
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid synthesis of cerium-UiO-66 MOF nanoparticles for photocatalytic dye degradation†
Ehsan Ezzatpour
Ghadim
a,
Marc
Walker
 b and
Richard I.
Walton
*a
b and
Richard I.
Walton
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: r.i.walton@warwick.ac.uk
bDepartment of Physics, University of Warwick, Coventry CV4 7AL, UK
First published on 18th July 2023
Abstract
An unprecedented synthesis method is used to form a series of Ce-UiO-66-X (X = NH2, OH, H, NO2, COOH) metal–organic frameworks by precipitation from mixed solvents, with instantaneous crystallisation on combining separate solutions of ligands and metal precursors. This allows the first direct synthesis of Ce-UiO-66-OH. Powder X-ray diffraction (PXRD) shows that all materials are pure phase with a broadened profile that indicates nano-scale crystallite domain size. The effect of different functional groups on the benzene-1,4-dicarboxylate linker within the UiO-66 structure has been investigated on degradation of two cationic (methylene blue and rhodamine B) and two anionic (Congo red, and Alizarin red S) dyes under UV and visible light irradiation at room temperature. Analysis of the dye adsorption in the absence of light is accounted for using pseudo-first order kinetics, and the Ce-UiO-66-NH2, Ce-UiO-66-OH, and Ce-UiO-66-H materials display a considerable photocatalytic activity to degrade Alizarin red S and Congo red rapidly between 1 and 3 minutes. The materials show excellent photostability and recyclability under UV and visible light, with no loss of crystallinity seen by PXRD and activity maintained over 5 cycles, with 16 hours photostability for Ce-UiO-66-NH2.
Introduction
Extensive efforts have been made during recent decades to develop a wide range of adsorbents, oxidising agents, and photocatalysts for the removal of pollutants from water that operate under different light illumination sources.1 A recent study on the products of the photodegradation shows that methylene blue (MB) is broken down into the non-toxic species CO2, H2O, Cl−, SO42−, and NO3−.2 Metal organic-frameworks (MOFs) are a class of porous materials developed since the early 1990s that provide good candidate materials to remove pollutants from the environment.3,4 The combination of different secondary building units (SBUs) and organic linkers forms an almost unlimited number of frameworks which can show considerable porosity with a diverse range of chemistry depending on the choice of metals and the functionality of the organic ligand.5 Around 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 different metal–organic frameworks (MOFs) had been recorded in the Cambridge Structural Databases by 2020.6 Porosity, high surface area, stability, and catalytic activities are the desirable characteristics of MOFs which make them excellent candidates for potential applications in gas storage, wastewater treatment, and CO2 conversion.7
000 different metal–organic frameworks (MOFs) had been recorded in the Cambridge Structural Databases by 2020.6 Porosity, high surface area, stability, and catalytic activities are the desirable characteristics of MOFs which make them excellent candidates for potential applications in gas storage, wastewater treatment, and CO2 conversion.7
One of the most well-known MOF structures is UiO-66 which has garnered a tremendous amount of interest in research and industry.8 The parent material is a zirconium benzene-1,4-dicarboxylate (BDC) that contains hexameric clusters linked to give a three-dimensional framework with considerable porosity and high stability, both thermal and chemical. The UiO-66 structure was reported by the Lillerud group in 2008 and it has since been synthesised from many different methods, using various metals, and modified linkers.9 The structure can be modified based on the needs of different applications by modification of the ligands or inclusion of other tetravalent metals and isoreticular versions of the structure are known, prepared with extended versions of the ligand.10,11 Among the tetravalent metals that can be used to form M-UiO-66 (M = Ti, Zr, Hf, Ce, U, Th),8 Ce(IV) has low-lying unoccupied 4f orbitals that can accelerate ligand-to-metal charge transfer (LMCT) with the ability of conversion between its oxidised (IV) and reduced (III) states.12 LMCT can facilitate the separation of photogenerated electrons rather than trapping them between ground and excited states.13 Furthermore, the energy of LMCT as well as the bandgap in UiO-66 structures are tuneable by using electron donating groups on the BDC-X ligand, such as X = –NH2, –OH, and –SH. These groups work as antennae to harvest photons and transfer them to metal clusters.14 MOFs are typically known as semiconductors with band gaps between 1 and 5 eV.15 The combination of Ce(IV) and electron donating groups on ligands can reduce the band gap as well as increasing the energy of absorption in the UV and visible regions of solar light.16
Ce(IV) is a strong oxidising agent, and it can oxidise the functional groups on benzene-1,4-dicarboxylic acids used in the synthesis, which makes means that the choice of conditions to form Ce-UiO-66-X (X = NH2, OH, H, NO2, COOH) frameworks needs careful consideration.17,18 The possibility of reduction of Ce(IV) to Ce(III) means that a short reaction time is needed to produce a pure phase of Ce-UiO-66.19,20 Other studies have focussed on using mixed-metal versions of UiO-66 such as Ce/Zr or Ce/Ti,21,22 and it is worth noting that the Ti oxo-cluster cannot easily be formed owing to the small size of Ti4+.23 Much focus has been recently devoted on the synthesis of Ce/Ti-UiO-66-X (X = NH2, Br, H, NO2) materials; however, they show no obvious changes to their photocatalytic performances compared to Zr-UiO-66-NH2.24,25 Other studies show there is a possibility of incorporating BDC-X (X = NH2, OH) in UiO-66. In the case of Zr-UiO-66-NH2, this was used to modify adsorptive properties,26 while for Ce-UiO-66-NH2, produced by post synthesis ligand exchange, redox catalysis was investigated for hydrolysis of a nerve agent simulant.27
Herein, we present a novel, rapid synthesis method to form Ce based UiO-66 with different ligand functional groups (NH2, OH, H, NO2, COOH) at room temperature with high yield. One significant outcome of developing a room temperature synthesis method is that it opens the possibility of scaling for industrial production of MOFs: this is important since most MOF materials must be prepared using heated solvents, in some cases above their boiling point, which implies high energy use, and may require complex reactor design. Notably the room temperature synthesis method allows the first direct synthesis of Ce-UiO-66-OH. The Ce-UiO-66-X materials as-prepared are phase pure and contain ultrafine crystal size domains. The effect of using electron donating groups on accelerating the photocatalytic activity have been investigated using two anionic and two cationic dyes, methylene blue (MB), rhodamine B (RhB), Congo red (CR), and Alizarin red S (AR) in this study. Some of the materials show strong photocatalytic activity as well as photostability and recyclability.
Experimental materials
Chemical reagents
Ammonium cerium nitrate ((NH4)2Ce(NO3)6) (Merck), 2-nitroterephthalic acid (Sigma-Aldrich) 99.99%, 2-aminoterephthalic acid (Fisher Scientific) 99%, benzene-1,4-dicarboxylic acid (Sigma-Aldrich) 98%, 2-hydroxy-1,4-benzenedicarboxylic acid (Merck) 97%, 1,2,4-benzenetricarboxylic acid (Alfa Aesar) 99%, potassium hydroxide (KOH, Sigma-Aldrich) 90% were used as provided.Solvents
Acetic acid (Merck) 100%, DMF (N,N-dimethylformamide Scientific Laboratory Supplies Ltd) 99.8%, and triethylamine (Sigma-Aldrich) 99.5% were used as supplied. Deionised water (18 MΩ cm) was obtained using a simplicity UV ultrapure water system.Dyes
Methylene blue (MB, Merck) 82%, rhodamine B (RhB, VWR International Ltd) 92%, Congo red (CR, Fisher Scientific) 35%, Alizarin red S (AR, VWR International UK Ltd) 99%, were purchased and used without further purification.Characterisation
Powder X-ray diffraction (PXRD) measurements were performed using a Panalytical Empyrean diffractometer operating with copper Kα1/2 radiation with an average wavelength of 1.5418 Å. Diffraction data were recorded between 5 and 50°2θ. The powdered sample was pressed into a flat sample silicon holder. The software GSAS-II was used to perform profile fits of the PXRD patterns in order to determine unit cell parameters.28 Fourier transform infrared spectroscopy (FT-IR, Bruker ALPHA platinum ATR spectrometer) was used to identify characteristic functional groups of organic species, and to investigate binding of adsorbates. Each measurement consisted of 8 scans combined, across a spectral range of 4000–600 cm−1. Thermogravimetric analysis (TGA) was carried out using a Mettler Toledo STARe instrument between 26 and 1000 °C under air with a flow rate of 50 ml per minute. Typically 10 mg of each sample was used held in a ceramic crucible. Scanning electron microscopy (SEM) was performed using a Zeiss Gemini with accelerating voltage 3 kV and resolution of 1 nm at 1 kV and 0.6 nm, UV-Vis diffuse reflectance spectroscopy (DRS) was measured using a Shimadzu UV-2600 spectrophotometer with an integrating sphere, and BaSO4 employed as a reference. The UV-Vis spectra were measured at room temperature between 200–800 nm.X-ray photoelectron spectroscopy (XPS) was used to identify the surface metal oxidation states and band gaps of the samples using a Kratos AXIS Ultra DLD (Manchester, UK) in a UHV system with a base pressure below 1 × 10−10 mbar. The sample was excited with X-rays from a monochromated Al Kα source (1486.7 eV), with the photoelectrons being detected at a 90° take-off angle with respect to the sample surface. Curve fitting was performed using the CasaXPS package, incorporating Voigt (mixed Gaussian–Lorentzian) line shapes and Shirley backgrounds for all regions.29
Photoluminescence (PL) spectroscopy was measured using a Renishaw in via Reflex Raman Microscope to determine excitation wavelength at 325 nm (PL only) at 0.1% power, nominally 0.006 mW. The analysis ran for 10 seconds for two scans. N2 adsorption analysis was determined by a Micromeritics ASAP2020 gas adsorption apparatus. The surface area was estimated at 77 K for 7 hours based on the relative pressure changes based on Brunauer–Emmett–Teller (BET) theory. Each sample was degassed between 150 and 200 °C for 12 hours according to its stability to remove any excess solvents captured within the pores. Zeta-potential measurements were performed on an Anton-Paar Litesizer 500 using Omega cuvettes. Samples were run at 25 °C with a 5 seconds temperature stabilisation time.
Preparation of MOFs
(NH4)2Ce(NO3)6 (0.15 g) was dissolved in 1.5 mL water (23 °C, solution 1) and 0.15 g of H2BDC-X (X = NH2, OH, H, NO2, COOH) were dissolved separately in 15 mL DMF (solution 2), using an ultra-sound bath (240 W) to obtain transparent solutions. A third solution, solution 3 was prepared: for Ce-BDC-X (X = NH2 and NO2), 3 mL acetic acid and 1 mL KOH (10 M) into 15 mL water at 55 °C; for Ce-BDC-OH, 1.5 mL acetic acid and 0.5 mL KOH (10 M) into 15 mL water at 53 °C; for Ce-BDC-H, 1 mL acetic acid and 9 drops of Et3N into 5 mL water (53 °C) for; for Ce-BDC-COOH, 3 mL acetic acid and 0.2 mL of Et3N into 10 mL water at 53 °C. The three solutions were added to each other and a precipitate immediately appeared. The colour and yield of precipitate for Ce-BDC-NH2, Ce-BDC-OH, Ce-BDC-H, Ce-BDC-NO2, and Ce-BDC-COOH were dark brown (58%), brown (37%), pale yellow (51%), pale yellow, yellow (48%), respectively. The solid samples were recovered by centrifugation (4000 rpm for 3 minutes) washed 3 times with acetone and dried at 110 °C in air.Photodegradation experiments
The photocatalytic activities of the Ce-UiO-66-X series were studied using degradation of methylene blue (MB), rhodamine B (RhB), Congo red (CR), and Alizarine Red S (AR) under UV and visible-light irradiation at room temperature. A 6 watt handheld UV lamp (UVGL-55) was used with 254 nm wavelength. A 400 W, portable site halogen light (SFD limited BA22) was employed as a source of visible light (8RT, R10W44). In each analysis 25 mg of Ce-UiO-66-X (X = H, NO2, NH2, OH, COOH) was used in 200 mL solutions of MB (4 mg L−1), RhB (6 mg L−1), CR (15 mg L−1), and AR (9 mg L−1). Before running the analysis, the photocatalysts were sonicated for 2 minutes using an ultra-sonic bath (240 W). During the photocatalytic measurement, MOFs and the dye solutions were stirred by a magnetic follower via a stirrer plate to achieve a homogeneous suspension. The analysis interval for both UV and visible light was typically 10 minutes although in some cases, reduced to every minute to detect fast degradation changes. An Eppendorf centrifuge was used to remove the MOF particles and the remaining solution was analysed. The absorbance (A) changes in photodegradation process were measured by UV-Vis spectroscopy (PerkinElmer Lambda XLS/XLS+) using eqn (1): | (1) |
C 0 and Ct are initial concentration and the concentration at time t, respectively.30
Table 1 shows the dye structures and their λmax.



The adsorption measurements were carried out in the dark under otherwise the same conditions to determine adsorption of the dyes by the materials. Pseudo-first and pseudo-second order kinetic model equations were tested to determine the rate and capacity of adsorption between dyes and the surface of UiO-66 series in dark measurement.35
The adsorption capacities (qe and qt) of the UiO-66 series towards dyes in solution were calculated using the following equations:
Adsorption capacity (qe) at equilibrium concentration:
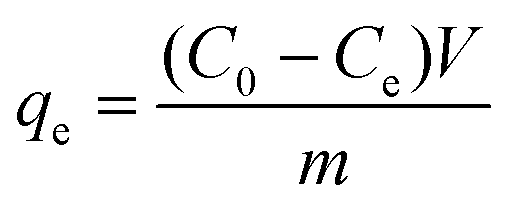 | (2) |
Adsorption capacity at time (qt):
 | (3) |
C 0 is an initial concentration at reaction time (t = 0), Ct is concentration at reaction time (t). Ce, V, and m are equilibrium concentration, the volume of solution (in L), the mass of adsorbent (in g), respectively.
The pseudo-first order expression used takes the form:
 | (4) |
q e and qt are the amount of dye adsorbed at equilibrium and at time t, respectively, (mg g−1) and k1 (min−1) is a rate constant. The value of k1 was determined from plots of log (qe − qt) versus t.
The pseudo-second order expression was used in the form:
 | (5) |
k 2 (g mg−1 min−1) is the rate constant for adsorption. A plot of t/qtversus t gives the values of qe and k2.
Results and discussion
The precipitation of the Ce-UiO-66-X (X = NH2, OH, H, NO2, COOH) materials can be compared to other aqueous-based room temperature methods recently reported in literature which have been performed for periods between 15 min and few hours.36 For example, Ce-UiO-66-NH2 was synthesised in 2 hours at room temperature with yield of 42%,37 which is lower than the yield of 58% by the method reported here. The synthesis of Ce-UiO-66-NH2 by Zaremba et al. in water and DMF with different modulators (acetic and formic acid) showed that using DMF required a time of synthesis from 30 to 15 min.36 Su et al. prepared Ce-UiO-66-NH2 at 100 °C for 3 hours, but the material shows poor crystallinity.38FT-IR spectroscopy, Fig. 1, confirms that BDC with different functional groups is introduced successfully in all five samples of Ce-UiO-66-X. The asymmetric stretching vibrations of the carboxylate group in BDC-NH2 and the symmetric vibrations of the amino group appear at 1374 and 1404 cm−1, and at 1502 and 1577 cm−1, respectively. The stretching vibrational bands in the fingerprint region are seen at 760–800, cm−1 in all five Ce-UiO-66 structures, and they are assigned to the μ3-O stretching, Ce(O–C), and μ3-OH stretching.39,40 The O–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bands of carboxylate group of BDC-NH2 appear at 3461 and 1663 cm−1, respectively, while NH2 stretching and bending peaks are observed at 3344 and 1576 cm−1. The broad peaks between 3000 and 3500 cm−1 in both Ce-UiO-66-OH and Ce-UiO-66-COOH are due to O–H stretching modes. Regarding NO2, C–N stretching and NO2 asymmetric bands appear at 1357 and 1533 cm−1, respectively. Based on other studies, there are some particular bands belonging to NO2 expected at 1290–1360 cm−1 (stretching) and 1537 cm−1 (asymmetric),41,42 although these are difficult to resolve because of overlap with neighbouring bands in the spectrum, Fig. 1.
O stretching bands of carboxylate group of BDC-NH2 appear at 3461 and 1663 cm−1, respectively, while NH2 stretching and bending peaks are observed at 3344 and 1576 cm−1. The broad peaks between 3000 and 3500 cm−1 in both Ce-UiO-66-OH and Ce-UiO-66-COOH are due to O–H stretching modes. Regarding NO2, C–N stretching and NO2 asymmetric bands appear at 1357 and 1533 cm−1, respectively. Based on other studies, there are some particular bands belonging to NO2 expected at 1290–1360 cm−1 (stretching) and 1537 cm−1 (asymmetric),41,42 although these are difficult to resolve because of overlap with neighbouring bands in the spectrum, Fig. 1.
 | ||
| Fig. 1 FT-IR spectra of Ce-UiO-66-X (X = NH2, OH, H, NO2, COOH) materials. | ||
Fig. 2 shows PXRD patterns of the Ce-UiO-66-X (X = NH2, OH, H, NO2, COOH) which confirm the phase purity and crystallinity of the samples. The unit cell parameters were determined by Pawley refinement analysis based on the expected UiO-66 crystal structure, as shown on Fig. 2. The analysis showed that the unit cell for all samples is cubic with space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. The broadness of the Bragg peaks in four patterns (Ce-UiO-66-X, X = NH2, OH, H, NO2) is likely due to the small crystal domain sizes, which are between 15 and 24 nm, as determined from Scherrer analysis of the diffraction profile. Ce-UiO-66-COOH, however, shows a crystalline domain size of 126 nm, with the Bragg peaks superimposed on an amorphous background. A previous study on Ce-UiO-66-COOH by Zaremba et al. showed the presence of an amorphous phase.36 The cubic unit cell parameter is similar for all five samples, between 21.534 and 21.621 Å, see Table 2. The unit cell parameters in the materials reported here are slightly larger than other reports on Ce-UiO-66 materials for example, Redfern et al. found a value of 21.488 Å (ref. 43) and Lomachenko et al. also found 21.488 Å.44
m. The broadness of the Bragg peaks in four patterns (Ce-UiO-66-X, X = NH2, OH, H, NO2) is likely due to the small crystal domain sizes, which are between 15 and 24 nm, as determined from Scherrer analysis of the diffraction profile. Ce-UiO-66-COOH, however, shows a crystalline domain size of 126 nm, with the Bragg peaks superimposed on an amorphous background. A previous study on Ce-UiO-66-COOH by Zaremba et al. showed the presence of an amorphous phase.36 The cubic unit cell parameter is similar for all five samples, between 21.534 and 21.621 Å, see Table 2. The unit cell parameters in the materials reported here are slightly larger than other reports on Ce-UiO-66 materials for example, Redfern et al. found a value of 21.488 Å (ref. 43) and Lomachenko et al. also found 21.488 Å.44
 | ||
| Fig. 2 Fitted powder XRD patterns of Ce-UiO-66-X series showing the experimental pattern (red line), Pawley refinement (black line), difference plot (green line), and Bragg peak positions (blue). (a) Ce-BDC-NH2, (b) Ce-BDC-OH, (c) Ce-BDC-H, (d) Ce-BDC-NO2, and (e) Ce-BDC-COOH. | ||
m
| Structures | Crystal domain size (nm) | a (Å) | Unit cell volume (Å3) | GOF |
|---|---|---|---|---|
| Ce-UiO-66-NH2 | 18.93 (±0.2) | 21.548(7) | 10006.118 (±3.089) |
1.14 |
| Ce-UiO-66-OH | 20.82 (±0.1) | 21.542(8) | 9997.851 (±3.592) | 1.18 |
| Ce-UiO-66-H | 15.11 (±0.1) | 21.621(4) | 10107.759 (±3.448) |
1.08 |
| Ce-UiO-66-NO2 | 24.67 (±0.1) | 21.568(7) | 10034.010 (±1.917) |
1.23 |
| Ce-UiO-66-COOH | 126.45 (±0.38) | 21.534(9) | 9986.894 (±1.604) | 1.42 |
SEM images of the Ce-UiO-66 series are shown in Fig. 3. The size of Ce-UiO-66-NH2 particles is between 20 and 60 nm and the primary particles appear to be agglomerated, which may be due to the sample preparation used to make the measurements.
 | ||
| Fig. 3 SEM images of (a), Ce-UiO-66-NH2, (b) Ce-UiO-66-COOH, (c) Ce-UiO-66-NO2, (d) Ce-UiO-66-OH, (e) Ce-UiO-66-H. | ||
As can be seen in Fig. 3b, the size of particles in Ce-UiO-66-COOH is between 50 and 90 nm, and although is different from crystal domain size determined by PXRD, of 110 nm, PXRD is probably sensitive to the largest crystallites present. The shape of particles is similar for Ce-UiO-66-NH2. The morphology of Ce-UiO-66-NO2 is quite different from other members as it shows stacked plate-like particles. Based on Fig. 3c, the particle size of Ce-UiO-66-NO2 is almost 200 nm which is rather different from the PXRD analysis (24.67 nm), suggesting that the plates are agglomerates of primary crystallites, see Table 2. The size of particles in Ce-UiO-66-OH is between 10 and 30 nm as can be observed from Fig. 3d, which is similar to the value from PXRD refinement (20 nm). Regarding the shape and phase of particles seen in Fig. 3e, Ce-UiO-66-H shows agglomerated particles in addition to the more isotropic shapes seen in the other samples.
DR/UV-Vis spectra were measured over wavelengths between 200 and 800 nm, as shown in Fig. 4a. It is confirmed that all five photocatalysts can harvest the photons in both visible and UV regions; however, Ce-UiO-66-NH2 and OH show narrower band gaps than the rest of samples. The charge transfer between organic linkers and metal nodes in the region 200 to 400 nm is attributed to ligand to metal charge transfer (LMCT).45 The flat plateau between 200 and 400 nm arises from the strong charge transfer between 2p of O2− on carboxylic groups of organic linkers and Ce 4f orbitals.24 A negative energy of LMCT (meaning low-lying unoccupied metal orbitals with an energy lower than the donor level), as well as suppressing the recombination of photogenerated electrons among excitation states causes the enhancement of efficiency of electron transitions between the cluster and linker.46 Ce(IV) with low lying and empty 4f orbitals enhances the energy of LMCT (ELMCT) and extends the lifetime of photoexcited states leading to a narrower band gap as well as covering the energy of absorption in both the UV and visible regions.47 The ELMCT can be tuned by introduction of electron donating functional groups such as NH2 or OH into the MOF ligands, which cause a bathochromic shift.48 Although all five Ce-based UiO-66 should be able to harvest visible light, the Ce-UiO-66-NH2 and OH materials cover all regions of visible light wavelengths. The Tauc plot method was used to estimate the band gap with the curve as in eqn (6):
| αhν = A(hν − Eg)n/2 | (6) |
 | ||
| Fig. 4 (a) DR/UV-Vis spectra of the Ce-UiO-66-X series, (b) baseline-correction of Tauc plots, (c) Tauc plot analysis for determining band gaps with the extrapolated lines shown and (d) PL analysis. | ||
α, h, ν, A, Eg, and n consecutively are the adsorption coefficient of photocatalysts, Planck constant, light frequency (or photon energy), constant, band gap, and factor (n) which depends on the type of optical transition in photocatalysts.49 As can be seen in Fig. 4b and c, the band gaps are estimated after baseline correction from the intercept of the linear region of the curve on the x-axis. Ce-UiO-66-NH2 (2.36 eV ± 0.022), Ce-UiO-66-COOH (2.53 ± 0.024 eV), Ce-UiO-66-H (2.55 ± 0.023 eV), Ce-UiO-66-NO2 (2.88 ± 0.027 eV), and Ce-UiO-66-OH (2.92 ± 0.028 eV). Campanelli et al. reported 2.86 eV for Ce-UiO-66-H and 2.95 eV for Ce-UiO-66-NO2;50 however, Chen et al. found 2.63 and 2.60 eV for Ce-UiO-66-H and Ce-UiO-66-NO2, respectively. Zhao et al. reported the band gap of Ce-UiO-66-H as 2.81 eV.51 Another study by Mu et al. showed that the band gaps of Zr-UiO-66-H and Zr-UiO-66-NH2 were 3.91 and 2.83 eV, respectively.52 Wu et al. used density functional theory to calculate the band gap of UiO-66(Ce)-H as 2.66 eV.12 The range of values reported by different authors for materials of nominally the same compositions is likely to be due to the presence of different levels of defects (either missing linkers, missing clusters, or the presence of modulators) which may arise when different synthesis methods are used. It is presently difficult to rationalise theoretically this complex dependency of electronic structure on defect chemistry.
Photoluminescent (PL) curves of the Ce-UiO-66 series are shown in Fig. 4c. This analysis allows an estimate of the trapping of electrons among excited states.53,54 Ce-UiO-66-X (X = NH2, H, OH) do not show any significant emissions attributed to the repressed recombination of charges. Only a minor hole/electron recombination feature appears at 430 nm for Ce-UiO-66-H. The spectra of Ce-UiO-66-NO2 and Ce-UiO-66-COOH reveal an emission peak centered at 430 nm which is attributed to the maximum electron–hole recombination, especially for Ce-UiO-66-COOH. Based on recent studies, missing clusters reduce the photocatalytic enhancement.55 The ELMCT response is dominated by missing clusters leading to hole–electron recombination; therefore, the intensity of peaks in the PL spectra can be relevant to the density of defects.
XPS was used to provide details about the electronic properties of the metal cations in the Ce-UiO-66-X series (X = NH2, OH, H, NO2, COOH), see Fig. 5 and Table S1a, b,† with the analysis based on previous interpretations in the literature.56–59 The XPS spectra of Ce(IV) 3d5/2 and 3d3/2 regions for Ce-UiO-66-NH2 are deconvoluted into six components and are assigned to Ce(IV) oxo-cluster formation, appearing between 884.6 and 917.3 eV. The signatures of Ce(III) 3d5/2 and 3d3/2 are observed between 882 and 911 eV. Information about the Ce oxidation state is shown in Table 3. It can be observed that the surface is dominated by the presence of Ce(III) with the ratio of Ce(III)/Ce(IV) largest at 2.2 for Ce-UiO-66-NH2. This may be due to the sample preparation from DMF and warm water leading to reduction of surface Ce(IV).60,61 Ce-UiO-66-X (NO2 and COOH) have a smaller amount of Ce(III), with Ce(III)/Ce(IV) ratios of 1.3 and 1.7. The presence of mixed Ce(III)/Ce(IV) plays a significant role in enhancing the ELMCT, harvesting the photon energy, and moving Eabs to the visible light region24 and the reduction of Ce(III) to Ce(IV) under light irradiation can provide an interior redox cycle.47 Another report on the synthesis of Ce-UiO-66-NH2 by a solvothermal method showed that most Ce(IV) is reduced to Ce(III), as shown by the XPS spectrum missing the Ce(IV) 3d3/2 feature 920.1 eV.38 As can be seen in Fig. 4c, Ce-UiO-66-X (NO2 and COOH) have a maximum rate of hole–electron recombination among all the materials as determined by PL. As can be seen in Fig. S1,† the oxygen 1s features in the XPS are observed between 532.1 and 535 eV, and lattice oxygen (Olatt) and surface-active oxygen (Osur) appear at 532.5 and 533.2 eV, respectively.62 Oxygen vacancies allow the internal transfer of electrons between the oxidised and reduced states to produce (˙OH) and superoxide (˙O2−) free radicals.62 The intensity of Olatt is almost constant in all samples. The role of Osur is important to increase the surface adsorption because they can facilitate photocatalytic degradation at the MOF surface.63 The uncoupled N (1s) appears at 399 and 402 eV, and 402 eV, assigned to C–N64 which is related to the NH2 and NO2 functional groups of in Ce-UiO-66-X and is consistent with the FT-IR results in Fig. 1. The C 1s region of Ce-UiO-66-X(NH2 and NO2) shows features at 285.4 (C–C/C–H), 286.3 (C–O), 287.2 (CO), 288 (CO–O), and 290.6 eV (carbonates), see Fig. S1.† The C–O/C–N signals in the spectrum confirms the presence of amine and hydroxy functional groups, which may be derived from the solvent DMF, and acetic acid used for the synthesis of all samples.65,66
 | ||
| Fig. 5 Ce 3d5/2 and 3d3/2 region of the XPS spectra of the Ce-UiO-66-X series, (a) NH2, (b) NO2, (c) OH, (d) H, (e) COOH. | ||
| Sample | Ce 3d fraction (%) | Ce(III)/Ce(IV) | |
|---|---|---|---|
| Ce(III) | Ce(IV) | ||
| Ce-UiO-66-NH2 | 69.1 | 30.9 | 2.2 |
| Ce-UiO-66-NO2 | 57 | 43 | 1.3 |
| Ce-UiO-66-OH | 74.3 | 25.5 | 2.9 |
| Ce-UiO-66-H | 72.8 | 27.2 | 2.7 |
| Ce-UiO-66-COOH | 63.1 | 36.9 | 1.7 |
The photocatalytic activities of the Ce-UiO-66-X series was determined by examining decolourisation of the dyes MB, RhB, CR, and AR under UV and visible light. Ce-UiO-66-X (X = NH2, OH, H) show a notable photocatalysis performance particularly for anionic dyes (CR and AR). The photocatalytic activities of porous materials must also consider strong adsorption due to various possible interactions, including high surface area, pore-filing/breathing, electrostatic attraction between cluster nodes and functional groups on dyes, and π–π interactions of unsaturated bonds between organic linkers with dyes, acid base interaction, and hydrogen bonding connections.67,68 Therefore, adsorption must be investigated to define properly the nature of photocatalytic enhancement. Some of these characteristics that might affect adsorption are included in Table 4. The surface charge (zeta potential) shows that four MOFs are almost neutral and only Ce-UiO-66-NO2 has a significant negative charge of −2.221 mV. Ce-UiO-66-NH2 is the only material with a small positive surface charge (+0.161 mV). The values of zeta potential are closed to zero which demonstrates that all these MOFs have a potential capacity to adsorb both anionic and cationic dyes.69 It should be noted that the large molecular size of the dyes that have been studied means that they are highly unlikely to be adsorbed into the pores of the UiO-66, and so any adsorption will take place on the surface.
| Samples | Zeta (mV) | BET surface area (m2 g−1) | Band gap (eV) | Ligand (TGA) |
|---|---|---|---|---|
| Ce-BDC-NH2 | +0.161 | 180.13 | 2.36 | 5.76 |
| Ce-BDC-OH | −0.355 | 308.25 | 2.92 | 5.38 |
| Ce-BDC-H | −0.176 | 385.13 | 2.54 | 5.44 |
| Ce-BDC-NO2 | −2.221 | 245.02 | 2.88 | 5.91 |
| Ce-BDC-COOH | −0.970 | 100.49 | 2.53 | 5.16 |
Pseudo-first and second order kinetic models were investigated to determine the nature and rate of adsorption (i.e. in the absence of light) among these photocatalysts in the same chemical environment and concentrations. All samples show a potential ability to adsorb dyes due to their high surface areas and also likely their surface charges, as shown Fig. S2, and Table S2.† Based on previous studies, defective UiO-66 structures can enhance the adsorption capacity in aqueous solution and increase the rate of dye degradation.70 The rate constant for adsorption in the pseudo-first order model (k1) for Ce-UiO-66-X (X = NH2, OH, H) is found to be between 3.3 and 9.7 × 10−2 min−1, see Fig. 6 and Table S2.† Considering all the materials studied, the pseudo first-order model gave consistently better fits to the adsorption data compared to the pseudo second-order model (see Fig. S2 and Table S2†). Zhao et al. studied the capacity and rate of adsorption of various dyes by Ce-UiO-66 (with BDC as linker) and found an adsorption capacity of 168.5 mg g−1 for RhB and 144.8 mg g−1 for MB, rather higher than for our samples, and their calculated rate constants by the pseudo-first order model were 0.1071 min−1 and 0.0879 min−1, respectively.71 Another study found the equilibrium adsorption capacity of MB by Ce-UiO-66 to be somewhat lower at 49 mg g−1 with k1 also lower at 1.4 × 10−2 min−1, while for CR the equilibrium adsorption capacity of 123 mg g−1 with k1 = 1.3 × 10−2 min−1 were reported.72
 | ||
| Fig. 6 The pseudo-first order kinetic fit of dye adsorption in the dark by UiO-66-X (X = OH, NO2, COOH, H, NH2) for (a) MB, (b) RhB, (c) CR, (d) AR. | ||
Having established the adsorption capacity of the materials towards the dyes, the results of photodegradation measurements are shown in Fig. 7. This shows a considerable effect towards removal of the dyes from aqueous solution. The favourable photocatalytic activity of Ce-UiO-66-X (X = NH2, OH, H), may be due to their lower band gaps, measured between 1.4 and 2.2 eV, coupled with the lowest rate of hole and electron recombination seen by DR/UV-Vis and PL, as well as the surface adsorption. In addition, NH2 is an electron donating group that can charge the cluster to increase the photocatalytic activity. Although H is a neutral functional group,73 Ce-UiO-66-H shows an exceptional performance in photodegradation, almost comparable to both X = NH2 and X = OH, which may be due to its defective structure. TGA results in Table 4 shows that all samples are defective with respect to ligand content, with ligand amount per Ce6 cluster between 5.16 and 5.91 compared to 6 expected for the perfect UiO-66 structure. Based on previous studies, structural defects may have some enhancements on energy of absorbance (Eabs) and energy of LMCT (ELMCT), although the mechanism is still unresolved.74,75 The ideal UiO-66 structure is highly coordinated therefore missing linker defects likely makes the clusters more accessible for photocatalytic activities.76
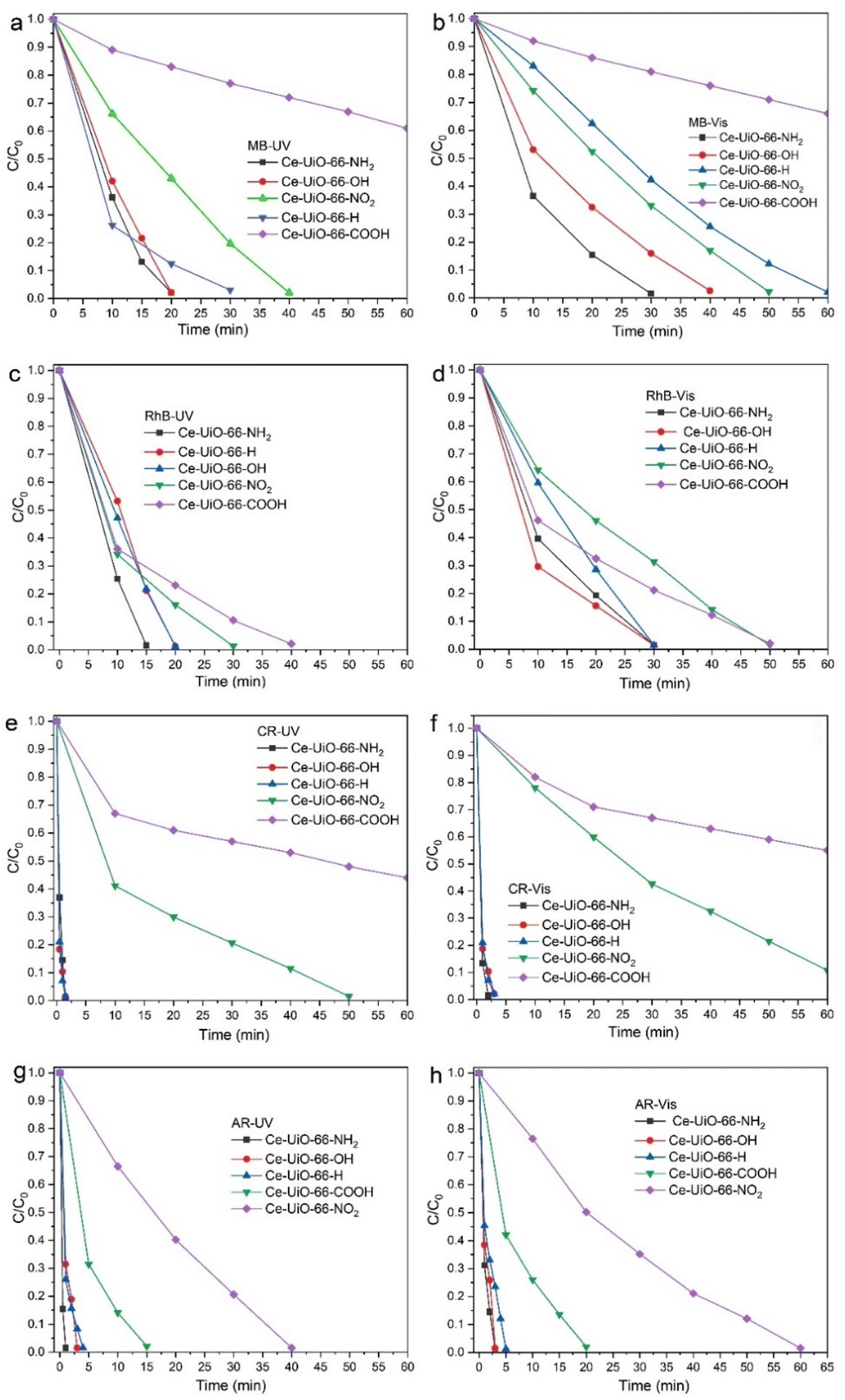 | ||
| Fig. 7 Photocatalytic measurements of Ce-UiO-66-X (X = NH2, OH, H, COOH, NO2) under UV and visible light towards four dye solutions (MB, RhB, AR, CR), (a) MB-UV, (b) MB-Vis, (c) RhB-UV, (d) RhB-Vis, (e) AR-UV, (f) AR-Vis, (g) CR-UV, (h) CR-Vis. | ||
To identify the potential enhancement of adsorption by the high surface area of the Ce-UiO-66-X series, the surface adsorption was also measured in the dark with similar conditions as for UV and visible light analyses, see Fig. S4a–q.† As can be seen in Table 4 and Fig. S3,† the surface area of the Ce-UiO-66-X (X = NH2, OH, H, COOH, NO2) is comparatively lower than materials reported by conventional synthesis methods.77 The materials with modified linkers all have lower surface areas than the parent material (X = H), which is expected when a bulky side group occupies some of the pore space, but even the parent has a lower measured surface area than other samples of Ce-UiO-66-H reported in the literature. For example, Lammert et al. reported a value of 1282 m2 g−1.20 The lower surface areas of the materials we have prepared may be due to incomplete removal of unreacted ligand precursors and/or solvent.
As can be seen at Fig. S4,† Ce-UiO-66-X (X = NH2, OH, H) in the dark showed at least 30 and 100 percent of surface adsorption between 30 and 60 min for almost all dyes. The Ce-UiO-66-H results in 100 percent of dye removal (in the dark) for AR in just 40 min, likely due to the strong hydrogen bond formation between the surface of Ce-UiO-66-H and water soluble groups on AR.78 The surface areas for Ce-UiO-66-X (H, OH, and NH2) are 385.1, 308.2, and 180.1 m2 g−1, respectively, and these have the highest rate of surface adsorption with active sites, see Table S2.† Furthermore, Ce-UiO-66-X (NH2 and H) have a strong surface adsorption, fading the red colour of AR in just 40 min and the rate of adsorption in Ce-UiO-66-H is 2.1 × 10−4 g mg−1 min−1. The amount of those MOFs was chosen to be 25 mg to control the adsorption while measuring the photocatalytic performance. The strong dye adsorption is likely due to hydrogen bonding as well as electrostatic interactions between clusters and the dyes soluble functionalities (Fig. S4†). RhB can be removed by Ce-UiO-66-H in 50 min which may be because of п-п stacking between organic linkers and dyes unsaturated bonds.79,80
In contrast, Ce-UiO-66-NO2 and COOH show weaker photodegradation primarily because of trapping of electrons among the excited states. The crystal size domain of Ce-UiO-66-COOH is 110 nm with a surface area of 100.49 m2 g−1 and these reduce the adsorption as well as the photocatalytic activity, see Table 4. However, it is still strong enough to decolourise the AR in just 15 min in UV and 20 min in visible light, see Fig. S4t.† The Ce-UiO-66-NO2 displays the weakest photocatalytic behavior in the series. Fig. S4i–l.† shows that adsorption plays a pivotal role in this structure towards both cationic and anionic dyes rather than showing any photocatalytic activities because NO2 is a withdrawing electron group.
The effect of photocatalytic activities of Ce-UiO-66-X (X = NH2, OH, H, COOH, NO2) was investigated for anionic and cationic dyes under UV and visible light, Fig. 7. As can be seen in Fig. 7a and b, Ce-UiO-66-X (X = NH2, OH) show a decolourisation in 20 min under UV irradiation; however, they have a weaker performance under visible light emission. Ce-UiO-66-H has a weaker photoactivity especially under visible light. Based on dark measurements, Ce-UiO-66-X (X-COOH, NO2) show a high level of surface adsorption in MB solution along with their photocatalytic activity under both light sources (see, Fig. S4i and q†); therefore, their activities in Fig. 7a and b, arise from their adsorption properties. Ce-UiO-66-COOH shows the poorest photocatalytic and surface adsorption, although this might be due to large crystal size domains and smaller surface area. Ce-UiO-66-X (X = NH2, OH, H, COOH, NO2) towards RhB have a different behaviour compared to MB as shown in Fig. 7c and d. Ce-UiO-66-NH2 has a good photocatalytic activity to decolourise RhB in 15 min and Ce-UiO-66-X (X = OH, H) can fade the colour of RhB in 20 min. Although there are some differences between Ce-UiO-66-X (X = OH, H) in UV light, they have similar behaviour in visible light and decolourise RhB in 30 min. Ce-UiO-66-X (X = COOH, NO2) show weaker photocatalytic activities in both UV and visible light towards RhB in 50 min. Adsorption is likely playing a significant role in those MOFs rather than their photocatalytic activities, based on the dark measurements, see Fig. S4i, r† and Fig. 7c, d.
The photocatalytic activities of Ce-UiO-66-X (X = NH2, OH, H, COOH, NO2) show similar results towards AR and in both UV and visible light, see Fig. 7e and f. Ce-UiO-66-NH2 shows the fastest photodegradation rates in both UV and visible light, 1 min and 3 min in UV and visible light, respectively. Ce-UiO-66-X (X = OH, H) show similar and fast rate between 3 and 5 min in both UV and visible light, respectively. In contrast to the behavior of Ce-UiO-66-COOH towards MB and RhB decolourisation, it shows a good photodegradation of AR in both UV and visible light (see Fig. 7e and f); however, Ce-UiO-66-NO2 still has the weakest photocatalytic activity, see ESI Fig. S4k and t.†
Ce-UiO-66-X (X = NH2, OH, H) displays a good photocatalytic activity and decolourise CR between 1.5 and 5 min in both UV and visible light irradiation (see Fig. 7g and h); however, Ce-UiO-66-X (X = COOH, NO2) have a similar behaviour in photodegradation to MB and RhB, see Fig. 7. The dark measurements show that for Ce-UiO-66-X (X = COOH, NO2) adsorption dominates their photocatalytic activities, see Fig. S4p and s.† Total organic carbon content analysis of the clear solution remaining after the Congo red dye was degraded by Ce-UiO-66-NH2 showed around 25% of the initial carbon remained in solution (see ESI†), proving that mineralisation of the dye has occurred.
For all photocatalysts, photostability, reusability, and water stability are necessary, and MOFs are often very sensitive towards pH or prolonged exposure to solution.81 The three best photocatalysts Ce-UiO-66-X (X = NH2, OH, H) were selected for measurement of photostability and recyclability. All three MOFs are stable under UV irradiation over 5 cycles and for Ce-UiO-66-NH2 the measurement was continued until 16 hours. FT-IR spectroscopy and PXRD confirm that there are not any considerable changes to Ce-UiO-66-NH2 after different times of UV exposure, see ESI, Fig. S5a and b.† Ce-UiO-66-OH and Ce-UiO-66-H show a promising photostability after 5 cycles continuously under UV light emission, see ESI, Fig. S6a and b.† The PXRD reveals that the Ce-UiO-66-H structure has a slightly distorted (222) Bragg peak (at 8.15°2θ) after UV light exposure which may be due to partially reduction of Ce(IV) to Ce(III). With the absence of electron donating groups on the BDC organic linker, the metal oxo-cluster cannot be charged enough to reverse Ce(III) to Ce(IV).82,83 In addition, the colour of Ce-UiO-66-H under UV irradiation changes from yellow to light brown, consistent with a permanent reduction of the material Fig. S7a.† The DR/UV-Vis shows that the band gap of reduced Ce-UiO-66-H is changed from 2.25 eV to 1.47 eV which can cause more light absorption during the photocatalytic activities, see ESI Fig. S7b and c.† It might explain why Ce-UiO-66-H shows good photocatalytic activities towards both anionic and cationic dyes. The change in colour of Ce-UiO-66-H after light irradiation may be because BDC is transformed to BDC-OH, as has been reported in the literature by Guo et al.84 It is likely that the transformation took place partially during the photocatalytic testing which is evidenced by the (002) peak in the XRD pattern being slightly distorted; however, the UiO-66 structure is still maintained, Fig. S6b.†
The recyclability experiments show that these three photocatalysts have a good capacity under both UV and visible light emission towards CR and AR, see Fig. 8a–c and Fig. S8a–l.† However, the photocatalytic activity of Ce-UiO-66-OH decreased from the 3rd cycles onwards. Multiple washing strategies were employed to remove any adsorbed dyes on these MOFs, with acetone, ethanol, and 0.1 mol of NaOH. The photocatalytic activity of Ce-UiO-66-NH2 and Ce-UiO-66-OH under both UV and visible light are the same for CR and AR showing that the photocatalytic activity in both MOFs is strong enough to be accelerated by any sources of light.
 | ||
| Fig. 8 Recyclability measurements of Ce-UiO-66-X (X = NH2, OH, H) in 5 cycles towards AR and CR decomposition, under visible light irradiation, and at room temperature, (a) Ce-UiO-66-NH2 with CR, (b) Ce-UiO-66-OH with AR under UV light, (c) Ce-UiO-66-H with AR. | ||
Among the MOF photocatalysts reported in the literature, only a few of them show exceptional photocatalytic activity as well as stability and recyclability, such as g-C3N4/MIL-125, g-C3N4/UiO-66 (Zr), UiO-66(NH2), BiOBr@UiO-66,85–87 see ESI Table S3.† It should be noted that three of these materials are composites, for which sample homogeneity may be an issue to achieve reproducibility. The Ce-UiO-66-X (X = NH2, OH, H) materials are stable and recyclable towards both cationic and anionic dyes and various sources of light. In addition, Ce-UiO-66-NO2 and Ce-UiO-66-COOH show good surface adsorption. The methods of synthesis of MOFs already reported in the literature for photocatalysis are exclusively solvothermal reactions for at least 12 hours; in contrast the Ce-UiO-66 materials are prepared by a simple precipitation at room temperature. The band gaps are typically between 2 and 3.80 eV except Ag/AgCl/ZIF-8 with exceptionally small band gap (0.5 eV) and this MOF can be a good rival with the Ce-UiO-66 materials because it can completely decolourise RhB in 16 min, see Table S3.† However, the preparation of this composite material is very time consuming, with several steps requiring at least 20 h, and the product must be dried at vacuum oven in 60 °C for 2 hours in each section of the preparation. It can also be noted that the photocatalytic activities as well as surface adsorption were not reported in visible light to determine fully the performance of this material as a photocatalyst.
Conclusions
This study reports a method to form a series of cerium MOFs UiO-66-X (X = NH2, OH, H, NO2, COOH) by a rapid synthetic process at room temperature, with high yield, and high phase purity. In terms of photocatalytic activity, the combination of Ce and donating electron groups on BDC provides versatile photocatalysts as well as strong surface adsorption. The band gap of the Ce-UiO-66 series is decreased dramatically to between 2.3 and 3.0 eV compared to the parent material Zr-UiO-66 which is reported between 3.5–4.2 eV. The MOFs show a good UV and visible light stability as well as recyclability. The most effective and stable MOF in this group is Ce-UiO-66-NH2, which possesses the smallest band gap (2.36 eV) and the lowest electron–hole recombination rate. It can perform strong dye decolourisation under UV and visible light irradiation in 1 and 3 min for the anionic dyes AR and CR, respectively, with 16 hours UV photostability, and five times recyclability proven. The UiO-66-X (X = NH2, OH, H) materials can be proposed as feasible photocatalysts for dye photodegradation. Their rapid synthesis by precipitation offers the prospect of scalable production and future work must consider long-term stability under environmental conditions. The mechanism of photocatalytic degradation of dyes over UiO-66 materials is complex, and as well as the interaction of the dyes with the MOF and the band positions of the MOF to allow electron transfer, the presence of defects is likely to be of significance in their activity. Further work is needed to provide a theoretical rationalisation of the role of defects on the reactivity of these materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
EEG is grateful to the University of Warwick for provision of a PhD scholarship and CARA for support. MW acknowledges financial support from the EPSRC-funded Warwick Analytical Science Centre (EP/V007688/1). Some of the equipment used in this research was provided by the University of Warwick Research Technology Platforms, and we thank Dr Rajan Randev, Dr David Walker, Dr Daniel Lester, Dr Ben Breeze, and Dr Chris Waldron, Katie Pickering, Jasmine Clayton, Dr Ryan Mushinski and James Coe for assistance with data collection. The research data supporting this publication can be accessed at: https://wrap.warwick.ac.uk/177554.References
- S. Dutta, B. Gupta, S. K. Srivastava and A. K. Gupta, Mater. Adv., 2021, 2, 4497–4531 RSC.
- I. Khan, K. Saeed, I. Zekker, B. Zhang, A. H. Hendi, A. Ahmad, S. Ahmad, N. Zada, H. Ahmad, L. A. Shah, T. Shah and I. Khan, Water, 2022, 14, 242 CrossRef CAS.
- S. Rojas and P. Horcajada, Chem. Rev., 2020, 120, 8378–8415 CrossRef CAS PubMed.
- S. Dhaka, R. Kumar, A. Deep, M. B. Kurade, S.-W. Ji and B.-H. Jeon, Coord. Chem. Rev., 2019, 380, 330–352 CrossRef CAS.
- S. M. Moosavi, A. Nandy, K. M. Jablonka, D. Ongari, J. P. Janet, P. G. Boyd, Y. Lee, B. Smit and H. J. Kulik, Commun. Chem., 2020, 11, 4068 CAS.
- A. Li, R. Bueno-Perez, S. Wiggin and D. Fairen-Jimenez, CrystEngComm, 2020, 22, 7152–7161 RSC.
- H. Li, L. Li, R.-B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, Energy Chem., 2019, 1, 100006 CrossRef.
- J. Winarta, B. Shan, S. M. McIntyre, L. Ye, C. Wang, J. Liu and B. Mu, Cryst. Growth Des., 2020, 20, 1347–1362 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- S. J. Garibay and S. M. Cohen, Angew. Chem., Int. Ed., 2010, 46, 7700–7702 CAS.
- I. Abánades Lázaro, C. J. R. Wells and R. S. Forgan, Angew. Chem., Int. Ed., 2022, 59, 5211–5217 CrossRef PubMed.
- X.-P. Wu, L. Gagliardi and D. G. Truhlar, J. Am. Chem. Soc., 2018, 140, 7904–7912 CrossRef CAS PubMed.
- J. G. Santaclara, F. Kapteijn, J. Gascon and M. A. van der Veen, CrystEngComm, 2017, 19, 4118–4125 RSC.
- H. Chen, C. Liu, W. Guo, Z. Wang, Y. Shi, Y. Yu and L. Wu, Catal. Sci. Technol., 2022, 12, 1812–1823 RSC.
- A. Kuc, A. Enyashin and G. Seifert, J. Phys. Chem. B, 2007, 111, 8179–8186 CrossRef CAS PubMed.
- J. Jacobsen, A. Ienco, R. D'Amato, F. Costantino and N. Stock, Dalton Trans., 2020, 49, 16551–16586 RSC.
- Y.-F. Zhang, Q. Wang, D.-X. Xue and J. Bai, Inorg. Chem., 2020, 59, 11233–11237 CrossRef CAS PubMed.
- M. Ronda-Lloret, I. Pellicer-Carreño, A. Grau-Atienza, R. Boada, S. Diaz-Moreno, J. Narciso-Romero, J. C. Serrano-Ruiz, A. Sepúlveda-Escribano and E. V. Ramos-Fernandez, Adv. Funct. Mater., 2021, 31, 2102582 CrossRef CAS.
- M. Lammert, C. Glißmann, H. Reinsch and N. Stock, Cryst. Growth Des., 2017, 17, 1125–1131 CrossRef CAS.
- M. Lammert, M. T. Wharmby, S. Smolders, B. Bueken, A. Lieb, K. A. Lomachenko, D. D. Vos and N. Stock, Chem. Commun., 2015, 51, 12578–12581 RSC.
- S. Payra, S. Ray, R. Sharma, K. Tarafder, P. Mohanty and S. Roy, Inorg. Chem., 2022, 61, 2476–2489 CrossRef CAS PubMed.
- L. Jin, H. Liu, A. Xu, Y. Wu, J. Lu, J. Liu, S. Xie, Y. Yao, L. Dong, M. Zhang, S. Kai and M. Fan, Microporous Mesoporous Mater., 2021, 317, 110997 CrossRef CAS.
- J. G. Santaclara, A. I. Olivos-Suarez, A. Gonzalez-Nelson, D. Osadchii, M. A. Nasalevich, M. A. van der Veen, F. Kapteijn, A. M. Sheveleva, S. L. Veber, M. V. Fedin, A. T. Murray, C. H. Hendon, A. Walsh and J. Gascon, Chem. Mater., 2017, 29, 8963–8967 CrossRef CAS.
- P. Parnicka, W. Lisowski, T. Klimczuk, A. Mikolajczyk and A. Zaleska-Medynska, Appl. Catal., B, 2022, 310, 121349 CrossRef CAS.
- S. Payra, S. Ray, R. Sharma, K. Tarafder, P. Mohanty and S. Roy, Inorg. Chem., 2022, 61, 2476–2489 CrossRef CAS PubMed.
- S. N. Tambat, P. K. Sane, S. Suresh, N. Varadan O, A. B. Pandit and S. M. Sontakke, Adv. Powder Technol., 2018, 29, 2626–2632 CrossRef CAS.
- F. A. Son, A. Atilgan, K. B. Idrees, T. Islamoglu and O. K. Farha, Inorg. Chem. Front., 2020, 7, 984–990 RSC.
- B. H. Toby and R. B. Von Dreele, Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan and J. Baltrusaitis, Appl. Surf. Sci., 2021, 5, 100112 CrossRef.
- J. Zhang, F. Li and Q. Sun, Appl. Surf. Sci., 2018, 440, 1219–1226 CrossRef CAS.
- A. Mills and N. Wells, Chem. Commun., 2015, 51, 4161–4163 RSC.
- N. Wu, P. Bai, T. Yang, H. Li, J. Zhang, Z. Chai and X. Wang, Photochem. Photobiol. Sci., 2020, 19, 1042–1053 CrossRef CAS PubMed.
- S. Chatterjee, N. Guha, S. Krishnan, A. K. Singh, P. Mathur and D. K. Rai, Sci. Rep., 2020, 10, 111 CrossRef CAS PubMed.
- L. Legan, K. Retko and P. Ropret, Microchem. J., 2016, 127, 36–45 CrossRef CAS.
- E. D. Revellame, D. L. Fortela, W. Sharp, R. Hernandez and M. E. Zappi, Cleaner Eng. Technol., 2020, 1, 100032 CrossRef.
- O. Zaremba, J. Andreo and S. Wuttke, Inorg. Chem. Front., 2022, 9, 5210–5216 RSC.
- S. Dai, E. Montero-Lanzuela, A. Tissot, H. G. Baldovi, H. García, S. Navalón and C. Serre, Chem. Sci., 2023, 14, 3451 RSC.
- Y. Su, W. Wang, W. Wang, A. Wang, Y. Huang and Y. Guan, J. Electrochem. Soc., 2022, 169, 030528 CrossRef CAS.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- A. Lin, A. A. Ibrahim, P. Arab, H. M. El-Kaderi and M. S. El-Shall, ACS Appl. Mater. Interfaces, 2017, 9, 17961–17968 CrossRef CAS PubMed.
- R. S. Andriamitantsoa, J. Wang, W. Dong, H. Gao and G. Wang, RSC Adv., 2016, 6, 35135–35143 RSC.
- S. Bernt, V. Guillerm, C. Serre and N. Stock, Chem. Commun., 2011, 47, 2838–2840 RSC.
- L. R. Redfern, M. Ducamp, M. C. Wasson, L. Robison, F. A. Son, F.-X. Coudert and O. K. Farha, Chem. Mater., 2020, 32, 5864–5871 CrossRef CAS.
- K. A. Lomachenko, J. Jacobsen, A. L. Bugaev, C. Atzori, F. Bonino, S. Bordiga, N. Stock and C. Lamberti, J. Am. Chem. Soc., 2018, 140, 17379–17383 CrossRef CAS PubMed.
- L. Ding, F. Bai, B. Borjigin, Y. Li, H. Li and X. Wang, J. Chem. Eng., 2022, 446, 137102 CrossRef CAS.
- S. Shirase, S. Tamaki, K. Shinohara, K. Hirosawa, H. Tsurugi, T. Satoh and K. Mashima, J. Am. Chem. Soc., 2020, 142, 5668–5675 CrossRef CAS PubMed.
- J.-X. Gu, H. Chen, Y. Ren, Z.-G. Gu, G. Li, W.-J. Xu, X.-Y. Yang, J.-X. Wen, J.-T. Wu and H.-G. Jin, ChemSusChem, 2022, 15, e202102368 CrossRef CAS PubMed.
- X.-P. Wu, L. Gagliardi and D. G. Truhlar, J. Am. Chem. Soc., 2018, 140, 7904–7912 CrossRef CAS PubMed.
- P. Makuła, M. Pacia and W. Macyk, J. Phys. Chem. Lett., 2018, 9, 6814–6817 CrossRef PubMed.
- M. Campanelli, T. Del Giacco, F. De Angelis, E. Mosconi, M. Taddei, F. Marmottini, R. D'Amato and F. Costantino, ACS Appl. Mater. Interfaces, 2019, 11, 45031–45037 CrossRef CAS PubMed.
- D. Zhao and C. Cai, Dyes Pigm., 2021, 185, 108957 CrossRef CAS.
- X. Mu, J. Jiang, F. Chao, Y. Lou and J. Chen, Dalton Trans., 2018, 47, 1895–1902 RSC.
- Y. L. Wang, S. Zhang, Y. F. Zhao, J. Bedia, J. J. Rodriguez and C. Belver, J. Environ. Chem. Eng., 2021, 9, 106087 CrossRef CAS.
- N. Kolobov, M. G. Goesten and J. Gascon, Angew. Chem., Int. Ed., 2021, 60, 26038–26052 CrossRef CAS PubMed.
- S.-Q. Wang, X. Gu, X. Wang, X.-Y. Zhang, X.-Y. Dao, X.-M. Cheng, J. Ma and W.-Y. Sun, Chem. Eng. J., 2022, 429, 132157 CrossRef CAS.
- C.-C. Hsieh, A. Roy, Y.-F. Chang, D. Shahrjerdi and S. K. Banerjee, Appl. Phys. Lett., 2016, 109, 223501 CrossRef.
- M. N. Revoy, R. W. J. Scott and A. P. Grosvenor, J. Phys. Chem. C, 2013, 117, 10095–10105 CrossRef CAS.
- E. Paparazzo, Mater. Res. Bull., 2011, 46, 323–326 CrossRef CAS.
- J. Fonseca de Lima, F. V. S. Moreno, B. A. T. Menezes, J. da Silva Barbosa, M. C. Waddington, S. A. Franklin, G. J. Clarkson, M. Walker, O. A. Serra and R. I. Walton, Dalton Trans., 2022, 51, 145–155 RSC.
- T. Caruso, E. Bedini, C. De Castro and M. Parrilli, Tetrahedron, 2006, 62, 2350–2356 CrossRef CAS.
- D. Hong, M. Murakami, Y. Yamada and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 5708–5716 RSC.
- Y. Zhang, H. Chen, Y. Pan, X. Zeng, X. Jiang, Z. Long and X. Hou, Chem. Commun., 2019, 55, 13959–13962 RSC.
- F. Wang, R. Xue, Y. Ma, Y. Ge, Z. Wang, X. Qiao and P. Zhou, RSC Adv., 2021, 11, 32955–32964 RSC.
- X. Fang, S. Wu, Y. Wu, W. Yang, Y. Li, J. He, P. Hong, M. Nie, C. Xie, Z. Wu, K. Zhang, L. Kong and J. Liu, Appl. Surf. Sci., 2020, 518, 146226 CrossRef CAS.
- S.-M. You, W. M. A. El Rouby, L. Assaud, R.-A. Doong and P. Millet, Hydrogen, 2021, 2, 58–75 CrossRef CAS.
- I. Simonsson, P. Gärdhagen, M. Andrén, P. L. Tam and Z. Abbas, Dalton Trans., 2021, 50, 4976–4985 RSC.
- R. M. Rego, G. Sriram, K. V. Ajeya, H.-Y. Jung, M. D. Kurkuri and M. Kigga, J. Hazard. Mater. Lett., 2021, 416, 125941 CrossRef CAS PubMed.
- F. Ahmadijokani, H. Molavi, M. Rezakazemi, S. Tajahmadi, A. Bahi, F. Ko, T. M. Aminabhavi, J.-R. Li and M. Arjmand, Prog. Mater. Sci., 2022, 125, 100904 CrossRef CAS.
- A. H. Ibrahim, W. A. El-Mehalmey, R. R. Haikal, M. E. A. Safy, M. Amin, H. R. Shatla, S. G. Karakalos and M. H. Alkordi, Inorg. Chem., 2019, 58, 15078–15087 CrossRef CAS PubMed.
- Y. Feng, Q. Chen, M. Jiang and J. Yao, Ind. Eng. Chem. Res., 2019, 58, 17646–17659 CrossRef CAS.
- D. Zhao and C. Cai, Dyes Pigm., 2021, 185, 108957 CrossRef CAS.
- R. M. Rego, G. Sriram, K. V. Ajeya, H.-Y. Jung, M. D. Kurkuri and M. Kigga, J. Hazard. Mater., 2021, 416, 125941 CrossRef CAS PubMed.
- R. Gupta, G. Kumar and R. Gupta, Inorg. Chem., 2022, 61, 7682–7699 CrossRef CAS PubMed.
- S.-Q. Wang, X. Gu, X. Wang, X.-Y. Zhang, X.-Y. Dao, X.-M. Cheng, J. Ma and W.-Y. Sun, J. Chem. Eng., 2022, 429, 132157 CrossRef CAS.
- X. Feng, J. Hajek, H. S. Jena, G. Wang, S. K. P. Veerapandian, R. Morent, N. De Geyter, K. Leyssens, A. E. J. Hoffman, V. Meynen, C. Marquez, D. E. De Vos, V. Van Speybroeck, K. Leus and P. Van Der Voort, J. Am. Chem. Soc., 2020, 142, 3174–3183 CrossRef CAS PubMed.
- M. A. Nasalevich, M. G. Goesten, T. J. Savenije, F. Kapteijn and J. Gascon, Chem. Commun., 2013, 49, 10575–10577 RSC.
- M. R. DeStefano, T. Islamoglu, S. J. Garibay, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 1357–1361 CrossRef CAS.
- J. Liu, X.-Y. Zhang, J.-X. Hou, J.-M. Liu, X. Jing, L.-J. Li and J.-L. Du, J. Solid State Chem., 2019, 270, 697–704 CrossRef CAS.
- M. S. Khan, M. Khalid and M. Shahid, Mater. Adv., 2020, 1, 1575–1601 RSC.
- J. Qiu, Y. Feng, X. Zhang, M. Jia and J. Yao, J. Colloid Interface Sci., 2017, 499, 151–158 CrossRef CAS PubMed.
- G. Tan, R.-Q. Jia, W.-L. Wu, B. Li and L.-Y. Wang, Cryst. Growth Des., 2022, 22, 323–333 CrossRef CAS.
- K. Hendrickx, D. E. P. Vanpoucke, K. Leus, K. Lejaeghere, A. Van Yperen-De Deyne, V. Van Speybroeck, P. Van Der Voort and K. Hemelsoet, Inorg. Chem., 2015, 54, 10701–10710 CrossRef CAS PubMed.
- J. Gascon, M. D. Hernández-Alonso, A. R. Almeida, G. P. M. van Klink, F. Kapteijn and G. Mul, ChemSusChem, 2008, 1, 981–983 CrossRef CAS PubMed.
- Z. Guo, Q.-C. Luo, L. Qin, Z. Tian, Y.-Z. Zheng, Y. Ma and Y. Qu, Inorg. Chem. Front., 2022, 61, 9557–9563 CrossRef CAS PubMed.
- V. Muelas-Ramos, M. J. Sampaio, C. G. Silva, J. Bedia, J. J. Rodriguez, J. L. Faria and C. Belver, J. Hazard. Mater., 2021, 416, 126199 CrossRef CAS PubMed.
- X. Zhang, Y. Yang, W. Huang, Y. Yang, Y. Wang, C. He, N. Liu, M. Wu and L. Tang, Mater. Res. Bull., 2018, 99, 349–358 CrossRef CAS.
- R. Bibi, Q. Shen, L. Wei, D. Hao, N. Li and J. Zhou, RSC Adv., 2018, 8, 2048–2058 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt00890h |
| This journal is © The Royal Society of Chemistry 2023 |