
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVery efficient organo-zinc scorpionates for CO2 fixation into a variety of cyclic carbonates: synthesis, coordination ability and catalytic studies†‡
Marta
Navarro
 a,
Andrés
Garcés
*a,
Luis F.
Sánchez-Barba
*a,
David
González-Lizana
a and
Agustín
Lara-Sánchez
b
a,
Andrés
Garcés
*a,
Luis F.
Sánchez-Barba
*a,
David
González-Lizana
a and
Agustín
Lara-Sánchez
b
aDepartamento de Biología y Geología, Física y Química Inorgánica, Universidad Rey Juan Carlos, Móstoles, 28933-Madrid, Spain. E-mail: luisfernando.sanchezbarba@urjc.es; andres.garces@urjc.es
bUniversidad de Castilla-La Mancha, Departamento de Química Inorgánica, Orgánica y Bioquímica- Centro de Innovación en Química Avanzada (ORFEO-CINQA), Campus Universitario, 13071-Ciudad Real, Spain
First published on 29th March 2023
Abstract
The fixation of CO2 mediated by metal-based catalysts for the production of organic molecules of industrial interest such as cyclic carbonates is urgently required under green and eco-friendly conditions. Herein, we describe the easy preparation of sterically demanding scorpionate ligands bearing different electron-withdrawing groups, and their coordination ability for the preparation of robust zinc-based mononuclear complexes of the type [ZnMe(κ3-NNN′)] (4–6). These complexes, in combination with co-catalysts comprising larger ionic radius-based halides such as tetra-n-butylammonium, functioned as very active and selective catalysts for CO2 fixation into five-membered cyclic carbonates. These studies have led to the development of sustainable, inexpensive, and low-toxicity systems formed by 4–5 and Bu4NBr for the cycloaddition of CO2 into epoxides under very mild and solvent-free conditions, reaching very good to excellent conversions (TOF = 260 h−1). Moreover, these bicomponent systems show a broad substrate scope and functional group tolerance, including mono- and di-substituted epoxides, as well as bio-renewable diepoxides. Very interestingly, these are the first zinc-based systems reported to date for the successful transformation of the very challenging tri-substituted terpene-derived cis/trans-limonene oxide, whose reaction proceeds with high stereoselectivity to the formation of the bicyclic trans-limonene carbonate. Additionally, these bicomponents can be efficiently used up to six times without significant loss of activity. Kinetic investigations confirmed that the reaction shows an apparent first-order dependence on the catalyst and co-catalyst concentrations, which indicates an intramolecular monometallic mechanism.
Introduction
Two critical challenges in this century1,2 for the sustainability of our planet, included in the “Twelve Principles of Green Chemistry”,3 are the rational use of natural resources and the efficient management of waste materials.In this context, the valorisation of CO2 as an attractive C-1 renewable building block4–6 is being intensively explored by many research groups in this decade. This unsaturated molecule has high abundance in nature, low cost, non-toxic features, as well as a lack of colour and redox activity. Particularly, this low reactivity molecule finds interesting chemical applications. For instance, the 100% atom-economical production of cyclic carbonates (CCs)7–14 through the cycloaddition of CO2 to epoxides (see Scheme 1) and that of polycarbonates (PCs)15,16via ring opening copolymerization (ROCOP) of CO2 with epoxides are currently two highly competitive areas in the scientific community.
 | ||
| Scheme 1 Cycloaddition of CO2 to epoxides for the preparation of 5-membered cyclic carbonates. | ||
Particularly, CCs have important applications as electrolytes, engineering plastics, solvents, fuel additives, and precursors of fine chemicals.17,18 For this reason, a variety of very active and selective metal-based catalysts have been recently reported, with chromium,19,20 cobalt,21 iron,22–24 magnesium,25,26 zinc27–30 (see Chart 1) or aluminum31–36 as leading metals in this field, with the assistance of a nucleophile as a co-catalyst.
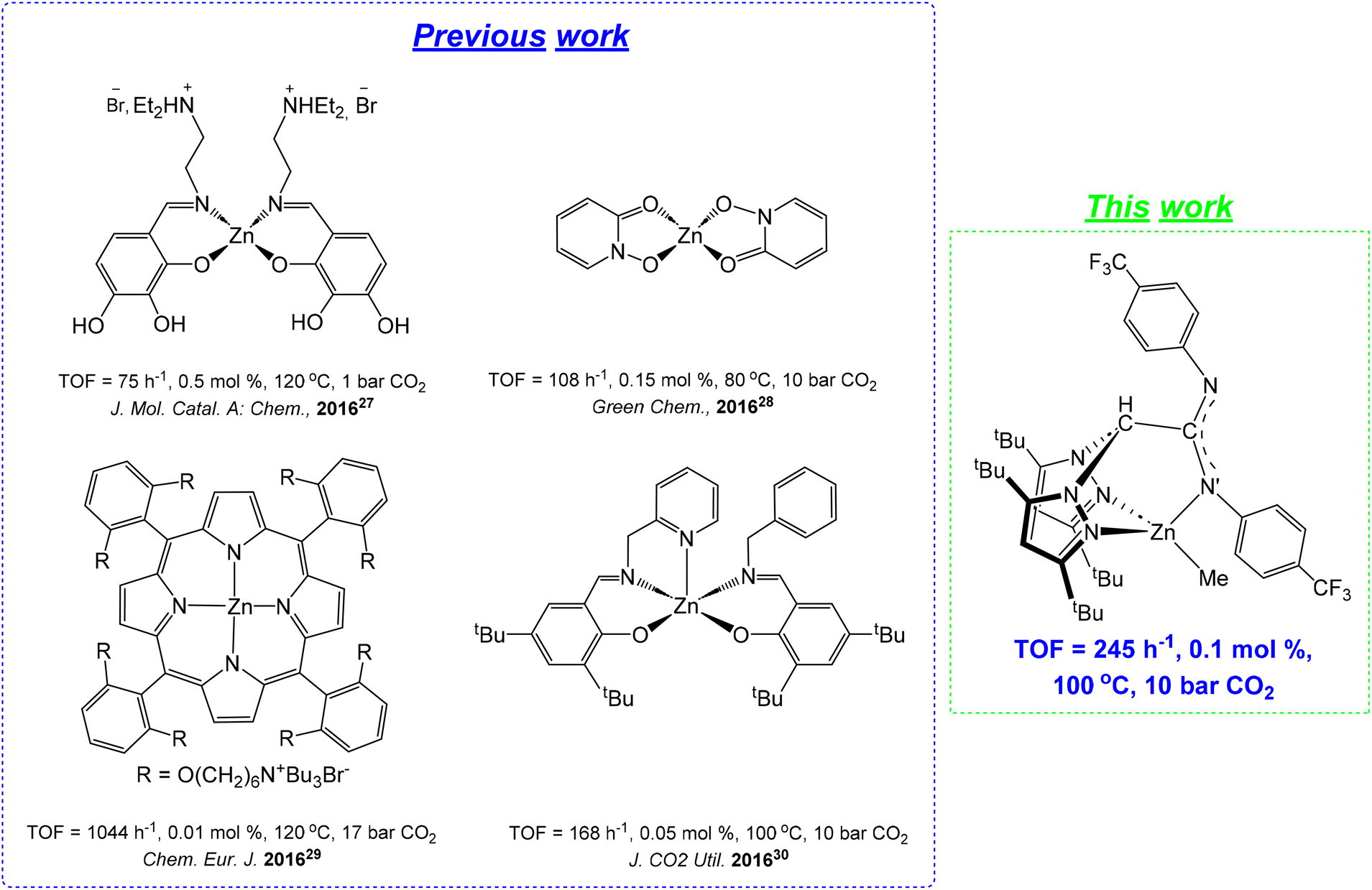 | ||
| Chart 1 Representative zinc-based catalysts for the cycloaddition of CO2 to styrene oxide using bifunctional or bicomponent complex/Bu4NBr systems. | ||
However, the use of abundant, economical and biocompatible metals such as zinc37–39 is highly desirable to make this process much cleaner and greener, and to avoid potential health issues related to the toxicity of several metal-based residues in the isolated materials.40–42
In this sense, recently our research group has also successfully developed efficient zinc-based scorpionate catalysts for cyclic carbonates43 through the cycloaddition of CO2 with a wide range of terminal and internal epoxides, showing broad substrate scope and functional group tolerance under mild and solvent-free conditions.
Nevertheless, the search for robust and efficient zinc-based catalysts with wider substrate scope capable of functioning under much milder conditions in this process still remains poorly explored (see Chart 1). On the basis of our previous expertise,43 now we endeavour the challenging aim to develop novel sustainable, inexpensive, low-toxicity and versatile zinc-based44 systems very efficient in this industrially demanding process. For this purpose, we have successfully developed new sterically hindered acetamidinate-based scorpionates,44,45 containing electron-withdrawing groups as ancillary ligands, as an alternative to others from our extended library.45–47
We report hereby the preparation of a new family of robust mononuclear zinc-based complexes supported by a series of sterically hindered scorpionate ligands with different electronic and steric features, and their detailed assessment as catalysts for efficient CO2 fixation into five-membered cyclic carbonates. These catalysts, in combination with Bu4NBr, exhibit excellent performance and display very broad substrate scope, including terminal, internal and bio-renewable diepoxides and tri-substituted terpene-derived substrates such as limonene oxide.
Results and discussion
Synthesis and characterization of the electron-withdrawing carbodiimide 1 and sterically demanding scorpionate ligands 2–3
The reaction of p-tolylisocyanate, p-trifluoromethylphenylisocyanate and 3,5-bis(trifluoromethyl)phenylisocyanate, in the presence 3-methyl-1-phenyl-2-phospholene 1-oxide (MPPO) as the catalyst,48 at 0 °C afforded bis-p-tolylcarbodiimide,48 bis(p-trifluoromethylphenyl)carbodiimide48 and new bis(3,5-ditrifluoromethylphenyl)carbodiimide (1), respectively, as pale yellow or white solids in high yields (>90%) (see Scheme 2a).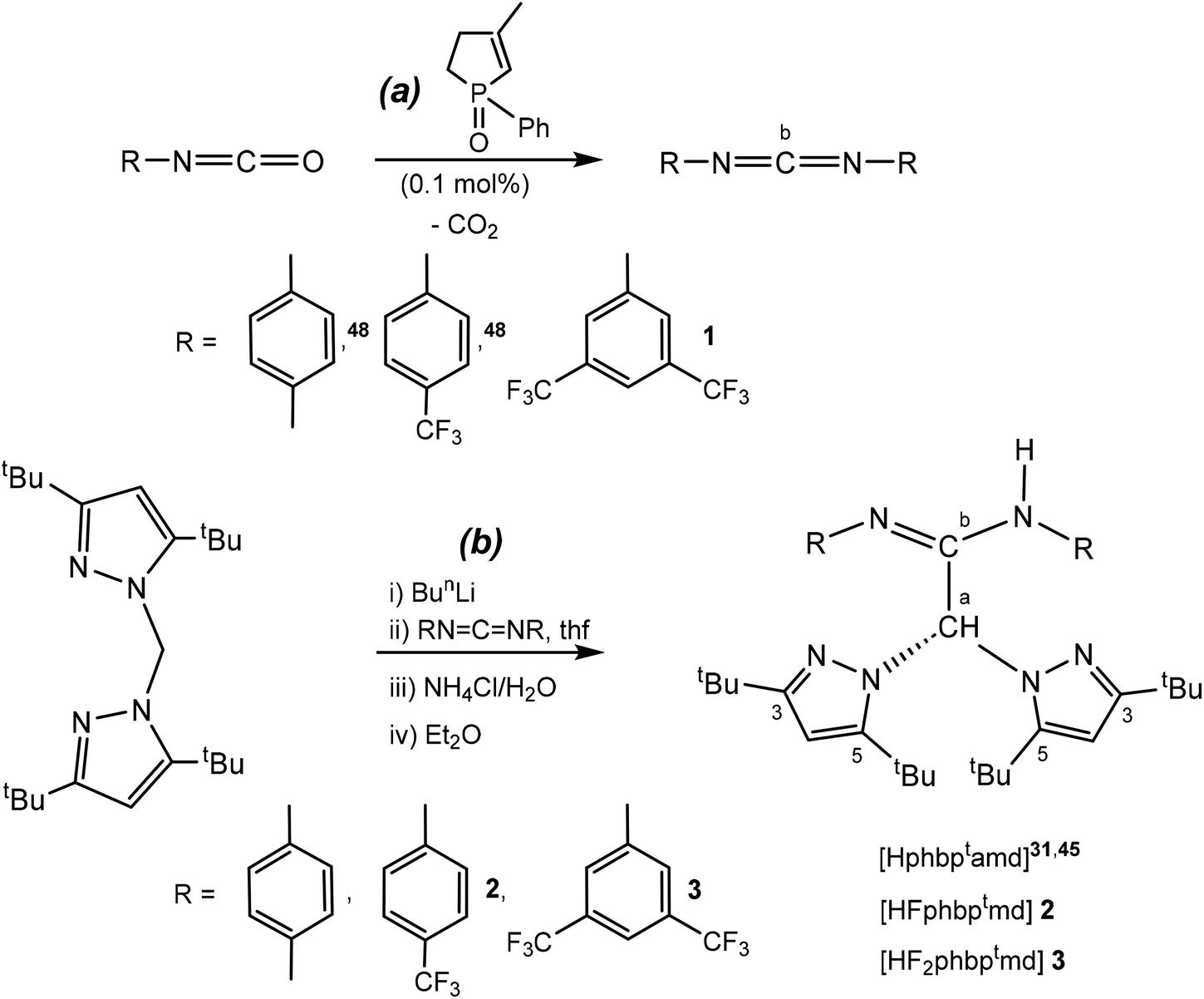 | ||
| Scheme 2 Preparation of carbodiimide 1 and the sterically demanding scorpionate acetamidine-based protioligands 2–3. | ||
In a second step, a mixture of a cooled (−70 °C) solution of bis(3,5-di-tert-butylpyrazol-1-yl)methane (bdtbpzm) in THF and 1 equiv. of BunLi, was treated with the carbodiimides bis-p-tolylcarbodiimide,48 bis(p-trifluoromethylphenyl)carbodiimide48 and 1, respectively, and subsequently hydrolysed with NH4Cl/H2O in diethyl ether to finally give rise to the corresponding amidine protioligands Hphbptamd,45 HFphbptamd (2) [HFphbptamd = N,N′-di-p-trifluoromethyphenylbis(3,5-di-tert-butylpyrazol-1-yl)acetamidine] and HF2phbptamd (3) [HF2phbptamd = N,N′-bis(3,5-ditrifluoromethyl)phenylbis(3,5-di-tert-butylpyrazol-1-yl)acetamidine] in very good yields (>90%) (see Scheme 2b).
The 1H and 13C{1H} NMR spectra of the new carbodiimide 1 in chloroform-d at room temperature (see Fig. S1 in the ESI‡) display a single set of resonances indicating the symmetry of the molecule, with a characteristic signal in a very low field, corresponding to the sp carbon (Cb, ∼140 ppm), indicating a highly electrophilic centre (see Scheme 2a). In addition, the 1H and 13C{1H} NMR spectra of the amidine heteroscorpionate compounds 2 and 3 in benzene-d6 at room temperature (see Fig. S2 and S3 in the ESI‡) show a single set of resonances for the pyrazolyl rings, indicating that both rings are equivalent and two set of resonances for the substituents in the amidine fragment (see Scheme 2b). The structures proposed for compounds 2 and 3 were further verified by X-ray diffraction studies (see below Fig. 1).
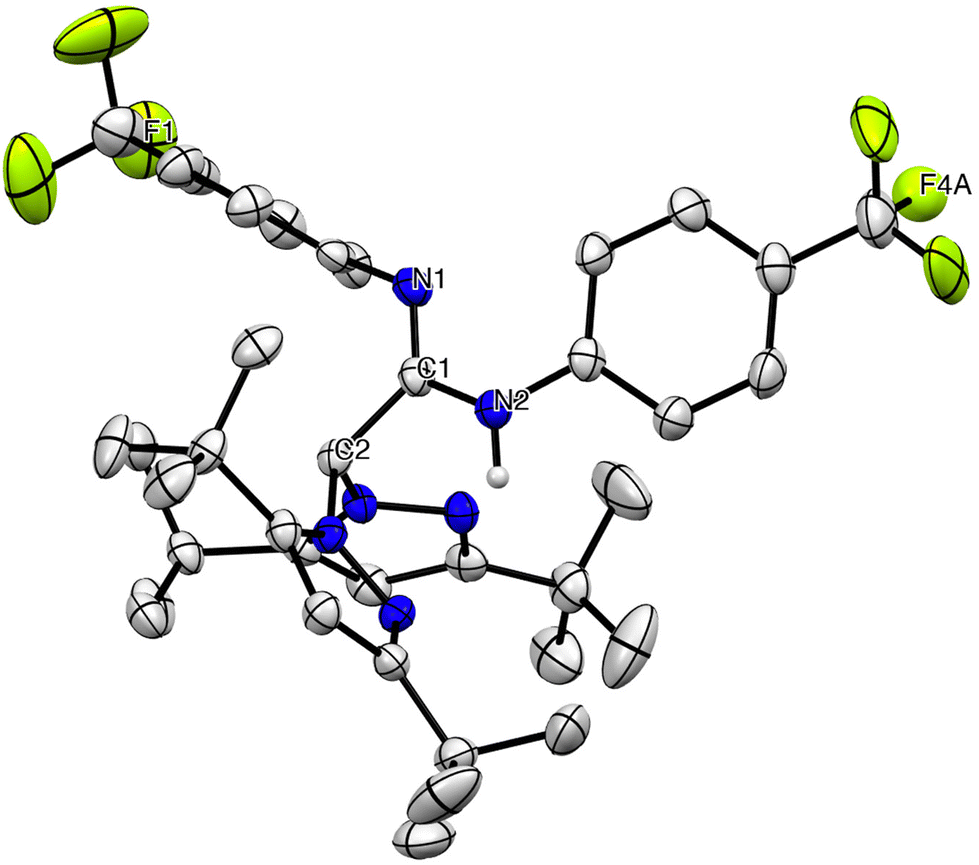 | ||
| Fig. 1 ORTEP view of [HFphbptamd] (2). Hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at the 30% probability level. | ||
Synthesis and characterization of scorpionate alkyl zinc complexes 4–6
The reaction of the sterically demanding acetamidine-based scorpionate protioligands Hphbptamd,45 HFphbptamd (2) and HF2phbptamd (3) with one equivalent of ZnMe2, efficiently afforded the mononuclear scorpionate zinc alkyl complexes of type [ZnMe(κ3-NNN′)], (κ3-NNN′ = phbptamd 4,44 Fphbptamd 5, and F2phbptamd 6), in very high yields (>85%) (see Scheme 3). All complexes have low air-sensitivity, tolerate up to 5 hours of air exposure, and can be dissolved in chlorinated solvents as CDCl3 during 6 h without hydrolysis. This robustness makes them ideal for application in this catalytic process. | ||
Scheme 3 Preparation of the acetamidinate-based NNN′-scorpionate zinc complexes 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 44 and 5–6. 44 and 5–6. | ||
The 1H and 13C{1H} NMR spectra of the new zinc complexes 5 and 6 in benzene-d6 at room temperature (see Fig. S4 and S5 in the ESI‡ display a single set of resonances for the two pyrazoles, indicating that both rings are equivalent, and two sets of resonances for the amidinate substituents, showing a monodentate coordination mode to the metal. These data confirm a tetrahedral disposition of the zinc atom with NNN′-coordination for the scorpionate ligand, where a plane of symmetry exists and contains the acetamidinate group, the zinc metal and the methyl ligand (see Scheme 3). 1H NOESY-1D experiments were also performed in order to confirm the assignment of the signals to the tBu3, tBu5, and H4 groups. Furthermore, 1H-13C heteronuclear correlation (gHSQC) experiments were carried out and allowed us to assign the resonances corresponding to C4, tBu3, and tBu5 of the pyrazole rings.
The structures proposed for complexes 5 and 6 were further confirmed by X-ray molecular analysis (see below Fig. 2).
 | ||
| Fig. 2 ORTEP view of [ZnMe(κ3-Fphbptamd)] (5). Hydrogen atoms are omitted for clarity. Thermal ellipsoids are drawn at the 30% probability level. | ||
X-ray diffraction studies
Single crystals of compound 2 and complex 5 suitable for X-ray diffraction analysis were easily grown from a toluene solution at −26 °C. The molecular structures are depicted in Fig. 1 and 2, respectively. Selected bond lengths and angles are collected in Table 1, and the crystallographic details are reported in Table S1 in the ESI.‡ The molecular structure of both compounds consists of a monomeric arrangement in the solid state. Compound 2 shows a C(1)–N(1) distance [1.266(8) Å] typical of C–N double bonds, as in Schiff bases and oximes (ca. 1.26 Å), whereas the C(1)–N(2) (1.374(8) Å) bond length is typical of a C–N single bond. The hydrogen atom is located on the N(2) atom. The essentially sp2 hybridization and planar nature of the imino carbon atom C(2) are further confirmed by the summations of the angles (∼120°) around it (360°).| HFphbptamd (2) | |||||||
|---|---|---|---|---|---|---|---|
| 2a | 2b | ||||||
| Distances (Å) | Angles (°) | Distances (Å) | Angles (°) | ||||
| C(1)–N(1) | 1.266(8) | N(2)–C(1)–N(1) | 122.61(18) | C(39)–N(7) | 1.273(8) | N(8)–C(39)–N(7) | 122.85(18) |
| C(1)–N(2) | 1.374(8) | N(2)–C(1)–C(2) | 113.85(18) | C(39)–N(8) | 1.368(8) | N(8)–C(39)–C(40) | 113.10(18) |
| C(1)–C(2) | 1.542(8) | C(2)–C(1)–N(1) | 123.53(6) | C(39)–C(40) | 1.542(8) | C(40)–C(39)–N(7) | 123.91(6) |
| [ZnMe(κ3-Fphbptamd)] (5) | |||
|---|---|---|---|
| Distances (Å) | Angles (°) | ||
| N(1)–Zn(1) | 2.038(5) | C(39)–Zn(1)–N(1) | 126.60(20) |
| N(4)–Zn(1) | 2.104(5) | C(39)–Zn(1)–N(4) | 129.86(20) |
| N(6)–Zn(1) | 2.182(5) | C(39)–Zn(1)–N(6) | 122.65(20) |
| C(39)–Zn(1) | 1.967(7) | N(4)–Zn(1)–N(6) | 81.86(20) |
| C(1)–N(1) | 1.341(8) | N(4)–Zn(1)–N(1) | 89.85(18) |
| C(1)–N(2) | 1.299(8) | N(6)–Zn(1)–N(1) | 92.99(18) |
| C(1)–C(2) | 1.525(8) | N(1)–C(1)–N(2) | 134.66(6) |
| N(1)–C(1)–C(2) | 114.14(4) | ||
| N(2)–C(1)–C(2) | 111.19(4) | ||
On the other hand, the zinc metal in complex 5 exhibits a distorted tetrahedral geometry, with the scorpionate ligand in a κ3-NNN′ coordination mode. The N(4)–Zn and N(6)–Zn bond lengths [2.104(5) Å and 2.182(5) Å, respectively] are balanced and compared well with those observed in the analogous acetamidinate-based scorpionate magnesium49 and zinc44 alkyls, but are considerably longer than the N(1)–Zn bond length [2.038(5) Å]. The solid-state structure also confirms that the acetamidinate is coordinated in a monodentate fashion with the Zn atom, and delocalisation is also evidenced in the N–C–N moiety of the acetamidinate, with the bond lengths C(1)–N(1) and C(1)–N(2) ranging from 1.341(8) Å to 1.299(8) Å. In addition, the C(1)–C(2) bond lengths in complex 5 and in ligand 2 [1.525(8) Å and 1.542(8) Å, respectively] are consistent with a C–C single bond (∼1.455 Å). Finally, the Zn–Me bond distance is also in agreement with that of analogous alkyl derivatives [C(39)–Zn(1) = 1.967(7) Å].44,49
Studies on the catalytic cycloaddition of CO2 to epoxides using complexes [ZnMe(κ3-phbptamd)] (4), [ZnMe(κ3-Fphbptamd)] (5) and [ZnMe(κ3-F2phbptamd)] (6)
Complexes 4–6 were used as freshly prepared materials and firstly screened as catalysts for the formation of styrene carbonate 8a by the coupling reaction of CO2 with styrene oxide 7a as a benchmark reaction (see Scheme 4). The process was initially assessed at 25 °C and 10 bar CO2 pressure and under solvent free conditions for 18 hours in a 1:1 molar ratio, using a catalyst loading of 5 mol% in the presence of tetrabutylammonium bromide (Bu4NBr or TBAB). The results are presented in Table 2.
 | ||
| Scheme 4 Cyclic carbonate synthesis catalysed by complexes 4–6. | ||
a
| Entry | Catalyst | [Cat]:[co-cat] |
Conversion [%] | |
|---|---|---|---|---|
| [mol%] | 25b °C | 50b °C (TOF, h−1)c | ||
| a Reactions carried out at 10 bar CO2 pressure during 18 h, using 5 mol% of complexes 4–6/5 mol% of TBAB as the co-catalyst unless specified otherwise. b Determined by 1H NMR spectroscopy of the crude reaction mixture. c TOF (turnover frequency) = number of moles of styrene oxide consumed/(moles of catalyst × time of reaction). d Reaction carried out during 8 h. e Reaction carried out at 100 °C during 4 h. f Not determined. | ||||
| 1 | 4 | 5.0:5.0 |
85 | ndf |
| 2 | 5 | 5.0:5.0 |
95 | nd |
| 3 | 6 | 5.0:5.0 |
76 | nd |
| 4 | 5 | 5.0:5.0 (TBAF) |
7 | nd |
| 5 | 5 | 5.0:5.0 (TBAC) |
54 | nd |
| 6 | 5 | 5.0:5.0 (TBAI) |
59 | nd |
| 7 | 5 | 5.0:5.0 (NMI) |
17 | nd |
| 8 | 5 | 5.0:5.0 (DMAP) |
6 | nd |
| 9 | 5 | 2.5:2.5 |
72 | 100 |
| 10 | 5 | 1.5:1.5 |
51 | 100 |
| 11 | 5 | 1.0:1.0 |
30 | 100 |
| 12 | 5 | 0.5:0.5 |
15 | 100 |
| 13 | 5 | 0.2:0.2 |
nd | 100 (28) |
| 14 |
5d |
0.2:0.2 |
nd | 75 (47) |
| 15 | 5 | 0.2:0 |
nd | 0 |
| 16 | — | 0:0.2 |
2 | 5 |
| 17 | HFphbp t amd | 0.2:0.2 |
nd | 4 |
| 18 |
5e |
0.1:0.1 |
nd | 98 (245) |
Styrene oxide 7a conversion into styrene carbonate 8a was determined by 1H NMR without any further purification (see Fig. S6 in the ESI‡). Not surprisingly, the formation of styrene polycarbonate was not detected under the aforementioned conditions (selectivity >99%). Complex 5 displayed very high catalytic activity with almost complete conversion for the synthesis of 8a, while derivative 4 showed lower conversion under identical experimental conditions, possibly due to the presence of the two electron-withdrawing groups in the amidinate fragment in 5, which increase the Lewis acidity of the zinc metal centre and promote the initial epoxide coordination and further enhance the catalytic performance, as a result. In the case of 6, this higher activation effect in the metal centre, given the existence of two additional electron-attracting groups, is partially counteracted by the greater steric environment of this ligand, resulting in a lower conversion than 4 and 5 (Table 2, entries 1–3). Therefore, we selected complex 5 as the most efficient catalyst for further cycloaddition reactions under these experimental conditions.
The effect of halide counter ions on the catalyst system was next inspected for complex 5 by employing different onium salts at 25 °C and 10 bar CO2 pressure for 18 hours employing this catalyst:co-catalyst loading. Interestingly, whereas the fluoride counter ion led to lower catalytic activity than the chloride and iodide counter ions, the bromide anion displayed the highest activity (Table 2, entries 1 and 4–6), indicating that this counter ion performs as both a good nucleophile to ring-open the epoxide and a good leaving group for cyclic carbonate formation. Furthermore, 1-methylimidazole (NMI) and 4-dimethylaminopydine (DMAP) were also assessed as co-catalysts, resulting in poorly active systems (Table 2, entries 7 and 8, respectively). Therefore, we identified Bu4NBr as the most efficient co-catalyst for complex 5 under these reaction conditions.
Catalyst and co-catalyst loadings were also inspected at 25 °C and 50 °C, and they could be reduced down to 0.2 mol% to reach complete conversion in 18 hours at 50 °C (Table 2, entries 9–13); therefore, it was identified as the optimal loading for the bicomponent system 5/Bu4NBr under these experimental conditions for further catalytic studies. Interestingly, very high conversion (75%) was reached at 50 °C employing a combination of 0.2 mol% of complex 5 and 0.2 mol% of Bu4NBr at 10 bar CO2 pressure after only 8 hours (Table 2, entry 14).
Consistently, a control experiment for 5 in the absence of Bu4NBr revealed no catalytic activity, whereas the use of Bu4NBr without the presence of 5 produced nearly zero conversion (2%) at 25 °C and minimal conversion (5%) at 50 °C, respectively, using 10 bar CO2 pressure after 18 h of reaction, confirming the necessity of both catalytic components in the reaction mixture to succeed in this cycloaddition process. Also, the corresponding ancillary sterically hindered protioligand, HFphbptamd in complex 5, displayed very poor conversion in the presence of Bu4NBr (4%) under otherwise identical conditions (Table 2, entries 15–17).
It is also worth noting that under these conditions, the bicomponent system 5/Bu4NBr was much more active (Table 2, entry 14, TOF = 37 h−1) than the NNO′-scorpionate zinc-based bicomponent mononuclear (TOF = 2.33 h−1), the dinuclear (TOF = 3.0 h−1) and the bifunctional (TOF = 2.9 h−1) analogs also under mild conditions (50 °C), previously reported by our group.43 In addition, this bicomponent system can operate more efficiently for the production of styrene carbonate 8a under softer and comparable experimental conditions (Table 2, entry 14, TOF = 47 h−1, 50 °C and entry 18, TOF = 245 h−1, 100 °C, respectively) than very well-performed zinc-based catalysts previously reported.27–30
In view of the promising results achieved by 5/TBAB, a variety of terminal substrates such as alkyl, aryl and functionalized terminal epoxides 7b–7j were additionally assessed using this bicomponent system (see Scheme 4), at 50 °C and 10 bar CO2 pressure, with 0.2 mol% of catalyst/co-catalyst loading in a 1:1 molar ratio under solvent free conditions (see Fig. S7–S15 in the ESI‡). Notably, under these conditions, very good to excellent conversions were achieved in 12 hours, including those substrates bearing alcohol or ether functionalities with phenyl or alkyl chains (see Fig. 3).
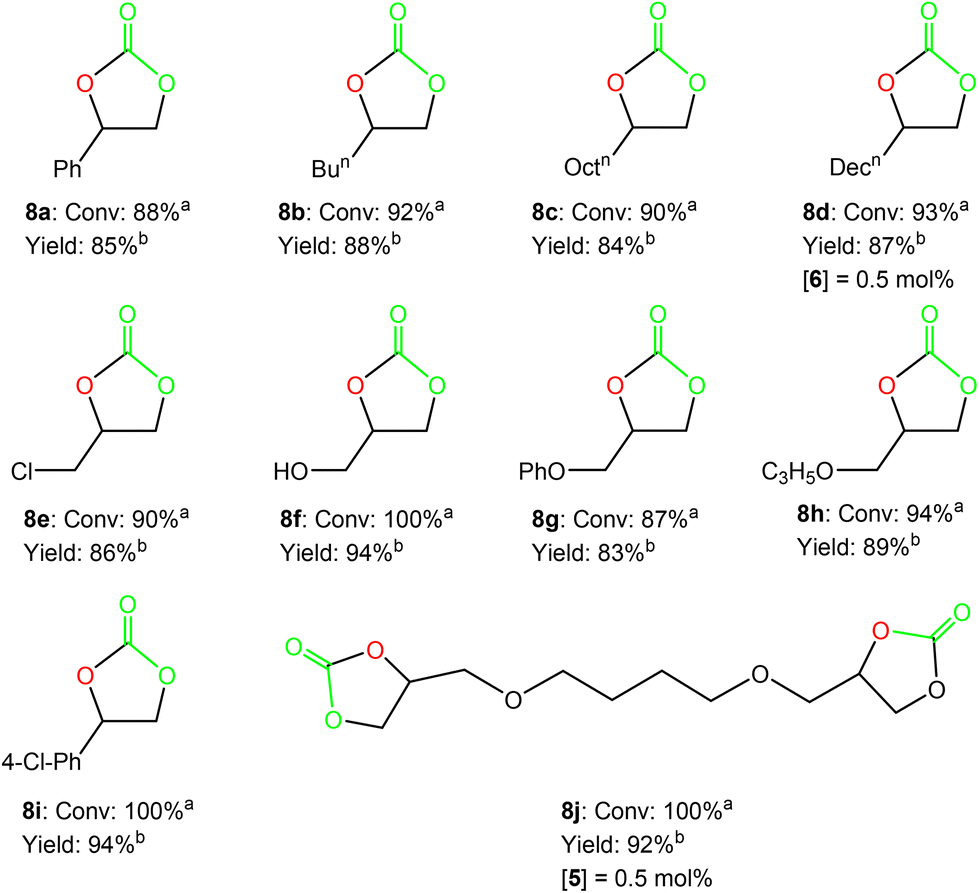 | ||
| Fig. 3 Synthesis of cyclic carbonates 8a–8j from epoxides 7a–7j using 0.2 mol% of the bicomponent system formed by complex 5/TBAB at 50 °C and 10 bar CO2 pressure for 12 hours, unless specified otherwise. aConversion and selectivity were determined by 1H NMR. bIsolated yield after column chromatography. | ||
Considering the high activity displayed by the bicomponent system 5/TBAB, we additionally extended the substrate scope to catalyst 5, and assessed the conversion of internal epoxides 9a–9b, and bio-based derived substrates 11a–11d, into the corresponding cyclic carbonates 10a–10b and 12a–12d, respectively (see Fig. 4). Important progress has been reported in very recent years employing Fe(II)-,50,51 Ca(II)-52 and Al(III)-31,53,54 based catalyst systems, despite the lower reactivity of these epoxides.55,56 However, a few examples of Zn(II)-based complexes43,57 have been reported for the efficient and selective synthesis of these cyclic carbonates (5 mol%, 20 bar of CO2, 80 °C, 24 h),43,55,56 but not under current milder conditions and using these lower catalyst/cocatalyst loadings (see Fig. 4).
 | ||
| Fig. 4 Synthesis of cyclic carbonates 10a–10b and 12a–12d from epoxides 9a–9b and 11a–11d, respectively, using equimolecular amounts (0.2 mol%) of the system 5/TBAB at 50 °C and 10 bar CO2 pressure for 18 hours, unless specified otherwise. aConversion and selectivity were determined by 1H NMR. bIsolated yield after column chromatography. | ||
Thus, we increased the reaction temperature at 70 °C and catalyst:co-catalyst loadings up to 0.5 and 1 mol% for cyclohexene oxide (CHO) 9a and cyclopentene oxide (CPO) 9b, respectively, but maintained 10 bar CO2 pressure. To our delight, a 1:1 proportion of the binary system 5/TBAB displayed excellent activities (conv. >97%) for both internal substrates in 18 hours, under these mild and solvent-free conditions (see Fig. 4), showing the efficiency of this system. The NMR spectra of cyclic carbonates 10a–10b confirmed the retention in the epoxide stereochemistry, as only the cis-isomer was formed for cyclohexene carbonate58,59 with a selectivity higher than 99%, and for cyclopentene carbonate,60–62 as in the last case it was the only stereoisomer thermodynamically permitted (Fig. S16 and S17, respectively, in the ESI‡).
In addition, we were interested in the synthesis of bioresourced cyclic carbonates 12a–12d, considering their potential as non-toxic feedstocks to produce NIPUs (non-isocyanate poly(hydroxy)urethanes).63,64 Therefore, we initially explored the synthesis of the bio-based furan-derived cyclic carbonate 12a, and complete conversion was obtained after 18 hours at 50 °C and 10 bar CO2 pressure, using only 0.2 mol% loading of the 5/TBAB system. Encouraged by these results, we finally decided to extend this study to transform other bio-based diepoxide derivatives that include the fumaryl, succinyl and glutaryl platforms, 11b–11d. We were also pleased to find that cyclic carbonates 12b–12d were obtained in almost complete yields under identical conditions using only 0.2 mol% of this bicomponent system (see Fig. 4 and Fig. S18–S21 in the ESI‡).
Very interestingly, we investigated the production of another bio-renewable cyclic carbonate namely limonene carbonate 14 obtained from limonene oxide 13, a highly substituted monocyclic unsaturated terpene derived from biomass,65,66 (extracted from the peel of citrus waste) (see Scheme 5). It is worth noting that a few metal-based catalysts have been reported for the successful cycloaddition reaction of CO2 to limonene oxide (LO),31,32,54,67–69 and, as far as we are aware, no examples of well-defined zinc-based catalysts70 that can operate under mild conditions have been reported to date.
 | ||
| Scheme 5 Synthesis of cyclic carbonate 14 from (R)-(+)-limonene oxide using a combination of 0.5 mol% of complex 4/5 and 1.5 mol% TBAC at 70 °C and 10 bar CO2 pressure for 72 hours. | ||
Thus, we investigated the bicomponent system formed by a combination of complex 5 and TBAB in a very low 0.5:1.5 catalyst:co-catalyst loading ratio for the preparation of bicyclic limonene carbonate 14 (see Table 3), employing commercially available limonene oxide 13 as a mixture of cis/trans isomers (43:57), under 10 bar CO2 pressure and at 70 °C temperature. Expectedly, a rather low conversion was reached after 72 h of reaction, in accordance with the lower reactivity and the higher steric hindrance of this tri-substituted epoxide 13, and also in agreement with our previous findings with this challenging substrate.31,32 Importantly, the use of TBAC as the co-catalyst did not allow any progress on the catalytic activity, which indicates that the higher steric demand of the ligand in complex 5 counters the possible enhanced catalytic performance as a result of the greater Lewis acidity of the metal centre with this ligand, as mentioned above (Table 3, entries 1 and 2). More interestingly, bicomponent 4/TBAC allowed this cycloaddition reaction proceeding with both high yield and stereoselectivity to the bicyclic trans-limonene carbonate, affording a 39% of conversion after 72 h, possibly due to the lower steric demand of this ligand (Table 3, entry 3), (see Fig. S22 and S23 in the ESI‡). Therefore, we selected complex 4 as the most efficient catalyst against this challenging substrate. Increasing of the catalyst:co-catalyst loadings did not produce a relevant beneficial effect (Table 3, entry 4).
| Entry | Catalyst | [Cat]/[TBAC] (mol%) | P(CO2)(bar) | T (°C) | Time (h) | Conv.a (%) | Yieldb (%) (dr, trans) |
|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR spectroscopy of the crude reaction mixture. b Isolated yield after column chromatography. | |||||||
| 1 | 5 | 0.5:1.5 (TBAB) |
10 | 70 | 72 | 9 | nd |
| 2 | 5 | 0.5:1.5 |
10 | 70 | 72 | 10 | nd |
| 3 | 4 | 0.5:1.5 |
10 | 70 | 72 | 39 | 34 (1:99) |
| 4 | 4 | 1:3 |
10 | 70 | 72 | 43 | 38 (1:99) |
| 5 | 4 | 0:1.5 |
10 | 70 | 72 | 7 | nd |
| 6 | 4 | 0.5:1.5 (PPNCl) |
10 | 70 | 72 | 4 | nd |
| 7 | 4 | 0.5:1.5 (TBAI) |
10 | 70 | 72 | 6 | 9 (1:99) |
| 8 | 4 | 0.5:1.5 |
10 | 100 | 72 | 35 | 31 (6:94) |
| 9 | 4 | 0.5:1.5 |
20 | 70 | 72 | 34 | 30 (1:99) |
A control experiment using only TBAC produced a poor 7% of conversion under otherwise identical conditions (Table 3, entry 5). In addition, the employment of bis(triphenylphosphoranylidene)ammonium chloride (PPNCl) as the co-catalyst under equal conditions produced a drastic loss of activity (4% conv.), possibly as a consequence of the lower co-catalyst solubility in this terpene-derived epoxide (Table 3, entry 6). As expected, the use of co-catalysts comprising larger ionic radius-based halides such as TBAI, also produced a detrimental effect on conversion (Table 3, entry 7). Unexpectedly, neither the increase of the reaction temperature up to 100 °C nor the CO2 pressure up to 20 bar did produce a significant change on conversion, with the important loss of reaction stereoselectivity with temperature (Table 3, entries 8 and 9).
Multi-feed studies of catalyst 5 with propylene oxide 15
Finally, we turned our attention to the usability of these catalytic systems. Thus, a combination of 0.16 mol% of complex 5 and 0.16 mol% of TBAB as the co-catalyst displayed an excellent activity (83%) for the cycloaddition of CO2 and propylene oxide (PO) 15 into cyclic propylene carbonate 16 at 100 °C and 10 bar CO2 pressure after 2 h (TON = 520; TOF = 260 h−1) (see Fig. S24 in the ESI‡).Very interestingly, this developed binary catalytic system could be used in up to six successive multi-feed experiments under these conditions (see Experimental details in the ESI‡), without a significant loss in catalytic activity (see Fig. 5). Very few examples of organo-zinc catalysts have been reported for the efficient cycloaddition of CO2 with terminal epoxides under multi-feed conditions;29 however, as far as we know, no examples of zinc-based catalysts supported by scorpionate ligands have been described until now.
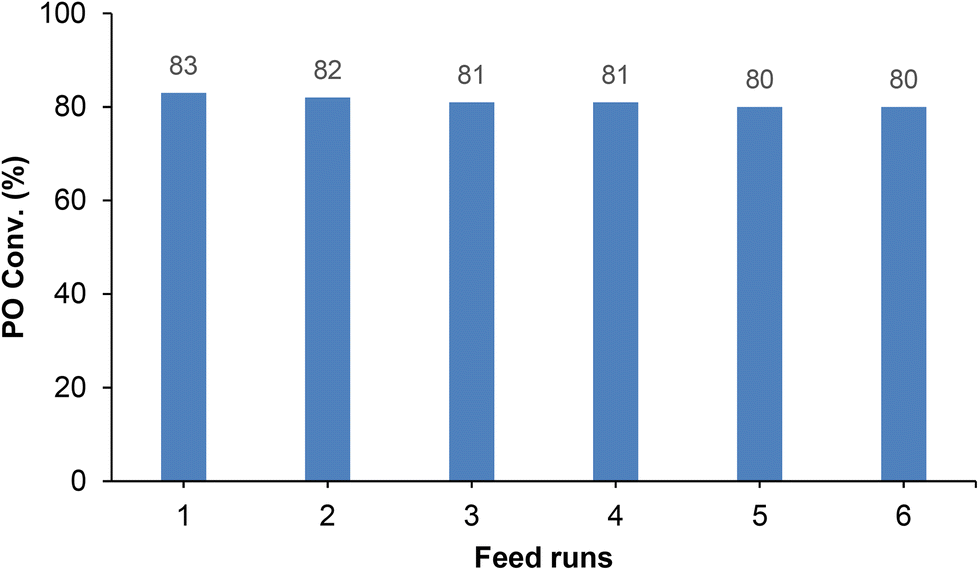 | ||
| Fig. 5 Multi-feed experiments for the cycloaddition of CO2 and propylene oxide (PO) 15 into cyclic propylene carbonate 16, using equimolecular amounts (0.16 mol%) of the bicomponent system 5/TBAB at 100 °C and 10 bar CO2 pressure for 2 hours. | ||
Kinetic and mechanism investigations
With the aim to understand how the [ZnMe(κ3-NNN′)] (4–6)/TBAB systems operate during the catalytic process and to elucidate a plausible reaction mechanism, a series of kinetic studies were conducted employing the bicomponent 4/TBAB.Initially, an experiment was performed at 40 °C and 1 bar CO2 with 8.77 M in styrene oxide (SO, 7a) in the presence of an equimolecular combination of complex 4 as the catalyst and TBAB as the co-catalyst at a concentration of 0.351 M in order to determine the reaction order with respect to the epoxide. Aliquots were taken at regular time intervals until almost full conversion was reached (ca. 95%). As shown in Fig. 6, the plot of ln[7a] vs. time clearly exhibited a linear correlation, indicating a first-order dependence of the reaction rate with [7a].
 | ||
| Fig. 6 Plot of ln[7a] vs. time (h) showing a linear fit. [7a]0 = 8.77 M, [4] = [TBAB] = 0.351 M at 40 °C, 1 bar CO2, up to 95% conversion. | ||
Accordingly, the general rate equation for this reaction, shown in eqn (1), can be rewritten in the form of eqn (2) as follows:
| Rate = k1[7a]a[CO2]b[4]c[TBAB]d | (1) |
| Rate = k1,obs.[7a], where k1,obs. = k1[CO2]b[4]c[TBAB]d | (2) |
Additional kinetic measurements were performed at early stages of the reaction in order to determine the reaction order with respect to the catalyst and co-catalyst concentrations. Under these conditions, the general rate law formula, expressed by eqn (1), can be simplified to:
| Rate0 = k0,obs.[4]c[TBAB]d, where k0,obs = [7a]a[CO2]b | (3) |
That simplification is based on the fact that during the early stages of the reaction (between 5% and 20% conversion) both [CO2] and [7a] may be considered pseudo-constant. Employing eqn (3), we can estimate the initial rate of the reaction at different catalyst and co-catalyst concentrations. By keeping one of them constant, it will be possible to determine the reaction order with respect to the other species (see complete analysis of kinetic experiments in Fig. S25 and Tables S2 in the ESI‡). Thus, kinetics experiments allowed one to demonstrate apparent first-order with respect to both catalyst [4] and co-catalyst [TBAB].
Finally, considering that the systems 4–6/TBAB were very active in the synthesis of cyclic carbonates 8a–8j, 10a–10b, 12a–12d and 16 with the retention of the epoxide stereochemistry, a plausible mechanism for cyclic carbonate production catalysed by these bicomponent zinc-based systems is presented in Fig. 7. The reaction could be followed on CDCl3 at 50 °C for 3 hours considering the robustness of complex 4 in this chlorinated polar solvent. This mechanism follows a monometallic binary pathway, in agreement with the kinetic investigations employing 4/TBAB and 7a described above. This behaviour is similar to that previously proposed for analog mononuclear NNO′-scorpionate zinc complexes used for coupling CO2 and epoxides into cyclic carbonates43 reported by our group. The proposal is consistent with the initial coordination of the epoxide to the zinc centre, with the expansion of the coordination sphere,71 subsequent nucleophilic attack of the bromide to the less sterically hindered carbon atom of the epoxide, CO2 insertion into the Zn–O bond, and final ring-closure of the cyclic carbonate with stereochemistry retention (see Fig. S26 in the ESI‡).
 | ||
| Fig. 7 Plausible mechanism for the conversion of epoxides and CO2 into cyclic carbonates catalysed by the bicomponent system 4/TBAB. | ||
Conclusions
Herein, we describe the easy preparation of sterically hindered scorpionate ligands containing different electron-withdrawing groups, and their utility in the preparation of robust zinc-based mononuclear complexes of the type [ZnMe(κ3-NNN′)] (4–6). These complexes, in combination with tetra-n-butylammonium bromide/chloride, behave as highly efficient and selective systems for the cycloaddition of CO2 to epoxides into five-membered cyclic carbonates.Very interestingly, these bicomponent systems formed by 4–6/TBAB showed very broad substrate scope and functional group tolerance, including not only terminal and internal epoxides but also bio-renewable diepoxides, and terpene-derived tri-substituted substrates such as limonene oxide, under very mild and solvent-free conditions, achieving TOF values up to 260 h−1. As far as we know, these are the first zinc-based systems that successfully transform the biomass-derived limonene oxide to the bicyclic trans-limonene carbonate with high stereoselectivity. Interestingly, these successful systems were suitable for up to six-times feed experiments without apparent loss of activity. Kinetic investigations at early stages of the reaction confirmed an apparent first-order dependence on the catalyst and co-catalyst concentrations, in agreement with an intramolecular monometallic binary pathway mechanism. The effect of these sterically hindered scorpionate ligands on the zinc metal centre, which compromises the right balance between electronic and steric properties in complexes 4–5, and suppresses the symmetrical equilibrium,72 is possibly responsible for such good catalytic performances.
Although several zinc-based catalytic systems have been reported for CO2 fixation into 5-membered cyclic carbonates in the last few years,27–30 we consider that these results represent an important further step forward in the search of more sustainable, inexpensive, and low-toxicity metal-based catalysts capable of functioning efficiently under mild conditions and at very low catalyst loadings for the transformation of a wide range of challenging bio-resourced substrates in this industrially demanding process.
Author contributions
M. N., A. G. and L. F. S–B. carried out the synthesis and characterisation of the complexes. A. G. and L. F. S–B. wrote the manuscript. M. N. and D. G.-L. performed the catalytic studies. A. L.-S. supervised all results. All authors helped in the conceptualization and critical reading of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge the financial support: grant PID2020-117788RB-I00 funded by Ministerio de Ciencia e Innovación, grant CTQ2017-84131-R funded by Ministerio de Ciencia e Innovación and grant SBPLY/21/180501/000132 funded by Junta de Castilla-La Mancha.References
- J. Amouroux, P. Siffert, J. P. Massué, S. Cavadias, B. Trujillo, K. Hashimoto, P. Rutberg, S. Dresvin and X. Wang, Prog. Nat. Sci.: Mater. Int., 2014, 24, 295–304 CrossRef CAS.
- E. MacArthur, Completing the picture: How the circular economy tackles climate change, 2019, https://ellenmacarthurfoundation.org/completing-the-picture, (Accessed 15/01/2023).
- P. T. Anastas and J. C. Warner, Green chemistry: theory and practice, Oxford University Press, Oxford [England], New York, 1998 Search PubMed.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21, 406–448 RSC.
- A. W. Kleij, M. North and A. Urakawa, ChemSusChem, 2017, 10, 1036–1038 CrossRef CAS PubMed.
- H. Büttner, L. Longwitz, J. Steinbauer, C. Wulf and T. Werner, Top. Curr. Chem., 2017, 375, 50 CrossRef PubMed.
- L. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC.
- V. Aomchad, À. Cristòfol, F. Della Monica, B. Limburg, V. D'Elia and A. W. Kleij, Green Chem., 2021, 23, 1077–1113 RSC.
- D. Prasad, K. N. Patil, N. K. Chaudhari, H. Kim, B. M. Nagaraja and A. H. Jadhav, Catal. Rev., 2022, 64, 356–443 CrossRef CAS.
- M. Liu, X. Wang, Y. Jiang, J. Sun and M. Arai, Catal. Rev., 2019, 61, 214–269 CrossRef CAS.
- R. R. Shaikh, S. Pornpraprom and V. D'Elia, ACS Catal., 2018, 8, 419–450 CrossRef CAS.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- C. M. Kozak, K. Ambrose and T. S. Anderson, Coord. Chem. Rev., 2018, 376, 565–587 CrossRef CAS.
- G. Trott, P. K. Saini and C. K. Williams, Philos. Trans. R. Soc., A, 2016, 374, 20150085 CrossRef PubMed.
- W. Guo, J. E. Gómez, À. Cristòfol, J. Xie and A. W. Kleij, Angew. Chem., Int. Ed., 2018, 57, 13735–13747 CrossRef CAS PubMed.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 11, 1312–1330 RSC.
- J. Kiriratnikom, N. Laiwattanapaisarn, K. Vongnam, N. Thavornsin, P. Sae-ung, S. Kaeothip, A. Euapermkiati, S. Namuangruk and K. Phomphrai, Inorg. Chem., 2021, 60, 6147–6151 CrossRef CAS PubMed.
- J. A. Castro-Osma, K. J. Lamb and M. North, ACS Catal., 2016, 6, 5012–5025 CrossRef CAS.
- X. Jiang, F. Gou, F. Chen and H. Jing, Green Chem., 2016, 18, 3567–3576 RSC.
- F. Della-Monica, A. Buonerba and C. Capacchione, Adv. Synth. Catal., 2019, 361, 265–282 CrossRef CAS.
- E. Y. Seong, J. H. Kim, N. H. Kim, K.-H. Ahn and E. J. Kang, ChemSusChem, 2019, 12, 409–415 CrossRef CAS.
- F. Chen, N. Liu and B. Dai, ACS Sustainable Chem. Eng., 2017, 5, 9065–9075 CrossRef CAS.
- C. Maeda, T. Taniguchi, K. Ogawa and T. Ema, Angew. Chem., Int. Ed., 2015, 54, 134–138 CrossRef CAS PubMed.
- T. Ema, Y. Miyazaki, J. Shimonishi, C. Maeda and J.-Y. Hasegawa, J. Am. Chem. Soc., 2014, 136, 15270–15279 CrossRef CAS.
- L. Cuesta-Aluja, A. Campos-Carrasco, J. Castilla, M. Reguero, A. M. Masdeu-Bultó and A. Aghmiz, J. CO2 Util., 2016, 14, 10–22 CrossRef CAS.
- C. Maeda, J. Shimonishi, R. Miyazaki, J.-Y. Hasegawa and T. Ema, Chem. – Eur. J., 2016, 22, 6556–6563 CrossRef CAS PubMed.
- R. Ma, L.-N. He and Y.-B. Zhou, Green Chem., 2016, 18, 226–231 RSC.
- X.-D. Lang, Y.-C. Yu and L.-N. He, J. Mol. Catal. A: Chem., 2016, 420, 208–215 CrossRef CAS.
- M. Navarro, L. F. Sánchez-Barba, A. Garcés, J. Fernández-Baeza, I. Fernández, A. Lara-Sánchez and A. M. Rodríguez, Catal. Sci. Technol., 2020, 10, 3265–3278 RSC.
- F. de la Cruz-Martínez, M. Martínez de Sarasa Buchaca, J. Martínez, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Rodríguez-Diéguez, J. A. Castro-Osma and A. Lara-Sánchez, ACS Sustainable Chem. Eng., 2019, 7, 20126–20138 CrossRef.
- Y. Kim, K. Hyun, D. Ahn, R. Kim, M. H. Park and Y. Kim, ChemSusChem, 2019, 12, 4211–4220 CrossRef CAS PubMed.
- F. de la Cruz-Martínez, J. Martínez, M. A. Gaona, J. Fernández-Baeza, L. F. Sánchez-Barba, A. M. Rodríguez, J. A. Castro-Osma, A. Otero and A. Lara-Sánchez, ACS Sustainable Chem. Eng., 2018, 6, 5322–5332 CrossRef.
- J. Rintjema and A. W. Kleij, ChemSusChem, 2017, 10, 1274–1282 CrossRef CAS PubMed.
- M. Cozzolino, T. Rosen, I. Goldberg, M. Mazzeo and M. Lamberti, ChemSusChem, 2017, 10, 1217–1223 CrossRef CAS PubMed.
- S. W. X.-F. Enthaler, Zinc catalysis : applications in organic synthesis, 2015 Search PubMed.
- R. J. Wood, P. M. Suter and R. M. Russell, Am. J. Clin. Nutr., 1995, 62, 493–505 CrossRef CAS PubMed.
- J. R. Turnlund, A. A. Betschart, M. Liebman, M. J. Kretsch and H. E. Sauberlich, Am. J. Clin. Nutr., 1992, 56, 905–910 CrossRef CAS PubMed.
- E. Castro-Aguirre, F. Iñiguez-Franco, H. Samsudin, X. Fang and R. Auras, Adv. Drug Delivery Rev., 2016, 107, 333–366 CrossRef CAS PubMed.
- T. Nakanishi, J. Health Sci., 2007, 53, 1–9 Search PubMed.
- K. E. Appel, Drug Metab. Rev., 2004, 36, 763–786 CrossRef CAS PubMed.
- S. Sobrino, M. Navarro, J. Fernández-Baeza, L. F. Sánchez-Barba, A. Garcés, A. Lara-Sánchez and J. A. Castro-Osma, Dalton Trans., 2019, 48, 10733–10742 RSC.
- M. Navarro, A. Garcés, L. F. Sánchez-Barba, F. de la Cruz-Martínez, J. Fernández-Baeza and A. Lara-Sánchez, Polymers, 2021, 13, 2356 CrossRef CAS PubMed.
- A. Garcés, L. F. Sánchez-Barba, J. Fernández-Baeza, A. Otero, I. Fernández, A. Lara-Sánchez and A. M. Rodríguez, Inorg. Chem., 2018, 57, 12132–12142 CrossRef PubMed.
- A. Otero, J. Fernández-Baeza, A. Lara-Sánchez and L. F. Sánchez-Barba, Coord. Chem. Rev., 2013, 257, 1806–1868 CrossRef CAS.
- L. F. Sánchez-Barba, C. Alonso-Moreno, A. Garcés, M. Fajardo, J. Fernández-Baeza, A. Otero, A. Lara-Sánchez, A. M. Rodríguez and I. López-Solera, Dalton Trans., 2009, 38, 8054–8062 RSC.
- T. W. Campbell and J. J. Monagle, Org. Synth., 1963, 43, 31–33 CrossRef CAS.
- A. Garcés, L. F. Sánchez-Barba, J. Fernández-Baeza, A. Otero, A. Lara-Sánchez and A. M. Rodríguez, Organometallics, 2017, 36, 884–897 CrossRef.
- F. Chen, Q.-C. Zhang, D. Wei, Q. Bu, B. Dai and N. Liu, J. Org. Chem., 2019, 84, 11407–11416 CrossRef CAS PubMed.
- H. Büttner, C. Grimmer, J. Steinbauer and T. Werner, ACS Sustainable Chem. Eng., 2016, 4, 4805–4814 CrossRef.
- L. Longwitz, J. Steinbauer, A. Spannenberg and T. Werner, ACS Catal., 2018, 8, 665–672 CrossRef CAS.
- L. Peña Carrodeguas, À. Cristòfol, J. M. Fraile, J. A. Mayoral, V. Dorado, C. I. Herrerías and A. W. Kleij, Green Chem., 2017, 19, 3535–3541 RSC.
- G. Fiorani, M. Stuck, C. Martín, M. M. Belmonte, E. Martin, E. C. Escudero-Adán and A. W. Kleij, ChemSusChem, 2016, 9, 1304–1311 CrossRef CAS PubMed.
- C. J. Whiteoak, E. Martin, M. M. Belmonte, J. Benet-Buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469–476 CrossRef CAS.
- D. J. Darensbourg, A. Horn Jr. and A. I. Moncada, Green Chem., 2010, 12, 1376–1379 RSC.
- C.-Y. Li, Y.-C. Su, C.-H. Lin, H.-Y. Huang, C.-Y. Tsai, T.-Y. Lee and B.-T. Ko, Dalton Trans., 2017, 46, 15399–15406 RSC.
- C. Beattie, M. North, P. Villuendas and C. Young, J. Org. Chem., 2013, 78, 419–426 CrossRef CAS PubMed.
- D. J. Darensbourg, S. J. Lewis, J. L. Rodgers and J. C. Yarbrough, Inorg. Chem., 2003, 42, 581–589 CrossRef CAS PubMed.
- D. J. Darensbourg, W.-C. Chung and S. J. Wilson, ACS Catal., 2013, 3, 3050–3057 CrossRef CAS.
- B. Gabriele, R. Mancuso, G. Salerno, L. Veltri, M. Costa and A. Dibenedetto, ChemSusChem, 2011, 4, 1778–1786 CrossRef CAS PubMed.
- T. Itaya, T. Iida, I. Natsutani and M. Ohba, Chem. Pharm. Bull., 2002, 50, 83–86 CrossRef CAS PubMed.
- J. Datta and M. Włoch, Polym. Bull., 2016, 73, 1459–1496 CrossRef CAS.
- G. Rokicki, P. G. Parzuchowski and M. Mazurek, Polym. Adv. Technol., 2015, 26, 707–761 CrossRef CAS.
- K. A. Maltby, M. Hutchby, P. Plucinski, M. G. Davidson and U. Hintermair, Chem. – Eur. J., 2020, 26, 7405–7415 CrossRef CAS PubMed.
- A. Rehman, A. M. López Fernández, M. F. M. Gunam Resul and A. Harvey, J. CO2 Util., 2019, 29, 126–133 CrossRef CAS.
- A. Rehman, F. Saleem, F. Javed, A. Ikhlaq, S. W. Ahmad and A. Harvey, J. Environ. Chem. Eng., 2021, 9, 105113 CrossRef CAS.
- J. Chen, X. Wu, H. Ding, N. Liu, B. Liu and L. He, ACS Sustainable Chem. Eng., 2021, 9, 16210–16219 CrossRef CAS.
- J. Fernández-Baeza, L. F. Sánchez-Barba, A. Lara-Sánchez, S. Sobrino, J. Martínez-Ferrer, A. Garcés, M. Navarro and A. M. Rodríguez, Inorg. Chem., 2020, 59, 12422–12430 CrossRef PubMed.
- G. N. Bondarenko, O. G. Ganina, A. A. Lysova, V. P. Fedin and I. P. Beletskaya, J. CO2 Util., 2021, 53, 101718 CrossRef CAS.
- I. Karaméa, S. Zahera, N. Eida and L. Christ, Mol. Catal., 2018, 456, 87–95 CrossRef.
- J. T. B. H. Jastrzebski, J. Boersma and G. van Koten, Structural Organozinc Chemistry, in The Chemistry of Organozinc Compounds, ed. S. Patai, Z. Rappoport and I. Marek, 2006 Search PubMed.
Footnotes |
| † Dedicated to Dr Juan Fernández-Baeza on the occasion of his retirement. |
| ‡ Electronic supplementary information (ESI) available: Details of materials and methods, and experimental and spectroscopic data as well as kinetic and mechanistic studies for the synthesis of cyclic carbonates. CCDC 2240438 and 2240439. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00510k |
| This journal is © The Royal Society of Chemistry 2023 |