straightforward comparison with our results is not possible due to the different conditions used. We note, though, that an as-proposed direct conversion of 1c˙+ to 2c˙+ with the evolution of hydrogen is not possible for thermodynamic reasons (unless an effective purging was used); thus, the calculated ΔG for 1c = 2c +H2 is +17.5 kcal mol−1, and +12.9 kcal mol−1 for 1c˙+ = 2c˙+ + H2. If 2c˙+ was indeed formed, it was most likely through disproportionation reactions, for example the reaction 2 × (PCP)IrHCl+˙ (1c˙+) = (PCP)Ir+ + (PCP)Ir˙ + H2 has a more favourable ΔG = −33.8 kcal mol−1. (PCP)Ir+, as will be shown below, can further convert to 2c+.
Mechanism of conversion of 1 to 2
The known cyclometallations of an internal tert-butyl group in iridium pincer complexes proceed very slowly or under harsh conditions. Some examples include deuterium scrambling between tert-butyls and hydridic positions in (p-H-POCOP)IrH2 (slow at rt, with complete deuteration upon prolonged heating at 120 °C and above),26 dehydrogenative metallation of the tert-butyl group in (PCyP)IrH2 at 120 C° and at 200 °C in (PCyP)IrHCl,27 metallation of the p-MeO–PCP pincer ligand at 80 °C for 24 h, that gives a mixture of (p-MeO–PCP)IrHCl and a complex with metallated tert-butyl group 2e.22 Mechanistic studies27 suggested oxidative addition of the C–H bond to Ir(III) IrH2 or IrHCl centres to give Ir(V) as a plausible pathway.Given the mild conditions of the 1 to 2 conversion reported here, it is likely that another mechanism of C–H activation is involved. It is logical to propose that the reaction begins with oxidation of 1a to give 1a˙+. Then, an oxidant anion can abstract H+ from 1a˙+ with the formation of Ir(II) complex i˙ (Scheme 4). From that point, there are two possible pathways. One option is that i˙ can undergo oxidation to the Ir(III) complex ii, from which a low-energy 1a-TS1 (15.9 kcal mol−1) leads to a product with a cyclometallated tert-butyl group, iii (Scheme 4, red). Subsequently, abstraction of H+ from iii or the energetically close iv with an oxidant anion leads to the product 2a. The same mechanism is energetically viable for the 1c to 2c conversion as well, with 1c-TS1 and 1c-iii located at +2.4 and −6.7 kcal mol−1versus1c-ii. This mechanism may be relevant to the above-mentioned reaction of 1a with acids as well, with ii being generated through the protonation of hydrides or H+ serving as an oxidant. Alternatively, i˙ can undergo C–H activation directly through 1a-TS3 to give complex v (Scheme 4, blue). Oxidation of v would then lead to iii. We slightly favour the pathway through ii and 1a-TS1, because we expect i to be oxidized readily (E1/2calc = 0.079 V); however more studies are needed to make a reliable choice.
 | ||
| Scheme 4 Possible mechanism for the oxidation-induced C–H bond activation in iridium pincer complexes. “Ox” denotes oxidant. DFT calculated Gibbs free energies are given in kcal mol−1versus complexes i and ii. | ||
If the proposed mechanism is correct, one could envision the possibility that 14e cationic complexes similar to ii mediate oxidation-driven C–H activations and dehydrogenations in a manner similar to the mediation of 14e (pincer)Ir species in the dehydrogenation of alkanes. Scheme 5 depicts a comparison of the hypothetical catalytic cycle of ethane dehydrogenation through (Me4POCOP)IrCl+ (that employs two equivalents of oxidant and base as a driving force) with the traditional catalytic cycle through (Me4POCOP)Ir (that employs a sacrificial hydrogen acceptor as the driving force). A ligand with methyl groups replacing tert-butyls was taken as a model. From the electronic point of view, it can be seen that the key transition states for both pathways are energetically accessible; the slowest steps are β-elimination for the (Me4POCOP)IrCl+ cycle (+23.2 kcal mol−1) and ethylene dissociation for the (Me4POCOP)Ir cycle (+16.0 kcal mol−1). To enable the cycle through (pincer)IrCl+, possibly pincer ligands that are more reluctant to intramolecular C–H activation would be required, for example, fluorinated ligands.28 Also, important is that H+ would be effectively removed from the oxidized species, while the hydride in the parent IrHCl compound would remain unaffected.
 | ||
| Scheme 5 DFT modelled comparison of the dehydrogenation of ethane through 14e (Me4POCOP)IrCl+ that can be driven by oxidation (top), with the traditional dehydrogenation through 14e (Me4POCOP)Ir that is driven by a sacrificial hydrogen acceptor (bottom). | ||
Recently, a conceptually similar dehydrogenation was reported. Thus, Goldman and Miller15 generated (pincer)Ir from (PCP)IrH2 and (POCOP)IrH2 using two equivalents of oxidant and base, through two consecutive proton removals. The (pincer)Ir species were then reacted with alkanes to give alkenes, with recovery of (pincer)IrH2. While perhaps being a step towards something bigger, the reported process uses the traditional pathway through electron-rich (pincer)Ir and (pincer)IrH2, and thus is sensitive to over-oxidation.15 Hence, more electron-deficient species are desired to enable the use of cheap and powerful oxidants, such as oxygen. It could be that the pathway through (pincer)IrCl+ could offer some alternatives in that respect. For example, 1a is reasonably air-stable, and oxidation with CF3COOAg to give 2a can be performed in air.
Conclusions
To sum up, we have reported oxidations of iridium pincer complexes 1a and 1c with CF3COOAg and [FcAc]PF6, respectively, to give compounds 2a-COOCF3 and 2c with cyclometallated tert-butyl groups. This transformation thus represents oxidation-induced C–H activation. An electrochemical study and DFT calculations provided a mechanistic guess that involves (pincer)IrCl+ (or –COOCF3) as the compound responsible for C–H activation. Interestingly, some analogies could be drawn between the reactivity of (pincer)IrCl+ and (pincer)Ir, with the latter being a key compound in the catalytic cycle for alkane dehydrogenation. Thus, (pincer)IrCl+ or related species hold some promise to substitute (pincer)Ir in oxidation-driven dehydrogenation reactions, since the electron-poor (pincer)IrCl+ species are expected to be more stable with respect to undesired over-oxidation.Experimental part
General considerations
All the manipulations were conducted under an argon atmosphere using standard Schlenk techniques unless otherwise stated. All the solvents (including deuterated) were distilled under an argon atmosphere from the appropriate drying agents. Commercially available reagents were used as received. Compounds 1a,261b′,291c,30 and 1g26 were prepared according to literature procedures. NMR spectra were recorded on Bruker Avance 400 MHz and Bruker Avance 500 MHz spectrometers. 1H NMR chemical shifts are reported in parts per million downfield from tetramethylsilane; the residual signals of the deuterated solvent were used as a reference. 31P{1H} NMR chemical shifts are reported relative to an external 85% solution of phosphoric acid in D2O. 19F{1H} NMR chemical shifts are reported relative to external CFCl3. Elemental analyses were performed at the A. N. Nesmeyanov Institute of Organoelement Compounds of RAS.
Reaction of complex 1a with one equivalent of CF3COOAg. Formation of complex 1-COOCF3
To a solution of complex 1a (0.063 g, 0.101 mmol) in THF (15 ml) in a light-protected flask a solution of CF3COOAg (0.023 g, 0.104 mmol) in THF (5 ml) was added dropwise. This was accommodated by a colour change from red-orange to yellow-orange and the formation of a precipitate. The reaction mixture was stirred for an additional 2.5 hours, the volatiles were removed in vacuum and the residue was extracted with CH2Cl2 and filtered through a thin layer of Celite™. After evaporation of the volatiles and drying in a vacuum, complex 1-COOCF3 (0.069 g, 97%) was obtained as a yellow-orange powder. Calculated for C24H40F3IrO4P2: C, 40.96; H, 5.73. Found: 41.17; H, 5.64. 1H NMR (400 MHz, CDCl3): δ 6.73 (t, 1H, 3JHH = 8.0 Hz, Ar–H), 6.48 (d, 2H, 3JHH = 8.0 Hz, Ar–H), 1.36–1.31 (m, 36 H, 4 C(CH3)3), −38.98 (t, 1H, 3JPH = 12.7 Hz). 31P{1H} NMR (1612 MHz, CDCl3): δ 172.0 (s). 19F{1H} NMR (377 MHz, CDCl3): δ −75.0 (s). 13C NMR (126 MHz, C6D6) δ 167.14 (vt, J = 5.9 Hz, 2-C and 6-C), 164.90 (q, 2JC–F = 37.4 Hz, CO–CF3), 125.92 (s, 4-C), 118.06 (q, 2JC–F = 287 Hz, CO–CF3) 108.86 (m, 1-C), 105.22 (vt, J = 5.3 Hz, 3-C and 5-C), 42.76 (vt, J = 12.1 Hz, C(CH3)3), 39.83 (vt, J = 12.4 Hz, C(CH3)3), 27.78 (vt, J = 3.2 Hz, C(CH3)3), 27.30 (vt, J = 3.0 Hz, C(CH3)3).
Reaction of complex 1a with an excess of CF3COOAg. Formation of complex 2a
To a solution of complex 1a (0.046 g, 0.074 mmol) in THF (15 ml) in a light-protected flask a solution of CF3COOAg (0.066 g, 0.299 mmol) in THF (5 ml), was added dropwise. This was accommodated by a colour change from red-orange to yellow-orange and the formation of a precipitate. The reaction mixture was stirred for an additional 30 min, the volatiles were removed in a vacuum and the residue was extracted several times with pentane and filtered through a thin layer of Celite™. After evaporation of the volatiles and drying in a vacuum complex, 2a (0.047 g, 90%) was obtained as a yellow-orange powder. Calculated for C24H38F3IrO4P2: C, 41.08; H, 5.46. Found: C, 40.89; H, 5.47. 1H NMR (400 MHz, CDCl3): δ 6.84 (apparent t, 1H, J = 8.0 Hz, Ar–H), 6.67 (d, 2H, 3JHH = 8.0 Hz, Ar–H), 6.64 (d, 2H, 3JHH = 8.0 Hz, Ar–H), 2.79 (ddd, 1H, J1 = 9.7 Hz, J2 = 6.0 Hz, J3 = 1.6 Hz, Ir–CH2), 2.08–2.05 (m, 1H, Ir–CH2), 1.42 (d, 9H, 3JPH = 15.7 Hz, –C(CH3)3), 1.42 (overlapping d, 3H, 3JPH ≈ 15.7 Hz, Ir–CH2–C(CH3)2), 1.24 (d, 9H, 3JPH = 14.7 Hz, –C(CH3)3), 1.16 (d, 9H, –C(CH3)3), 3JPH = 14.1 Hz, 0.95 (d, 3H, 3JPH = 12.4 Hz, Ir–CH2–C(CH3)2). 31P{1H} NMR (161.98 MHz, CDCl3): δ 159.1 (d, 2JPP = 367.4 Hz), 113.5 (d, 2JPP = 367.4 Hz). 19F{1H} NMR (376.50 MHz, CDCl3): δ −74.9 (s). 13C NMR (126 MHz, C6D6) δ 168.10 (dd, 2JC–P = 8.0 Hz, 3JC–P = 5.2 Hz, 2-C or 6-C), 165.73 (dd, 2JC–P = 7.2 Hz, 3JC–P = 3.3 Hz, 2-C or 6-C), 164.93 (q, 2JC–F = 36.7 Hz, CO–CF3), 126.84 (s, 4-C), 116.74 (apparent t, 2JC–P1 = 2JC–P2 = 3.3 Hz, 1-C), 117.79 (q, 2JC–F = 288 Hz, CO–CF3), 105.95 (d, 3JC–P = 11.0 Hz, 3-C or 5-C), 105.64 (d, 3JC–P = 11.0 Hz, 3-C or 5-C), 66.10 (dd, 1JC–P = 18.6 Hz, 3JC–P = 5.0 Hz, P–C(CH3)2–CH2–), 45.09 (dd, 1JC–P = 17.5 Hz, 3JC–P = 5.4 Hz, C(CH3)3), 39.81–39.47 (m, overlapping, 2 × C(CH3)3), 27.23–27.09 (m, overlapping, 2 × C(CH3)3), 26.12 (dd, 2JC–P = 4.4 Hz, 4JC–P = 1.1 Hz, C(CH3)3), 24.08 (d, 2JC–P = 3.9 Hz, CH3), 22.96 (d, 2JC–P = 4.7 Hz, CH3), −3.90 (dd, 2JC–P = 25.4 Hz, 2JC–P = 2.3 Hz, –CH2–Ir).
Reaction of complex 1c with acetyl–ferrocenium hexafluorophosphate. Formation of complex 2c
To a solution of complex 1c (0.030 g, 0.048 mmol) in THF (15 ml), a solution of acetyl–ferrocenium hexafluorophosphate (0.036 g, 0.096 mmol) in THF (5 ml) was added. The reaction mixture was stirred overnight, the volatiles were removed in a vacuum and the residue was purified by flash chromatography on silica gel using a hexane–benzene mixture as the eluent. After evaporation of the volatiles, the residue was dissolved in hexane and crystallized. Complex 2c (0.014 g, 47%) was obtained as a red solid. Calculated for C24H42ClIrP2: C, 46.48; H, 6.83. Found: C, 46.57; H, 6.70. 1H NMR (500 MHz, C6D6) δ 7.22–7.14 (m, overlapping, 2H, 3-C and 5-C), 7.03 (t, 3JHH = 7.3 Hz, 1H, 4-H), 4.36–4.26 (m, 1H, –CH2–P), 3.53 (ddd, J1 = 8.9 Hz, J2 = 5.1 Hz, J3 = 2.0 Hz, 1H, Ir–CH2), 3.20–3.00 (m, 3H, –CH2–P), 2.14–2.09 (m, 1H, Ir–CH2), 1.49 (d, 3JPH = 13.7 Hz, 3H, Ir–CH2–C(CH3)2), 1.34 (d, 3JPH = 13.7 Hz, 9H, –C(CH3)3), 1.17 (d, 3JPH = 12.8 Hz, 9H, –C(CH3)3), 1.11 (d, 3JPH = 12.7 Hz, 9H, –C(CH3)3), 0.72 (d, 3JPH = 12.8 Hz, 3H, Ir–CH2–C(CH3)2). 31P NMR (162 MHz, C6D6) δ 48.93 (d, 2JPP = 349.2 Hz), 7.85 (d, 2JPP = 349.2 Hz). 13C NMR (126 MHz, C6D6) δ 152.22 (dd, 2JC–P = 13.7 Hz, 3JC–P = 5.1 Hz, 2-C or 6-C), 151.09 (s, 1-C), 149.15 (dd, 2JC–P = 10.4 Hz, 3JC–P = 4.2 Hz, 2-C or 6-C), 123.54 (apparent t, 4JC–P1 = 4JC–P2 = 1.2 Hz, 4-C), 122.43 (d, 3JC–P = 16.3 Hz, 3-C or 5-C), 121.86 (d, 3JC–P = 15.9 Hz, 3-C or 5-C), 59.43 (dd, 1JC–P = 20.4 Hz, 3JC–P = 2.2 Hz, P–C(CH3)2–CH2–), 40.90 (dd, 1JC–P = 16.5 Hz, 3JC–P = 3.1 Hz, C(CH3)3), 36.65 (apparent t, 1JC–P = 3JC–P = 8.1 Hz, C(CH3)3), 34.97 (dd, 1JC–P = 14.7 Hz, 3JC–P = 4.8 Hz, C(CH3)3), 34.26 (dd, 1JC–P = 15.2 Hz, 3JC–P ≈ 1.2 Hz, P–CH2–Ir), 34.05 (dd, 1JC–P = 13.9 Hz, 3JC–P ≈ 1.2 Hz, P–CH2–Ir), 30.44 (dd, 2JC–P = 3.8 Hz, 4JC–P = 1.5 Hz, C(CH3)3), 29.17 (dd, 2JC–P = 2.7 Hz, 4JC–P = 1.7 Hz, C(CH3)3), 29.02 (dd, 2JC–P = 3.3 Hz, 4JC–P = 1.6 Hz, C(CH3)3), 27.06 (dd, 2JC–P = 3.9 Hz, 4JC–P ≈ 0.9 Hz, CH3), 26.08 (s, CH3), −6.00 (dd, 2JC–P = 23.3 Hz, 2JC–P = 2.3 Hz, –CH2–Ir).
X-ray crystallography
Intensity data were collected with an Oxford Diffraction Excalibur 3 system, using ω-scans and Mo Kα (λ = 0.71073 Å) radiation.31 The data were extracted and integrated using Crysalis RED.32 The structures were solved and refined by full-matrix least-squares calculations on F2 using JANA2006.33 Molecular graphics were generated using Mercury 3.10.3.34Cyclic voltammetry
Cyclic voltammetry measurements were performed on an Autolab PGSTAT101 potentiostat for 10−3 M solutions in rigorously dried and degassed THF (or CH2Cl2) in a standard three-electrode cell equipped with a glassy carbon working electrode (S = 7 mm2), platinum wire as the counter electrode, and Ag/Ag+ as the reference electrode. NBu4PF6 was used as the supporting electrolyte at the 0.1 M concentration. The scan rate was 0.02–1 V s−1. Ferrocene–ferrocenium (Fc/Fc+) pair was applied as the external standard.Computational details
DFT calculations were performed with ORCA 4.1.1,35 using the PBE functional36 with a D3BJ dispersion correction.37 For geometry optimization and frequencies, the def2-SVP basis set38 was used, while the def2-TZVPP38 basis set was used for single-point energies. The RI algorithm with appropriate fitting basis sets was exploited. Solvent effects were incorporated using the CPCM solvation model39 with DCM used as the solvent. Redox potentials were calculated using the E° = ΔG(ox)/−nF expression, where ΔG is the Gibbs free energy difference between the neutral and oxidized species, F is the Faraday constant, and n is number of electrons. Some of the cation-radical species reveal very small negative frequencies; these were not given any special treatment due to the expected negligible effect on E°. Computed redox potentials were referred to the ferrocene–ferrocenium (Fc/Fc+) pair. NMR spectra were calculated using the ZORA40 two-component approximation to relativistic effects, with the SARC–ZORA–TZVPP41 basis set used for Ir and the ZORA–def2–TZVPP basis set for other atoms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received no dedicated external funding. We gratefully acknowledge Dr S. M. Peregudova for her kind assistance in the initial attempts of the electrochemical studies.References
- (a) B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154–162 CrossRef CAS; (b) K. I. Goldberg and A. S. Goldman, Acc. Chem. Res., 2017, 50, 620–626 CrossRef CAS PubMed.
- For selected reviews on pincer complexes, see: (a) L. Piccirilli, D. L. J. Pinheiro and M. Nielsen, Catalysts, 2020, 10, 773 CrossRef CAS; (b) H. Valdés, E. Rufino-Felipe and D. Morales-Morales, J. Organomet. Chem., 2019, 898, 120864 CrossRef; (c) M. A. W. Lawrence, K.-A. Green, P. N. Nelson and S. C. Lorraine, Polyhedron, 2018, 143, 11–27 CrossRef CAS; (d) H. Valdés, M. A. García-Eleno, D. Canseco-Gonzalez and D. Morales-Morales, ChemCatChem, 2018, 10, 3136–3172 CrossRef; (e) E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC; (f) Pincer Compounds: Chemistry and Applications, ed. D. Morales-Morales, Elsevier, The Netherlands, 2018 Search PubMed; (g) H. Valdés, L. González-Sebastián and D. Morales-Morales, J. Organomet. Chem., 2017, 845, 229–257 CrossRef; (h) M. Asay and D. Morales-Morales, Dalton Trans., 2015, 44, 17432–17447 RSC; (i) The Chemistry of Pincer Compounds, ed. D. Morales-Morales and C. Jensen, Elsevier, The Netherlands, 2007 Search PubMed.
- For dehydrogenation by iridium pincer complexes, see: (a) J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed; (b) A. Kumar, T. M. Bhatti and A. S. Goldman, Chem. Rev., 2017, 117, 12357–12384 CrossRef CAS PubMed; (c) A. V. Polukeev and O. F. Wendt, J. Organomet. Chem., 2018, 867, 33–50 CrossRef CAS; (d) D. Morales-Morales, R. Redón, C. Yung and C. M. Jensen, Inorg. Chim. Acta, 2004, 357, 2953–2956 CrossRef CAS; (e) C. M. Jensen, Chem. Commun., 1999, 2443–2449 RSC; (f) F. Liu, E. B. Pak, B. Singh, C. M. Jensen and A. S. Goldman, J. Am. Chem. Soc., 1999, 121, 4086–4087 CrossRef CAS.
- T. Tian, Z. Li and C.-J. Li, Green Chem., 2021, 23, 6789–6862 RSC.
- For selected reviews, see: (a) A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed; (b) J. A. Labinger, Chem. Rev., 2017, 117, 8483–8496 CrossRef CAS PubMed; (c) N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521–8573 CrossRef CAS PubMed.
- N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086–7103 CrossRef CAS.
- See, for example: (a) M. C. Lehman, D. R. Pahls, J. M. Meredith, R. D. Sommer, D. M. Heinekey, T. R. Cundari and E. A. Ison, J. Am. Chem. Soc., 2015, 137, 3574–3584 CrossRef CAS PubMed; (b) A. J. Ingram, A. B. Wolk, C. Flender, J. Zhang, C. J. Johnson, U. Hintermair, R. H. Crabtree, M. A. Johnson and R. N. Zare, Inorg. Chem., 2014, 53, 423–433 CrossRef CAS PubMed; (c) M. Zhou, D. Balcells, A. R. Parent, R. H. Crabtree and O. Eisenstein, ACS Catal., 2012, 2, 208–218 CrossRef CAS; (d) E. V. Sackville, G. Kociok-Köhn and U. Hintermair, Organometallics, 2017, 36, 3578–3588 CrossRef CAS; (e) M. C. Lehman, P. D. Boyle, R. D. Sommer and E. A. Ison, Organometallics, 2014, 33, 5081–5084 CrossRef CAS; (f) S. K. Gupta and J. Choudhury, Dalton Trans., 2015, 44, 1233–1239 RSC; (g) S. Hohloch, S. Kaiser, F. L. Duecker, A. Bolje, R. Maity, J. Košmrljb and B. Sarkar, Dalton Trans., 2015, 44, 686–693 RSC; (h) M. E. Kerr, I. Ahmed, A. Gunay, N. J. Venditto, F. Zhu, E. A. Ison and M. H. Emmert, Dalton Trans., 2016, 45, 9942–9947 RSC; (i) Y. Yan, Y. Chen, M. Yan, X. Li and W. Zeng, Catal. Commun., 2013, 35, 64–67 CrossRef CAS; (j) M. Zhou, U. Hintermair, B. G. Hashiguchi, A. R. Parent, S. M. Hashmi, M. Elimelech, R. A. Periana, G. W. Brudvig and R. H. Crabtree, Organometallics, 2013, 32, 957–965 CrossRef CAS; (k) M. Zhou, N. D. Schley and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 12550–12551 CrossRef CAS PubMed.
- J.-I. Ito, T. Kaneda and H. Nishiyama, Organometallics, 2012, 31, 4442–4449 CrossRef CAS.
- K. E. Allen, D. M. Heinekey, A. S. Goldman and K. I. Goldberg, Organometallics, 2013, 32, 1579–1582 CrossRef CAS.
- D. R. Pahls, K. E. Allen, K. I. Goldberg and T. R. Cundari, Organometallics, 2014, 33, 6413–6419 CrossRef CAS.
- K. E. Allen, D. M. Heinekey, A. S. Goldman and K. I. Goldberg, Organometallics, 2014, 33, 1337–1340 CrossRef CAS.
- M. Zhou, S. I. Johnson, Y. Gao, T. J. Emge, R. J. Nielsen, W. A. Goddard III and A. S. Goldman, Organometallics, 2015, 34, 2879–2888 CrossRef CAS.
- A. G. Walden, A. Kumar, N. Lease, A. S. Goldman and A. J. M. Miller, Dalton Trans., 2016, 45, 9766–9769 RSC.
- B. M. Lindley, A. G. Walden, A. M. Brasacchio, A. Casuras, N. Lease, C.-H. Chen, A. S. Goldman and A. J. M. Miller, Chem. Sci., 2019, 10, 9326–9330 RSC.
- A. D. R. Shada, A. J. M. Miller, T. J. Emge and A. S. Goldman, ACS Catal., 2021, 11, 3009–3016 CrossRef CAS.
- A. V. Polukeev, S. A. Kuklin, P. V. Petrovskii, F. M. Dolgushin, M. G. Ezernitskaya and A. A. Koridze, Russ. Chem. Bull., Int. Ed., 2010, 59, 745–749 CrossRef CAS.
- A. V. Polukeev, PhD Thesis, A. N. Nesmeyanov Institute of Organoelement Compounds, RAS, 2012.
- For heterobimetallic pincer complexes, see: (a) H. Valdés, J. M. Germán-Acacio, G. van Koten and D. Morales-Morales, Dalton Trans., 2022, 51, 1724–1744 RSC; (b) N. Á. Espinosa-Jalapa, S. Hernández-Ortega, D. Morales-Morales and R. Le Lagadec, J. Organomet. Chem., 2012, 716, 103–109 CrossRef CAS; (c) S. A. Kuklin, A. M. Sheloumov, F. M. Dolgushin, M. G. Ezernitskaya, A. S. Peregudov, P. V. Petrovskii and A. A. Koridze, Organometallics, 2006, 25, 5466–5476 CrossRef CAS.
- M. Rimoldi, A. Nakamura, N. A. Vermeulen, J. J. Henkelis, A. K. Blackburn, J. T. Hupp, J. F. Stoddart and O. K. Farha, Chem. Sci., 2016, 7, 4980–4984 RSC.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS PubMed.
- Y. Garcıa-Rodeja, F. Feixas, E. Matito and M. Sola, Phys. Chem. Chem. Phys., 2022, 24, 29333–29337 RSC.
- H. A. Y. Mohammad, J. C. Grimm, K. Eichele, H.-G. Mack, B. Speiser, F. Novak, M. G. Quintanilla, W. C. Kaska and H. A. Mayer, Organometallics, 2002, 21, 5775–5784 CrossRef CAS.
- A. Arunachalampillai, D. Olsson and O. F. Wendt, Dalton Trans., 2009, 8626–8630 RSC.
- M. Isegawa, F. Neese and D. A. Pantazis, J. Chem. Theory Comput., 2016, 12, 2272–2284 CrossRef CAS PubMed.
- F. Novak, B. Speiser, H. A. Y. Mohammad and H. A. Mayer, Electrochim. Acta, 2004, 49, 3841–3853 CrossRef CAS.
- I. Gottker-Schnetmann and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 9330–9338 CrossRef PubMed.
- A. V. Polukeev, R. Marcos, M. S. G. Ahlquist and O. F. Wendt, Chem. Sci., 2015, 6, 2060–2067 RSC.
- B. C. Gruver, J. J. Adams, S. J. Warner, N. Arulsamy and D. M. Roddick, Organometallics, 2011, 30, 5133–5140 CrossRef CAS.
- N. T. Mucha and R. Waterman, Organometallics, 2015, 34, 3865–3872 CrossRef CAS.
- C. J. Moulton and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1976, 1020–1024 RSC.
- Crysalis CCD, Oxford Diffraction Ltd., Abingdon, Oxfordshire, UK, 2005 Search PubMed.
- Crysalis RED, Oxford Diffraction Ltd., Abingdon, Oxfordshire, UK, 2005 Search PubMed.
- V. Petříček, M. Dušek and L. Palatinus, Crystallographic Computing System JANA2006: General features, Z. Kristallogr., 2014, 229, 345–352 Search PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- (a) F. Neese, Mol. Sci., 2012, 2, 73–78 CrossRef CAS; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 8, e1327 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- (a) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed; (b) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- M. Cossi and V. Barone, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef.
- R. Bouten, E. J. Baerends, E. van Lenthe, L. Visscher, G. Schreckenbach and T. Ziegler, J. Phys. Chem. A, 2000, 104, 5600–5611 CrossRef CAS.
- D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional electrochemical data, table of coordinates for compounds used in DFT calculations. CCDC 2251444. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00903c |
| This journal is © The Royal Society of Chemistry 2023 |