
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermally processed Ni-and Co-struvites as functional materials for proton conductivity†
Stephanos
Karafiludis
*ab,
Biswajit
Bhattacharya
 a,
Ana Guilherme
Buzanich
a,
Friedrich
Fink
a,
Ines
Feldmann
a,
Johan E.
ten Elshof
c,
Franziska
Emmerling
ab and
Tomasz M.
Stawski
*a
a,
Ana Guilherme
Buzanich
a,
Friedrich
Fink
a,
Ines
Feldmann
a,
Johan E.
ten Elshof
c,
Franziska
Emmerling
ab and
Tomasz M.
Stawski
*a
aFederal Institute for Materials Research and, Testing, Unter den Eichen 87, 12205 Berlin, Germany. E-mail: tomasz.stawski@bam.de; stephanos.karafiludis@bam.de
bDepartment of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Straße 2, 12489 Berlin, Germany
cMESA+ Institute for Nanotechnology, University of Twente, P.O. Box 217, 7500 AE Enschede, the Netherlands
First published on 26th May 2023
Abstract
Here, we describe how to synthesise proton-conductive transition metal phosphates (TMPs) by direct thermal processing of precursor M-struvites, NH4MPO4·6H2O, with M = Ni2+, Co2+. In the as-derived TMP phases their thermal history and bulk proton conductivity were linked with the structural information about the metal coordination, phosphate groups, and volatile compounds. These aspects were investigated with vibrational and synchrotron-based spectroscopic methods (FT-IR, FT-RS, XAS). We elucidated the structures of amorphous and crystalline Ni- and Co phosphate phases in association with different coordination changes and distortion degrees of the metal polyhedra as they developed upon heating. Ni-struvite transformed to a stable amorphous phase over a broad range of temperatures (90 °C < T < 600 °C), in which it remained in an octahedral coordination environment, but the degree of distortion changed with T. In contrast, heating of Co-struvite led to several successive crystalline phases with only unstable transitional and short-lived amorphous components. Among the as-occurring phases, a highly functional layered M-dittmarite NH4MPO4·H2O obtained at low temperatures (T < 200 °C) demonstrated high proton conductivity values of 4.2 × 10−5 S cm−1 for Ni-dittmarite and Co-dittmarite > 10−4 S cm−1 at room temperature. Even at low humidity, these values are comparable with those found for Nafion, MOFs, some perovskites or composite materials. Coprecipitation of phosphates and transition metal cations in the form of struvite is potentially a viable method to extract these elements from wastewater. Thus, we propose that recycled M-struvites could be potentially further directly upcycled into crystalline and amorphous TMPs useful for electrochemical applications.
Introduction
Transition metal phosphates (TMPs) have attracted considerable attention due to their favourable properties for electro-, photo- or organic catalysis and proton exchange membranes.1–7 For example, TMPs are known as cost-effective cathode materials in lithium/sodium battery technologies,8–11 for their adequate catalytical performance in the oxygen evolution reaction12–14 or in organic reactions15–17 such as the selective oxidation of methane.18TMPs are highly suitable for this broad range of applications due to their ability to form thermally stable diverse crystalline and amorphous structures, including transition metals at various oxidation states and in several coordination environments. Each of these structures can exhibit beneficial properties for specific applications. For instance, the distorted phosphate tetrahedrons in TMPs promote the adsorption or incorporation of water molecules,19,20 advantageous for proton conductivity. TMPs are known to exhibit relatively high proton conductivity of the order of >10−2–10−8 S cm−1 from 25 °C to temperatures as high as 400 °C.21–24 For instance, Zr- and Sn-phosphates demonstrate a broad range of applications due to their sufficiently high proton conductivities at ambient conditions and high temperature.22,25–27
Importantly for mass applications, phosphates are readily inexpensive compounds providing potentially high mechanical strength,28 high thermal stability of the associated stable hydrogen-bonded network, and chemical compatibility with fuel cell device components (electrodes, catalysts, seals, etc.). In addition, TMPs with tuneable mesoporous frameworks can be synthesized either by using template-based3,14,20 or template-free methods.29 Thus, these materials can be obtained in many form factors such as bulk ceramics, thin films, or membrane materials.30–32 Due to the different condensation degrees of the phosphate units and variable exchangeable metal cations, TMPs provide a wide range of chemical compositions.
Moreover, TMPs can be made through a variety of synthesis procedures such as hydrothermal, mechanochemical, molten salt, or precipitation routes, resulting in highly crystalline to purely amorphous materials, depending on specific requirements.19,20 Even for near-identical chemical composition, crystalline and amorphous compounds could exhibit completely different physicochemical properties such as solubility, conductivity, or adsorption.33,34 Nevertheless, due to the lack of long-range order, the structure–property relationships in amorphous solid phases of TMPs are still relatively poorly explored in contrast to their crystalline counterparts.
The necessary challenge in the research on the applicational use of TMPs in all their varieties is an ability to obtain, elucidate and optimise different compositions and structures. This goal can be achieved by processing a universal TMP precursor with a single yet flexible crystal structure. Such potential TMP compound is M-struvite NH4MPO4·6H2O, where M2+ is usually Mg2+, but other divalent cations such as Ni2+, Co2+, Zn2+, Cu2+ readily substitute Mg2+ in the struvite structure due to similar ionic radii.35–38 In this work, we focus on Ni- and Co-containing phosphate materials as these are the most promising for electrochemistry.3,14,39 In our previous study, we showed how to precipitate Co- and Ni-struvites of various morphologies and sizes from aqueous solutions at concentrations similar to those found in wastewaters.40 Building on this previous work, we present how these recycled M-struvite materials could be potentially further upcycled for electrochemical applications by undergoing a direct thermal treatment (Fig. 1). Consequently, to upcycle and modify these materials for electrochemical applications, a complete understanding of the phase transition sequence during thermal heating and resolving of the complex structures is required. Amorphous and crystalline phases are formed upon the thermal decomposition of Ni- and Co-struvites. We show that some of such as-derived from M-struvite phases, namely M-dittmarites, exhibit high proton conductivity.
 | ||
| Fig. 1 The precursor M-struvites are obtained by a simple one-pot synthesis and further thermally treated, which results in degassing of H2O + NH3 and the associated occurrence of many phases, e.g. Ni- and Co-dittmarite, amorphous phases, HCPH, ACP and finally M2P2O7 (MPY) with M = Ni, Co. Arrows indicate thermal history, lower arrow (Ni), upper arrow (Co). The synthesis leads to TMP materials potentially suitable for proton exchange membranes (PEMs) in fuel cells. A balance between high thermal stability and significant proton conductivity is the key to their promising applicational use. | ||
Synthesis
(NH4)2HPO4 (DAP) (ChemSolute, 99%), NiSO4·6H2O (ChemSolute, 99%) and CoSO4·7H2O (Alfa Aesar, 98%) were used to synthesise precursor M-struvites (M = Ni or Co). A detailed description of the synthesis of the precursor transition metal struvites can be found elsewhere.40 For further analysis, the precursor M-struvites were thermally treated in a temperature series (T-series = 25 °C, 90 °C, 120 °C, 150 °C, 180 °C, 210 °C, 240 °C, 270 °C, 300 °C, 400 °C, 500 °C, 600 °C, 700 °C and 800 °C) with a constant dwell time of t = 24 h to evaluate the thermal decomposition products and related crystalline or amorphous phase transitions. The long dwell time was used to assure equilibration. The following abbreviations are used for the occurring phases: nickel struvite NH4NiPO4·H2O (NIS), amorphous phase (am), nickel pyrophosphate Ni2P2O7 (NPY), cobalt struvite NH4CoPO4·6H2O (COS), cobalt dittmarite NH4CoPO4·H2O (COD), hydrogen cobalt(II) phosphate monohydrate HCoPO4·H2O (HCPH), ammonium cobalt phosphate NH4CoPO4 (ACP), cobalt pyrophosphate Co2P2O7 (CPY) or M-pyrophospahte (MPY) with M = Ni or Co.| NH4MPO4·6H2O(s) → 0.5 M2P2O7(s) + NH3(g) + 6.5H2O(g) | (1) |
Upon the heat treatment, M-struvites converted sequentially to amorphous and crystalline phases through many phase transitions on a pathway to a final crystalline M-pyrophosphate, which formed at high temperatures, >500 °C.
The thermal treatment of Co-struvite resulted in the formation of a crystalline Co-dittmarite phase, while Ni-struvite converted completely to an amorphous phase. The Ni-dittmarite analogue was not found directly in the heating experiments. Therefore, in an additional series of experiments, the ∼5 g of Ni-struvite precursor powder was put into a hydrothermal autoclave reactor with a small amount of water (<50 ml) and heated up to T = 90 °C to obtain directly crystalline Ni-dittmarite, allowing for a comparison of its properties with those of Co-dittmarite.
Complete technical descriptions of the used characterisation methods, including X-ray diffraction (XRD) for identification of the phase composition, scanning electron microscopy (SEM), combined thermogravimetric analysis, and differential scanning calorimetry (TGA/DSC) for imaging and tracking phase transitions, infrared spectroscopy (IR), Raman spectroscopy (RS), X-ray absorption spectroscopy (XAS) for determination of the coordination environment, proton conductivity (PC) and dynamic vapour sorption (DVS) measurements for evaluation of the applicational use can be found in the ESI (Methods†).
Results and discussion
Composition and morphology of crystalline and amorphous phases
For a clear presentation of the occurring complex phase transitions, we summarise the most important aspects of our synthesis and phases in Fig. 1. Highly crystalline, phase-pure, transition metal M-struvite powders with M = Ni and Co were obtained as precursor materials through a simple one-pot synthesis (Fig. 1),40 as was evidenced by the X-ray diffraction (Fig. 2A–C). | ||
| Fig. 2 Diffractograms of T-series of (A) Ni- struvite and (B and C) Co-struvite from 25 °C to 800 °C; 90 °C* = hydrothermal synthesis at RH ≈ 100% at 90 °C; simulated XRD patterns were always added at the beginning and end of the diffractogram; The colour of the powder in the circles are placed next to the diagram; selected SE images of (D) Ni- and (E) Co- phosphates heated at different temperatures from 25 to 800 °C. SE images of the complete T-series of Ni and Co can be found in the ESI (Fig. S2 and S3†). Ni-struvite (NIS) reference database ICSD 403058; Ni-dittmarite (NID) reference database ICSD 424553; α-Ni2P2O7 (NPY) reference ICSD 403058; Co-struvite reference ICSD 170042; Co-dittmarite (COD) with COD reference database 2008122; α -NH4CoPO4 (ACP) reference PDF 00-018-0402; Hydrogen cobalt(II)phosphate hydrate (HCPH) reference database ICSD 33828; α -Co2P2O7 (CPY) reference database ICSD 280959. | ||
After treating the precursor Ni- and Co-struvites at different temperatures (T = 90–800 °C, T-series) in air with a dwell time of t = 24 h, chemical and phase composition changes occurred and involved transformations to amorphous and crystalline compounds. The changes were also associated with colour changes of the powders. In the T-series of Ni-struvite, the crystal decomposed to an amorphous phase already at 90 °C (Fig. 2A). Simultaneously, the colour turned from an emerald green (Ni-struvite precursor) to yellow-green at 90 °C to brown at 120 °C. Up to 600 °C, the amorphous phase remained stable. At temperatures >600 °C Ni2P2O7 began to crystallise, accompanied by a colour change of the powders from brown to pure yellow (Fig. 2A).
In contrast to Ni-struvite, Co-struvite NH4CoPO4·6H2O transformed to crystalline Co-dittmarite NH4CoPO4·H2O already after the first heating step at 90 °C (Fig. 2B and C). Up to 210 °C Co-dittmarite remained present until it converted to a mixture of hydrogen cobalt(II)phosphate hydrate HCoPO4·H2O, ammonium cobalt(II)phosphate NH4CoPO4, and an amorphous phase (Fig. 1). The amount of NH4CoPO4 increased with higher temperatures up to 300 °C while the reflections of HCoPO4·H2O disappeared. At T = 300 °C, a binary mixture of majorly NH4CoPO4 and minorly the amorphous phase(s) was present (Fig. 2B and C). At T = 400 °C Co2P2O7 crystallised, although the amorphous phase was still present. Heating to T ≥ 500 °C, resulted in higher intensities of peaks in the diffractogram. At the same time, the proportion of the amorphous phase decreased successively, meaning that the crystalline phase evolved at the expense of the amorphous component.
Thus, Ni- and Co-struvite systems differ significantly in their thermal decomposition behaviour (Fig. 1). The TGA/DSC analysis was carried out to elucidate the mentioned thermally induced phase transitions, and SE images of the heated samples were collected (ESI: Fig. S2–S4†). A detailed description of the TGA/DSC data can be found in the ESI (Supplementary Data Analysis 1 and Fig. S4†). The thermally induced phase transitions and resulting mass losses due to degassing of volatile compounds are in agreement with the phases detected by XRD measurements. Based on the mass losses and DSC signals, the range of possible chemical compositions of the amorphous phase at given temperatures could be significantly narrowed down (ESI: Supplementary Data Analysis 1†).
The SE images show the presence of a “relict” Ni-struvite crystal morphology in the thermally treated amorphous Ni samples, overall indicating a gradual crystalline-amorphous phase transition and an associated pseudomorphism of the amorphous phases after the crystalline Ni-struvite (Fig. 2D and ESI: Fig. S2†). Even at higher temperatures T ≥ 600 °C, the relict Ni-struvite morphology can still be observed (Fig. 2D, ESI: Fig. S2†). In this case, the pseudomorphism merely signifies a formation of the amorphous phase(s) without any crystalline intermediate step (Fig. 1A). Crystalline-crystalline pseudomorphism involving usually low volume changes is possible,41 but is unlikely to occur for these transformations, as the decomposition of M-struvite demonstrates significant volume dilatation due to loss of water and ammonia. Notably, the amorphous inhabited Ni-struvite structure is mechanically weak and can be easily distorted during sample manipulation (ESI: Fig. S2†).
In contrast, the transformation of Co-struvite (Fig. 2E and ESI: Fig. S3†) to other Co-phosphate phases is directly visible as changes in the morphology of the crystals. Here, partial crystalline-crystalline pseudomorphism also occurs i.e. of Co-dittmarite after Co-struvite, although the formation of other crystalline phases quickly extinguishes these structures (ESI: Fig. S3†). At T > 90 °C, Co-dittmarite replaces Co-struvite as a pseudomorph at progressing temperatures (Fig. 2E and ESI: Fig. S3†). This is remarkably well-visible as the habit of the former original X-shaped Co-struvite crystals can still be identified (Fig. 2E and ESI: Fig. S3†), although Co-dittmarite covers the whole surface. The crystals’ thin 2D sheet morphology indicates a crystalline-crystalline phase transition from Co-dittmarite to mainly HCoPO4·H2O and NH4CoPO4 (Fig. 1, 2E and ESI: Fig. S3†). Above 270 °C, the NH4CoPO4 exhibits its final crystal habit as two-dimensional plates similar to Co-dittmarite. When Co-pyrophosphates begin to crystallise at 400 °C (Fig. 2E and ESI: Fig. S3†), the crystal habit changes again to an elongated prism or a tabular habit. The single crystals form massive aggregates of tens of microns.
Tracking the removal of volatile components
As the phase composition and structure changed due to degassing (see eqn (1) and Fig. 1), we wanted to track the evaporation of the volatile components, mainly NH3/H2O, and the condensation of the phosphate environment, by performing infrared (IR) and Raman spectroscopy (ESI: Supplementary Note 2 and Fig. S5†).The IR and Raman bands correlate well with the phase compositions and the degrees of crystallinity of the Ni and Co samples42,43. The degree of crystallinity decreases in the T-series up to 300–400 °C due to the decomposition of the highly crystalline precursor M-struvite. It increases up to 800 °C during the crystallisation of Ni- and Co-pyrophosphates (Fig. 1). This trend is reflected in the progressive broadening and later narrowing/crystal field splitting of the phosphate stretching bands. Surprisingly, ammonium and water bands are still present at elevated temperatures of T = 400 °C, implying that these two species remain incorporated in the compounds.
The thermal effect on the coordination environment in the amorphous and crystalline Ni- and Co-phases
We further investigated the structures of amorphous and crystalline phases by following the changes/distortion in the metal coordination environment using X-ray absorption spectroscopy (XAS). Since XAS probes local coordination, it is particularly well-suited for establishing any potential differences among the structures of amorphous phases. Especially, the signal in the pre-peak region (X-ray absorption near-edge structure, XANES) contains information about the 1s-3d orbital transitions and expresses the degree of centrosymmetry of the first metal coordination sphere. Near-ideal centrosymmetric coordination shows very low pre-peak intensities, while more distorted geometries exhibit higher intensities. As only 3d-4p hybridisation enables parity-allowed transitions, the extent of those is strongly correlated with the coordination geometry. Due to an inversion centre, ideal octahedral coordination sites show lower pre-peak intensities than tetrahedral symmetries (App = area of the pre-peak, Atetrahedralpp > Aoctahedralpp) (Fig. 3). | ||
| Fig. 3 XAS spectra of the thermally processed TMPs plotted in the real space and the R-space of (A and B) Ni-phosphates and (C and D) Co-phosphates; 90 °C* = hydrothermal synthesis at RH ≈ 100% at 90 °C. Inset in (B and D): pre-peak region and integration from Gaussian fits, O = octahedron, T = tetrahedron. | ||
The signal beyond the absorption edge in the EXAFS region (extended X-ray absorption fine structure) depicts the excited photoelectron's interactions with the compound's next neighbour atoms. Here, the degree of crystallinity directly influences the signal's intensity and width. Since amorphous phases do not have an established long-range order, they show broader, and less intense peaks in the R-space mainly in the low R-region due to an irregular arrangement of the lattice ions. The XAS spectra (Fig. 4) prove that Co and Ni are present exclusively as divalent M2+ species in all the samples (due to absorption edge at around 7730 and 8350 eV), as it is evidenced by comparing them with several M2+ standards (only metal foils are shown for the sake of clarity). Quantitative pre-peak integration results were calculated to compare the distortion in the first coordination sphere (ESI: Table S3†). The thermally induced change in the pre-peak area, indicative of a higher degree of distortion, differs significantly between the Ni- and Co-phosphate systems (Fig. 1). Up to 600 °C, crystalline Ni-struvite decomposes to an amorphous phase with a higher degree of distortion in remanent octahedral coordination [Ni: A25 °Cpp = 0.028(2) (precursor), A300 °Cpp = 0.034(2) and A600 °Cpp = 0.041(2)]. In contrast, at low T < 200 °C the Co-system comprises crystalline octahedrally coordinated Co-phases, exhibiting a higher degree of distortion than the precursor Co-struvite [Co: A25 °Cpp = 0.026(1), A90 °Cpp = 0.032(1) and A150 °Cpp = 0.041(2)]. In the further heating process due to the occurrence of a tetrahedrally coordinated Co-phase (NH4CoPO4) at T > 180 °C, the pre-peak intensity rises significantly [A210 °Cpp = 0.071(1) and A300 °Cpp = 0.075(3)]. When the crystallisation to M2P2O7 (M = Ni, Co) occurs at T > 400 °C (for Ni at T = 600 °C, for Co at 400 °C), the pre-peak intensities in both systems decrease again. Here, the metal octahedra are still more distorted than those in the near-ideal octahedrally coordinated precursor M-struvites, which we associate with the presence of twisted and linked pyrophosphate chains (A800 °Cpp (Ni and Co) = 0.033(1)).
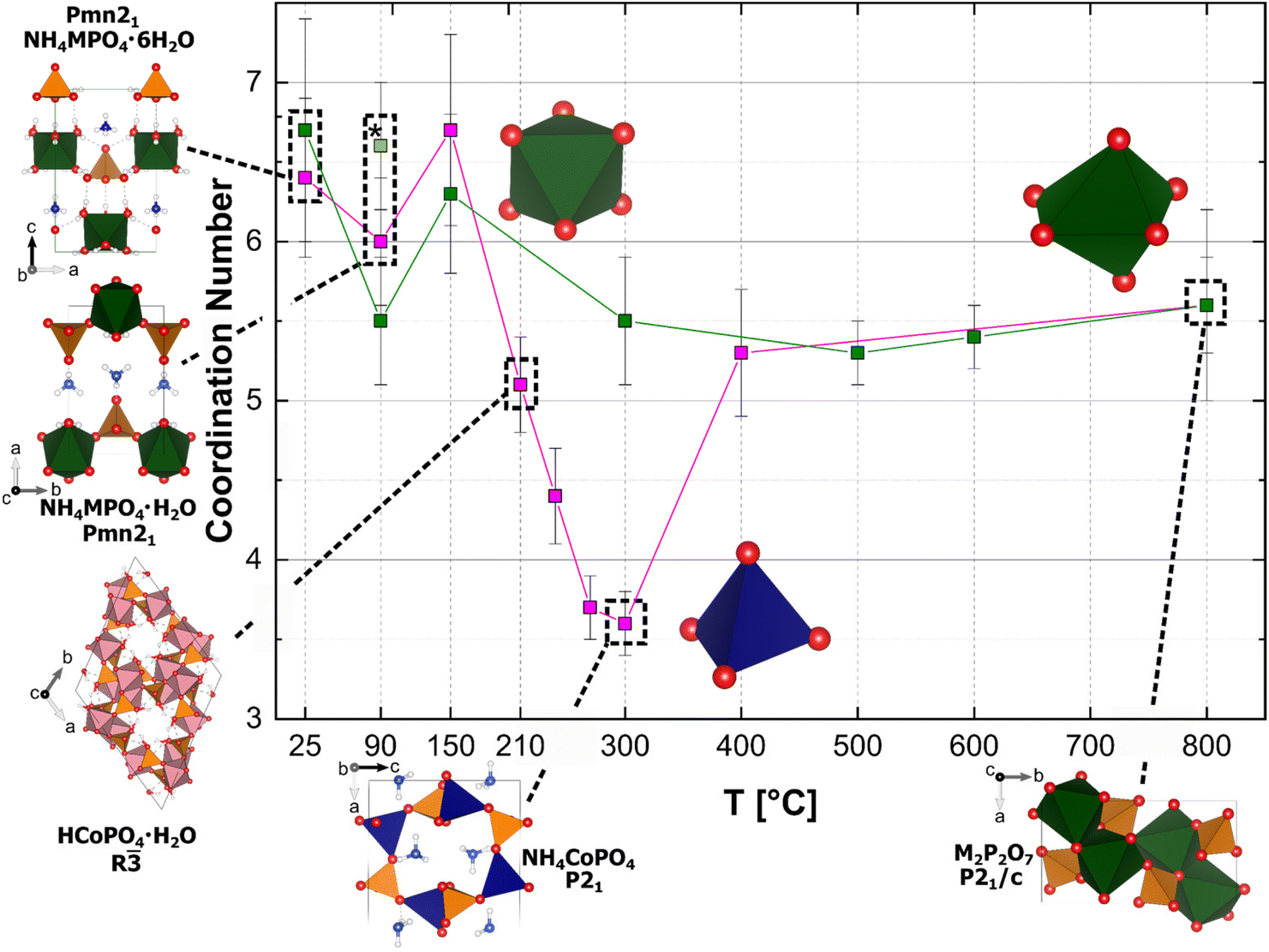 | ||
| Fig. 4 Calculated coordination numbers through the thermal history of Ni (green squares)- and Co (pink squares)-phosphate compounds; Occurring phases with their crystal structure in tetrahedral and octahedral coordination are depicted. The asterisk marks the hydrothermal synthesised Ni-dittmarite compound. | ||
Based on the fitting of the spectra in the R-space, radial distances from the absorber metal atom and average coordination numbers of the first coordination sphere could be calculated (ESI: Fig. S6–S8 and Tables S4–S20†). In the Ni-system, the calculated average coordination number, CN (i.e. degeneracy of oxygen), changes slightly from CN = 6.7(7) in the crystalline precursor Ni-struvite (T = 25 °C) to CN = 6.3(5) − 5.3(2) in the amorphous phase(s) (T = 90 °C–500 °C). Finally, Ni exhibits average coordination numbers of CN = 5.6(3) in the crystallised pyrophosphates, proving a remanent octahedral coordination with a slight degree of distortion in all occurring phases. The occurrence of several amorphous phases (polyamorphism) cannot be entirely ruled out. If this were the case, they all would have to exhibit similar local structures, as we do not observe any peak splitting in the Fourier-transformed spectra in R-space. In the Co-system the calculated average coordination number varies strongly from 6.4(5) in the precursor Co-struvite, to CN = 6.0(4) in Co-dittmarite heated at T = 90 °C to CN = 5.1(3) in the phase mixture of NH4CoPO4, HCoPO4·H2O, an amorphous phase and residues of Co-dittmarite heated at 210 °C. Finally, at 300 °C, the average coordination number of Co decreases to a value of CN = 3.6(2), indicative for a tetrahedral coordination. When the crystallisation of Co2P2O7 begins at ≥400 °C, the coordination number increases again to a value near 6 (CN = 5.3(4) at 400 °C and CN = 5.6(6) at 800 °C), which suggests octahedral coordination. In this way, the anticipated phase-dependent coordination transitions from octahedral (NH4CoPO4·6H2O, NH4CoPO4·H2O, HCoPO4·H2O) to tetrahedral (NH4CoPO4), and back to octahedral (Co2P2O7) could be successfully derived by fitting EXAFS spectra in the R-space.
Our EXAFS characterisation and observations from other methods paint a coherent image of changes in metal coordination, correlating with the phase compositions. These results are summarised in Fig. 4. Here the degeneracy of oxygen, i.e. the distinct metal's calculated coordination number (CN), is plotted against the processed temperature. A change in the coordination geometry causes the intense colour change of Co in the heated samples from an octahedral one in [CoO6] (NH4CoPO4·6H2O, NH4CoPO4·H2O, HCoPO4·H2O, amorphous phase, see also Fig. 1) to a tetrahedral one in [CoO4] (NH4CoPO4) and again to an octahedral coordination environment (Co2P2O7). Due to an increasing Jahn–Teller effect as Co2+ is no longer coordinated by six O2− of H2O molecules but by five O2− of PO4 and one remaining O2− from H2O, a color change can be observed from pink (Co-struvite) to purple (Co-dittmarite). When heated at 210 °C, Co-dittmarite transforms into HCoPO4·H2O and secondly into NH4CoPO4 accompanied by an intense colour change from purple to “cobalt blue”. The analogous colour change caused by an octahedral-tetrahedral coordination change was also observed in the related potassium compound of KCoPO4·H2O and KCoPO4.44 Here, the degree of distortion increases even more indicated by an increase in pre-peak area due to a Co2+ coordination change from an octahedron to a tetrahedron. The associated change in d-orbital splitting with a change in coordination significantly affects the colour of the Co-compounds. When the coordination switches again to an octahedral configuration accompanied by the phase transformation of NH4CoPO4 to Co2P2O7, the CoO6 octahedron in Co-pyrophosphate exhibits a lower degree of distortion than the CoO4 tetrahedron in NH4CoPO4. Therefore, the pre-peak area decreases while the intensity of the white line increases again (Fig. 4). Based on the calculations on the degeneracy, Co2+ has a near octahedral coordination in the amorphous compound. Due to their presence in the T-range of 210–500 °C, a low amount of Co is always bound in octahedral coordination.
From the crystal field stabilisation energy (CFSE) perspective, Ni2+ as a d8 ion strongly prefers octahedral coordination. The difference in CFSE between tetrahedral and octahedral geometries is significantly higher (|ΔCFSEO–T| = 0.84 with CFSEO = −1.2 ΔO, CFSET ≈ −0.36 ΔO assuming ΔT = 4/9 ΔO45) than the difference between both geometries in the Co-system. Co2+ as a d7 ion exhibits only a slight difference between the CFSEO and CFSET in high spin configurations (|ΔCFSEO–T| = 0.27 with CFSEO = −0.8 ΔO and CFSET ≈ −0.53 ΔO). This difference in energy/stability in comparison with Ni2+ promotes an additional occurrence of tetrahedral coordination in the Co-system.46,47
Summarised order of phase transitions with increasing temperature
Based on the combined multi-modal characterisation, we established the order of phase transitions in our M-struvite systems. At first, Ni-struvite decomposes to an amorphous phase while Co-struvite transforms through a crystalline-crystalline phase transition to Co-dittmarite at T = 90 °C (Fig. 1). The pseudomorphic phase transition of the amorphous Ni-PO4 phase formed from Ni-struvite, leads to a relict “Ni-struvite crystal skeleton” visible in the SE images (ESI: Fig. S2†). Although pseudomorphism indicates a weak degree of densification and structural reconfiguration (otherwise precursor crystal framework would collapse), the relict crystal morphology is not in every case preserved as in the 500 °C sample (ESI: Fig. S2K†). Looking at the degassing events of heated Co-struvite based on XRD and DSC, an amorphous phase formed in a second and third thermal event at 140 °C and 250 °C, associated with the partial decomposition of Co-dittmarite and HCoPO4·H2O (Fig. 1). The amorphous phase is proposed to be akin to HCoPO4 based on the TGA/DSC, as is also suggested by other studies in the literature48,49 Surprisingly, ammonia is still present in the amorphous phases up to 400 °C in both metal systems, which is important for potential proton carrier molecules. Based on the EXAFS fits, the spectra of the amorphous phases T > 400 °C resemble mostly those of crystalline Ni-pyrophosphate with less pronounced long-range order above R distances > 4 Å. This fact indicates a beginning of pyrophosphatisation already at 400 °C in agreement with FT-IR and FT-RS. Further heating leads to the actual crystallisation of M-pyrophosphate. At 400 °C Co2P2O7 forms, although a major proportion of amorphous phases is present. In contrast, the amorphous nickel phosphate phase remains stable up to 600 °C until it converts to Ni2P2O7. The humidity and heating time/rate can significantly influence the phase stability and crystallinity, especially in the low T-region below 400 °C. This ability of M-struvite to sequentially convert into various amorphous and crystalline transition metal phosphate phases opens the space for possible crystal and phase engineering and upcycling of the waste-derived struvite29,40,50,51 into tailored TMPs, as we indeed explore below.Proton conducting properties of selected Ni- and Co-phosphates
The structural results presented above point to the potential presence of extensive hydrogen-bonded frameworks involving the coordination of water H2O and ammonia NH3 molecules in some of the amorphous or crystalline TMP phases of Ni and Co. Such frameworks indicate a potential for good proton conductive properties of these compounds. To characterise our processed TMP phases as functional proton conductive materials, proton conductivity (PC) measurements were performed on the pressed powder pellets at different relative humidity values and temperatures (Fig. 5, see Methods Proton conductivity measurements for more details, Fig. S9–S11†). Before and after each PC measurement, a diffraction pattern was recorded to demonstrate the phase stability (Fig. S12†). The impedance spectra were fitted with a minimal circuit model consisting of a parallel constant phase element (CPE) and resistor (R) in series with two parallel Warburg elements (WS and WO, inset: Fig. 5B). | ||
Fig. 5 Nyquist plots with real and imaginary impedance of (A) Ni- and (B) Co-dittmarite, NH4MPO4·H2O with M = Ni, Co at RH = 68%–98% at T = 25 °C; inset in B: equivalent circuit of the fitting model; (C) Arrhenius plots of Ni- and Co-dittmarite with ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (σT) vs. 1000/T [K−1] (T = 25 °C–80 °C), red dashed line = fit of the data; inset: DVS isotherms of Ni- and Co-dittmarite; (D) view on the (0k0) plane of the crystal structure of M-dittmarite Pmn21 from different perspectives; dashed black arrows and blue marked (0k0) planes indicate enhanced proton migration pathways. (σT) vs. 1000/T [K−1] (T = 25 °C–80 °C), red dashed line = fit of the data; inset: DVS isotherms of Ni- and Co-dittmarite; (D) view on the (0k0) plane of the crystal structure of M-dittmarite Pmn21 from different perspectives; dashed black arrows and blue marked (0k0) planes indicate enhanced proton migration pathways. | ||
The high-frequency R-CPE circuit represents the electronic resistivity of the metal phosphate phase. In our model, the low-frequency WS–WO circuit models the effects of the proton transport through grain boundaries. The Warburg short element expresses the transmissive, while the Warburg open element points to the reflective character of the grain boundary. No singular Warburg element (either a general, short or open W), fitted the impedance spectra sufficiently well. Including two Warburg elements resulted in much more accurate fitting results. From the intersection of the fits and the abscissa axis (real part of impedance Z′), the proton conductivity was calculated (more details in ESI: Methods proton conductivity measurements†). As the current is measured perpendicularly to the pellet base, the proton conductivity is interpreted as bulk proton conductivity under the assumption of randomly orientated grain boundaries.52 The activation energy (EA) of proton migration is the amount of energy required for a proton to jump from one ionic domain to another, an important value to distinguish between Grotthuss and vehicle mechanism.
Selected samples of the Ni- and Co T-series (Fig. 1) were analysed. For Co: Co-dittmarite processed at T = 90 °C, NH4CoPO4 at T = 300 °C and Co-pyrophosphate at T = 800 °C. For Ni: Ni-dittmarite at T = 90 °C in 100% H2O, amorphous Ni-PO4 at T = 500 °C and Ni-pyrophosphate at T = 800 °C. All data and fits associated with the proton conductivity are summarised in the ESI (Fig. S9–S11 and Tables S22, S23†). The electrochemical impedance data were plotted in Nyquist diagrams with the imaginary part of the impedance on the y-axis Zimaginary and the real part of the impedance on the x-axis Zreal. All Nyquist plots demonstrate semicircles in the high-frequency region and an elongated tail in the low-frequency region. The M-dittmarite compounds of Ni and Co, heated at T = 90 °C and stable up to 200 °C, exhibit the highest proton conductivities of all analysed phases at 1.4 × 10−4 S cm−1 and 4.2 × 10−5 S cm−1 for Co- and Ni-dittmarite, respectively, at RH = 98% and T = 25 °C. Considering the low-frequency region, the Warburg coefficients from the fits show a slight increase of the WS/WO ratio with higher RH, visible in both M-dittmarite components from nearly near zero to 2.5(1) for Ni and 6.5(9) for Co (ESI: Table S23†). This could indicate a more transmissive nature of the grain boundaries at high RH. From a crystallographic point of view, protons can "jump" in the hydrogen-bond network mainly in the (0k0) plane in two directions, where ammonium NH4 and the coordinating water of the metal octahedron are located.
Since protons are most likely transported through 2D layers, build up by the ammonium sites (Fig. 5) in the M-dittmarite structure Pmn21, the compound probably demonstrates a significant anisotropic proton conductivity in directions in the (010) plane. Perpendicularly to this direction, the proton conductivity is most likely decreasing. As we measured the bulk proton conductivity of a pressed pellet of the material, grain boundaries, microstructure and the orientation of the different grains effect the overall bulk properties. Due to these anisotropic features, the overall bulk proton conductivity could be influenced. Furthermore, to evaluate the water affinity and adsorption, a major factor influencing the proton conductivity, dynamic vapour sorption (DVS) measurements were carried out (inset in Fig. 6C and ESI: Fig. S11†). Co-dittmarite displays a final water uptake of 0.0263 cm3 g−1 at RH = 98%, corresponding to the sorption of 0.27 H2O molecules per formula unit. Ni-dittmarite exhibits lower values of 0.0164 cm3 H2O/g at 98% RH equating to 0.17 molecules of H2O per formula unit. However, for all the RH values and T = 25 °C, the Co compound exhibits proton conductivities around one order of magnitude higher than those found for the Ni analogue. This discrepancy disappears with higher temperatures up to 80 °C (Fig. 6C). Although both compounds share the same structure, Co2+ exhibits higher proton conductivity at T = 25 °C in Co-dittmarite, which can be correlated with slight structural differences compared to Ni-dittmarite, as indicated by our XAS measurements. The differences in bond lengths, especially of the first coordination sphere Co–O and Ni–O could cause different local polarities resulting in a changed proton mobility (ESI: Tables S11 and S13†). The adsorbed amount of water could only promote an increase of proton conductivity at high RH > 80%, but not at lower RH ≤ 80%. The dependency of the proton conductivity on the water partial pressure (associated with the RH) is similar for both dittmarite phases, because their log(σ) vs. log(pH2O) slope values are relatively close to each other (16 ± 2 (Ni) vs. 13 ± 1 (Co), Table S22†). When examining other processed phosphate phases, NH4CoPO4 shows a significant proton conductivity of 6.42 × 10−5 S cm−1 at RH = 98% and T = 80 °C. The other phases, Ni2P2O7, Co2P2O7 and the amorphous Ni-PO4 phase of heated at 500 °C, demonstrate low to medium proton conductivities in the range of 10−6–10−7 S cm−1 (ESI: Fig. S8 and S9†). The M-dittmarite and M2P2O7 phases did not change its phase composition after the PC measurement, while the amorphous Ni-phase and NH4CoPO4 crystallised. In addition, the water partial pressure-dependent proton conductivity behaviour of the M2P2O7 compounds is significantly different from that of Ni-, Co-dittmarite, and NH4CoPO4. Due to a highly condensed structure, the adsorbed water in the pyrophosphates, even at high RH (>85%), does not provide a significant jump in proton conductivity (ESI: Fig. S10 and Table S22†).
This work's goal was to obtain a TMP material with high thermal stability and significant proton conductivity optimal for their potential applicational use, e.g., proton exchange membranes in fuel cells (Fig. 1). For this purpose, the mechanism of ionic conductivity/proton migration in the material had to be investigated by determining its activation energy. In general, proton conductivity is assumed to rely on two major transport mechanisms, the Grotthuss mechanism and the vehicle mechanism.53 The Grotthuss mechanism i.e. proton jumping, points to the migration processes of protons through translation from an H2n+1On+ ion to a water molecule by tunnelling in a hydrogen bond with subsequent molecule rotation. This diffusing proton in the newly formed H2n+1On+ ion can attach to the next neighbour water molecules resulting in a successive motion of H+ over small molecular distances through the compound.53,54 Recent O–K-edge XAS results could prove the dominant occurrence of an H7O3+ proton complex in contrast to the conventional explanation of Zundel or Eigen ions, H5O2+ and H9O4+.55 In the vehicle mechanism, molecules such as water H2O, ammonia NH3, or alkali metals Na, K, etc. act as proton acceptors/Brønsted bases and transport the protons as carriers through the compound over large molecular distances.53 The activation energy of proton migration EA, calculated from the Arrhenius plots, is a key parameter to distinguish between those mechanisms.53,56 A value between 0.1–0.4 eV per proton indicates proton jumping, meaning the transfer of protons is accomplished between the proton donor and acceptor. A value greater than 0.4 eV per particle implies a vehicle mechanism where a proton transport proceeds via the diffusion of protonated carriers.52,53 A low content of ammonia and water in the compound resulting from heating at elevated temperatures would decrease the proton conductivity performance as a weaker hydrogen-bond network and a lower number of potential carriers of protons are available. The crystal structure determines the possibilities and pathways for preferential proton transport e.g. through 1D channels, 2D layers, or 3D frameworks, and the binding strength of the hydrogen network (e.g. “free” crystal water vs. bound hydroxyl groups). Therefore, the absolute number of protons and carrier molecules in the structure and the internal structure significantly influence the overall PC performance. The dittmarite structure Pmn21 seems to provide a beneficial structure for enhanced proton conduction as protons can migrate through the weakly bound ammonium layers in two dimensions. The increase of the Warburg ratio WS (transmissive boundary)/WO (reflective boundary) with increasing RH could represent a more transmissive behaviour of the grain boundaries as interpreted by other studies.57–59 More water units are adsorbed at the interfaces providing an enhanced proton transfer due to a higher density of potential transporting molecules. The calculated values for the activation energy EA infer a Grotthuss mechanism (<0.4 eV) in most compounds except for the Ni- and Co-pyrophosphates. In these compounds, no structural water or ammonia are present, and carrier molecules only accomplish proton conduction, indicated by the high activation energy EA and the slopes of log(σ) vs. log(pH2O) (ESI: Table S22†). The similar increase of proton conductivity of Ni- and Co-dittmarite at higher RH/partial pressures of water majorly agrees with the uptake of water visible in the DVS isotherms and the slopes of log(σ) vs. log(pH2O) (Fig. 6 and ESI: Fig. S9–S11, Table S22†). The activation energies for proton migration of 0.16 eV for Co-dittmarite and 0.36 eV for Ni-dittmarite are relatively low compared to other oxides and phosphate compounds (Table 1 and ESI: Table S23†).
| Material | Chemical composition | T [°C]/RH [%] | σ [S cm−1] | E a (eV) | Ref. |
|---|---|---|---|---|---|
| Ionomer | NAFION® | 25/98 | 7.80 × 10−2 | — | 63 |
| Na-Al-PO4 | Na6[(AlPO4)8(OH)6]·9H2O | 20/98 | 3.60 × 10−3 | 0.21 | 64 |
| Doped Sn2P2O7 | In0.1Sn0.9P2O7 | 300/— | 0.195 | — | 65 |
| Sn-PO4 | Sn(HPO4)2 3H2O | 100/— | 1.00 × 10−2 | — | 27 |
| ZrP2O7 | Zr(P2O7)0.81(O3POH)0.38 | 20/98 | 1.30 × 10−3 | — | 25 |
| TiP2O7 | TiP2O7 | 100/— | 4.40 × 10−3 | 0.14 | 66 |
| Ti-PO4 | Ti2(HPO4)4 | 20/95 | 1.20 × 10−3 | 0.13 | 67 |
| org./inorg. Mn-PO4-comp. | (C2H10N2) [Mn2(HPO4)3](H2O) | 20/99 | 1.60 × 10−3 | 0.22 | 68 |
| org./inorg. Fe-PO4-comp. | (C4H12N2)1.5[Fe2(OH)(H2PO4)(HPO4)2(PO4)] 0.5(H2O) | 40/98 | 5.10 × 10−4 | — | 69 |
| org./inorg. Co-PO4-comp. | (C2N2H10)0.5CoPO4 | 56/98 | 2.00 × 10−3 | 1.01 | 70 |
| org./inorg. Zn-PO4-comp. | NMe4Zn[HPO4][H2PO4] | 30/98 | 1.30 × 10−2 | 0.92 | 71 |
| K-V-PO4 | K2[(VO)2(HPO4)2(C2O4)] | 40/95 | 1.15 × 10−2 | — | 72 |
| NH4-Ni-PO4 | NH4NiPO4 H2O | 25/98 | 4.20 × 10−5 | 0.36 | o.w. |
| NH4-Co-PO4 | NH4NiPO4 H2O | 25/98 | 1.40 × 10−4 | 0.16 | o.w. |
In addition, based on recent research,60 the dittmarite structure shows high suitability as a framework crystal for high-entropy phosphate materials (HEMPs). HEMPs are known to be electrochemically more active than their pure endmembers as the high configurational entropy of mixing promotes their reactivity.61,62 In such a way, a high-entropy transition metal dittmarite compound derived from a high-entropy struvite could show even better performance than Ni- or Co in proton conductivity, but this aspect has to be further evaluated.
Finally, we showed in our previous study,40 that M-struvite could be precipitated from industrial wastewater. Here we demonstrate that through a simple thermal treatment, these reclaimed TMP materials could be upcycled for applicational purposes (Fig. 1). The processed M-dittmarite compounds of Ni- and Co demonstrate high to superprotonic conductive behaviour comparable to ionomers, i.e. Nafion 117,63,73,76 MOFs,74 perovskites,75or other oxide/phosphate compounds.
A summary of selected high-performing proton conductive transition metal phosphate compounds can be found in Table 1 in further detail in the ESI (Table S23†).While most of the reported compounds require a complex multistep synthesis, our as-obtained materials could be precipitated in a facile one pot way directly out of wastewater. For example, ceramic oxide proton conductors, often perovskite-type, require in most cases an energy intensive, and complex solid state or solvothermal syntheses. Even if the absolute bulk proton conductivity values of the as-obtained Ni-and Co-dittmarites are not among the highest measured, they are still sufficiently conductive considering that we are discussing recycled waste materials. Due to their high thermal and chemical stability up to 210 °C, the M-dittmarite compounds could endure the optimal operating temperatures in conventional fuel cells. These compounds may be upcycled directly or could be reused indirectly by processing them even further to an advanced proton conductive material. In combination with their high compatibility with other cell components (electrodes, electrolytes, seals) and low cost, M-dittmarites are promising proton-conducting materials.77,78
Conclusions
In this study, we followed the thermal history of Ni- and Co-struvite decomposition all the way to their crystallisation into M-pyrophosphates. Ni-struvite forms an amorphous phase during the thermal treatment, which preserves octahedral metal coordination with slight distortion at all temperatures. The complex amorphous structure increasingly resembles a local structure of a Ni-pyrophosphate with progressive temperatures. In contrast, heated Co-struvite exhibits several crystalline-crystalline phase transitions with an associated change of metal coordination from octahedral to tetrahedral, and back to octahedral. In both systems, volatile compounds such as H2O or NH3 are still present in the solids up to 400 °C, potentially making transition metal phosphates promising high-temperature water-retaining materials. The metal and phosphate coordination environment associated with the amount of bound water and ammonia in the thermally treated TMPs could be directly linked to their proton conductivity performance. Indeed, with values of >10−4 S cm−1 at room temperature, a low activation energy of 0.16 eV, and high thermal stability above 200 °C, the layered Ni- and Co-dittmarites are the most promising candidates as proton exchange material, among all the obtained phases. Importantly, we showed that such functional materials could be directly made by thermally processing M-struvites, which might be precipitated from various wastewater streams.Conflicts of interest
There are bo conflicts do declare.Acknowledgements
We acknowledge Dr Martin Radtke (BAM) for his help at the BAMline beamline of BESSY II. We thank BAM and Helmholtz-Zentrum Berlin (HZB) for providing us with the beamtime at BESSY II. We thank Dr Thomas Schmidt for providing us access to the Raman microscope.References
- Y. Zhao, Z. Chen, D. B. Xiong, Y. Qiao, Y. Tang and F. Gao, Hybridized Phosphate with Ultrathin Nanoslices and Single Crystal Microplatelets for High Performance Supercapacitors, Sci. Rep., 2016, 6, 17613 CrossRef CAS PubMed.
- W.-X. Lu, B. Wang, W.-J. Chen, J.-L. Xie, Z.-Q. Huang, W. Jin and J.-L. Song, Nanosheet-like Co3(OH)2(HPO4)2 as a Highly Efficient and Stable Electrocatalyst for Oxygen Evolution Reaction, ACS Sustainable Chem. Eng., 2019, 7, 3083–3091 CrossRef CAS.
- J. Zhang, Y. Yang, Z. Zhang, X. Xu and X. Wang, Rapid synthesis of mesoporous NixCo3−x(PO4)2hollow shells showing enhanced electrocatalytic and supercapacitor performance, J. Mater. Chem. A, 2014, 2, 20182–20188 RSC.
- X. Li, P. Xu, M. Chen, G. Zeng, D. Wang, F. Chen, W. Tang, C. Chen, C. Zhang and X. Tan, Application of silver phosphate-based photocatalysts: Barriers and solutions, Chem. Eng. J., 2019, 366, 339–357 CrossRef CAS.
- W. Zhang, P. Oulego, S. K. Sharma, X. L. Yang, L. J. Li, G. Rothenberg and N. R. Shiju, Self-Exfoliated Synthesis of Transition Metal Phosphate Nanolayers for Selective Aerobic Oxidation of Ethyl Lactate to Ethyl Pyruvate, ACS Catal., 2020, 10, 3958–3967 CrossRef CAS PubMed.
- D. Zhang, H. Qiu, L. Jiang, F. Lv, C. Ma and W. Hu, Enantioselective palladium(II) phosphate catalyzed three-component reactions of pyrrole, diazoesters, and imines, Angew. Chem., Int. Ed. Engl., 2013, 52, 13356–13360 CrossRef CAS PubMed.
- M. Chen, X. Fu, Z. Chen, J. Liu and W. H. Zhong, Protein–Engineered Functional Materials for Bioelectronics, Adv. Funct. Mater., 2021, 31, 2006744 CrossRef CAS.
- N. Omar, M. A. Monem, Y. Firouz, J. Salminen, J. Smekens, O. Hegazy, H. Gaulous, G. Mulder, P. Van den Bossche, T. Coosemans and J. Van Mierlo, Lithium iron phosphate based battery – Assessment of the aging parameters and development of cycle life model, Appl. Energy, 2014, 113, 1575–1585 CrossRef CAS.
- B. Senthilkumar, Z. Khan, S. Park, I. Seo, H. Ko and Y. Kim, Exploration of cobalt phosphate as a potential catalyst for rechargeable aqueous sodium-air battery, J. Power Sources, 2016, 311, 29–34 CrossRef CAS.
- Q. Liu, L. Zhou, D. Zhao, C. Wan, P. Sun, X. Lv, X. Sun and L. Fang, Constructing aqueous rechargeable Zn//Co battery with hierarchical structural cobalt phosphate octahydrate for high-performance energy storage, J. Power Sources, 2022, 533, 231344 CrossRef CAS.
- J. Hassoun, F. Bonaccorso, M. Agostini, M. Angelucci, M. G. Betti, R. Cingolani, M. Gemmi, C. Mariani, S. Panero, V. Pellegrini and B. Scrosati, An Advanced Lithium-Ion Battery Based on a Graphene Anode and a Lithium Iron Phosphate Cathode, Nano Lett., 2014, 14, 4901–4906 CrossRef CAS PubMed.
- N. L. W. Septiani, Y. V. Kaneti, K. B. Fathoni, K. Kani, A. E. Allah, B. Yuliarto, Nugraha, H. K. Dipojono, Z. A. Alothman, D. Golberg and Y. Yamauchi, Self-Assembly of Two-Dimensional Bimetallic Nickel–Cobalt Phosphate Nanoplates into One-Dimensional Porous Chainlike Architecture for Efficient Oxygen Evolution Reaction, Chem. Mater., 2020, 32, 7005–7018 CrossRef CAS.
- L. Xie, R. Zhang, L. Cui, D. Liu, S. Hao, Y. Ma, G. Du, A. M. Asiri and X. Sun, High-Performance Electrolytic Oxygen Evolution in Neutral Media Catalyzed by a Cobalt Phosphate Nanoarray, Angew. Chem., Int. Ed., 2017, 56, 1064–1068 CrossRef CAS PubMed.
- M. Pramanik, C. Li, M. Imura, V. Malgras, Y. M. Kang and Y. Yamauchi, Ordered Mesoporous Cobalt Phosphate with Crystallized Walls toward Highly Active Water Oxidation Electrocatalysts, Small, 2016, 12, 1709–1715 CrossRef CAS PubMed.
- K. Otsuka and Y. Wang, Direct conversion of methane into oxygenates, Appl. Catal., A, 2001, 222, 145–161 CrossRef CAS.
- N. Brodt and J. Niemeyer, Chiral organophosphates as ligands in asymmetric metal catalysis, Org. Chem. Front., 2023 10.1039/D3QO00206C.
- J. De Maron, L. Bellotti, A. Baldelli, A. Fasolini, N. Schiaroli, C. Lucarelli, F. Cavani and T. Tabanelli, Evaluation of the Catalytic Activity of Metal Phosphates and Related Oxides in the Ketonization of Propionic Acid, Sustain. Chem., 2022, 3, 58–75 CrossRef CAS.
- A. Matsuda, H. Tateno, K. Kamata and M. Hara, Iron phosphate nanoparticle catalyst for direct oxidation of methane into formaldehyde: effect of surface redox and acid–base properties, Catal. Sci. Technol., 2021, 11, 6987–6998 RSC.
- R. Lin and Y. Ding, A Review on the Synthesis and Applications of Mesostructured Transition Metal Phosphates, Materials, 2013, 6, 217–243 CrossRef CAS PubMed.
- H. Zhao and Z. Yuan, Insights into Transition Metal Phosphate Materials for Efficient Electrocatalysis, ChemCatChem, 2020, 12, 3797–3810 CrossRef CAS.
- H. Mohammed, A. Al-Othman, P. Nancarrow, Y. Elsayed and M. Tawalbeh, Enhanced proton conduction in zirconium phosphate/ionic liquids materials for high-temperature fuel cells, Int. J. Hydrogen Energy, 2021, 46, 4857–4869 CrossRef CAS.
- J. Pandey, M. M. Seepana and A. Shukla, Zirconium phosphate based proton conducting membrane for DMFC application, Int. J. Hydrogen Energy, 2015, 40, 9410–9421 CrossRef CAS.
- Y. Kawakami and M. Miyayama, Proton Conducting Properties of Layered Metal Phosphate Hydrates, Key Eng. Mater., 2006, 320, 267–270 CAS.
- K. P. Ramaiyan, S. Herrera, M. J. Workman, T. A. Semelsberger, V. Atanasov, J. Kerres, S. Maurya, Y. S. Kim, C. R. Kreller and R. Mukundan, Role of phosphate source in improving the proton conductivity of tin pyrophosphate and its composite electrolytes, J. Mater. Chem. A, 2020, 8, 16345–16354 RSC.
- G. Alberti, M. Casciola, S. Cavalaglio and R. Vivani, Proton conductivity of mesoporous zirconium phosphate pyrophosphate, Solid State Ionics, 1999, 125, 91–97 CrossRef CAS.
- P. V. Valsaraj and C. Janardanan, Comparative studies on Proton conducting nature of Copper selective tin Zirconium phosphate Cation exchanger with its single Salts counter parts, Res. J. Chem. Sci., 2014, 4, 84–90 Search PubMed.
- W. Huang, S. Komarneni, Y. D. Noh, J. Ma, K. Chen, D. Xue, X. Xue and B. Jiang, Novel inorganic tin phosphate gel: multifunctional material, Chem. Commun., 2018, 54, 2682–2685 RSC.
- D. Shakhvorostov, M. A. Nicholls, P. R. Norton and M. H. Müser, Mechanical properties of zinc and calcium phosphates, Eur. Phys. J. B, 2010, 76, 347–352 CrossRef CAS.
- S. Karafiludis, A. G. Buzanich, C. Heinekamp, A. Zimathies, G. J. Smales, V.-D. Hodoroaba, J. E. ten Elshof, F. Emmerling and T. M. Stawski, Template-free synthesis of mesoporous, amorphous transition metal phosphate materials, Nanoscale, 2023, 15, 3952–3966 RSC.
- G. Renaudin, S. Gomes and J. M. Nedelec, First-Row Transition Metal Doping in Calcium Phosphate Bioceramics: A Detailed Crystallographic Study, Materials, 2017, 10, 92 CrossRef PubMed.
- E. Mahboubi, A. Yourdkhani and R. Poursalehi, Liquid phase deposition of iron phosphate thin films, CrystEngComm, 2018, 20, 5256–5268 RSC.
- R. M. P. Colodrero, P. Olivera-Pastor, A. Cabeza and M. Bazaga-Garcia, Properties and Applications of Metal Phosphates and Pyrophosphates as Proton Conductors, Materials, 2022, 15, 1292 CrossRef CAS PubMed.
- P. P. Prosini, L. Cianchi, G. Spina, M. Lisi, S. Scaccia, M. Carewska, C. Minarini and M. Pasquali, Synthesis and Characterization of Amorphous 3Fe2O3·2P2O5·10H2O and Its Electrode Performance in Lithium Batteries, J. Electrochem. Soc., 2001, 148, A1125 CrossRef CAS.
- O. Delmer, P. Balaya, L. Kienle and J. Maier, Enhanced Potential of Amorphous Electrode Materials: Case Study of RuO2, Adv. Mater., 2008, 20, 501–505 CrossRef CAS.
- K. S. Le Corre, E. Valsami-Jones, P. Hobbs and S. A. Parsons, Phosphorus Recovery from Wastewater by Struvite Crystallization: A Review, Crit. Rev. Environ. Sci. Technol., 2009, 39, 433–477 CrossRef CAS.
- A. Uysal, Y. D. Yilmazel and G. N. Demirer, The determination of fertilizer quality of the formed struvite from effluent of a sewage sludge anaerobic digester, J. Hazard. Mater., 2010, 181, 248–254 CrossRef CAS PubMed.
- X. Hao, C. Wang, M. C. van Loosdrecht and Y. Hu, Looking beyond struvite for P-recovery, Environ. Sci. Technol., 2013, 47, 6–4965 Search PubMed.
- M. Sena, M. Seib, D. R. Noguera and A. Hicks, Environmental impacts of phosphorus recovery through struvite precipitation in wastewater treatment, J. Cleaner Prod., 2021, 280, 124222 CrossRef CAS.
- Z. Song, K. C. Wang, Q. Sun, L. Zhang, J. Li, D. Li, P. W. Sze, Y. Liang, X. Sun, X. Z. Fu and J. L. Luo, High-Performance Ammonium Cobalt Phosphate Nanosheet Electrocatalyst for Alkaline Saline Water Oxidation, Adv. Sci., 2021, 8, 2100498 CrossRef CAS PubMed.
- S. Karafiludis, A. G. Buzanich, Z. Kochovski, I. Feldmann, F. Emmerling and T. M. Stawski, Ni- and Co-Struvites: Revealing Crystallization Mechanisms and Crystal Engineering toward Applicational Use of Transition Metal Phosphates, Cryst. Growth Des., 2022, 22, 4305–4315 CrossRef CAS.
- E. T. Pedrosa, C. V. Putnis and A. Putnis, The pseudomorphic replacement of marble by apatite: The role of fluid composition, Chem. Geol., 2016, 425, 1–11 CrossRef CAS.
- L. Popovi, D. de Waal and J. C. A. Boeyens, Correlation between Raman wavenumbers and P-O bond lengths in crystalline inorganic phosphates, J. Raman Spectrosc., 2005, 36, 2–11 CrossRef.
- K. D. Litasov and N. M. Podgornykh, Raman spectroscopy of various phosphate minerals and occurrence of tuite in the Elga IIE iron meteorite, J. Raman Spectrosc., 2017, 48, 1518–1527 CrossRef CAS.
- Q. Che, X. Xie, Q. Ma, J. Wang, Y. Zhu, R. Shi and P. Yang, Coordination environment evolution of Co(ii) during dehydration and re-crystallization processes of KCoPO4·H2O towards enhanced electrocatalytic oxygen evolution reaction, RSC Adv., 2020, 10, 14972–14978 RSC.
- E. Larsen and G. N. L. Mar, The angular overlap model. How to use it and why, J. Chem. Educ., 1974, 51, 8 Search PubMed.
- H. Irving and R. Williams, Order of stability of metal complexes, Nature, 1948, 162, 746–747 CrossRef CAS.
- H. Irving and R. Williams, The stability of transition-metal complexes, J. Chem. Soc., 1953, 3192–3210, 10.1039/JR9530003192.
- D. Choi, X. Li, W. A. Henderson, Q. Huang, S. K. Nune, J. P. Lemmon and V. L. Sprenkle, LiCoPO4 cathode from a CoHPO4.xH2O nanoplate precursor for high voltage Li-ion batteries, Heliyon, 2016, 2, e00081 CrossRef PubMed.
- H. Zhang, Y. Lu, X. Jin, B. Wang, T. Yang, H. Xu, H. Liu and C. Chao, A facile and gentle method to fabricate NH4CoPO4 coating with superhydrophobic character study, Mater. Res. Express, 2019, 6, 105044 CrossRef CAS.
- X. Lu, Z. Huang, Z. Liang, Z. Li, J. Yang, Y. Wang and F. Wang, Co-precipitation of Cu and Zn in precipitation of struvite, Sci. Total Environ., 2021, 764, 144269 CrossRef CAS.
- R. Zhangchen, W. G. Liu, Q. H. Ji, Y. Li, J. Zhang, J. Song, Y. Zhong and L. Zhou, Struvite as seed materials for treatment of heavy metals in wastewater, IOP Conf. Ser.: Earth Environ. Sci., 2021, 770, 1–5 Search PubMed.
- J. Escorihuela, R. Narducci, V. Compañ and F. Costantino, Proton Conductivity of Composite Polyelectrolyte Membranes with Metal–Organic Frameworks for Fuel Cell Applications, Adv. Mater. Interfaces, 2018, 6, 1801146 CrossRef.
- K.-D. Kreuer, A. Rabenau and W. Weppner, Vehicle Mechanism, A New Model for the Interpretation of the Conductivity of Fast Proton Conductors, Angew. Chem., Int. Ed. Engl., 1982, 21, 208–209 CrossRef.
- N. Agmon, The Grotthuss mechanism, Chem. Phys. Lett., 1995, 244, 456–462 CrossRef CAS.
- M. Ekimova, C. Kleine, J. Ludwig, M. Ochmann, T. E. G. Agrenius, E. Kozari, D. Pines, E. Pines, N. Huse, P. Wernet, M. Odelius and E. T. J. Nibbering, From Local Covalent Bonding to Extended Electric Field Interactions in Proton Hydration, Angew. Chem., Int. Ed., 2022, 61, e202211066 CAS.
- K.-D. Kreuer, Proton Conductivity: Materials and Applications, Chem. Mater., 1996, 8, 610–641 CrossRef CAS.
- T. Q. Nguyen and C. Breitkopf, Determination of Diffusion Coefficients Using Impedance Spectroscopy Data, J. Electrochem. Soc., 2018, 165, E826–E831 CrossRef CAS.
- Z. Xie, H. Wu, Q. Wu and L. Ai, Synthesis and performance of solid proton conductor molybdovanadosilicic acid, RSC Adv., 2018, 8, 13984–13988 RSC.
- S. Haghighat, S. Ostresh and J. M. Dawlaty, Controlling Proton Conductivity with Light: A Scheme Based on Photoacid Doping of Materials, J. Phys. Chem. B, 2016, 120, 1002–1007 CrossRef CAS PubMed.
- J. Fan, T. Wang, Y. Yuan, C.-L. Do-Thanh, X. Suo, Z. Yang, H. Chen and S. Dai, Mechanochemically Assisted Synthesis of High-Entropy Layer-Structured Dittmarite Analogues, ACS Appl. Energy Mater., 2022, 5, 3290–3297 CrossRef CAS.
- A. Amiri and R. Shahbazian-Yassar, Recent progress of high-entropy materials for energy storage and conversion, J. Mater. Chem. A, 2021, 9, 782–823 RSC.
- Y. Sun and S. Dai, High-entropy materials for catalysis: A new frontier, Sci. Adv., 2021, 7, eabg1600 CrossRef CAS PubMed.
- Y. Sone, P. Ekdunge and D. Simonsson, Proton Conductivity of Nafion 117 as Measured by a Four–Electrode AC Impedance Method, J. Electrochem. Soc., 1996, 143, 1254 CrossRef CAS.
- Y. Sun, Y. Yan, Y. Wang, Y. Li, J. Li and J. Yu, High proton conduction in a new alkali metal-templated open-framework aluminophosphate, Chem. Commun., 2015, 51, 9317–9319 RSC.
- K. Scott, C. Xu and X. Wu, Intermediate temperature proton-conducting membrane electrolytes for fuel cells, Wiley Interdiscip Rev Energy Environ, 2014, 3, 24–41 CAS.
- W. H. J. Hogarth, S. S. Muir, A. K. Whittaker, J. C. Diniz da Costa, J. Drennan and G. Q. Lu, Proton conduction mechanism and the stability of sol–gel titanium phosphates, Solid State Ionics, 2007, 177, 3389–3394 CrossRef CAS.
- P. G. M. Mileo, T. Kundu, R. Semino, V. Benoit, N. Steunou, P. L. Llewellyn, C. Serre, G. Maurin and S. Devautour-Vinot, Highly Efficient Proton Conduction in a Three-Dimensional Titanium Hydrogen Phosphate, Chem. Mater., 2017, 29, 7263–7271 CrossRef CAS.
- H.-R. Zhao, C. Xue, C.-P. Li, K.-M. Zhang, H.-B. Luo, S.-X. Liu and X.-M. Ren, A Two-Dimensional Inorganic–Organic Hybrid Solid of Manganese(II) Hydrogenophosphate Showing High Proton Conductivity at Room Temperature, Inorg. Chem., 2016, 55, 8971–8975 CrossRef CAS PubMed.
- K.-M. Zhang, Y.-L. Lou, F.-Y. He, H.-B. Duan, X.-Q. Huang, Y. Fan and H.-R. Zhao, The water-mediated proton conductivity of a 1D open framework inorganic-organic hybrid iron phosphate and its composite membranes, Inorg. Chem. Commun., 2021, 134, 109032 CrossRef CAS.
- M. Wang, H.-B. Luo, S.-X. Liu, Y. Zou, Z.-F. Tian, L. Li, J.-L. Liu and X.-M. Ren, Water assisted high proton conductance in a highly thermally stable and superior water-stable open-framework cobalt phosphate, Dalton Trans., 2016, 45, 19466–19472 RSC.
- J.-W. Yu, H.-J. Yu, Q. Ren, J. Zhang, Y. Zou, H.-B. Luo, L. Wang and X.-M. Ren, Humidity-sensitive irreversible phase transformation of open-framework zinc phosphate and its water-assisted high proton conduction properties, Dalton Trans., 2021, 50, 8070–8075 RSC.
- H.-X. Sun, H.-N. Wang, Y.-H. Zou and X. Meng, Tuning proton conduction by different particle sizes in open-framework metal phosphates, Inorg. Chem. Commun., 2021, 124, 108322 CrossRef CAS.
- S. Matsumura, R. H. Antisar, C. Lepiller, J. Gaudet, D. Guay, Z. Shi, S. Holdcroft and A. S. Hay, Ionomers for Proton Exchange Membrane Fuel Cells with Sulfonic Acid Groups on the End Groups: Novel Branched Poly(ether-ketone)s, Macromolecules, 2008, 41, 281–284 CrossRef CAS.
- D. W. Lim and H. Kitagawa, Rational strategies for proton-conductive metal-organic frameworks, Chem. Soc. Rev., 2021, 50, 6349–6368 RSC.
- J. Kim, S. Sengodan, S. Kim, O. Kwon, Y. Bu and G. Kim, Proton conducting oxides: A review of materials and applications for renewable energy conversion and storage, Renewable Sustainable Energy Rev., 2019, 109, 606–618 CrossRef CAS.
- R. Sigwadi, M. S. Dhlamini, T. Mokrani, F. Nemavhola, P. F. Nonjola and P. F. Msomi, The proton conductivity and mechanical properties of Nafion(R)/ZrP nanocomposite membrane, Heliyon, 2019, 5, e02240 CrossRef CAS PubMed.
- A. Çalı, A. Şahin and İ. Ar, Experimental Investigation of boron phosphate Incorporated speek/pvdf blend membrane for proton exchange membrane fuel cells, Int. J. Hydrogen Energy, 2022, 47, 40476–40490 CrossRef.
- O. Zholobko, J. Hurley, X.-F. Wu, T. Aulich and J. Thakare, Intermediate-Temperature Proton Exchange Membranes Based on Cerium Ultraphosphate Composited with Polybenzimidazole, J. Electrochem. Soc., 2022, 169, 094505 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray diffraction (XRD), scanning electron microscopy (SEM), infrared spectroscopy (IR), Raman spectroscopy (RS), thermal gravimetric analysis with differential scanning calorimetry (TGA/DSC), X-ray absorption spectroscopy (XAS), proton conductivity (PC) and dynamic vapor sorption (DVS) measurements, Figs. S1–S12; Tables S1–S23. See DOI: https://doi.org/10.1039/d3dt00839h |
| This journal is © The Royal Society of Chemistry 2023 |