
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The π-interactions of ammonia ligands evaluated by ab initio ligand field theory†
Moritz
Buchhorn
 and
Vera
Krewald
*
and
Vera
Krewald
*
TU Darmstadt, Department of Chemistry, Theoretical Chemistry, Alarich-Weiss-Straße 4, 64287 Darmstadt, Germany. E-mail: vera.krewald@tu-darmstadt.de
First published on 11th April 2023
Abstract
Ammonia and amine ligands are commonly assumed to be σ-only ligands in coordination chemistry, i.e. they are not expected to interact significantly with a metal via a π path. Ligand field analyses employing the Angular Overlap Model resulted in good fits to experimental data without a π parameter for ammonia ligands, thereby supporting this assumption. In this work, we challenge this assumption and suggest that it is an oversimplification. We use complete active space calculations for electronic structure analyses of copper ammine complexes that are in good agreement with the transitions observed in experimental UV-vis spectra. These findings lead to a reinterpretation of the experimental spectra that necessitates a significant π interaction of the ammonia ligands. The strength of the ammonia π interaction is evaluated by parameterizing the ligand field splittings of a series of metal hexammine complexes ([M(NH3)6]n+ with M = Cr, Mn, Fe, Co, Ni, Ru, Os and n = 2, 3) and selected tetrammine complexes ([M(NH3)4]n+ with M = Cr, Mn, Fe, Co, Ni and n = 2 or 3) with the Angular Overlap Model. The resulting π parameters show that ammonia is a π donor of similar strength as chloride.
Introduction
In coordination chemistry, it is frequently assumed that the interaction between a metal centre and an ammonia ligand is characterised exclusively by σ character and thus that any π character is negligible.1–12 This can be rationalized in an orbital picture: ammonia does not have any molecular orbitals of π character; only the σ/σ* molecular orbitals of the N–H bonds are partly oriented such that the metal-ammonia binding axis can lie in the nodal plane of a metal d orbital. Ammonia is therefore commonly classified as a σ-only ligand.Metal–ligand interactions can be evaluated with ligand field theory. Besides global ligand field descriptors like the ligand field splitting Δ or Racah parameters, the Angular Overlap Model (AOM) provides a ligand-specific parameterisation that conforms to the familiar interpretation of chemistry in terms of functional groups. The AOM quantifies the metal–ligand interaction via σ and π overlaps, with an additional parameter for d–s mixing.2,13,14 The destabilisation of each pure metal d orbital with respect to the situation in the free ion is associated with a spherical component E and a directional component that is expressed with a specific number and magnitude of eσ and eπ parameters depending on the coordination environment, see Fig. 1. The orbital splitting of tetrahedral and octahedral coordination spheres does not allow for a distinction between eσ and eπ.15 This means that neglecting eπ comes with the convenience of having an unambiguous relationship between eσ and Δ. Lower symmetry coordination environments result in fewer orbital degeneracies which should provide a sufficient number of states to fit eσ and eπ parameters simultaneously.
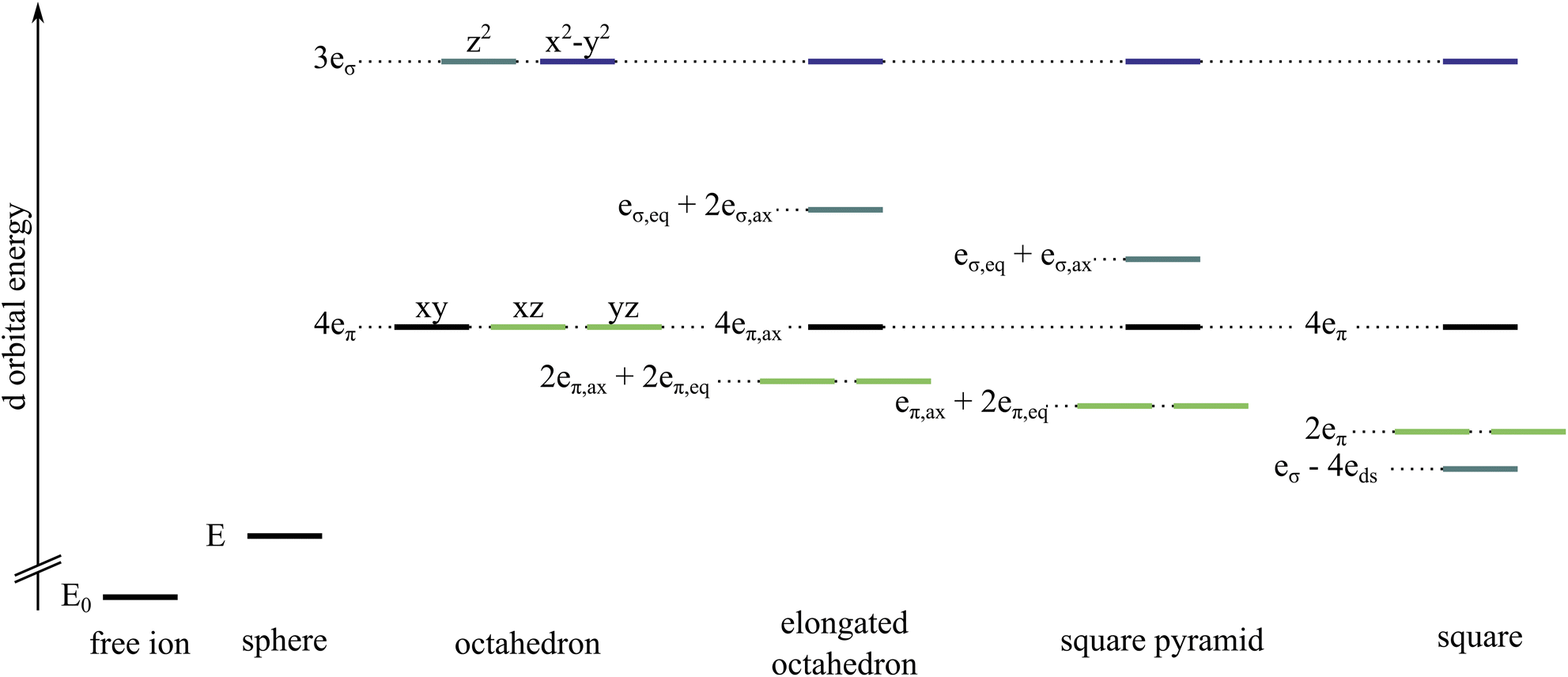 | ||
| Fig. 1 Comparison of d orbital energy splittings for a free ion, a spherical potential and different coordination environments with the respective AOM parameters eλ. The orbital levels are coloured as follows, dz2 teal, dx2−y2 dark blue, dxy black, dxz and dyz light green. For an elongated octahedron and a square pyramid, the ligands on the z-axis have parameters labelled ax, while the others in the xy-plane are labelled eq. For symmetries with at least one d orbital in the totally symmetric representation, d–s mixing must be considered which affects the orbital energy by an additional parameter eds. More details regarding d–s mixing are provided in the ESI.† | ||
We recently developed an AOM parameter fitting procedure16 based on ab initio ligand field theory17–19 as a tool that provides insights into metal–ligand bonding situations. Notably, it is able to obtain AOM parameters for complexes for which previously the ligand field equation system would have been underdetermined. We showed that the method yields qualitatively correct parameters and reproduces expected chemical trends like the eλ values of halide ligands being associated with their donor capacity and position in the spectrochemical series.16
In this paper, we present some incentives to rethink the assumption of amines being σ-only ligands. Firstly, we revisit the experimental UV-vis spectra of a square planar copper tetrammine and a pyramidal copper pentammine complex. They had been interpreted to not contain d–d transitions that would be expected if π interactions were present.5 Their analysis with ab initio ligand field theory calculations demonstrates the need for an ammine π interaction. Building on this, we present and discuss AOM parameters for a series of octahedral and tetrahedral ammine complexes obtained with our recently presented AOM parameter fitting procedure.16 We find that ammonia should be viewed as capable of significant π interactions (ranging from about 400 cm−1 to more than 1000 cm−1), which raises the question of whether a true σ-only ligand can exist.
Methodology and computational details
The ORCA 4.2.1 quantum chemistry package20,21 was used for all quantum chemical calculations. Geometries were optimized using the unrestricted Kohn–Sham formalism with the BP86 functional,22,23 the def2-SVP basis set,24 and the def2/J auxiliary basis.25 The resolution of identity approximation for the Coulomb term was used.26,27 Convergence criteria were NormalSCF for all self-consistent field calculations and TightOpt for geometry optimizations. The geometry optimizations employed the default integration grid (Accuracy 2: Lebedev 110 points) for optimization steps and the final SCF at the optimized geometry (Accuracy 4: Lebedev 302 points). Geometry optimizations of halide complexes of the type [MX6]3−/4− with large negative charges additionally employed the CPCM/SMD solvation model with the parameters of water and an increased solvent radius of 3.0 Å to aid convergence.28,29For square planar [Cu(NH3)4]2+ and square pyramidal [Cu(NH3)5]2+, the heavy atom positions were taken from the crystal structures (ICSD entries 14372 and 201229) and only the hydrogen atom positions were optimised with the settings stated above, but without employing a solvent model. The electronic states corresponding to the d orbitals were calculated using CASSCF30,31 in the ab initio ligand field theory variant17–19 with the def2-TZVP basis set. The calculations on [Cu(NH3)4]2+ and [Cu(NH3)5]2+ also employed a subsequent perturbation theory treatment (NEVPT2).32–35 The active space was chosen to contain the five valence d orbitals and n d electrons, or in shorthand notation a CAS(n,5) space. The ab initio ligand field theory module17 was employed to construct the effective ligand field Hamiltonian from the calculated states. For selected examples, spin–orbit coupling was considered using the spin–orbit mean field approach as implemented in ORCA.36
The AOM was used to fit the one-electron part of the ligand field Hamiltonian13,37 according to our fitting procedure.16 The ligand field matrix contains up to 15 unique equations, which quickly results in underdetermined problems. For instance for six-coordinate complexes, one needs to fit 13 parameters of the ligand field potential: E, six eσ and six eπ. Such a fit is in theory possible but in practice, linear dependencies can occur in the equation system so that the actual number of equations can be lower than 13, resulting in an underdetermined problem.
We address this restriction by grouping chemically equivalent ligands at similar bond lengths (e.g. in Jahn–Teller distorted [Mn(NH3)6]3+, ligands are grouped into axial and equatorial parameter sets). In practice, grouping is achieved by adding additional equations to the system that require 0 = eσ,L − eσ,L′. From a formal perspective, one could use these equations to require parameters to be strictly equal and thereby reduce their number. For solving the least squares problem of the overdetermined system, the addition of more equations has different consequences than the reduction of the number of parameters. Additional equations allow the parameters eσ,L and eσ,L′ to deviate, even if they are expected to be equal. This flexibility is important, since the asymmetric distortions generated during the fitting procedure render the ligands not perfectly equivalent so that small deviations are expected. If not stated otherwise, all ligands in the complexes studied are put into a single group. Complexes with significant differences in bond lengths due to Jahn–Teller distortions have two or more ligand groups as indicated by labels.
Results
Pyramidal copper pentammine
An explicit assessment of π interactions is only possible for symmetries lower than Oh or Td, see Fig. 1. The [CuII(NH3)5]2+ subunit in K[Cu(NH3)5][PF6]3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38 and NH4[Cu(NH3)5][PF6]339 is approximately square pyramidal (point group: C2v) and thus represents a suitable test case. Another convenient feature is its d9 electronic configuration that results in four d–d transitions. The interelectronic repulsion within each electronic state is equal, and therefore the energy differences of these states can be equated with the energy differences of the d orbitals.
38 and NH4[Cu(NH3)5][PF6]339 is approximately square pyramidal (point group: C2v) and thus represents a suitable test case. Another convenient feature is its d9 electronic configuration that results in four d–d transitions. The interelectronic repulsion within each electronic state is equal, and therefore the energy differences of these states can be equated with the energy differences of the d orbitals.
The electronic spectrum of [Cu(NH3)5]2+ (see Fig. 2a) shows a band at 15300 cm−1 with a weak shoulder at 14000 cm−1 and a second band at 11000 cm−1, see Table 1. The shoulder is assigned to a dxy → dx2−y2 transition by Duggan et al.,38 implying a state splitting of 1300 cm−1. Within the AOM, this splitting results in an eπ value of 1300 cm−1 in a perfect square pyramid or even a slightly larger value in a C2v distorted square pyramid (see ESI† for details). Since the shoulder is not a very pronounced feature, the signal was interpreted later as “essentially unsplit”, ruling out a π interaction.5 With this interpretation, ammonia is viewed as a σ-only ligand. This assumption was also made for other complexes.6–12,40
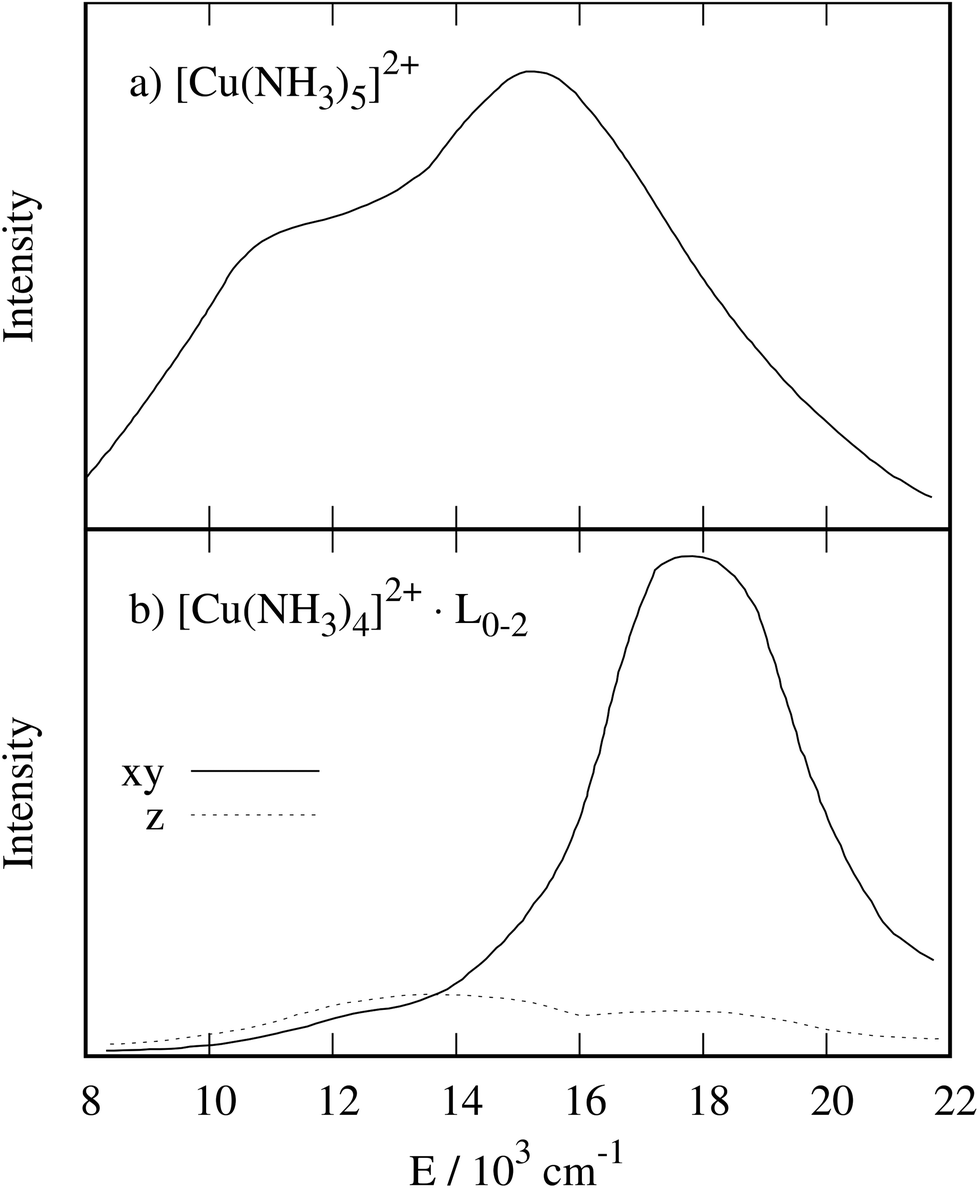 | ||
| Fig. 2 Electronic spectrum of (a) NH4[Cu(NH3)5][PF6]3, reprint from ref. 38 with permission from the Royal Society of Chemistry. Polarized electronic spectrum of (b) Na4[CuII(NH3)4·L][CuI(S2O3)2]2, reprint from ref. 41 with permission from the Royal Society of Chemistry. Please note the structural uncertainty of (b) discussed in the main text. | ||
| dxz,dyz → dx2−y2/cm−1 | dxy → dx2−y2/cm−1 | dz2 → dx2−y2/cm−1 | |
|---|---|---|---|
| CASSCF | 11549–11673 |
10523 |
8 635 |
| NEVPT2 | 16145–16308 |
15341 |
11687 |
| Exp. | 15300 |
ca. 14000 |
11000 |
CASSCF/NEVPT2 calculations on the [Cu(NH3)5]2+ subunit from the crystal structure yield an orbital ordering and d–d transitions in very good agreement with the measured ones, see Table 1. We note that while spin–orbit coupling (SOC) may be of importance for copper complexes (single-electron SOC parameter of ca. 830 cm−1 for free Cu2+),15 inclusion of SOC does not qualitatively alter the picture, see ESI.† The calculations thus fully support the spectral assignments of Duggan et al., and hence the need for an ammonia π interaction of about 1300 cm−1.
Square planar copper tetrammine
As a second example, we selected the [CuII(NH3)4]2+ subunit in the crystal structure of Na4[CuII(NH3)4][CuI(S2O3)2]2.42 While initially, the crystal structure was thought to contain copper ions in a square-planar environment,42 later studies suggested that one or two axial ammonia ligands may weakly coordinate.43 Although the precise structure is therefore not clear, there are two independently reported UV-vis spectra.41,44 Tomlinson et al. assigned the d–d transitions for the presumed square-planer complex using polarised electronic spectra, see Fig. 2b, leading to a one-electron orbital sequence of dx2−y2 > dz2 > dxy ≥ dxz,dyz.41To evaluate the electronic structure with CASSCF/NEVPT2 calculations as for the square-pyramidal system, hydrogen atoms were added to complete the ammonia ligands of the [Cu(NH3)4]2+ subunit and their positions were optimized (see computational details). The influence of one or two axial ammonia ligands was evaluated explicitly, see below. Using CASSCF calculations that facilitate a direct assignment of configurations to states, we arrive at a different energetic ordering for the square planar complex, namely dx2−y2 > dxy > dxz, dyz ≈ dz2 in agreement with other computational studies.45–47 Giner et al. found this orbital ordering, notably including the pronounced energy difference between the dxy and dxz, dyz levels, at different levels of theory that capture electron correlation adequately, e.g. CCSD(T).47 Atanasov et al. assigned an eπ of roughly 1200 cm−1 to NH3 from an ab initio ligand field theory analysis.48
There is some uncertainty surrounding the exact composition of the ligand field experienced by the copper ion in the crystal structure,42,43 where the gaps between the square planar [Cu(NH3)4]2+ subunits are large enough to host ammonia molecules. These additional molecules would result in axial Cu–N distances of 2.88 Å. We investigated these different possibilities with one or two ammonia ligands approaching the copper ion along the z-axis, see Fig. 3. In these scans, the position of the dxy orbital is taken as the reference value for the d orbital energies. As expected, the energy difference dxy → dx2−y2 remains constant to a good approximation, and the position of the dz2 orbital is influenced significantly by the additional ligands on the z-axis. The energies of the dxz and dyz orbitals are lower in energy than the dxy orbital. In the extreme case of a square planar [Cu(NH3)4]2+ (Fig. 3, middle panel), the dz2 orbital is found coincidentally at about the same energy as the dxz and dyz orbitals. Here, too, SOC does not qualitatively alter the picture, see ESI.† Tomlinson et al. probably investigated a mixture of structures, leading to the large line broadening of the lower intensity peak.41 It is not possible to determine a dominant composition with the present data.
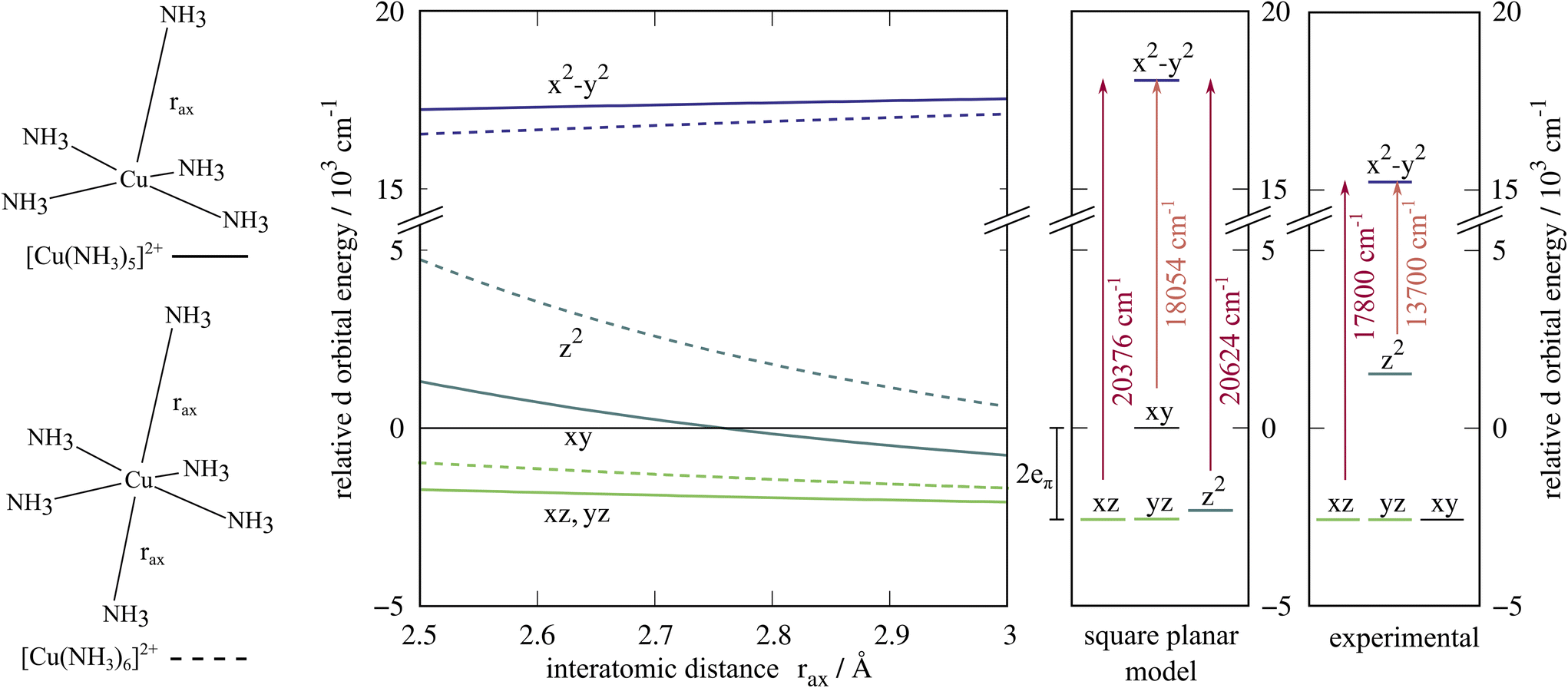 | ||
| Fig. 3 Energy levels of the d orbitals in approximately square-pyramidal and octahedral ligand environments where the axial ligand positions rax are varied (left), the square planar case (middle), and the interpretation by Tomlinson, Hathaway et al. based on the observed UV-vis transitions (right).41 The gaps in the crystal structure41 might be filled with ammonia molecules, yielding a square pyramidal, elongated octahedral or square-planar case. The orbital energies are referenced to the dxy orbital energy. | ||
With regard to a possible ammonia π interaction, the ligand field splitting of the square planar complex can be discussed as follows. If there was no π interaction at all, the dxy(b2g) and dxz/yz(eg) orbital energies ε would be degenerate (see also ESI, Fig. 3†):
| εdxz = εdyz = 2eπ = 0 |
| εdxy = 4eπ = 0 |
The dz2 orbital energy depends on the extent of d–s mixing, and thus could be lower or roughly equal to the energy of the aforementioned orbitals. If there is a donating π interaction, the dxz and dyz orbitals are shifted up by 2eπ and the dxy orbital is shifted up by 4eπ. Indeed, the CASSCF calculation shows that the energy of the dxy orbital is significantly higher than that of the dyz and dxz orbitals, while the energy of the dz2 orbital is accidentally equal to the ones of the dyz and dxz orbitals. A similar orbital energy sequence (dx2−y2 ≫ dxy > dz2 > dxz,dyz) was assigned to the D4h [Cu(H2O)4]2+ subunits in meta-zeunerite (Cu(UO2)2(AsO4)2·8H2O) by Billing et al.49 For this system, a significant π interaction from the equatorial water ligands was expected.49 Ten years later, this sequence was still considered to be plausible only for strong π donors.50
The energy difference between dxy and dyz,dxz from the CASSCF calculation is independent of any specific AOM fitting routine and too large to be a computational artefact. Increasing the basis set size does not lead to a qualitatively different result, see ESI.† Additionally, the computational studies mentioned above45–48 find the same pronounced difference between the dxy and dxz, dyz orbital energies using different levels of theory. In the AOM, the higher energy of the dxy orbital can only be explained if eπ > 0. Therefore, we interpret this ligand field splitting as strong support for the existence of a π interaction for ammonia ligands.
Equilibrium AOM parameters
Having seen that a π interaction is relevant for ammonia ligands with experimental evidence and examples that do not rely on our previously introduced sampling procedure, we now turn to a broader scope of complexes. Using our ab initio AOM sampling procedure, we evaluated a series of octahedral and tetrahedral complexes where all orbital energies affected by π interactions are degenerate so that the ammonia π interactions cannot be isolated. To this end, we chose complexes of the type [M(NH3)6]n+, with M = Cr, Mn, Fe, Co, Ni, Ru, Os and n = 2, 3. We note that some of the structures are Jahn–Teller distorted and therefore have different axial and equatorial AOM parameters. For strongly distorted structures, the ligands were grouped such that those at similar bond lengths are set to have equal parameters.The average ab initio AOM parameters based on CASSCF calculations are listed in Table 2 for selected equilibrium structures. Note that the standard deviations result from the data points of five distinct calculations at similar structures. While the CASSCF calculation itself may carry a systematic error that could not be avoided even with a larger number of samples, the order of magnitude was shown to be reliable.16,18,51,52
| M | 2S + 1 | ΔE/kJ mol−1 | r/Å | e σ/cm−1 | e π/cm−1 |
|---|---|---|---|---|---|
| M(II) | |||||
| Mn | 6 | 0 | 2.34 | 3613(213) | 894(159) |
| Fe | 1 | 5 | 2.05 | 5491(324) | 710(243) |
| Fe | 3 | 43 | 2.03ax | 6011(352) | 1051(263) |
| Fe | 3 | 43 | 2.25eq | 3772(385) | 826(265) |
| Fe | 5 | 0 | 2.28 | 3598(229) | 851(171) |
| Co | 4 | 0 | 2.23 | 3471(62) | 770(19) |
| Co | 2 | 7 | 2.39ax | 1818(379) | 591(284) |
| Co | 2 | 7 | 2.02eq | 5857(379) | 1024(285) |
| Ni | 3 | 0 | 2.18 | 3024(355) | 412(266) |
| Ru | 1 | 0 | 2.16 | 9755(203) | 799(152) |
| Os | 1 | 0 | 2.18 | 11271(156) |
594(120) |
| M(III) | |||||
| Cr | 4 | 0 | 2.14 | 6743(341) | 1027(255) |
| Mn | 5 | 0 | 2.36ax | 3285(360) | 576(270) |
| Mn | 5 | 0 | 2.12eq | 6707(359) | 1046(270) |
| Fe | 6 | 56 | 2.23 | 6532(915) | 2052(683) |
| Fe | 4 | 47 | 2.32ax | 3909(907) | 972(679) |
| Fe | 4 | 47 | 2.08eq | 7452(909) | 1433(682) |
| Fe | 2 | 0 | 2.06 | 7042(92) | 1087(74) |
| Co | 1 | 0 | 2.02 | 6788(408) | 698(307) |
| Ni | 4 | 0 | 2.15a | 7515(1628) | 1971(1238) |
| Ni | 4 | 0 | 2.18b | 7500(1420) | 2349(1060) |
| Ni | 4 | 0 | 2.22c | 6804(1058) | 2665(792) |
| Ru | 2 | 0 | 2.16 | 10749(417) |
649(311) |
| Os | 2 | 0 | 2.18 | 11686(170) |
194(149) |
The ab initio AOM parameters identified with this procedure show a substantial π interaction for all complexes studied here. The data set is consistent with expectations and follows common trends, such as larger parameter values for higher oxidation states. Our findings for this more generalised data set thus contradict the widespread assumption of ammonia having only σ interactions.
Comparison to tetrahedral complexes
To rule out any conceivable sources of error for the chemically relevant magnitude of the ammonia π interactions we have identified above, we expanded the data set to tetrahedral complexes. This tests the possibility that artefacts arise due to the overlap of two adjacent σ potentials in an octahedral, square-planar or other setting with closely spaced ligands. This overlap would occur at a position which would be covered by a π parameter. In such cases, both σ and π would be artificially increased, while the spherical contribution E would be decreased correspondingly. Therefore, it should be tested whether ammonia π parameters also appear in complexes where the ligands are spaced further apart and thus overlapping σ potentials can be ruled out.We chose complexes of the type [M(NH3)4]2+/3+ with M = Cr, Mn, Fe, Co, Ni. These complexes are hypothetical; their only purpose is to provide more room for the ligands. The fits yield ab initio AOM parameters that are even larger than the ones obtained for the octahedral complexes, even though some scattering is observed. We note that d–s mixing needed to be included for some of the complexes where structural deviations from ideal tetrahedral symmetry were apparent (see ESI†). In conclusion, overlapping σ potentials can be ruled out as the origin for the observed ammonia π interaction in both the octahedral and tetrahedral complexes.
Scan and comparison to halido complexes
In order to verify that the order of magnitude obtained for the ammonia π parameters is reasonable, we replaced the ammonia ligands in [Cr(NH3)6]3+ with chloride ligands in exactly the same positions. Since it is widely accepted that halides and metals interact via a considerable π path, this data set allows a meaningful comparison with the metal–ammine parameters.Indeed, the chloride ligands show eπ values that are even smaller than those of the ammonia ligands (eπ chloride: 313(524) cm−1, eπ ammonia: 1027(255) cm−1). However, the M–NH3 bonds in these complexes are much shorter than M–Cl bonds would be in fully relaxed complexes. For instance, the Co–NH3 bond length in [Co(NH3)6]Cl2 is 2.11 Å,53 whereas the Co–Cl bond length in CoCl2 is 2.51 Å.54 The metal–chloride distance when placing the chloride ions at the nitrogen atom positions of the relaxed [Cr(NH3)6]3+ complexes is thus unnaturally short, which presumably leads to the broad scattering of the parameters and the unexpectedly small eπ parameters. We observed this behaviour already in our previous study on tetrahedral halido metalates, where eπ decreases at shorter bond lengths.16 Calculations on relaxed [MCl6]3−/4− complexes yield bond lengths in the range of 2.43 Å to 2.53 Å and eπ parameters around 500 cm−1 with significantly less scatter (see ESI†). Scanning the metal–ligand distances in these examples from the equilibrium bond length of Cr–NH3 to that of Cr–Cl, see Fig. 4, shows the similar order of magnitude for the eπ parameters in these two scenarios. The comparison thus confirms that the π interaction of ammonia is unlikely to be an artefact, since the chloride interactions predicted at the same bond lengths match expectations and are qualitatively correct.
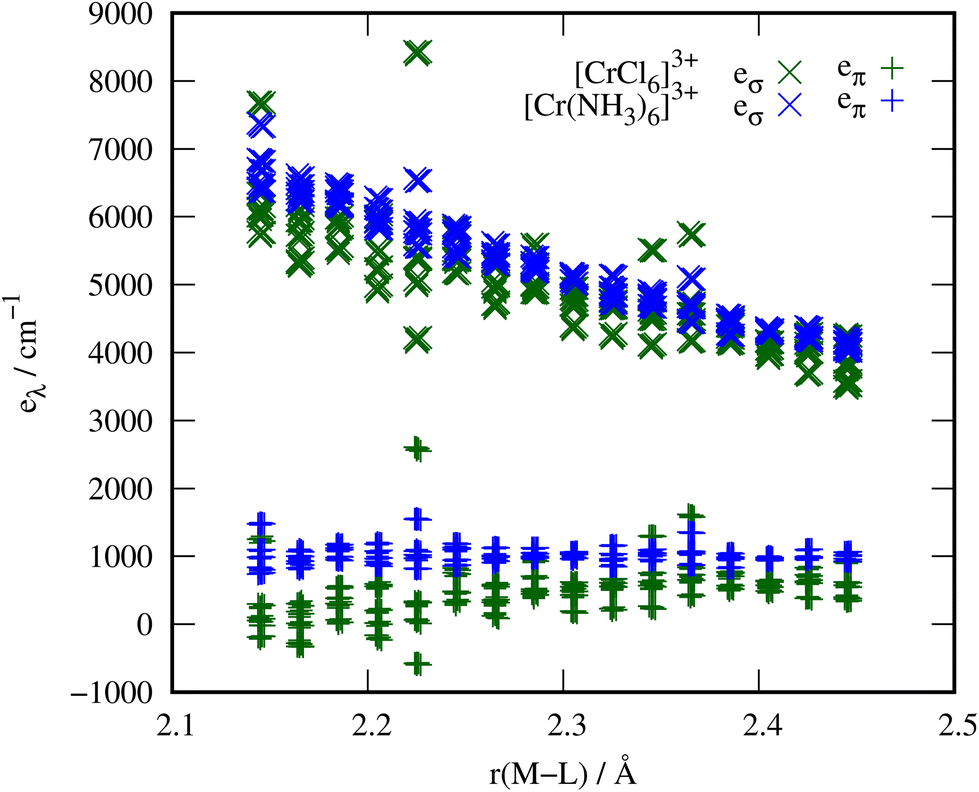 | ||
| Fig. 4 Bond length scans of [Cr(NH3)6]3+ and [CrCl6]3− from the optimized metal–ligand bond length of the ammine complex to that of the chloride complex. The upper data points (>3000 cm−1) are eσ parameters and the lower set of data points are eπ parameters. | ||
Monodentate amine ligands
To better understand the origin of the π interaction of ammonia, we studied the influence of the substituent R in –NR3 on the π interaction, specifically comparing –NH3 and –NMe3. Complexes with different amine ligands can be compared in terms of their ligand field splitting or via AOM parameterization. A problem arising for such comparisons is that the bond lengths vary and hence the observed splitting is influenced by the bond length and the ligand field strength of the amine itself. While these effects cannot be disentangled fully in experimental data, it appears that the bond length is the more dominant factor in a study by Lever et al.4Due to the bulkiness of the methylamine ligand, it is not possible to obtain reasonable structures for [M(NMe3)6]n+ complexes. Therefore, we chose to study heteroleptic complexes of the type [M(NH3)4(NMe3)2]n+, with the methylamine ligands positioned trans to each other, see Table 3. For all complexes, the eσ parameters of the ammonia ligands are larger than those of the amine ligands, whereas the magnitudes of the eπ parameters are comparable or larger.
| Complex | A = NH3 | B = NMe3 | |||||
|---|---|---|---|---|---|---|---|
| Composition | Bond lengths | r(M–L)/Å | e σ/cm−1 | e π/cm−1 | r(M–L)/Å | e σ/cm−1 | e π/cm−1 |
| a The NH3 ligands were moved with no subsequent geometry optimization. b The NMe3 ligands were replaced with NH3 with subsequent optimization of the positions of the new hydrogen atoms. | |||||||
| 4[CrA4B2]3+ | Optimized | 2.13 | 8115(842) | 1944(632) | 2.34 | 6291(847) | 1917(634) |
| [CrA4B2]3+ | Fixed | 2.34 | 5228(222) | 1378(167) | 2.34 | 6425(223) | 1746(167) |
| [CrA6]3+ | Fixed | 2.34 | 5143(93) | 1193(71) | |||
| 6[MnA4B2]2+ | Optimized | 2.33 | 3946(247) | 1023(185) | 2.53 | 2793(247) | 905(185) |
| [MnA4B2]2+ | Fixed | 2.53 | 2667(63) | 866(47) | 2.53 | 2992(63) | 975(47) |
| [MnA6]2+ | Fixed | 2.53 | 2667(21) | 849(17) | |||
| 5[FeA4B2]2+ | Optimized | 2.25 | 4740(185) | 1548(137) | 2.48 | 3360(185) | 1410(138) |
| [FeA4B2]2+ | Fixed | 2.48 | 2981(104) | 1152(77) | 2.48 | 3320(106) | 1268(79) |
| [FeA6]2+ | Fixed | 2.48 | 2864(64) | 1044(47) | |||
| 1[CoA4B2]3+ | Optimized | 2.00 | 8353(623) | 1518(469) | 2.29 | 5018(636) | 1290(471) |
| [CoA4B2]3+ | Fixed | 2.29 | 4439(38) | 992(28) | 2.29 | 6032(39) | 1365(29) |
| [CoA6]3+ | Fixed | 2.29 | 4531(84) | 853(63) | |||
To evaluate the influence of the M–N bond length on the AOM parameters, we set the M–N bond lengths with ammonia ligands to the value of the M–NMe3 distances. As expected, the eσ and eπ parameters of the ammonia ligands decrease so that the AOM parameters of –NMe3 are consistently larger than those of –NH3. In the hexammine reference complex with all ammonia ligands at the same distance as the amine ligands in the relaxed structure, the ammonia ligands have very similar AOM parameters as in the mixed complex with fixed ammonia bond lengths. We can therefore conclude that amine ligands have intrinsically higher eπ parameters than ammonia ligands. This comparison furthermore points towards hyperconjugation of the N–R bonds as a possible origin of the π interaction for both amines and ammonia.
From a molecular orbital perspective, the chemical origin and a possible explanation for the considerable π interaction of ammonia ligands may lie in the hyperconjugation of the N–H bonds with the respective d orbitals. Hyperconjugation was already observed and interpreted by Mulliken55–57 and became an important tool for rationalizing formation and stabilization energies58 and chemical shifts in NMR experiments.59 Experimental NMR studies of Ru(NH3)x complexes show that there is an interaction between N–H bonds and the metal d orbitals, leading to measurable hyperfine interactions. This interaction is explained by hyperconjugation of the N–H bonding orbital with the ruthenium d orbital.60–62
An alternative view is offered by the interpretation of ligand field splittings in terms of electrostatic potentials as developed by Gerloch and Woolley.63–65 This picture is completely independent of molecular orbital theory and treats the influence of the ligands on the d orbital energies purely electrostatically. With this approach, the π interaction could be explained by the electron density of the N–H bond and the negative partial charge on the nitrogen atom exerting an influence on the metal d orbital energies.
Both interpretations are supported by the results for the methylamine complexes. The larger N–C bonding orbital of –NMe3 compared to the N–H bonding orbitals of –NH3 suggest a stronger hyperconjugation with the metal d orbitals. Likewise, the methyl groups are electron donating, leaving a higher electron density at the nitrogen and thus supporting the picture of an electrostatic ligand field interaction.
Chelating amine ligands
When applying the aiLFT procedure on ethylenediamine (en) and diethylenetriamine (dien), it is apparent that it is not possible to obtain a good fit to the d orbital energies with just eσ and eπ. We attribute this to an effect called “misdirected valency” by Deeth et al.48,66–69 Misdirected valency is caused by bent bonding and non-bonding lone pairs where the centroid of the bond is not aligned with the metal–ligand axis, as depicted in ref. 66. For the chelating en and dien ligands, the M–N bond is bent because of the orientation of the carbon backbone. We note that methylamine, although not chelating, also shows slight off-axis bonding since the bulky –CH3 groups prevent full alignment. The parameters found for chelating amines (shown in the ESI†) should therefore be interpreted with some caution.If the metal–ligand interaction is asymmetric with respect to the bonding axis, it cannot be fully described by the set of parameters employed. It is in principle possible to include off-diagonal eσπ parameters in the AOM parameterisation.48 Naturally, this would aggravate the underdetermination problem and hinder a clear interpretation of the results.
Conclusions
To summarise, we presented indications that the widespread assumption of ammonia being a σ-only ligand might be incorrect. Our findings question the reliability of published AOM parameters for ammonia complexes from previous fitting procedures applied to experimental and computational data where eπ was neglected, noting of course that this was often done to reduce the number of AOM parameters. Without this assumption, many cases would not have been solvable. Our work furthermore raises the question whether any ligand can be considered a σ-only ligand.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank R. J. Deeth for many fruitful discussions and valuable input. The reviewers’ comments were greatly appreciated. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – CRC 1487, “Iron, upgraded!” – project number 443703006.References
- D. W. Smith, J. Chem. Soc. A, 1969, 1708–1712 RSC.
- C. E. Schäffer, Pure Appl. Chem., 1970, 24, 361–392 CrossRef.
- M. Gerloch, J. H. Harding and R. G. Woolley, Inorganic Chemistry, Berlin, Heidelberg, 1981, pp. 1–46 Search PubMed.
- A. B. P. Lever, I. M. Walker, P. J. McCarthy, K. B. Mertes, A. Jircitano and R. Sheldon, Inorg. Chem., 1983, 22, 2252–2258 CrossRef CAS.
- R. J. Deeth and M. Gerloch, Inorg. Chem., 1984, 23, 3846–3853 CrossRef CAS.
- M. Sano, T. Maruo, Y. Masuda and H. Yamatera, Inorg. Chem., 1984, 23, 4466–4469 CrossRef CAS.
- A. Bencini, C. Benelli and D. Gatteschi, Coord. Chem. Rev., 1984, 60, 131–169 CrossRef CAS.
- P. V. Bernhardt and P. Comba, Inorg. Chem., 1993, 32, 2798–2803 CrossRef CAS.
- T. Schönherr, M. Itoh and A. Urushiyama, Bull. Chem. Soc. Jpn., 1995, 68, 2271–2276 CrossRef.
- M. Atanasov, P. Comba, S. Helmle, D. Müller and F. Neese, Inorg. Chem., 2012, 51, 12324–12335 CrossRef CAS PubMed.
- D. Schweinfurth, M. G. Sommer, M. Atanasov, S. Demeshko, S. Hohloch, F. Meyer, F. Neese and B. Sarkar, J. Am. Chem. Soc., 2015, 137, 1993–2005 CrossRef CAS PubMed.
- P. Comba, G. Nunn, F. Scherz and P. H. Walton, Faraday Discuss., 2022, 234, 232–244 RSC.
- C. E. Schäffer and C. K. Jørgensen, Mol. Phys., 1965, 9, 401–412 CrossRef.
- M. Suta, F. Cimpoesu and W. Urland, Coord. Chem. Rev., 2021, 441, 213981 CrossRef CAS.
- B. N. Figgis and M. A. Hitchman, Ligand Field Theory and Its Applications, Wiley-VCH, New York, 1st edn, 2000 Search PubMed.
- M. Buchhorn, R. J. Deeth and V. Krewald, Chem. – Eur. J., 2022, 28, e202103775 CAS.
- M. Atanasov, D. Ganyushin, K. Sivalingam and F. Neese, Molecular Electronic Structures of Transition Metal Complexes II, Springer Berlin/Heidelberg, Berlin, Heidelberg, 2011/2012, pp. 149–220 Search PubMed.
- J. Jung, M. Atanasov and F. Neese, Inorg. Chem., 2017, 56, 8802–8816 CrossRef CAS PubMed.
- S. K. Singh, J. Eng, M. Atanasov and F. Neese, Coord. Chem. Rev., 2017, 344, 2–25 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- E. J. Baerends, D. E. Ellis and P. Ros, Chem. Phys., 1973, 2, 41–51 CrossRef CAS.
- O. Vahtras, J. Almlöf and M. W. Feyereisen, Chem. Phys. Lett., 1993, 213, 514–518 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- P. Siegbahn, A. Heiberg, B. Roos and B. Levy, Phys. Scr., 1980, 21, 323–327 CrossRef CAS.
- B. O. Roos, P. R. Taylor and P. E. Siegbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, Chem. Phys. Lett., 2001, 350, 297–305 CrossRef CAS.
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS.
- C. Angeli, R. Cimiraglia and J.-P. Malrieu, J. Chem. Phys., 2002, 117, 9138–9153 CrossRef CAS.
- C. Angeli and R. Cimiraglia, Theor. Chem. Acc., 2002, 107, 313–317 Search PubMed.
- F. Neese, J. Chem. Phys., 2005, 122, 34107 CrossRef PubMed.
- R. G. Woolley, Mol. Phys., 1981, 42, 703–720 CrossRef CAS.
- M. Duggan, N. Ray, B. Hathaway, G. Tomlinson, P. Brint and K. Pelin, J. Chem. Soc., Dalton Trans., 1980, 1342 RSC.
- B. J. Hathaway and A. Tomlinson, Coord. Chem. Rev., 1970, 5, 1–43 CrossRef CAS.
- R. J. Deeth and C. M. Kemp, J. Chem. Soc., Dalton Trans., 1992, 2013–2017 RSC.
- A. A. G. Tomlinson, B. J. Hathaway, D. E. Billing and P. Nichols, J. Chem. Soc. A, 1969, 65 RSC.
- A. Ferrari, A. Braibanti and A. Tiripicchio, Acta Crystallogr., 1966, 21, 605–610 CrossRef CAS.
- B. Morosin and A. C. Larson, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 1417–1419 CrossRef CAS.
- B. J. Hathaway and F. Stephens, J. Chem. Soc. A, 1970, 884 RSC.
- F. Neese, Magn. Reson. Chem., 2004, 42, 187–198 CrossRef PubMed.
- S. Vancoillie and K. Pierloot, J. Phys. Chem. A, 2008, 112, 4011–4019 CrossRef CAS PubMed.
- E. Giner, D. P. Tew, Y. Garniron and A. Alavi, J. Chem. Theory Comput., 2018, 14, 6240–6252 CrossRef CAS PubMed.
- M. Atanasov, P. Comba, C. A. Daul and F. Neese, Models, mysteries, and magic of molecules, Springer, Dordrecht, The Netherlands, 2008, pp. 411–445 Search PubMed.
- D. E. Billing, B. J. Hathaway and P. Nicholls, J. Chem. Soc. A, 1969, 316 RSC.
- Y. Nishida and S. Kida, Coord. Chem. Rev., 1979, 27, 275–298 CrossRef CAS.
- D. Aravena, M. Atanasov and F. Neese, Inorg. Chem., 2016, 55, 4457–4469 CrossRef CAS PubMed.
- L. Lang, M. Atanasov and F. Neese, J. Phys. Chem. A, 2020, 124, 1025–1037 CrossRef CAS PubMed.
- M. T. Barnet, B. M. Craven, H. C. Freeman, N. E. Kime and J. A. Ibers, Chem. Commun., 1966, 307–308 RSC.
- H. Grime and J. A. Santos, Z. Kristallogr. - Cryst. Mater., 1934, 88, 136–141 CrossRef CAS.
- R. S. Mulliken, Phys. Rev., 1933, 43, 279–302 CrossRef CAS.
- R. S. Mulliken, J. Chem. Phys., 1939, 7, 339–352 CrossRef CAS.
- R. S. Mulliken, C. A. Rieke and W. G. Brown, J. Am. Chem. Soc., 1941, 63, 41–56 CrossRef CAS.
- J. I.-C. Wu and P. v. R. Schleyer, Pure Appl. Chem., 2013, 85, 921–940 CrossRef CAS.
- J. W. de M. Carneiro, J. F. Dias, J. G. R. Tostes, P. R. Seidl and C. A. Taft, Int. J. Quantum Chem., 2003, 95, 322–328 CrossRef CAS.
- M. L. Naklicki, C. A. White, V. V. Kondratiev and R. J. Crutchleya, Inorg. Chim. Acta, 1996, 242, 63–69 CrossRef CAS.
- B. R. McGarvey, N. C. Batista, C. W. B. Bezerra, M. S. Schultz and D. W. Franco, Inorg. Chem., 1998, 37, 2865–2872 CrossRef CAS.
- W. M. Laidlaw, R. G. Denning, J. C. Green, J. Boyd, J. Harmer and A. L. Thompson, Inorg. Chem., 2013, 52, 7280–7294 CrossRef CAS PubMed.
- R. J. Deeth and M. Gerloch, J. Chem. Soc., Dalton Trans., 1986, 1531–1534 RSC.
- R. G. Woolley, Int. Rev. Phys. Chem., 1987, 6, 93–141 Search PubMed.
- M. Gerloch, Understanding Molecular Properties, D. Reidel Publishing Company, Dordrecht, 1987, pp. 111–142 Search PubMed.
- R. J. Deeth, M. J. Duer and M. Gerloch, Inorg. Chem., 1987, 26, 2573–2578 CrossRef CAS.
- R. J. Deeth, M. J. Duer and M. Gerloch, Inorg. Chem., 1987, 26, 2578–2582 CrossRef CAS.
- R. J. Deeth and M. Gerloch, Inorg. Chem., 1987, 26, 2582–2585 CrossRef CAS.
- M. J. Duer, N. D. Fenton and M. Gerloch, Int. Rev. Phys. Chem., 1990, 9, 227–280 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt00511a |
| This journal is © The Royal Society of Chemistry 2023 |