
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvaluation of triphenylene-based MOF ultrathin films for lithium batteries†
Isabel
Ciria-Ramos
 ab,
Inés
Tejedor
ab,
Lucía
Caparros
ab,
Beatriz
Doñagueda
ab,
Oscar
Lacruz
ab,
Ainhoa
Urtizberea
ac,
Olivier
Roubeau
a,
Ignacio
Gascón
ab and
Marta
Haro
*ab
ab,
Inés
Tejedor
ab,
Lucía
Caparros
ab,
Beatriz
Doñagueda
ab,
Oscar
Lacruz
ab,
Ainhoa
Urtizberea
ac,
Olivier
Roubeau
a,
Ignacio
Gascón
ab and
Marta
Haro
*ab
aInstituto de Nanociencia y Materiales de Aragón (INMA), CSIC-Universidad de Zaragoza, Zaragoza, 50009, Spain. E-mail: mharo@unizar.es
bDepartamento de Química Física, Facultad de Ciencias, Universidad de Zaragoza, Plaza San Francisco, Zaragoza, 50009, Spain
cDepartamento de Ciencia y Tecnologia de Materiales y Fluidos, EINA, Universidad de Zaragoza, Zaragoza, 50018, Spain
First published on 10th May 2023
Abstract
Metal–organic frameworks (MOFs) are attractive candidates to meet the requirement of next-generation batteries, as functional materials with a high surface area, well-defined metal centers, and organic linkers through coordination bonds. Due to their great tunability, MOFs have been investigated as electrodes or electrolytes in lithium batteries and more recently as protective layers in anode-less batteries. Here, we synthesize a Ni3(HHTP)2 MOF directly at the air–liquid interface of a Langmuir trough and grow the electrode on a conductive substrate by the transference process. The characterization during Langmuir film formation shows that the addition of crystallization time during the compression process enhances the formation of 2D crystalline domains, as observed by in situ grazing-incidence X-ray diffraction. Next, the transferred Ni3(HHTP)2 ultrathin films were studied as working electrodes in Li batteries in a half-cell configuration and compared with bare copper. The results show that the Ni3(HHTP)2 film protects the Cu collector from oxidation, and the negative charge accumulates in the organic ligand during the lithiation process while NiII oxidizes to NiIII, unlike other triphenylene-based MOFs with CuII or CoII metal nodes. The galvanostatic plating–stripping cycles of the batteries show that the inclusion of the crystallization time improves the coulombic efficiency, especially significantly in the first cycles when the SEI is formed. This work shows the Langmuir technique as a useful tool to test MOF based materials for batteries with the advantages of using a low amount of raw materials and without the need to introduce additives (binder and electron conductor) in the electrodes. The electrochemical study of this type of electrode allows a first screening to synthesize electrodes based on MOFs and can be a tool for the preparation of protective coatings under optimized conditions.
1. Introduction
Metal–organic frameworks (MOFs) are a subset of coordination networks extending in either two or three dimensions, composed of inorganic nodes (clusters or metal ions) connected by organic ligands and containing potential voids.1 Although most MOFs are crystalline and exhibit permanent porosity,2 there are also amorphous MOFs, MOF liquids, MOF glasses, and non-porous MOFs.3 Due to their rational design, structural diversity, and tunable physical and chemical properties (including surface area, pore size, functionalities, etc.), MOFs have been intensively studied as advanced functional materials for a large variety of applications,4 including environmental remediation processes,5 CO2 capture,6 and energy storage.7 In the particular case of lithium batteries, MOFs are scarcely used compared to inorganic or graphitic materials and most of the studies employ them as the template or precursor of carbon based electrodes because of the low MOF electric conductivity.8,9 Also the fabrication of electrodes, usually made by a deposition of slurry of the synthesized powder, requires a long time of preparation and a large amount of precursor materials and MOFs, particularly if a second pyrolysis process is performed. However, particular properties of MOFs like their stable porous structure that can promote mass transport,10 high tolerance to the change of volume during charge/discharge,11 the redox activity of the metal node and/or organic linker,12,13 and their hydrophobic nature can improve the cycle life and safety of batteries and can enable them to advance beyond lithium ion battery technology.One such technology is the lithium metal battery, in which the anode is made of lithium metal. In this type of battery, the anode is no longer a lithium-ion host material but the lithium metal, which allows the energy and power density as well as the maximum theoretical open-circuit voltage to be increased.14 For this technology, the suppression of lithium dendrites is necessary, using an optimized solid electrolyte or a protective layer on the top of lithium.15,16 A further advancement is the anode-less battery, in which the Li+ ions from the cathode and electrolyte are reduced to the Li metal directly on the anodic collector forming the “anode”.17 These anode-free batteries increase both the gravimetric and volumetric energy density, while improving safety, since no extra metallic lithium is included in the cell. However, anode-less batteries have the same concern of lithium dendrite growth, and thus protective layers are also required. Protective layers should be electrochemically and mechanically stable and homogeneous to avoid the formation of preferential channels or lithium depletion areas, which facilitate dendritic lithium deposition.18,19 In this context, MOFs can be great candidates to fulfill all the requirements due to their ordered porosity and synthetic versatility.17,20,21
Among the different MOF families, 2D MOFs based on triphenylene22 are promising materials for batteries due to their high electric conductivity caused by the presence of π delocalized electrons in the planar structure and by π–π stacking out-of-plane.23 Although the fabrication of extended uniform 2D-MOF thin films is still challenging, in recent years there has been relevant progress in the creation of MOF nanosheets utilizing the interfacial reaction between the organic ligands and the metal ions at the air–liquid or liquid–liquid interfaces.22,24,25 According to these studies, after a certain lapse of time the 2D MOF film is spontaneously formed at the air–liquid interface. The Langmuir technique is based on the formation of monolayers at the air–liquid interface and allows the area per molecule to be controlled by a mechanical compression process while measuring the surface pressure. Next, transfer of the monolayers from the air–liquid interface to a solid substrate can be performed at a controlled surface pressure to maintain a similar film structure throughout the deposition cycles. This bottom-up synthesis technique allows high control of the structure and requires a minimum amount of precursor materials.
To the best of our knowledge, the Langmuir technique is used here for the first time as the synthesis procedure of a MOF to study its electrochemical response in a lithium battery. In particular, the MOF studied is nickel 2,3,6,7,10,11-hexahydroxytriphenylene, Ni3(HHTP)2 (Fig. 1a). The optimization of the MOF film at the air–liquid interface includes a crystallization time step at a specific ligand surface density during the traditional compression process. In situ UV-vis reflection and synchrotron X-ray fluorescence and grazing-incidence diffraction measurements reveal that Ni3(HHTP)2 is spontaneously formed at the air–liquid interface, but crystalline domains can only be detected after the additional crystallization time, as opposed to a continuous barrier compression process. The transferred Ni3(HHTP)2 films are studied as working electrodes in coin cell Li half-cell batteries, without adding any additive (a binder and electron conductor carbon) and without using any other additional solvent (usually electrode slurries are formed with N-methyl pyrrolidone). The CV plots and XPS spectra before and after cycling reveal the oxidation of Ni from +II to +III during Li+ intercalation while the negative charges are accumulated in the HHTP moieties. Despite the similarity of Ni3(HHTP)2 with analogous MOFs, such as Co3(HHTP)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 and Cu3(HHTP)2,27,28 the metal node exhibits different redox behavior during lithiation processes. Finally, galvanostatic plating–stripping cycles are performed using as working electrodes bare copper and copper covered with Ni3(HHTP)2. In the latter case, the incorporation of crystallization time is the determining factor in enhancing the coulombic efficiency of the Ni3(HHTP)2 covered copper, especially in the first cycles revealing that it can act as a protective layer on the top of the battery anode. Beyond being a useful tool to screen MOF-materials, the Langmuir technique can be coupled to roll-to-roll technology, which will allow the fabrication of large dimensions of protective layers on the top of flexible substrates.29
26 and Cu3(HHTP)2,27,28 the metal node exhibits different redox behavior during lithiation processes. Finally, galvanostatic plating–stripping cycles are performed using as working electrodes bare copper and copper covered with Ni3(HHTP)2. In the latter case, the incorporation of crystallization time is the determining factor in enhancing the coulombic efficiency of the Ni3(HHTP)2 covered copper, especially in the first cycles revealing that it can act as a protective layer on the top of the battery anode. Beyond being a useful tool to screen MOF-materials, the Langmuir technique can be coupled to roll-to-roll technology, which will allow the fabrication of large dimensions of protective layers on the top of flexible substrates.29
 | ||
| Fig. 1 (a) Surface pressure (π) vs. area per HHTP unit isotherms of the Langmuir films of HHTP on the water subphase (red), and HHTP on the NiII subphase following the cc (blue) and cssc (black) compression protocols. Inset: scheme of the HHTP molecule and of the Langmuir film when the subphase contains NiII. Although the compression process starts at 3.2 nm2 HHTP−1, the isotherms are represented from 2.1 nm2 HHTP−1 to show clearly the differences in including the cation in the subphase and the compression protocol. (b) Portions of the X-ray fluorescence spectra of the films formed by HHTP over the NiII aqueous subphase at 1.3 nm2 HHTP−1 (cc protocol, the blue line) and after 3 hours at 1.3 nm2 HHTP−1 (cssc protocol, the black line). The spectra of the NiII subphase are shown as a full green line. The grey star indicates the detection of Cu which is present at variable intensities in all spectra due to Cu in the experimental set-up. (c) Surface voltage (ΔV) vs. area per HHTP unit isotherms of the Langmuir films of HHTP on the Ni(II) subphase following the cc (blue) and cssc (black) compression protocols. (d) Raw in situ GIXD data for the Ni(II) subphase (the green line) and for the film formed by HHTP over the Ni(II) aqueous subphase after 3 hours at a constant area = 1.3 nm2 HHTP−1 (cssc protocol, black symbols). Red ticks indicate the calculated positions of the peaks for a 2D structural model of the Ni3(HHTP)2 planes. The full grey line shows a Lorentzian fit of the [30] reflection yielding q = 9.387 nm−1. Inset: cssc protocol GIXD data for the different portions of the detectors as indicated, showing the absence of modulation along qz and therefore pointing at a pure 2D film. | ||
2. Experimental
2.1. MOF film synthesis and characterization
Ni3(HHTP)2 was in situ synthesized at the air–liquid interface using a Langmuir Teflon trough (NIMA, Model 720) with a symmetrical double-barrier configuration and dimensions of 720 × 100 mm2. 1 mg of 2, 3, 6, 7, 10, 11-hexahydroxytriphenylene (HHTP, TCI > 95.0%) was first dissolved in 20.0 mL of methanol (POCH Basic, 99.8%) and then 80.0 mL of chloroform (Fluka ≥ 99.5%) was added under continuous stirring, for a final concentration of 0.01 mg mL−1. HHTP solution was spread on the aqueous subphase containing 5 mM of nickel acetate (Ni(Ac)2) (Sigma-Aldrich, ReagentPlus ≥ 99.0%) and 10 mM of sodium acetate (NaAc) (Sigma-Aldrich, ReagentPlus ≥ 99.0%) dissolved in ultrapure Milli-Q water in the Langmuir trough. This device registers the surface pressure vs. area (π–A) isotherms at 20 ± 1 °C during barrier compression movement using a Wilhelmy balance with a filter paper plate. Surface potential measurements were performed with a KSV Nima Spot Surface Potential Sensor. Brewster angle microscopy (BAM) images were recorded with a KSV Nima Micro BAM using a red laser (50 mW, 659 nm) as a light source with a fixed incidence angle of 53.1° and with a spatial resolution of the optical system of 6 mm per pixel. UV-vis reflection spectra were registered with a Nanofilm Technologie GmbH reflection spectrometer. Grazing-incidence X-ray diffraction (GIXD) in situ measurements at the air–liquid interface were performed at the SIRIUS beamline of Synchrotron SOLEIL (Saint Aubin, France), using an incident X-ray beam of 8 keV (λ = 0.155 nm), a beam size of 0.1 × 2.0 mm2 (V × H), and an incidence angle of 2.0 mrad with the aqueous surface below the total external reflection critical angle value of the air–liquid interface (2.7 mrad at 8 keV). The detection set-up consisting of a 2D PILATUS2 (Dectris, Switzerland) detector combined with a Soller collimator of 0.05 degree resolution was continuously scanned over the in-plane 2θ angle in order to record the horizontal and vertical intensity distributions. Peak adjustment was performed with the Qz-integrated intensity. Total reflection X-ray fluorescence (TRXF) measurements were performed simultaneously with GIXD with a one element silicon drift detector (XFlash 430 M, Brüker, Germany) equipped with a collimator and mounted at 30° with respect to the vertical direction towards the X-ray source in order to reduce the elastic peak that would otherwise saturate the detector. Monolayers were prepared in a dedicated Langmuir trough enclosed in a gas-tight chamber flushed by a water saturated Helium gas flow to reduce gas scattering and to avoid the damage of the monolayer by the beam. Temperature was kept constant thanks to a water circulating bath at 20 ± 1 °C.Langmuir–Schaeffer (LS) films were fabricated in a Teflon KSV-NIMA trough, model KN 2003 of dimensions 580 × 145 mm2. Different substrates were used in the function of the characterization technique. The Quartz crystal was the substrate for UV-vis characterization with a Varian Cary 50 Bio spectrophotometer. Mica was selected for atomic force microscopy (AFM) characterization. AFM images were obtained with a commercial instrument (Multimode 5 Nanoscope 7.3, Veeco) operated in tapping mode. Measurements were carried out under ambient conditions with NSC15/AlBS silicon cantilevers (MikroMasch) at a 325 kHz nominal resonance frequency. MOF mass deposited onto QCM substrates (5 MHz ATcut QCM crystals from Stanford Research Systems) was determined using a Stanford Research Systems (SRS) QCM200 system equipped with a QCM25 crystal oscillator working at 5 MHz. QCM crystals were placed in a O100RH Kynar crystal holder before and after each film transfer, and the mass increases were calculated from frequency changes using the Sauerbrey equation, Δf = −CfΔm, where Δf is the observed frequency change, in Hz, Cf is the sensitivity factor of the QCM crystal provided by the manufacturer (0.0566 Hz cm2 ng−1), and Δm is the change in mass per unit area.
2.2. MOF tests in Li semi-batteries
The substrate to deposit the MOF was Cu foil (Alfa-Aesar >99.9%). After MOF film deposition, the samples were dried during 12 h at 60 °C under vacuum and cut into discs of 8 mm in diameter (the electrode and separator disc cutting machine, TOB). These discs were saved in an Ar glovebox and used as electrodes in Li-ion batteries assembled in a half-cell configuration, in which Li foil (Sigma-Aldrich, thickness 1.5 mm) was used as the counter and reference electrodes. The electrolyte was 1 M LiPF6 ethylene:diethylene carbonate (EC:DEC 1:1, Sigma-Aldrich) embedded in a glass microfiber (Whatmann GF/C™). The Li-ion half-cell battery was crimped in a CR2032 coin cell with a hydraulic manual crimper (TMAX-JK-KF20-TC).
A M204 Autolab Methrom potentiostat/galvanostat was used for galvanostatic and cyclic voltammetry characterization. Galvanostatic plating/stripping cycles were performed at 0.15 mA cm−2 during 2 hours with cut-off limits of +0.6 V and −0.6 V vs. Li/Li+, except the first plating that was performed at 0.05 mAcm−2 for 6 hours to facilitate the formation of a more stable solid electrolyte interface (SEI).
Pristine and cycled electrodes were characterized by X-ray photoelectron spectroscopy (XPS) using an AXIS SupraTM ultra DLD spectrometer from Kratos equipped with a monochromatic Al (Kα) X-ray source (1486.6 eV) using a pass energy of 20 eV. The photoelectron take-off angle was 90° with respect to the sample plane. The XPS binding energies reported in this work were referenced to the maximum of the C 1s peak at 284.6 eV.30 After battery cycling, the reference applied was the same and the analysis was qualitative since the SEI was not fully removed from the top of the sample. The data treatment was performed using the CasaXPS software. For this study, a Swagelok cell was used to open it inside the glovebox after battery cycling and the samples were cleaned with ethylene carbonate.
3. Results and characterization
3.1. Synthesis of Ni3(HHTP)3 layers at the air–liquid interface and on solid substrates by the Langmuir–Schaeffer (LS) technique and transference process
Langmuir Ni3(HHTP)2 films were synthesized by dropping the HHTP solution over the aqueous solution containing NiII as a subphase in a Langmuir trough. Next, the compression process proceeds following two different protocols: (i) typical constant compression (cc) in which the compression of the barriers is constant at 10 cm2 min−1, and (ii) compression + crystallization step + compression (cssc) in which the compression of the barriers is first done at 10 cm2 min−1 from 3.2 nm2 HHTP−1 to 1.3 nm2 HHTP−1, then stopped for 3 hours at this constant area, and finally continued further at 10 cm2 min−1 until collapse of the film.The surface pressure-area isotherms (π–A), Fig. 1a, show that the NiII cations present in the subphase produce more expanded isotherms than those when the subphase is water, suggesting the incorporation of the cations at the interface. This is confirmed by the X-ray fluorescence spectra at the air–water interface (Fig. 1b). The KL and KM bands of Ni at 7460 and 8260 eV increase dramatically when the HHTP molecules are dispersed on the NiII subphase, and the intensity hardly varies during the compression process regardless of the protocol followed. Then, interfacial coordination reactions between the building molecular units of the air–liquid interface and the metal ions of the subphase are initiated spontaneously after dispersion of the organic ligand onto the subphase, in good agreement with the literature.31 However, the compression protocol affects the π–A isotherm plots (Fig. 1a), with the film being more expanded for the cssc protocol, which also shows higher collapse pressure. This is likely due to the inclusion of the crystallization step which shows an increase in the structural order distance, producing crystals with larger lateral domains. Another subtle observation is the decay of surface pressure with time when barriers are stopped in the cssc protocol (Fig. SI.1†), which can be fitted to an exponential equation of type π = π0exp(−t/t1).32 The obtained value for t1 is ∼10600 s, which is in the order of those reported for the rearrangement of nanoparticles33 and liquid crystal polymers34 at the air–liquid interface, and an order of magnitude larger than the surface fluctuation damping when the barriers change from mechanical compression to a steady state.
The effect of adding crystallization time during the compression process is observed in the surface potential values, ΔV, of the film. Fig. 1c shows similar ΔV isotherms from the initial value of the compression up to 1.3 nm2 HHTP−1. Next, an increase of ∼50 mV occurs when the barriers stop at 1.3 nm2 HHTP−1 in the cssc protocol, and again the same shape is observed between 1.3 nm2 and the final value of the compression, but showing a difference of ∼50 mV. The gain in ΔV has been previously associated with an increase of ordered domains as in polymer films,35 with the incorporation of water molecules into the Langmuir film,36 or with an increase in the contribution of the double layer, ψ0.37 In the current study, the ΔV increase with time also follows first order kinetics, ΔV = ΔV0exp (t/t′1) (Fig. SI.2†), similar to the decay observed for π, suggesting that domain upregulation and rearrangement are the most plausible phenomena. This is in good concordance with in situ grazing incidence synchrotron X-ray diffraction (GIXD). Detectable diffraction is only observed in the later stages of the 3 h crystallization time in the cssc protocol (Fig. 1d). A relatively intense diffraction peak appears at approximately q = 9.4 nm−1, corresponding to a Bragg bar, with intensity at all qz (the inset in Fig. 1d), demonstrating the 2D character of the objects giving rise to the diffraction. In addition, weaker diffraction is observed at ca. 19 nm−1, with intensity also detectable at all qz. Both GIXD peaks can reasonably be attributed to the [30] and [60] reflections of a structural model based on the Ni3(HHTP)2 structure (Fig. SI.3†), but without intercalated hydrated molecules.38 Therefore, the crystallization step aids the formation of (larger) crystalline domains with respect to the typical cc compression protocol, and these crystalline domains coalesce together upon compression.
The BAM images (Fig. 2), recorded in the cssc compression process, show the formation of domains of a few micrometers at larger areas (1.1 nm2 HHTP−1). These domains coalesce with decreasing surface area, forming worm-like domains and empty spaces at 0.7 nm2 HHTP−1 where π starts to increase. Upon further compression, lighter gray spots begin to appear indicating the regions of increased thickness, while dark areas (no molecular coating or very thin layer) are still observed at 0.5 nm2 HHTP−1. At 0.4 nm2 HHTP−1, worm-like domains are observed similar to those at 0.7 nm2 HHTP−1 but brighter. These images suggest that the Langmuir film consists of regions formed by multilayer domains over thinner or bare areas, rather than an appropriate monomolecular film at the air–water interface. A second brightness increase in the BAM images appears at 0.2 nm2 HHTP−1, where the π–A isotherm shows film collapse.
 | ||
| Fig. 2 BAM images of the Langmuir film of Ni3(HHTP)2 at different areas per HHTP during the cssc compression process. | ||
The UV-vis reflection (ΔR) spectra of Ni3(HHTP)2 at the air–liquid interface during the cssc compression process (Fig. 3a) show the characteristic strong absorption in the UV and blue visible regions (below ∼450 nm) corresponding to the π–π* transition of the catecholate linking moieties.25,39 The second peak reported for the HHTP-based MOFs containing different metal nodes (Ni, Co and Cu) appears at 620 nm and is attributed to the ligand-to-metal charge transfer transition.25,39 This peak shifts to longer wavelengths in the spectra of the MOF formed at the air–liquid interface due to the highly polar environment in the presence of the aqueous subphase.40 However, the UV-vis reflection spectrum of the Langmuir film formed with the cc protocol differs significantly from this description (Fig. SI.4†), probably due to the formation of smaller light-scattering domains. The characteristic UV-vis spectrum of Ni3(HHTP)2 with the broad band of charge transfer at ∼600 nm is observed in the transferred film (Fig. 3b), once the polar aqueous subphase is not present. This observation suggests that the film formed at the air–liquid interface, which consists of Ni3(HHTP)2 crystalline domains, can be transferred to a solid substrate without any appreciable change in the film. In addition, the mass of the films after several transfers was weighed with a QCM showing a linear increase in mass with the number of cycles, passing through the origin. This linear mass increase indicates that the films transferred from the air–liquid interface to the substrate are homogeneous when π is maintained at 10 mN m−1 even though the films are formed by coalescing crystalline domains. The XPS spectra of the LS film formed on the Cu substrate (Fig. 7) show the characteristic Ni3(HHTP)2 peaks,41–43 confirming that extended Ni3(HHTP)2 framework domains are formed. Further discussion is performed in the next section.
 | ||
| Fig. 3 (a) Reflection UV-vis spectra of Ni3(HHTP)2 Langmuir films at different areas per HHTP during the compression process with the cssc protocol. (b) UV-vis absorption spectrum of the Ni3(HHTP)2 LS film (4 transferences) transferred at 10 mN m−1. Inset: Mass density increase of the LS film with the number of transference cycles at 10 mN m−1. | ||
The LS films of 1, 3 and 5 transference cycles were analyzed by AFM microscopy (Fig. 4). A nearly full surface coverage composed of worm-like domains of about 1.5 nm height is already observed after 1 transference cycle. This height is larger than a monolayer thickness (∼ 3.3 Å)44 and suggests the formation of domains with 3–4 molecular Ni3(HHTP)2 planes lying parallel to the surface. The worm-like structures are of a few nanometers in thickness and form larger circular domains of a few micrometers, in good agreement with the film described at the air–liquid interface. This structure is similar to other LS films of MOFs.45 Despite the worm-like fine structure, a full surface coverage is obtained after 3 transference cycles. In the film of 5 transferences, the worm-like fine structure is less clearly observed, and new round domains of ∼5 nm appear.
 | ||
| Fig. 4 AFM images and representative height profiles for Ni3(HHTP)2 LS films with: (a) 1, (b) 3, and (c) 5 transferences from Langmuir films at 10 mN m−1 formed with the cssc protocol. | ||
3.2. Study of the electrochemical response of Ni3(HHTP)2 layers against Li metal
The preparation of Ni3(HHTP)2 electrodes was carried out directly on a Cu foil substrate without the addition of any binder or electronic conductor, performing 20 transference cycles. Fig. 5 shows the AFM images of these electrodes. The images show a complete coating of the substrate with small defects and round domains with a diameter in the range of 100–200 nm and 2–6 nm in height. This type of rounded domain was already observed in the 5-transfer samples (Fig. 4c), which reveals that this organization is the most prevalent after 5 transfers. While, the worm-like structure observed in 1 and 3 transferences cannot be observed with clarity, probably because of the roughness of the surface. In this case, the Cu substrate contributes to both the roughness of the sample and the presence of defects, which can have a height of ∼100 nm. The roughness and the presence of steps on the Cu surface are observed on bare copper (Fig. SI.5†), and are in agreement with the literature.46 | ||
| Fig. 5 (a) and (b) AFM images of the Ni3(HHTP)2 electrodes (20 transferences) on Cu foil and (c) the corresponding height profile. | ||
The electrochemical response of the Ni3(HHTP)2 film with lithium was recorded in an assembled coin cell. Fig. 6a shows the CVs of the half-cell batteries when the working electrode is a Ni3(HHTP)2 LS film (20 transferences, cssc protocol) on copper foil and when it is bare copper. In the case of bare Cu, the oxidation of Cu is observed above 3 V vs. Li/Li+ which returns in a loop in the cathodic direction that can be ascribed to the oxidation of the substrate and its posterior solution. This effect is efficiently avoided when the LS film covers the Cu film. In the case of the LS film, the 3 CV cycles match indicating the good reversibility of the electrochemical processes, with two broad reduction/oxidation peaks centered at ∼1.4 V and ∼0.5 V vs. Li/Li+ in the cathodic sweep and their respective anodic peaks at ∼2.0 V and ∼1.2 V. The broad peaks suggest a gradual multipass insertion of Li+, stabilized with the reduction of the O atoms in the organic ligands47 and by electrostatic interaction with the π-electron cloud of the phenyl rings.48 This behavior obeys a pseudocapacitive mechanism described for MOFs in Li batteries,49–51 in which both redox and electrostatic mechanisms are involved. Both contributions can be differentiated from the power law relation i = avb, where i is the measured current and v is the sweep speed at which the CVs are obtained (Fig. SI.6†). The slope of the logv vs. logi plot gives the b parameter, which has a value between b = 0.5 for the diffusion–dominant process (related to faradaic processes) and b = 1 for the completely capacitive effect (related to double layer capacitance).52 Here the b-value in the range of 0.5–2 V is between 0.72 and 0.85 (Fig. SI.7†), which confirms the pseudocapacitive mechanism. Fig. 6b shows the capacitive contribution in the CV obtained at 0.5 mV s−1, which represents a 33% contribution. After the CV cycles at different sweep rates (0.1 mV s−1, 0.5 mV s−1, 1 mV s−1, 5 mV s−1, 10 mV s−1, and 50 mV s−1), other 3 cycles were performed at 0.1 mV s−1 showing high reproducibility in the shape of the plot at the beginning and at the end of the measurement (Fig. SI.8†). This result indicates that no significant degradation process occurs in the MOF during battery cycling.
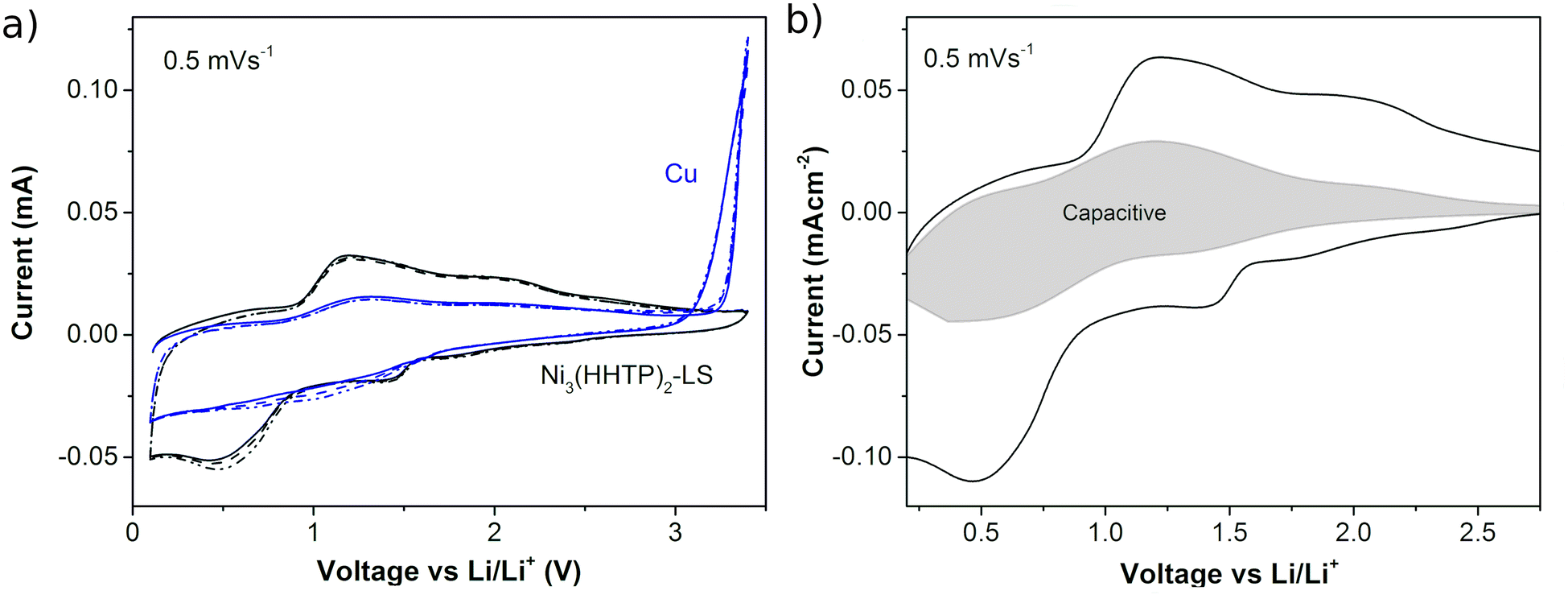 | ||
| Fig. 6 (a) 3 CV cycles of a Li half-cell battery using bare Cu foil or Cu foil covered with a LS film of 20 transferences of Ni3(HHTP)2 at 0.5 mV s−1. (b) Percentage of capacitive response within the CV registered at 0.5 mV s−1. | ||
The XPS spectra of the electrodes before and after being cycled in a Li half-battery at 0.1 V vs. Li/Li+ provide information on changes in the oxidation state or chemical environment of the Ni3(HHTP)2 components. The high resolution Ni 2p spectrum before cycling (Fig. 7a) shows two sets of peaks at 856.2 and 873.8 eV with their respective satellites, which correspond to the 2p3/2 and 2p1/2 levels, respectively, of the emission lines of NiII. Also, the 3p peaks of NiII are observed at 68.6 eV and 76.5 eV (Fig. 7b). These spectra prove that only one type of NiII is present within the LS film when it is deposited.41 The 2p peaks attributed to NiII are significantly reduced in the cycled sample, where two new peaks appear at 858.1 eV and 876.8 eV related to 2p3/2 and 2p1/2 of NiIII. The same behavior is observed in the high resolution spectrum of Ni 3p, a region in which the 1s LiI band appears for the cycled sample. Therefore, the intercalation of Li ions in the MOF presumably induces the oxidation of NiII to NiIII for the most part. The high resolution C1s and O1s XPS spectra (Fig. SI.9†) show the presence of traces of the SEI in the cycled samples; so it is not possible to perform a quantitative analysis. Qualitatively, the characteristic C1s and O1s bands described for the triphenylene-based MOFs are observed in the uncycled samples.42 Upon cycling, the bands become narrow indicating a preferential chemical environment of C and O, most likely due to a catecholate configuration53 that accumulates the negative charge upon cycling at 0.1 V vs. Li/Li+.
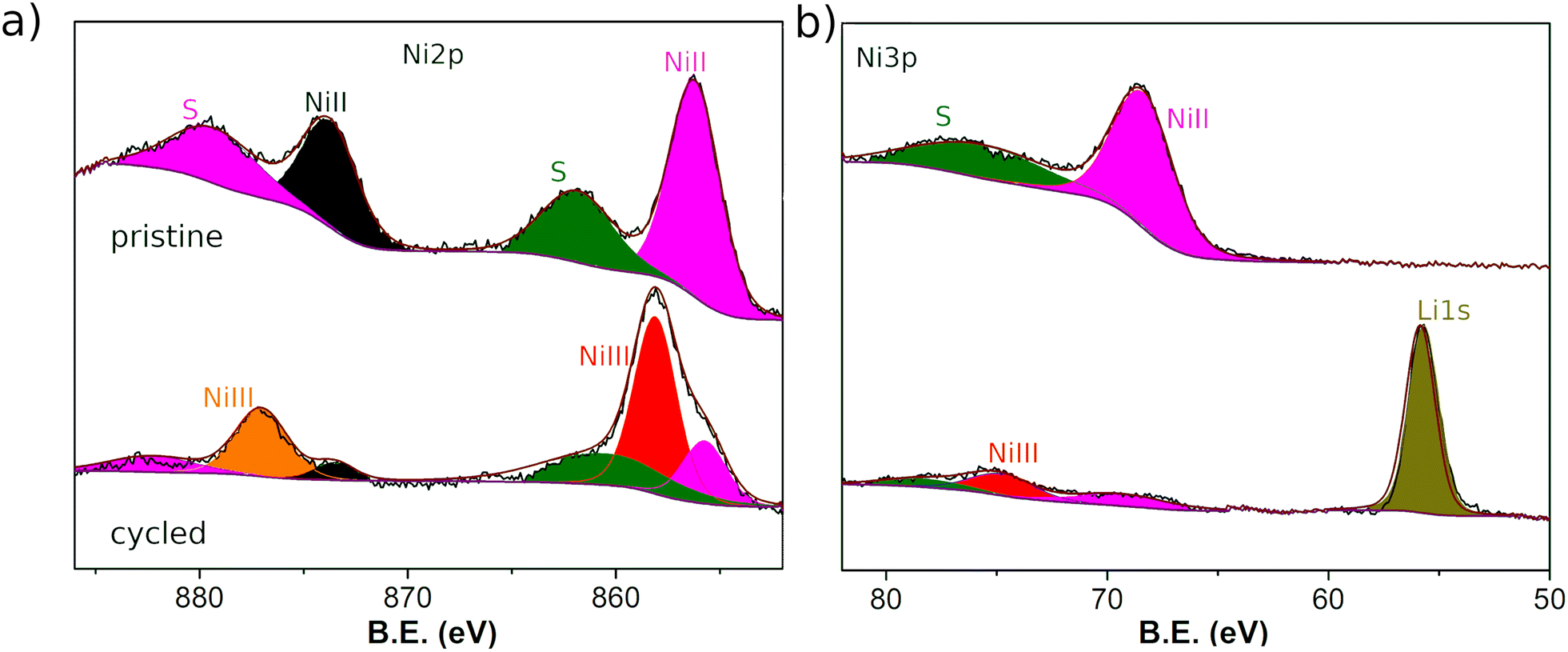 | ||
| Fig. 7 High resolution XPS spectra of: (a) Ni 2p, and (b) Ni3p and Li1s for the pristine Ni3(HHTP)2 LS film (top) and after being cycled as electrodes in a half-cell battery dissembled when the voltage was 0.1 V vs. Li/Li+ (bottom). | ||
Altogether, the XPS data suggest Ni oxidation that may stabilize the negative charge accumulation on the ligand, and may also explain the protection of the Cu foil from being oxidized, as observed in Fig. 6a. Taking all this into account, the electrochemical process we propose is:
| NiII3(HHTP)−III2 + 3e− + 3Li+ → LiI3NiIII3(HHTP)−VI2 | (1) |
This electrochemical behavior described for the Ni3(HHTP)2 film differs from those described for Cu3(HHTP)2 and Co3(HHTP)2 MOFs cyclized in Li batteries. In the former case, Cu is reduced from CuII to CuI, while in the latter CoII does not change its oxidation state.26–28 Thus, in principle, metal oxidation reduces the faradaic capacity of Ni3(HHTP)2 compared to those of other triphenylene-based MOFs. In particular, the study of the Co2(HHTP)3 MOF26 reported a capacity of 332 mA h g−1 at 0.1 A g−1vs. the value of 631 mA h g−1 at 0.2 A g−1 for Cu2(HHTP)3.27 However, it should be noted that the final organization of the MOFs in the electrodes determines the final capacity, being less than 100 mA h g−1 at 1C for Cu2(HHTP)3.28 Particularly, in the case of Ni3(HHTP)2, the capacitive contribution is significant, which could be favored and stabilized by the higher negative charge on the organic part. Noticeably, an important difference in our sample from these reported studies is the thickness of the films, being an order of magnitude smaller in the current study; so the effect of the film thickness cannot be completely discarded in terms of electrolyte embedding within the MOF that can facilitate Ni oxidation or even the depth of the XPS analysis.
The potential of the Ni3(HHTP)2 LS film as a protective layer for various Li half-cell batteries has been studied with the working electrodes: pristine copper (Cu) foil and LS films fabricated with the cssc protocol (20 transferences) and cc protocol (20 transferences). Fig. 8a shows the first galvanostatic cycles of plating and stripping for the three semi-batteries. During the Li plating process the Li+ ions from the electrolyte diffuse through the ultrathin MOF layer and are reduced to Li metal at the top of the Cu collector. Theoretically, the reduction should occur at 0 V vs. the Li/Li+ reference, but it occurs at a lower voltage which is the nucleation overpotential (μnuc); so the peak indicates the onset of Li deposition.54 Next, the plating process is controlled by mass transfer diffusion of Li+ ions through the deposited layer, which determines the plateau potential (μpla).54 The first plating plot (at a lower current density to facilitate the formation of a more stable SEI) is very similar for the three semi-batteries with a μpla value of ∼−13 mV and a μnuc value between −20 mV and −27 mV. After this first cycle, the plating plot obtained at a higher current density shows higher μnuc and μpla values due to the kinetic effect. In the case of the cssc sample, the plating plots are square in shape and with high reproducibility throughout the first eleven cycles, although μpla increases slightly after the 9th cycle. The two batteries fabricated with cc and Cu working electrodes show similar behavior. In these samples, μpla is slightly larger than that in the cssc sample and the plating plot loses the square shape: at the 10th cycle it increases with time instead of decreasing, and after the 11th cycle it first increases and then decreases. The fact that the plating does not occur in a plateau points that other effects besides the Li+ diffusion control the process, probably kinetic limitations or the formation of channels. This effect is not observed for the cssc sample (Fig. 8b), although an increase of overpotentials occurs during the first cycles, followed by cycles that show high reproducibility. On the other hand, the value of μnuc is similar in the three semi-batteries. These results suggest that the Ni3(HHTP)2 LS layer deposited using the cssc protocol aids to the diffusion of Li+ ions to the Cu substrate, but does not produce any significant effect on the reduction of the nucleation overpotential. However, when the Ni3(HHTP)2 LS layer is fabricated with the cc protocol, the positive effect of Li+ diffusion is not observed compared to the bare Cu substrate.
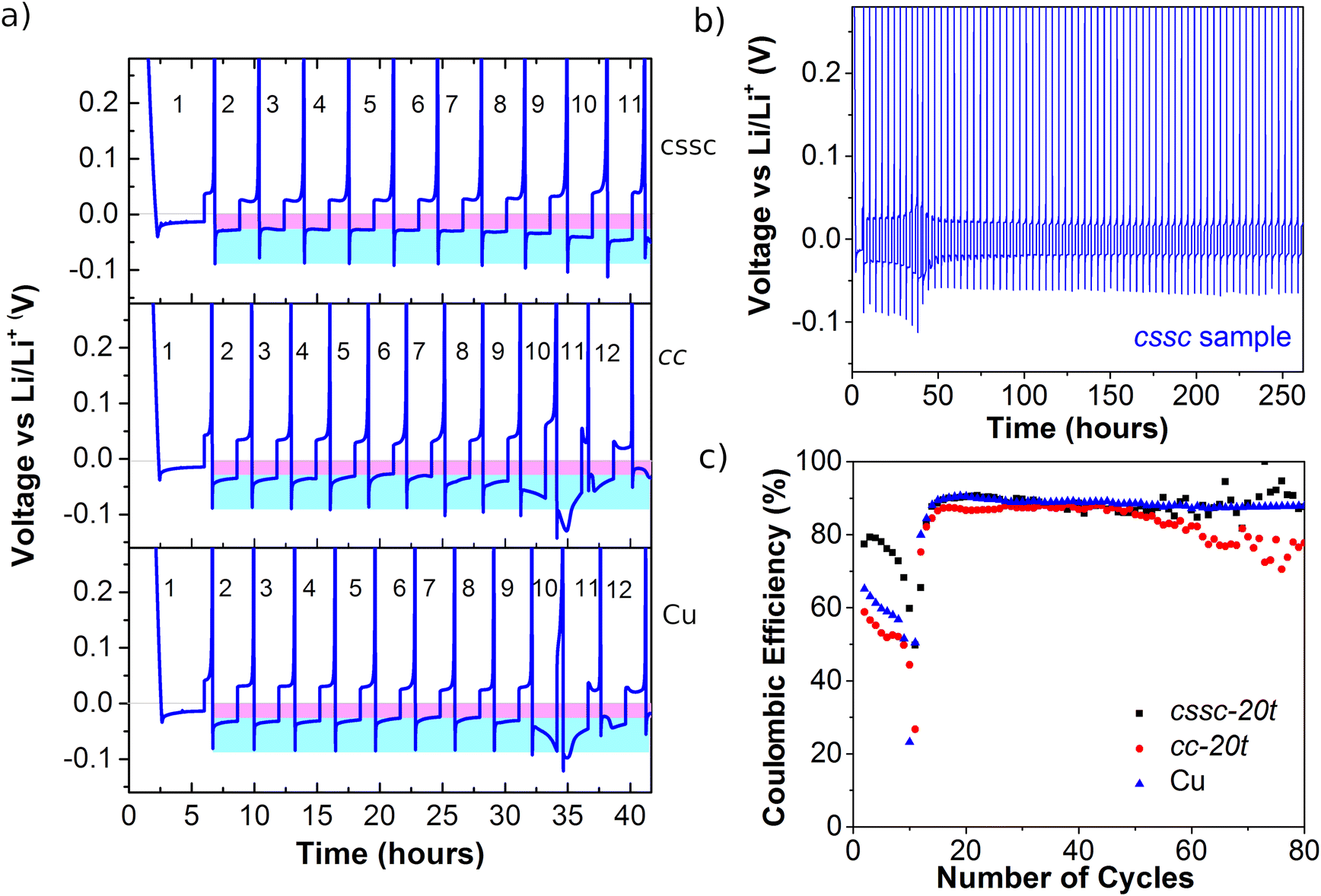 | ||
| Fig. 8 (a) Plating–stripping galvanostatic plots for the half-cell batteries fabricated with the working electrodes: Ni3(HHTP)2 LS film fabricated with the cssc and cc protocols on Cu foil, and bare Cu foil. For ease of comparison, the μpla (∼−28 mV) and μnuc (∼−55 mV) points of the second cycle are shaded in pink and blue for the cssc sample (elongated for the other cycles), and the same shading dimensions are superimposed on the plots obtained for the cc and Cu semi-batteries. (b) Plating–stripping galvanostatic plots for the cssc sample half-cell battery along 80 plating–striping cycles. (c) Coulombic efficiency (CE) of Li+ plating/stripping processes along 80 cycles. | ||
After the coating process, the stripping process begins at a positive voltage plateau. At the end of the ordered process, a rapid increase in the voltage is observed, due to an increase in the resistance associated with contact loss and the change in the open circuit voltage as the original Ni3(HHTP)2/Li cell is modified. Coulombic efficiency (CE) can be, then, calculated as a quantifiable indicator for the reversibility of plating/stripping processes, in which the electrons from irreversible reactions are also considered according to eqn (2):55
 | (2) |
The calculated CE values (Fig. 8c) are lower and decrease in the first 15 cycles from ∼80% to ∼50% for the sample cssc and from ∼60% to 20% for the Cu substrate and cc sample. The lower CE values are related to the occurrence of irreversible side reactions that take place during SEI formation, which may be prolonged during the first 20 cycles. However, the cssc sample is the one with a considerably higher CE value than the other two samples, indicating a lower electrolyte consumption in the secondary reactions probably because a more ordered and stable SEI layer is formed. After these preliminary cycles, the CE value increases to near 90% values for the cssc and Cu half-cell batteries and to somewhat lower values for the cc sample. This change in behavior can be attributed to the transition between two different regimes with Li+ ion capacity loss:56 the first being due to the formation of the SEI, and the second being due to the encapsulation of electronically isolated Li0. This transition is favored by the fact that the fabricated half-cells provide an “unlimited” Li system because the counter electrode is a metallic Li foil and the working electrode (bare Cu or covered by a nanometer-thick film) is “flooded” with the electrolyte; so that, despite the increase in the system impedance due to SEI formation, the introduction of the electrolyte is still sufficient to prevent battery termination.55 In this new regime, the first system to fail is the cc sample, indicating that in this case the presence of the Ni3(HHTP)2 LS film without large 2D crystalline domains introduces higher impedance than that obtained for the bare Cu foil. While, the presence of larger crystalline domains in the cssc sample shows scattered CE values after 50 cycles, in the range between 89% and 95%, principally. Considering the two contributions to the CE values, eqn (2), the higher values are probably due to the release of electrons in the electrochemical processes in the Ni3(HHTP)2 film, electrochemically active (eqn (1)), in contact with the SEI, since if it was due to an increase in the reversibility of the delithiation process itself one would expect a more homogeneous trend instead of such scattered values. To sum up, the fabricated Ni3(HHTP)2 LS films are not very efficient as protective layers for batteries without the anode, although the efficiency improves significantly with the film order. This deficit is presumably due to the ultra-thin nature of the fabricated films which are of a few nanometers compared to those found in the literature which are in the order of micrometers.20,57–59 However, the good response in the first plating-stripping cycles and the higher CE values observed for the Ni3(HHTP)2 LS film fabricated with the cssc protocol suggest that this film is promising as a protective layer in anodes to form a stable SEI facilitating the Li+ diffusion.
4. Conclusions
This study describes the benefit of including an additional crystallization time to enhance the 2D Ni3(HHTP)2 crystalline domain formation during the compression process at the air–liquid interface. This simple modification of the Langmuir technique provides a new parameter to control (crystallization time) in the in situ formation of MOFs at the air–liquid interface, which can enhance the extension of 2D domains and facilitate the integration of ultrathin films in practical devices. The electrochemical study of the Ni3(HHTP)2 LS film in a Li half-cell battery shows the oxidation of the Ni metal node from +II to +III, which can stabilize the accumulation of negative charge in the organic ligand besides protecting the Cu collector from oxidation. Related to the plating–stripping study, the CE plot previews that the Ni3(HHTP)2 LS film fabricated with the cssc protocol can reduce the side reactions related to the SEI formation and facilitate the Li+ diffusion through it, so it can be used as a protective layer of anodes. Despite the fact that under the current conditions it does not work properly as a protective layer to reduce/avoid the formation of lithium dendrites in anode-free batteries, the low amount of the material used and the thickness of the ultrathin film are two relevant properties that can be optimized by the Langmuir technique for the design of future protective layers for anode-less lithium or other metal batteries.Beyond the results obtained, the fabrication of Langmuir films of triphenylene-based MOFs provides a means to customize advanced materials, both through the nature of the metal ion node, with the Ni-based MOF having a different redox electrochemical behavior from the Cu or Co analogues, and the growth of 2D domains at the air–liquid interface, controlled by both the area per ligand and the crystallization time.
Author contributions
M. H. conceived the idea, designed the experiments, wrote the initial version of the manuscript, and supervised the work. I. C.-R. fabricated the half-cell batteries and performed and analysed the electrochemical study (CV and platting-stripping galvanostatic measurements). I. T. performed and analysed the in situ synchroton fluorescence and GIXD measurements. L. C. performed the characterization of Ni3(HHTP)2 Langmuir films by BAM, surface potential analysis, and UV-vis reflection spectroscopy and fabricated the electrodes and the half-cell batteries. B. D. performed preliminary studies for the optimization of the synthesis of Ni3(HHTP)2 at the air–liquid interface. O. L. performed the QCM tests and prepared the samples for AFM measurements. A. U. performed and analyzed the AFM measurements. O. R. proposed the material, performed the in situ synchroton fluorescence and GIXD measurements, and discussed the XPS data. I. G. conceived the idea of the addition of the crsytallization step in the compression process, designed the experiments, and supervised the work. All authors contributed to editing the paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the DGA/fondos FEDER (construyendo Europa desde Aragón) for funding the research group Platon (E31_20R) and the project LMP71_21. This work was also funded by MCIN/AEI/10.13039/501100011033 and ERDF “A way of making Europe” (PID2019-108247RA-I00 and PID2019-105881RB-I00). M. H. acknowledges the funding support from MCIN/AEI/10.13039/501100011033 for the Ramón y Cajal fellowship (RYC-2018-025222-I). I. T. gratefully acknowledges her DGA PhD fellowship. The authors would like to acknowledge the use of Servicio General de Apoyo a la Investigación-SAI, Universidad de Zaragoza. The authors thank the synchrotron SOLEIL for beamtime provision under projects 20191874 and 20210275 and are grateful to Prof. M. Goldmann and Dr P. Fontaine for their support during experiments.References
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O’Keeffe, M. P. Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- T. D. Bennett and S. Horike, Nat. Rev. Mater., 2018, 3, 431–440 CrossRef.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont and J. Goscianska, et al. , Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS PubMed.
- E. Barea, C. Montoro and J. A. Navarro, Chem. Soc. Rev., 2014, 43, 5419–5430 RSC.
- B. Seoane, J. Coronas, I. Gascon, M. E. Benavides, O. Karvan, J. Caro, F. Kapteijn and J. Gascon, Chem. Soc. Rev., 2015, 44, 2421–2454 RSC.
- B. Zhu, D. Wen, Z. Liang and R. Zou, Coord. Chem. Rev., 2021, 446, 214119 CrossRef CAS.
- S. Mubarak, D. Dhamodharan, P. N. Ghoderao and H.-S. Byun, Coord. Chem. Rev., 2022, 471, 214741 CrossRef CAS.
- W. Xia, A. Mahmood, R. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 RSC.
- Q. Gan, H. He, K. Zhao, Z. He and S. Liu, J. Colloid Interface Sci., 2018, 530, 127–136 CrossRef CAS PubMed.
- J. Jin, Y. Zheng, S.-Z. Huang, P.-P. Sun, N. Srikanth, L. B. Kong, Q. Yan and K. Zhou, J. Mater. Chem. A, 2019, 7, 783–790 RSC.
- K. Wada, K. Sakaushi, S. Sasaki and H. Nishihara, Angew. Chem., Int. Ed., 2018, 57, 8886–8890 CrossRef CAS PubMed.
- T. Mehtab, G. Yasin, M. Arif, M. Shakeel, R. M. Korai, M. Nadeem, N. Muhammad and X. Lu, J. Energy Storage, 2019, 21, 632–646 CrossRef.
- X. Shen, H. Liu, X.-B. Cheng, C. Yan and J.-Q. Huang, Energy Storage Mater., 2018, 12, 161–175 CrossRef.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 1–16 Search PubMed.
- D. Zhang, Z. Liu, Y. Wu, S. Ji, Z. Yuan, J. Liu and M. Zhu, Adv. Sci., 2022, 9, 2104277 CrossRef CAS.
- J. Qian, Y. Li, M. Zhang, R. Luo, F. Wang, Y. Ye, Y. Xing, W. Li, W. Qu and L. Wang, Nano Energy, 2019, 60, 866–874 CrossRef CAS.
- P. Bai, J. Li, F. R. Brushett and M. Z. Bazant, Energy Environ. Sci., 2016, 9, 3221–3229 RSC.
- J. Yun, B.-K. Park, E.-S. Won, S. H. Choi, H. C. Kang, J. H. Kim, M.-S. Park and J.-W. Lee, ACS Energy Lett., 2020, 5, 3108–3114 CrossRef CAS.
- Y. Xu, L. Gao, L. Shen, Q. Liu, Y. Zhu, Q. Liu, L. Li, X. Kong, Y. Lu and H. B. Wu, Matter, 2020, 3, 1685–1700 CrossRef.
- L. Fan, Z. Guo, Y. Zhang, X. Wu, C. Zhao, X. Sun, G. Yang, Y. Feng and N. Zhang, J. Mater. Chem. A, 2020, 8, 251–258 RSC.
- N. Contreras-Pereda, S. Pané, J. Puigmartí-Luis and D. Ruiz-Molina, Coord. Chem. Rev., 2022, 460, 214459 CrossRef CAS.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- R. Makiura, Coord. Chem. Rev., 2022, 469, 214650 CrossRef CAS.
- V. Rubio-Giménez, M. Galbiati, J. Castells-Gil, N. Almora-Barrios, J. Navarro-Sánchez, G. Escorcia-Ariza, M. Mattera, T. Arnold, J. Rawle and S. Tatay, Adv. Mater., 2018, 30, 1704291 CrossRef.
- P. Mao, H. Fan, C. Liu, G. Lan, W. Huang, Z. Li, H. Mahmoud, R. Zheng, Z. Wang and H. Sun, Sustainable Energy Fuels, 2022, 6, 4075–4084 RSC.
- L. Guo, J. Sun, W. Zhang, L. Hou, L. Liang, Y. Liu and C. Yuan, ChemSusChem, 2019, 12, 5051–5058 CrossRef CAS PubMed.
- S. Gu, Z. Bai, S. Majumder, B. Huang and G. Chen, J. Power Sources, 2019, 429, 22–29 CrossRef CAS.
- M. Parchine, J. McGrath, M. Bardosova and M. E. Pemble, Langmuir, 2016, 32, 5862–5869 CrossRef CAS PubMed.
- T. L. Barr and S. Seal, J. Vac. Sci. Technol., A, 1995, 13, 1239–1246 CrossRef CAS.
- T. Ohata, A. Nomoto, T. Watanabe, I. Hirosawa, T. Makita, J. Takeya and R. Makiura, ACS Appl. Mater. Interfaces, 2021, 13, 54570–54578 CrossRef CAS.
- X. Wang, X. Ma and D. Zang, Soft Matter, 2013, 9, 443–453 RSC.
- L. S. Boucheron, J. T. Stanley, Y. Dai, S. S. You, C. T. Parzyck, S. Narayanan, A. R. Sandy, Z. Jiang, M. Meron and B. Lin, Phys. Rev. E, 2018, 97, 052803 CrossRef.
- A. Maestro, H. M. Hilles, F. Ortega, R. G. Rubio, D. Langevin and F. Monroy, Soft Matter, 2010, 6, 4407–4412 RSC.
- A. Dhanabalan, D. T. Balogh, A. Riul Jr., J. A. Giacometti and O. Oliveira Jr., Thin Solid Films, 1998, 323, 257–264 CrossRef CAS.
- H. Morgan, D. M. Taylor and O. N. Oliveira Jr., Biochim. Biophys. Acta, Biomembr., 1991, 1062, 149–156 CrossRef CAS PubMed.
- J. Davies, J. Colloid Sci., 1956, 11, 377–390 CrossRef CAS.
- M. Hmadeh, Z. Lu, Z. Liu, F. Gándara, H. Furukawa, S. Wan, V. Augustyn, R. Chang, L. Liao and F. Zhou, Chem. Mater., 2012, 24, 3511–3513 CrossRef CAS.
- A. Mähringer, A. C. Jakowetz, J. M. Rotter, B. J. Bohn, J. K. Stolarczyk, J. Feldmann, T. Bein and D. D. Medina, ACS Nano, 2019, 13, 6711–6719 CrossRef PubMed.
- E. Sucre-Rosales, R. Fernandez-Teran, N. Urdaneta, F. E. Hernandez and L. Echevarria, Chem. Phys., 2020, 537, 110854 CrossRef CAS.
- M. Ko, L. Mendecki, A. M. Eagleton, C. G. Durbin, R. M. Stolz, Z. Meng and K. A. Mirica, J. Am. Chem. Soc., 2020, 142, 11717–11733 CrossRef CAS PubMed.
- H. Wu, W. Zhang, S. Kandambeth, O. Shekhah, M. Eddaoudi and H. N. Alshareef, Adv. Energy Mater., 2019, 9, 1900482 CrossRef.
- R. Dong, M. Pfeffermann, H. Liang, Z. Zheng, X. Zhu, J. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 12058–12063 CrossRef CAS.
- M. de Lourdes Gonzalez-Juarez, C. Morales, J. I. Flege, E. Flores, M. Martin-Gonzalez, I. Nandhakumar and D. Bradshaw, ACS Appl. Mater. Interfaces, 2022, 14, 12404–12411 CrossRef CAS PubMed.
- A. Urtizberea, E. Natividad, P. J. Alonso, M. A. Andrés, I. Gascón, M. Goldmann and O. Roubeau, Adv. Funct. Mater., 2018, 28, 1801695 CrossRef.
- L. Fan, Z. Li, Z. Xu, K. Wang, J. Wei, X. Li, J. Zou, D. Wu and H. Zhu, AIP Adv., 2011, 1, 032145 CrossRef.
- Q. Jiang, P. Xiong, J. Liu, Z. Xie, Q. Wang, X. Q. Yang, E. Hu, Y. Cao, J. Sun and Y. Xu, Angew. Chem., Int. Ed., 2020, 59, 5273–5277 CrossRef CAS PubMed.
- S. Maiti, A. Pramanik, U. Manju and S. Mahanty, Microporous Mesoporous Mater., 2016, 226, 353–359 CrossRef CAS.
- J. Park, M. Lee, D. Feng, Z. Huang, A. C. Hinckley, A. Yakovenko, X. Zou, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2018, 140, 10315–10323 CrossRef CAS PubMed.
- Z. Wu, D. Adekoya, X. Huang, M. J. Kiefel, J. Xie, W. Xu, Q. Zhang, D. Zhu and S. Zhang, ACS Nano, 2020, 14, 12016–12026 CrossRef CAS PubMed.
- S. Mutahir, C. Wang, J. Song, L. Wang, W. Lei, X. Jiao, M. A. Khan, B. Zhou, Q. Zhong and Q. Hao, Appl. Mater. Today, 2020, 21, 100813 CrossRef.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
- A. C. Gómez-Herrero, C. Sánchez-Sánchez, F. Chérioux, J. I. Martínez, J. Abad, L. Floreano, A. Verdini, A. Cossaro, E. Mazaleyrat and V. Guisset, Chem. Sci., 2021, 12, 2257–2267 RSC.
- M. S. Kim, J.-H. Ryu, D. Deepika, Y. R. Lim, I. W. Nah, K.-R. Lee, L. A. Archer and W. I. Cho, Nat. Energy, 2018, 3, 889–898 CrossRef CAS.
- J. Xiao, Q. Li, Y. Bi, M. Cai, B. Dunn, T. Glossmann, J. Liu, T. Osaka, R. Sugiura, B. Wu, J. Yang, J.-G. Zhang and M. S. Whittingham, Nat. Energy, 2020, 5, 561–568 CrossRef CAS.
- G. M. Hobold, J. Lopez, R. Guo, N. Minafra, A. Banerjee, Y. S. Meng, Y. Shao-Horn and B. M. Gallant, Nat. Energy, 2021, 6, 951–960 CrossRef CAS.
- J. Man, W. Liu, H. Zhang, K. Liu, Y. Cui, J. Yin, X. Wang and J. Sun, J. Mater. Chem. A, 2021, 9, 13661–13669 RSC.
- G. Wang, P. He and L. Z. Fan, Adv. Funct. Mater., 2021, 31, 2007198 CrossRef CAS.
- G. Jiang, K. Li, F. Yu, X. Li, J. Mao, W. Jiang, F. Sun, B. Dai and Y. Li, Adv. Energy Mater., 2021, 11, 2003496 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt00876b |
| This journal is © The Royal Society of Chemistry 2023 |