
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the influence of pH on the structural intricacies of uranium oxide hydrates containing both Cd(II) and K(I) ions †
Timothy A.
Ablott
,
Kimbal T.
Lu
,
Tao
Wei
and
Yingjie
Zhang
 *
*
Australian Nuclear Science and Technology Organisation, Locked Bag 2001, Kirrawee DC, NSW 2232, Australia. E-mail: yzx@ansto.gov.au
First published on 25th April 2023
Abstract
We report the synthesis of two new dual-cation uranium oxide hydrate (UOH) materials, containing both Cd2+ and K+ ions, along with their characterisation by means of single-crystal X-ray diffraction and a range of other structural and spectroscopic techniques. The materials were found to differ in structures, topology and uranium to cation ratios, with the layered UOH-Cd crystallising in a plate morphology and containing a U![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cd:K ratio of 3:1.5:1. Conversely, the framework-type UOF-Cd incorporates much less Cd, with a U:Cd:K ratio of 4.4:0.2:1 and is found as needle-like crystals. A common feature in both structures is the presence of β-U3O8 type layers with a distinct uranium centre which lacks the expected uranyl bonds, highlighting the importance of the β-U3O8 layer in the subsequent self-assembly and preferential formation of a variety of structural types. Most importantly, by exploiting the additional flexibility provided by monovalent cation species (i.e., K+) as secondary metal cations to synthesise these novel dual-cation materials, this work highlights the potential for broadening the scope of viable synthetic UOH phases towards furthering the understanding of these systems in their roles as alteration products in the surrounds of spent nuclear fuel in deep geological repositories.
:Cd:K ratio of 3:1.5:1. Conversely, the framework-type UOF-Cd incorporates much less Cd, with a U:Cd:K ratio of 4.4:0.2:1 and is found as needle-like crystals. A common feature in both structures is the presence of β-U3O8 type layers with a distinct uranium centre which lacks the expected uranyl bonds, highlighting the importance of the β-U3O8 layer in the subsequent self-assembly and preferential formation of a variety of structural types. Most importantly, by exploiting the additional flexibility provided by monovalent cation species (i.e., K+) as secondary metal cations to synthesise these novel dual-cation materials, this work highlights the potential for broadening the scope of viable synthetic UOH phases towards furthering the understanding of these systems in their roles as alteration products in the surrounds of spent nuclear fuel in deep geological repositories.
1. Introduction
The need to understand uranium oxide-based chemistry is ever present, principally due to its use as a nuclear fuel around the world.1,2 With an ever-increasing need for cleaner energy sources, many countries have turned to nuclear energy to achieve the goal of reducing carbon emissions.1,2 As a result, the study of uranium oxide compounds, both naturally occurring and synthetically derived, is of particular interest. With a drive to increased nuclear energy production comes an increase in the amount of spent nuclear fuel (SNF) being produced.3 The current methods of safe SNF disposal are varied, however the most straightforward and thus often the most widely utilised is the storage of the SNF in stable geological repositories.2 In these repositories, SNF is most commonly found in the form of UO2, which when stored under reducing environments are insoluble and thus should remain confined within the repository. However, if exposed to oxygen and air, there exists the possibility of alteration by oxidation and/or hydration, giving rise to the need to study these processes further.4,5When exposed to these oxidative conditions, UO2 species are known to oxidise from U4+ to U6+. In their 6+ state uranium species, existing as uranyl [(UO2)2+] cations, readily react with their environment to form uranyl oxide-containing compounds.6,7 Given the strongly bonded axial oxygen atoms, these compounds typically form layers extending along their equatorial positions in tetragonal, pentagonal and hexagonal bipyramidal geometries. This results in the formation of materials containing uranyl polyhedra layers (sheets), between which lie interlayer cation species which are present in the environment of the repository.8,9 Given their compositions, these compounds are given the name uranium oxide hydrates (UOH).10
The importance of understanding these materials has given rise to dozens of UOH-based minerals,1,10,11 with more than a dozen synthetic UOH compounds having also been identified.12–14 One of the main factors differentiating these materials is the secondary metal ion which lies between the uranyl oxide hydroxide layers, with a diverse range including alkali,15 alkaline earth,16 heavy metals,17 transition metals18 and lanthanides19 having been reported thus far.
Complicating matters further is the possible formation of a UOH sub-structure, wherein the UOH takes on a framework-like orientation with uranyl species acting as bridging ligands between the uranyl polyhedra layers to form uranium oxide frameworks (UOFs). This results in the creation of channels within which lie the secondary metal species, which have proven capable of incorporating a range of secondary cations including Cs+, Pb2+, Sr2+, Y3+, Er3+, Sm3+, Eu3+, Gd3+ and U4+.20–26 The additional complexity which UOFs introduce into understanding the chemistry of UO2 in the environs of these geological repositories exemplifies the need to study these materials further.
The driving force in this field is the possible cations which may be present in the surrounds of these geological repositories. Cadmium, being relatively rare in the environment, has accordingly been unexplored for these UOH materials. However, given cadmium is the most soluble of the heavy metals in water,27 there is a distinct possibility that the amount of cadmium in groundwater will increase due to ongoing anthropogenic activities.27,28 Whilst anthropogenic activities are likely to vary over the expected timeframe of SNF exposure to groundwater, in a worst case scenario where the contaminated groundwater is introduced into the surrounding environment of a geological repository earlier than expected, there is the possibility that Cd2+ ions may play a role in the phase formations or alterations of UO2 systems in these repositories.29
As of yet no Cd-based UOH systems, either synthetic or naturally occurring, have been reported in the literature, however the synthesis of UOH compounds containing 2+ transition metal species have proven successful (Table 1). Understanding whether Cd-based systems, given cadmium has a larger ionic radius compared to these 2+ transition metals, will also form under these synthetic conditions is therefore crucial to better understand its chemistry if introduced to the SNF environment.
| UOH-TM2+ | Chemical formula | Space group |
|---|---|---|
| UOH-Mn14 | [Mn(H2O)4][(UO2)3O3(OH)2]·H2O |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| UOH-Zn14 | [Zn(H2O)4][(UO2)3O3(OH)2]·H2O |
P |
| UOH-Ni14 | [Ni(H2O)4][(UO2)3O3(OH)2]·H2O |
P |
| UOH-Co14 | [Co(H2O)4][(UO2)3O3(OH)2]·H2O |
P |
| UOH-Ni30 | [Ni(H2O)4]3[U(OH,H2O)(UO2)8O12(OH)3] |
P |
| UOH-KNi18 | K4Ni(OH)3(H2O)9[(UO2)12O7(OH)13] | P31c |
| UOH-KCo18 | K4Co(OH)3(H2O)9[(UO2)12O7(OH)13] | P31c |
An additional consideration is the abundance of potassium in the earth's crust, which ensures that K+ ions will be present during any alteration or phase formation processes. Dual K+-metal systems have been reported in a small number of UOH minerals31,32 and synthetic phases.18 However the role that potassium plays in possibly stabilising these UOH/F structures needs to be better understood. The larger charge density of many of the previously identified secondary ions, such as lanthanides, excludes the possible inclusion of K+ into UOF structures. However, Cd2+ ion, having a smaller charge density, should also allow for potassium to be incorporated into the structure, which may aid in understanding its role in the possible alteration chemistry of SNF.
Herein we report the syntheses of two novel synthetic UOH/F compounds and explore the effect of synthetic conditions on their structure evolutions. The crystals isolated from the hydrothermal reaction of uranyl nitrate with Cd2+ and K+ ions were found to exist as either a layered UOH material or a framework-type structure. The U:Cd:K ratios, as determined through synchrotron single crystal X-ray diffraction analysis, was found to differ significantly between the two compounds. Their physical and spectroscopic properties were subsequently explored using scanning and transmission electron microscopy along with Raman and diffuse reflectance spectroscopy.
2. Experimental
2.1. Syntheses of materials
Uranyl nitrate hexahydrate with natural uranium was used in the synthesis of the materials. Compounds with uranium are radioactive and should be handled in the regulated laboratory. All other chemicals in A.R. grade were from Sigma-Aldrich (Merck).2.2. Characterisations
3. Results and discussion
3.1. Material syntheses and microstructures
UOH-Cd and UOF-Cd were successfully synthesized under hydrothermal conditions, with uranyl nitrate as the uranium precursor and the pH adjustments achieved using a diluted KOH solution. UOH-Cd, isolated as yellow-orange crystals (Fig. S1, ESI†), formed after dissolving equimolar amounts of Cd(NO3)2·4H2O and UO2(NO3)2·6H2O in H2O, adjusting the solution pH to 6.13 and heating the solution to 240 °C for 24 h. The formation of UOF-Cd, also existing as yellow-orange crystals (Fig. S1, ESI†), was accomplished in a similar manner, with an aqueous solution containing equimolar amounts of Cd(NO3)2·4H2O and UO2(NO3)2·6H2O in H2O instead adjusted to a solution pH of 7.15 prior to heating to 240 °C for 24 h.SEM-EDS analysis of UOH-Cd confirmed the presence of a single crystal morphology present as large plates (Fig. 1(a), left). EDS analysis confirmed the presence of U, Cd, K and O, with the U:Cd:K atomic ratios of 3:1.5:1 (Fig. 1(a), right; Fig. S2, ESI†). These ratios match those expected for layered UOH materials, with uranium to metal ratios of 2:1–4:1 most typically observed.19,39,40 The PXRD pattern matches the calculated pattern from the single crystal data (Fig. S3, ESI†) suggesting a pure phase compound was obtained.
 | ||
| Fig. 1 SEM analysis of UOH-Cd (a) and UOF-Cd (b): backscattered SEM images of the crystals on the left and their corresponding EDS spectra on the right, confirming the presence of U, Cd and K in the crystals. | ||
Immediately apparent in the SEM-EDS analysis of UOF-Cd (Fig. 1b) is the presence of two crystal morphologies, with the major phase existing as needle-like crystals (Fig. 1(b), left) and the minor phase as smaller plates (Fig. S4, ESI†). The presence of U, Cd and K was confirmed with EDS (Fig. 1(b), right; Fig. S4, ESI†), however the exact ratios could not be determined in this material due to the overlapping of major energies for U, Cd and K. In being unable to differentiate the energies of Cd and K from the U, which is suspected to exist as the major element in the material, this strongly suggests that the U:M (Cd/K) ratio is much higher than that observed in UOH-Cd and thus U dominates the EDS spectrum (Fig. S4, ESI†). With the needle-like morphology of the major phase matches those observed for previously reported UOF phases,24–26 the expected U:M ratio would be ∼5.5:1, which would explain the inability to obtain a definitive EDS spectrum for the trace amounts of Cd. The PXRD pattern of manually separated UOF-Cd matches the pattern calculated from its single crystal data (Fig. S5, ESI†), confirming its presence as the major phase. EDS analysis of the minor phase revealed U:Cd:K ratios akin to those observed in UOH-Cd ((Fig. S4(b), ESI†), confirming that the minor phase is that of a layered UOH material comparable to UOH-Cd. Given the relatively close solution pH between the two syntheses, UOF-Cd existing as a mixed phase is not unexpected. Some of the previously published UOFs have been reported as existing in a mixed phase system, with the UOF present as the major phase.23
In exploring the possible reason as to why the framework-type structure was favoured during the UOF-Cd synthesis, the most apparent conclusion is the solution pH. Past research has shown the influence the solution pH has on the formation of UOH compounds.12,24 Interestingly, in those examples the lower solution pH drove the formation of a UOF material with higher solution pH giving rise to a layered structure instead. For these Cd-based systems, the trend appears reversed, with the higher pH driving the framework assembly. One possible alternative explanation is the increased K+ concentration in solution during UOF-Cd formation, resulting from an increased amount of KOH required to increase the starting pH.
Given the EDS spectra shows that UOF-Cd contains a higher proportion of K+ compared to that of UOH-Cd, it is possible that this influenced the final structure by providing a means of charge balance without the need to incorporate further Cd2+ ions. Thus K+ ions appear to play a key role in the formation of UOF phase incorporating Cd2+ ions. Clearly illustrated is the elaborate chemistry for the formation of these UOH/F materials, hence the need to further the studies in this area.
3.2. Crystal structures and discussion
The crystal data and refinement details for UOH-Cd and UOF-Cd are summarised in Table 2. Selected bond lengths (Å) and angles (°) are provided in Tables 3 and 5, with calculated bond valence sums (BVSs)9 presented in Tables 4 and 6 with parameters for U6+, Cd2+ and K+ taken from the literature.9,41,42| Compound | UOH-Cd | UOF-Cd |
|---|---|---|
| a R 1 = Σ‖Fo| − |Fc‖/|Fo|; b wR2 = Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]1/2. | ||
| CCDC | 2244533† | 2244534† |
| Empirical formula | Cd3K2O23U6 | Cd0.5K2.5O39U11 |
| Formula weight | 2211.58 | 3396.28 |
| Crystal system | Orthorhombic | Monoclinic |
| Space group | Pnnm | C2/c |
| a (Å) | 12.398(3) | 11.589(2) |
| b (Å) | 7.0070(14) | 21.125(4) |
| c (Å) | 11.482(2) | 14.596(3) |
| α/(°) | 90 | 90 |
| β/(°) | 90 | 104.23(3) |
| γ/(°) | 90 | 90 |
| Volume (Å3) | 997.5(3) | 3463.8(13) |
| Z/μ (mm−1) | 2/52.169 | 4/6.513 |
| Min./Max. θ [°] | 4.836/51.998 | 3.856/49.992 |
| d calcd (g cm−3) | 7.363 | 6.513 |
| GOF | 1.081 | 1.114 |
| Final R1a [I > 2σ(I)] |
0.0294 | 0.0538 |
| Final wR2b [I > 2σ(I)] |
0.0814 | 0.1327 |
| UOH-Cd | |||||||
|---|---|---|---|---|---|---|---|
| 11/2 − X, −1/2 + Y, 1/2 + Z; 21/2 − X, −1/2 + Y, 1/2 − Z; 3−X, 1 − Y, 1 − Z; 4 +X, −1 + Y, +Z; 5 +X, +Y, 1 − Z; 61/2 − X, 1/2 + Y, 1/2 − Z;7 +X, 1 + Y, +Z; 81 − X, 2 − Y, 1 − Z; 91 − X, 1 − Y, 1 − Z; 101/2 + X, 3/2 − Y, 1/2 − Z; 111 − X, 2 − Y, +Z; 12−X, 2 − Y,1 − Z; 13−X, 1 − Y,+Z; 14−X, 2 − Y, +Z; 15–1/2 + X, 3/2 − Y, 1/2 − Z. | |||||||
| U1–O1 | 1.974(11) | U2–O46 | 2.350(7) | Cd1–O510 | 2.284(6) | K1–O13 | 2.864(8) |
| U1–O2 | 1.996(9) | U2–O4 | 2.289(8) | Cd1–O56 | 2.284(6) | K1–O17 | 2.864(8) |
| U1–O35 | 2.127(6) | U2–O5 | 1.885(6) | Cd1–O611 | 2.344(7) | K1–O37 | 2.743(7) |
| U1–O3 | 2.127(6) | U2–O6 | 1.858(7) | Cd1–O6 | 2.344(7) | K1–O313 | 2.743(7) |
| U1–O45 | 2.116(7) | U2–O7 | 2.132(2) | Cd2–O13 | 2.113(11) | K1–O5 | 2.830(7) |
| U1–O4 | 2.116(7) | O6![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) U2O5 U2O5 |
178.5(3) | Cd2–O1 | 2.113(11) | K1–O514 | 2.830(7) |
| U2–O37 | 2.301(7) | Cd1–O29 | 2.257(6) | Cd2–O5 | 2.544(7) | K1–O615 | 2.775(8) |
| U2–O36 | 2.397(7) | Cd1–O27 | 2.257(6) | Cd2–O513 | 2.544(7) | K1–O66 | 2.775(8) |
| U1 | U2 | Cd1 | Cd2 | K1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Occ. | 1 | 1 | 1 | 0.5 | 1 | |||||||
| Sym. | 2 | 1 | 1 | 2 | 2 | |||||||
| CN | 6 | 7 | 6 | 6 | 8 | Σ | ||||||
| O1 | 1.16 | 0.56 | 0.56 | 0.13 | 0.13 | 1.86 | ||||||
| O2 | 1.11 | 0.38 | 0.38 | 1.50 | ||||||||
| O3 | 0.86 | 0.86 | 0.62 | 0.51 | 0.18 | 0.18 | 2.18 | |||||
| O4 | 0.88 | 0.88 | 0.56 | 0.63 | 1.45 | |||||||
| O5 | 1.38 | 0.36 | 0.36 | 0.35 | 0.35 | 0.14 | 0.14 | 2.24 | ||||
| O6 | 1.45 | 0.30 | 0.30 | 0.17 | 0.17 | 1.92 | ||||||
| O7 | 0.85 | 0.86 | (OH) | |||||||||
| O8 | 0.00 | (H2O) | ||||||||||
| Σ | 5.77 | 6.01 | 2.09 | 1.84 | 1.25 | |||||||
| UOF-Cd | |||||||
|---|---|---|---|---|---|---|---|
| 13/2 − X, 3/2 − Y, 1 − Z; 21/2 − X, 3/2 − Y, 1 − Z; 31 + X, +Y, +Z; 4−X, +Y, 3/2 − Z; 5−X, 1 − Y, 1 − Z; 6–1 − X, 1 − Y, 1 − Z; 7–1 + X, +Y, +Z; 83/2 + X, 1/2 + Y, +Z; 92 − X, +Y, 3/2 − Z; 101/2 + X, 3/2 − Y, 1/2 + Z; 11–1/2 + X, 3/2 − Y, −1/2 + Z; 12−X, +Y, 1/2 − Z; 13 +X, 1 − Y, −1/2 + Z; 14 +X, 1 − Y, 1/2 + Z; 15–1 − X, +Y, 3/2 − Z; 161/2 − X, −1/2 + Y, 3/2 − Z; 17–3/2 + X, −1/2 + Y, +Z; 181 − X, +Y, 3/2 − Z. | |||||||
| U1–O11 | 2.271(19) | U2–O2 | 2.439(14) | U3–O7 | 2.208(14) | U4–O32 | 2.389(13) |
| U1–O1 | 2.271(19) | U2–O3 | 2.439(15) | U3–O8 | 1.932(16) | U4–O10 | 2.301(14) |
| U1–O2 | 2.005(14) | U2–O4 | 1.773(15) | U3–O9 | 1.813(14) | U4–O105 | 2.249(14) |
| U1–O21 | 2.005(14) | U2–O5 | 1.805(16) | U3–O10 | 2.200(14) | U4–O11 | 2.284(14) |
| U1–O3 | 1.982(14) | U2–O62 | 2.29(3) | U3–O11 | 2.186(14) | U4–O12 | 1.951(16) |
| U1–O31 | 1.982(14) | U2–O7 | 2.326(14) | U3–O195 | 2.200(13) | U4–O13 | 1.833(15) |
| U2–O112 | 2.296(14) | O9U3O8 |
176.2(7) | U4–O14 | 2.213(16) | ||
| O4U2O5 |
177.9(7) | O13U4O12 |
176.3(7) | ||||
| U5–O84 | 2.379(16) | U6–O27 | 2.438(14) | Cd1–O1 | 2.335(19) | K1–O411 | 2.984(16) |
| U5–O124 | 2.404(16) | U6–O77 | 2.359(14) | Cd1–O19 | 2.335(19) | K1–O42 | 2.984(16) |
| U5–O12 | 2.480(16) | U6–O14 | 2.236(16) | Cd1–O510 | 2.581(17) | K1–O9 | 2.919(15) |
| U5–O14 | 2.319(16) | U6–O17 | 1.798(14) | Cd1–O51 | 2.581(17) | K1–O912 | 2.919(15) |
| U5–O15 | 1.781(14) | U6–O18 | 1.883(15) | Cd1–O209 | 2.374(16) | K1–O1312 | 2.809(15) |
| U5–O16 | 1.777(14) | U6–O196 | 2.309(13) | Cd1–O20 | 2.374(16) | K1–O13 | 2.809(15) |
| U5–O18 | 2.437(14) | U6–O19 | 2.270(13) | K1–O155 | 2.947(15) | ||
| O16U5O15 |
177.4(7) | O17U6O18 |
176.0(6) | K1–O1513 | 2.947(15) | ||
| K2–O87 | 3.102(15) | K3–O418 | 2.852(17) | ||||
| K2–O84 | 3.102(15) | K3–O4 | 2.852(17) | ||||
| K2–O1714 | 2.726(15) | K3–O718 | 2.931(15) | ||||
| K2–O176 | 2.726(15) | K3–O7 | 2.931(15) | ||||
| K2–O18 | 2.685(16) | K3–O8 | 3.238(16) | ||||
| K2–O1815 | 2.685(16) | K3–O818 | 3.238(16) | ||||
| K2–O2016 | 2.714(16) | K3–O183 | 2.888(16) | ||||
| K2–O2017 | 2.714(16) | K3–O184 | 2.888(16) | ||||
| U1 | U2 | U3 | U4 | U5 | U6 | Cd1 | K1 | K2 | K3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Occ. | 1 | 1 | 1 | 1 | 1 | 1 | 0.5 | 1 | 0.75 | 0.75 | |||||||
| Sym. | 2 | 1 | 1 | 1 | 1 | 1 | 2 | 2 | 2 | 2 | |||||||
| CN | 6 | 7 | 6 | 7 | 7 | 7 | 6 | 8 | 8 | 8 | Σ | ||||||
| O1 | 0.65 | 0.65 | 0.31 | 0.31 | 0.97 | (OH) | |||||||||||
| O2 | 1.09 | 1.09 | 0.47 | 0.47 | 2.04 | ||||||||||||
| O3 | 1.14 | 1.14 | 0.47 | 0.52 | 2.14 | ||||||||||||
| O4 | 1.71 | 0.19 | 0.27 | 2.17 | |||||||||||||
| O5 | 1.61 | 0.16 | 0.16 | 1.77 | |||||||||||||
| O6 | 0.63 x2 | 1.26 | (OH) | ||||||||||||||
| O7 | 0.59 | 0.74 | 0.55 | 0.22 | 2.10 | ||||||||||||
| O8 | 1.26 | 0.53 | 0.14 | 0.10 | 2.02 | ||||||||||||
| O9 | 1.58 | 0.23 | 1.81 | ||||||||||||||
| O10 | 0.75 | 0.62 | 0.68 | 2.05 | |||||||||||||
| O11 | 0.62 | 0.77 | 0.64 | 2.03 | |||||||||||||
| O12 | 1.21 | 0.51 | 0.44 | 2.16 | |||||||||||||
| O13 | 1.52 | 0.30 | 1.83 | ||||||||||||||
| O14 | 0.73 | 0.60 | 0.70 | 2.03 | |||||||||||||
| O15 | 1.68 | 0.21 | 1.89 | ||||||||||||||
| O16 | 1.70 | 1.70 | |||||||||||||||
| O17 | 1.63 | 0.38 | 2.01 | ||||||||||||||
| O18 | 0.48 | 1.38 | 0.43 | 0.25 | 2.53 | ||||||||||||
| O19 | 0.75 | 0.61 | 0.66 | 2.01 | |||||||||||||
| O20 | 0.28 | 0.28 | 0.39 | 0.67 | (H2O) | ||||||||||||
| Σ | 5.78 | 6.11 | 5.85 | 5.93 | 5.93 | 6.00 | 1.51 | 0.93 | 1.34 | 0.83 | |||||||
UOH-Cd crystallises in the orthorhombic Pnnm space group, with the asymmetric unit containing two distinct U centres, two Cd centres (Cd1 in full and Cd2 in half occupancies) and a full occupancy K site. The U1, Cd2 and K1 sites are modelled with partial occupancies due to lying on Wyckoff positions. U1 exists in a distorted octahedral geometry and U2 in a pentagonal bipyramidal geometry. Both Cd1 and Cd2 exist in a 6-coordinate distorted octahedral geometry, with the K species present as an 8-coordinate square antiprism.
Exploring the full structure reveals that the material exists as a layered structure made up of 2D sheets constructed from the U polyhedra, with the secondary Cd2+ and K+ ions lying between these layers (Fig. 2(a)). The uranyl polyhedra layer exists as a β-U3O8-type structure, with the pentagonal bipyramidal U2 centres forming continual chains via O–O edge-sharing which are bridged by the U1 octahedra (Fig. 2(b)). The coordination sphere about U2 shows the expected UO bond lengths of 1.858(7) Å (O6) and 1.885(6) Å (O5) at the axial positions, with a nearly linear OU2O bond of 178.5(3)°. The equatorial U–O bonds (O3, O4 and O7), which participate in the O–O edge-sharing, fall in the range of 2.289(8) Å–2.397(7) Å, consistent with the observed values for uranyl polyhedra in these systems.8,30,43 The coordination environment about U1 is unusual in that it does not involve a uranyl species, with U–O bond lengths of 1.974(9) Å to 2.127(6) Å observed in the axial positions. Whilst uncommon, similar results have been found in other synthetic UOH systems.9,21,30,44
 | ||
| Fig. 2 Crystal structure of UOH-Cd: a polyhedral crystal structure along the b-axis (a), a polyhedral view of the uranyl oxide hydroxide layer with a β-U3O8 topology (b), and a polyhedral view of the Cd2+/K+ interlayer cations (c); U in yellow, Cd in blue and K in purple. | ||
BVS calculations (Table 4) using the parameters reported by Burns et al. (RU–O = 2.051; B = 0.519),9 confirm that both U centres are present as U6+, with the oxygen species existing majorly as O, with only O7 existing as OH which connects the adjacent U2-U2 chains. From this, the general formula for UOH-Cd was determined to be Cd3K2[(UO2)6O9(OH)2].
The formation of a β-U3O8 sheet topology is intriguing as it is rarer in synthetic UOH materials, with an α-U3O8 layer more commonplace.18,19,39,43,45 The β-U3O8 topology has been observed in a number of UOH minerals,46–50 but much less in layered synthetic structures.30,43 Interestingly, the β-U3O8 topology is overwhelming favoured in UOF materials,20,23–26,44 likely due to the fact that the β-U3O8 anion topology can accommodate multiple valence states of uranium, a necessity in UOFs which contain mixed U5+/U6+ sites within their structure.23–25 Moreover, in the studies by Zhang et al.43 and Rivenet et al.,30 wherein the uranyl polyhedra layers take on the β-U3O8 layout, the interlayer cations (Tb3+/Ni2+) act as pillars which separate the uranyl polyhedra sheets. Each cation ‘pillar’ exists as an isolated species with channels in between, creating a pseudo-UOF material. The same trend is not immediately apparent in UOH-Cd, wherein the Cd2+ ions form chains, linking the layers via the axial O2 and O5 oxygens, and which lie perpendicular to U2–U2 chains in the uranyl polyhedra sheets (Fig. 2(c)). However, this Cd–Cd coordination is vastly different from that seen for other interlayer cation species in the literature in that it exists as 1D chains (Fig. 3(a)) as opposed to the 2D topologies seen for many UOH materials (Fig. 3(b)).39,43 As a result, these Cd chains pillar the uranyl polyhedra sheets, with the chains themselves each linked through K+–K+ dimeric units (Fig. 2(c)), between which lies an isolated, unbound water molecule.
 | ||
| Fig. 3 A polyhedral view of the topology of the Cd–Cd chains in UOH-Cd (a) and the interlayer cation topology of U-La,39 representative of the more commonly observed topology (b). | ||
Interestingly, these Cd2+ pillars may also account for the different abundance of Cd:K compared to the rations reported in previous dual-cation systems. A study by Zhang and co-workers reported two dual-cation UOH materials containing both K+ and either Co+ or Ni+, however with reported K:Co/Ni ratios of 3.5:1. Also revealed was the Co2+ and Ni2+ ions lay close to one of the β-U3O8 layers and were only coordinated to one U centre, with the K+ ions linking the β-U3O8 sheets, which accounted for the observed K:Co/Ni ratio. The lower abundance of K+ in UOH-Cd can therefore be attributed to the capability of Cd2+, which has a larger ionic radius compared to both Co2+ and Ni2+, to coordinate adjacent β-U3O8 layers.
SC-XRD confirmed the structure of UOF-Cd existed as a framework (Fig. 4(a)), with the material crystallising in the monoclinic C2/c space group. The asymmetric unit was much larger, containing six unique U centres, one half occupancy Cd2+ site and three partial occupancy K+ sites. The uranium centres each exist in full occupancies, with U1 modelled in half occupancies as it lies in a special position. The U sites take on two geometries, with U1 and U3 as 6-coordinate octahedra and U2, U4, U5 and U6 present as pentagonal bipyramids. The Cd ion lies in the channels of the framework in a trigonal prismatic geometry, with two of the K+ centres taking on a bicapped trigonal prismatic geometry (K1 and K3), and one present as a square antiprism (K2).
 | ||
| Fig. 4 Crystal structure of UOF-Cd: a polyhedral crystal structure along the a-axis (a), a polyhedral view of the uranyl oxide hydroxide layer with a β-U3O8 topology (b), the uranyl oxide hydroxide layers linked by double U5 pentagonal bipyramids (c), the 8-fold coordinated K+ and 6-fold coordinated Cd2+ interlayer cations in the framework channels (d); U in yellow, Cd in blue and K in purple. | ||
The uranyl polyhedra sheets which make up the framework backbone is structurally similar to UOH-Cd, with the U centres taking on a β-U3O8-type layout. The U polyhedra are arranged to form chains of pentagonal bipyramids (U2, U4 and U6) through O–O edge sharing in a –(U2–U4–U4–U2–U6–U6–U2)– pattern (Fig. 4(b)). Each chain is antiparallel to the neighbouring sequence and are linked through U1 and U3 octahedra. Linking of the layers by discrete U5–U5 units through edge-sharing (U6/U4–U5) and corner-sharing (U2–U5) (Fig. 4(c)) completes the backbone of the framework-type structure.
Five of the six U centres (U2–U6) each have the expected pair of axial UO bonds (1.773(15) Å–1.951(16) Å) with near linear bond angles (176.0°–177.9°). Of these, the pentagonal bipyramids each also coordinate five oxygens in their equatorial planes, with the octahedral U3 centre coordinating four O species. These equatorial bond lengths ranging from 2.186(14) Å to 2.480(16) Å are typical of previously reported UOF materials, falling within the same ranges.23,24,26
Curiously, as with UOH-Cd, the octahedral U1 does not contain any uranyl bonds, with all six bonds in the range of 1.982(14) Å to 2.271(19) Å. As discussed above, whilst this type of uranium centre has been observed in other materials, the same U species present in both materials strongly suggests that a correlation exists between UOF-Cd and the layered UOH-Cd. BVS values were calculated (Table 6) and confirmed that, as with UOH-Cd, all U centres existed as U6+. Interestingly, this is divergent from many of the previously reported UOF structures which contain a mixed valence U5+/U6+ at the same position as the U1/U3 octahedral sites observed in UOF-Cd. The previously reported presence of U5+ at this U1/U3 site within these framework-type structures, along with the fact that the same octahedral U species is near identical in both UOH-Cd and UOF-Cd strongly implies a link between the chemistry about this site and the preferred structure of the UOH/F material.
The oxygen species exist primarily as O, with one H2O molecule (O20) shared between the Cd/K cations and two coordinated OH (O1 and O6), with the O6 site disordered in two positions. Based on the U and O types present, the material was formulated as Cd1K5(H2O)6[(UO2)22O23(OH)5].
A unique feature of UOF-Cd is the coordination of the secondary cations within the channels of the framework. The K+ and Cd2+ cations form a K1–K3–K2–Cd bonding motif, which lies across two channels of the framework, passing between the U5-U5 pillars (Fig. 4(d)). Each of the K centres is linked through shared uranyl–cation interactions via the axial oxygen atoms of the U centres, with the Cd terminus coordinating K2 via both uranyl–cation interactions and a shared H2O molecule (O20).
As a result of this secondary cation arrangement, UOF-Cd has a much higher abundance of K+ compared to that of UOH-Cd, with a K:Cd ratio of 5:1 in the framework, compared to 3:2 in the layered structure. This secondary cation arrangement is particularly intriguing when compared to other framework-type structures in the literature. UOF materials containing Eu, Gd and Er, which have ionic radii comparable to those of Cd2+, have all been reported as single-cation systems.23,25 The obvious difference is therefore the oxidation state of the secondary cation species. With the presence of the secondary cations also driven by charge balance, the monovalent K+ is therefore required to stabilise the framework structure. Whilst it has been hypothesised that the ionic radius of the secondary cations has an influence on the resultant structure, with the pore environment of UOF materials more readily impacted by the ionic radii of the secondary cations,12 this result clearly demonstrates that for framework-type structures to be favoured, charge balance of the anion layer and the ionic radii of the secondary cations must both be accommodated.
Thus, it stands to reason that monovalent cations such as K+, having comparable ionic radii to many of the cation species readily found in the environment (Ca2+, Sr2+, Zn2+) as well as species found in the surrounds of SNF in an underground repository (Pb2+, Ln3+), could offer additional stability to UOH/F materials by providing an additional means of balancing the charge of their structures.
3.3. TEM characterization
Both compounds were further examined by TEM. For UOH-Cd, a TEM bright field image (Fig. 5a) showed a crushed grain. TEM-EDS analysis confirmed the presence of U, Cd and K. The SAED pattern (Fig. 5b) collected on the grain in zone axis of [1 1 0] was indexed to the orthorhombic Pnnm space group, consistent with the SC-XRD result. A HRTEM image in zone axis [6 3 1] from edge of the UOH-Cd grain showed lattice fringes with an inserted FFT image (Fig. 5c). The d (1 −2 0) spacing value of 0.337 nm measured from the image is consistent with the crystal data from SC-XRD. For UOF-Cd, a bright field TEM image (Fig. 6a) showed thin needle crystals. A SAED pattern in zone axis of [0 −1 3] is indexed to the monoclinic C2/c space group (Fig. 6b). A HRTEM image in zone axis [0 −1 3] is showed in Fig. 6c with an inserted FFT image. The measured d (4 0 0) spacing value (0.281 nm) is consistent with the crystal data from SC-XRD.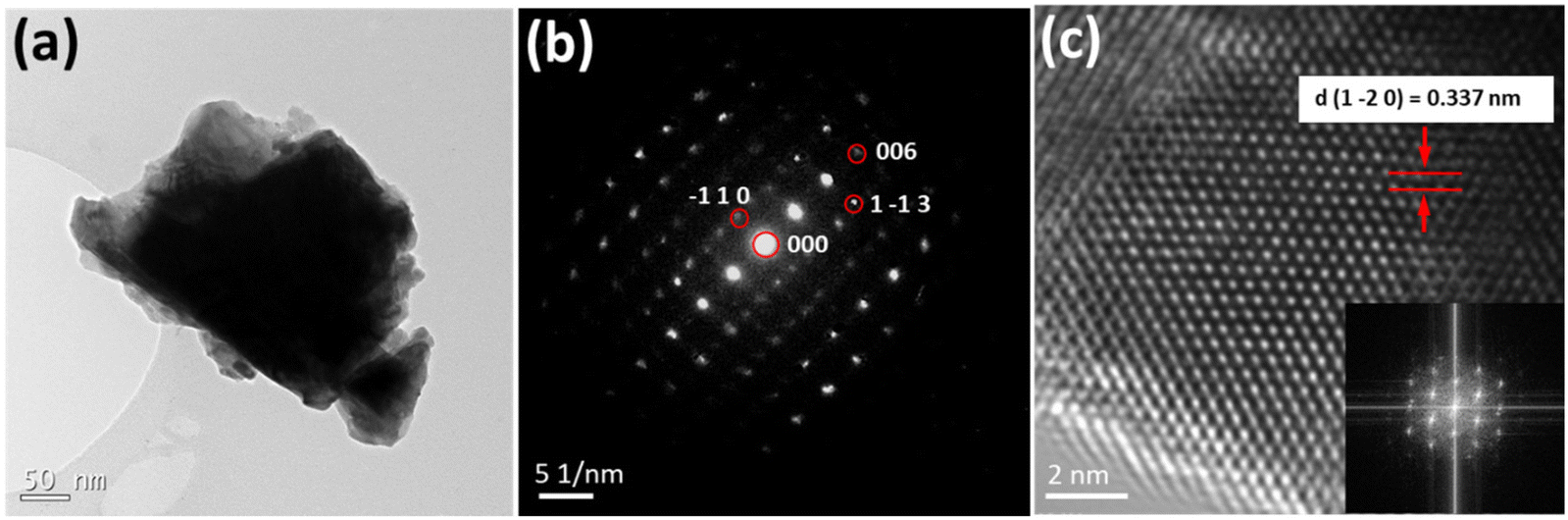 | ||
| Fig. 5 TEM of UOH-Cd: (a) a TEM bright field image of a grain; (b) a SAED pattern of the grain in zone axis of [1 1 0] indexed to the orthorhombic Pnnm space group; and (c) a HRTEM image in zone axis [6 3 1] from edge of the grain with inserted FFT image. | ||
 | ||
| Fig. 6 TEM of UOF-Cd: (a) a bright field TEM image; (b) a SAED pattern of a grain in zone axis of [0 −1 3] indexed to the monoclinic C2/c space group; and (c) a HRTEM image with inserted FFT image in zone axis [0 −1 3]. | ||
3.4. Electronic structures and uranium valences
With UOH/F structures previously reported to contain U5+ centres within their structures,22,23,26 DRS measurements were carried out to examine the uranium centres within both UOH-Cd and UOF-Cd. The presence of U5+ (5f1) is typically confirmed by the appearance of peaks in the near infrared region (1538–833 nm) resulting from crystal-field splitting of 2F5/2–2F7/2.26,51 Lacking any f electrons (5f0), U6+ can only be distinguished by charge-transfer bands in the UV and far UV regions.The most significant feature observed in both spectra (Fig. 7) is the broad, undefined absorption peaks in the UV region (220 nm–500 nm), correlating to the charge transfer bands of the U6+ centres in both materials. The only peaks of note in the NIR region are very weak and broad signals at ∼1460 nm, most likely arising from trace amounts of water in the samples. Of significance is the lack of any sharp absorption peaks correlating to the presence of U5+ in either material, confirming the U centres exist in the 6+ state, as indicated by the BVS calculations (Tables 4 and 6).
 | ||
| Fig. 7 The DRS spectra of UOH-Cd (a) and UOF-Cd (b) in the UV-vis region; inset is the same spectra in the near infrared region. | ||
3.5. Vibrational modes
Raman spectroscopy allows for the examination of the vibrational modes of UOH-Cd and UOF-Cd. As determined by past studies on UOH/F materials, peaks corresponding to the vibrational modes ν1(UO2)2+, ν(U3O)/γ[U3(OH)3] and ν2(UO2)2+ should be visible in the region of 900–700 cm−1 as strong, sharp peaks, 300 to 600 cm−1 as medium intensity, broad peaks and 200–300 cm−1 as weak, broad peaks, respectively.52–54The spectrum for UOH-Cd (Fig. 8a) reveals two peaks at ∼880 cm−1 and 860 cm−1, corresponding to the UO bonds of the bipyramidal U2. The bands at between 520–310 cm−1 are assigned to ν(U3O) bridge elongations and γ[U3(OH)3] bending vibrations, as well as possible ν(U–Oligand) vibrations. ν2(UO2)2+ bending vibrations account for the peaks between 295–200 cm−1, with lattice vibrations giving rise to the intense, sharp peak at ∼130 cm−1.
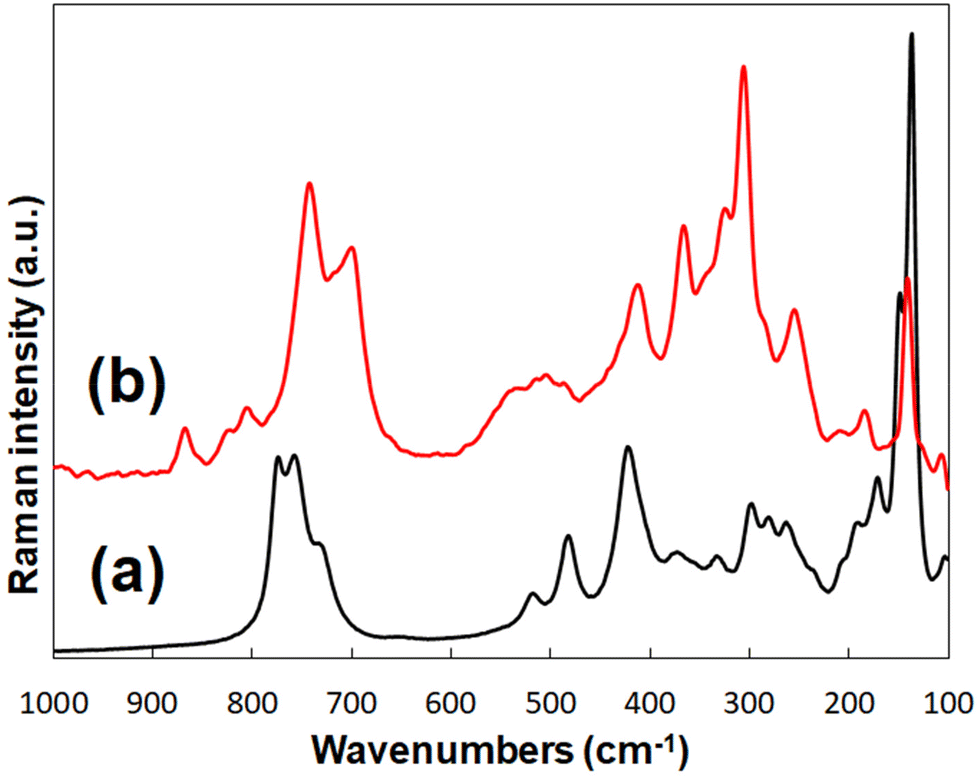 | ||
| Fig. 8 The Raman spectra of UOH-Cd (a) and UOF-Cd (b). | ||
Additional peaks are evident in the ν1(UO2)2+ region for UOF-Cd (Fig. 8b), due to the extra unique uranium centres present in the framework, with the peaks at 860, 810, 800, 750 and 705 cm−1 corresponding to the UO bond lengths between 1.773 Å–1.951 Å. The additional peaks below this region are attributed to the same vibrational modes as assigned for UOH-Cd.
3.6. Implications and perspectives
The successful synthesis of both a layered UOH and a related UOF material, containing the same secondary cation species but varying greatly in their relative abundances, clearly highlights the need to explore more dual-metal UOH systems. Many UOH minerals containing dual-cations have been documented previously, however despite this only a few synthetic UOH materials have been reported. The formation conditions, seemingly driven by the solution pH during synthesis, have been clearly demonstrated to be more tolerant than previously reported, with the highest solution pH resulting in the formation of a framework-type structure reported thus far. Past studies have established that solution pH heavily influences the structure of the final products, as a result of the increasing proportion of hydrolysed uranyl species such as [UO2(OH)]+, [(UO2)(OH)2], [(UO2)2(OH)2]2+ and [(UO2)3(OH)5]+ which form when solution pH is above 5. The apparent relationship between UOH and UOF structures, as evidenced by the near identical β-U3O8-type sheets present in both synthesised materials, highlights the seemingly delicate balance of reaction conditions which can lead to the preferred formation of one structure over the other. Future work towards exploring this balance, particularly targeting the entire pH range which gives rise to these hydrolysed species, is essential towards intuiting the conditions that drive the preferential formation of these UOH/F phases.The results of this work can be extended to encompass the alteration chemistry of UO2 under the conditions which may be found for disposal of SNF in geological repositories. The significance of studying dual-cation systems has been demonstrated within this work, as shown by the varying ratios of Cd:K incorporated within the two structures. Given the formation conditions of UOF structures have been found to be more restrictive, the additional flexibility provided by a mixed-valence cation system could have a significant impact on the final products of the UO2 alteration pathway under the repository conditions. Thus, these dual-metal systems warrant further study. In extending this work to other possible cations found within the surrounds of the SNF, a clear target is that of transition metals. Given their abundance in the environment, the understanding of the alteration chemistry of UO2 systems in the presence of transition metals is essential. With only a handful of transition metal-containing UOH materials reported,14,18,30 there exists a clear need for further study in this area.
Given the successful incorporation of K+ alongside the Cd2+ species, work exploring other monovalent cations in dual-metal systems is also crucial. The most apparent choice to explore is that of Cs+ ions, which will be present within the SNF and therefore a possible factor in the alteration of UO2. A small number of UOH-Cs structures have been reported,20,45 however exploring the Cs-system alongside other metals in a dual-cation system could prove insightful.
4. Conclusions
Two new UOH/F structures, each incorporating Cd2+ and K+ in differing ratios, have been synthesised under hydrothermal conditions with uranyl nitrate as the U source. The layered UOH structure, which crystallised in the Pnnm space group, had a U:Cd:K ratio of 3:1.5:1. Comparatively, the framework-type structure, crystallising in the C2/c space group, had a much higher abundance of K+, with a U:Cd:K ratio of 4.4:0.2:1. Both structures contained β-U3O8-type uranyl oxide hydroxide layers, consistent with previously reported framework-type structures but rare amongst layered structures. As both materials contained the near identical β-U3O8-type layers and a distinct octahedral U centre lacking the expected UO bonds, this strongly suggested a close correlation between the two materials. Significantly, the increased abundance of K+ in the UOF material suggests the inclusion of a second cation species with a different valence could be hugely impactful on the structure which eventuates from the synthesis.
These compounds further illustrate the sensitive, complex chemistry which drives the formation and alteration of UOH systems. Not only is the chemistry driven by such things as temperature, solution pH and time, the presence of multiple metal systems must also be considered. Therefore, further laboratory-based studies involving these dual-metal systems need to be carried out to better comprehend these factors.
Author contributions
T. A. Ablott: conceptualization, data curation, formal analysis, writing – original draft, writing – review & editing. K. T. Lu: data curation, formal analysis, writing – review & editing. T. Wei: Data curation, formal analysis, writing – review & editing. Y. Zhang: conceptualization, data curation, formal analysis, project administration, resources, supervision, writing – original draft, writing – review & editing.Conflicts of interest
The authors are not aware of any conflict of interest.Acknowledgements
We would like to thank I. Karatchevtseva for Raman measurement, the Nuclear Science and Technology (NST) at ANSTO for synthesis and characterization of materials. The crystallographic data for compounds UOH-Cd and UOF-Cd were collected on the MX2 beamline at the Australian Synchrotron, a part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector.References
- J. Plasil, J. Geosci., 2014, 59, 99–114 CrossRef CAS.
- R. J. Baker, Coord. Chem. Rev., 2014, 266–267, 123–136 CrossRef CAS.
- S. Alyokhina, Nucl. Eng. Technol., 2018, 50, 717–723 CrossRef CAS.
- J. Janeczek and R. C. Ewing, J. Nucl. Mater., 1992, 190, 157–173 CrossRef CAS.
- J. Janeczek and R. C. Ewing, J. Nucl. Mater., 1992, 190, 128–132 CrossRef CAS.
- D. J. Wronkiewicz, J. K. Bates, S. F. Wolf and E. C. Buck, J. Nucl. Mater., 1996, 238, 78–95 CrossRef CAS.
- R. J. Finch and R. C. Ewing, J. Nucl. Mater., 1992, 190, 133–156 CrossRef CAS.
- P. C. Burns, Structural Chemistry of Inorganic Actinide Compounds, ed. S. V. Krivovichev, P. C. Burns and I. G. Tananaev, Elsevier, 2007, pp. 1–30 Search PubMed.
- P. C. Burns, R. C. Ewing and F. C. Hawthorne, Can. Mineral., 1997, 35, 1551–1570 CAS.
- J. Plásil, Eur. J. Mineral., 2018, 30, 237–251 CrossRef.
- P. C. Burns, Can. Mineral., 2005, 43, 1839–1894 CrossRef CAS.
- Y. Zhang, K. T. Lu and R. Zheng, Dalton Trans., 2022, 51, 2158–2169 RSC.
- R. Vochten, L. Van Haverbeke and R. Sobry, J. Mater. Chem., 1991, 1, 637–642 RSC.
- N. G. Chernorukov, O. V. Nipruk, K. A. Klinshova, O. N. Tumaeva and D. V. Sokolov, New J. Chem., 2021, 45, 9922–9922 RSC.
- P. C. Burns and F. C. Hill, Can. Mineral., 2000, 38, 163–173 CrossRef CAS.
- R. E. Glatz, Y. Li, K.-A. Hughes, C. L. Cahill and P. C. Burns, Can. Mineral., 2002, 40, 217–224 CrossRef CAS.
- Y. Li and P. C. Burns, Can. Mineral., 2000, 38, 737–749 CrossRef CAS.
- Y. Zhang, J. Čejka, G. R. Lumpkin, T. T. Tran, I. Aharonovich, I. Karatchevtseva, J. R. Price, N. Scales and K. Lu, New J. Chem., 2016, 40, 5357–5357 RSC.
- Y. Zhang, R. Aughterson, I. Karatchevtseva, L. Kong, T. T. Tran, J. Čejka, I. Aharonovich and G. R. Lumpkin, New J. Chem., 2018, 42, 12386–12393 RSC.
- K.-A. Kubatko and P. C. Burns, Inorg. Chem., 2006, 45, 10277–10281 CrossRef CAS PubMed.
- Y. Li and P. C. Burns, Can. Mineral., 2000, 38, 1433–1441 CrossRef CAS.
- K. T. Lu, Y. Zhang, T. Wei, T. A. Ablott, T. H. Nguyen and R. Zheng, New J. Chem., 2022, 46, 1371–1371 RSC.
- T. A. Ablott, K. T. Lu, R. D. Aughterson and Y. Zhang, Dalton Trans., 2022, 51, 15965–15973 RSC.
- K. T. Lu, Y. Zhang, T. Wei, Z. Wang, D. T. Oldfield and R. Zheng, Inorg. Chem., 2021, 60, 13233–13241 CrossRef CAS PubMed.
- K. T. Lu, Y. Zhang, R. D. Aughterson and R. Zheng, Dalton Trans., 2020, 49, 15854–15863 RSC.
- Y. Zhang, T. Wei, T. T. Tran, K. T. Lu, Z. Zhang, J. R. Price, I. Aharonovich and R. Zheng, Inorg. Chem., 2020, 59, 12166–12175 CrossRef CAS PubMed.
- C. M. Hussain and R. Keçili, Modern Environmental Analysis Techniques for Pollutants, ed. C. M. Hussain and R. Keçili, Elsevier, 2020, pp. 1–36 Search PubMed.
- A. Kubier, R. T. Wilkin and T. Pichler, Appl. Geochem., 2019, 108, 1–16 CrossRef PubMed.
- R. C. Ewing, Nat. Mater., 2015, 14, 252–257 CrossRef CAS PubMed.
- M. Rivenet, N. Vigier, P. Roussel and F. Abraham, J. Solid State Chem., 2009, 182, 905–912 CrossRef CAS.
- F. Cesbron, W. L. Brown, P. Bariand and J. Geffroy, Mineral. Mag., 1972, 38, 781–789 CrossRef CAS.
- T. A. Olds, J. Plášil, A. R. Kampf, R. Škoda, P. C. Burns, J. Čejka, V. Bourgoin and J.-C. Boulliard, Eur. J. Mineral., 2017, 29, 129–141 CrossRef CAS.
- D. Aragão, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885–891 CrossRef PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 133–144 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, Empirical Absorption and Correction Software, University of Göttingen, Göttingen, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Y. Zhang, R. D. Aughterson, Z. Zhang, T. Wei, K. Lu, J. Čejka and I. Karatchevtseva, Inorg. Chem., 2019, 58, 10812–10821 CrossRef CAS PubMed.
- L. N. G. Chernorukov, O. V. Nipruk, K. A. Klin, G. N. Chernorukov and O. N. Tumaeva, Radiochemistry, 2021, 63, 110–120 CrossRef.
- N. E. Brese and M. O'Keefe, Acta Crystallogr., 1991, 47, 192–197 Search PubMed.
- R. M. Wood and G. J. Palenik, Inorg. Chem., 1999, 38, 1031–1034 CrossRef CAS PubMed.
- K. T. Lu, Y. Zhang, T. Wei, J. Čejka and R. Zheng, Dalton Trans., 2020, 49, 5832–5841 RSC.
- Y. Li, C. L. Cahill and P. C. Burns, Chem. Mater., 2001, 13, 4026–4031 CrossRef CAS.
- F. C. Hill and P. C. Burns, Can. Mineral., 1999, 37, 1283–1288 CAS.
- P. C. Burns, R. J. Finch, F. C. Hawthorne, M. L. Miller and R. C. Ewing, J. Nucl. Mater., 1997, 249, 199–206 CrossRef CAS.
- J. Plášil, R. Škoda, J. Čejka, V. Bourgoin and J.-C. Boulliard, Eur. J. Mineral., 2016, 28, 959–967 CrossRef.
- P. C. Burns and R. J. Finch, Am. Mineral., 1999, 84, 1456–1460 CrossRef CAS.
- F. C. Hawthorne, R. J. Finch and R. C. Ewing, Can. Mineral., 2006, 44, 1379–1385 CrossRef CAS.
- J. L. Brugger, S. V. Krivovichev, P. Berlepsch, N. Meisser, S. Ansermet and T. Armbruster, Am. Mineral., 2004, 89, 339–347 CrossRef CAS.
- G. L. Klunder, J. W. Plaue, P. E. Spackman, P. M. Grant, R. E. Lindvall and I. D. Hutcheon, Appl. Spectrosc., 2013, 67, 1049–1056 CrossRef CAS PubMed.
- Y. Zhang, M. Bhadbhade, I. Karatchevtseva, J. R. Price, H. Liu, Z. Zhang, L. Kong, J. Čejka, K. Lu and G. R. Lumpkin, J. Solid State Chem., 2015, 226, 42–49 CrossRef CAS.
- R. L. Frost, J. Čejka, G. A. Ayoko and M. L. Weier, Polyhedron, 2007, 26, 3724–3730 CrossRef CAS.
- R. L. Frost, J. Čejka and M. L. Weier, J. Raman Spectrosc., 2007, 38, 460–466 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM-EDS, supporting tables. CCDC 2244533 (UOH-Cd) and 2244534 (UOF-Cd). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00630a |
| This journal is © The Royal Society of Chemistry 2023 |