
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolarized Au(I)/Rh(I) bimetallic pairs cooperatively trigger ligand non-innocence and bond activation†
Macarena G.
Alférez
,
Juan J.
Moreno
,
Celia
Maya
and
Jesús
Campos
 *
*
Instituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Consejo Superior de Investigaciones Científicas (CSIC) and University of Sevilla, Avenida Américo Vespucio 49, 41092 Sevilla, Spain. E-mail: jesus.campos@iiq.csic.es; Web: https://jcamposgroup.iiq.us-csic.es/
First published on 22nd February 2023
Abstract
The combination of molecular metallic fragments of contrasting Lewis character offers many possibilities for cooperative bond activation and for the disclosure of unusual reactivity. Here we provide a systematic investigation on the partnership of Lewis basic Rh(I) compounds of type [(η5-L)Rh(PR3)2] (η5-L = (C5Me5)− or (C9H7)−) with highly congested Lewis acidic Au(I) species. For the cyclopentadienyl Rh(I) compounds, we demonstrate the non-innocent role of the typically robust (C5Me5)− ligand through migration of a hydride to the Rh site and provide evidence for the direct implication of the gold fragment in this unusual bimetallic ligand activation event. This process competes with the formation of dinuclear Lewis adducts defined by a dative Rh → Au bond, with selectivity being under kinetic control and tunable by modifying the stereoelectronic and chelating properties of the phosphine ligands bound to the two metals. We provide a thorough computational study on the unusual Cp* non-innocent behavior and the divergent bimetallic pathways observed. The cooperative FLP-type reactivity of all bimetallic pairs has been investigated and computationally examined for the case of N–H bond activation in ammonia.
Introduction
The area of bimetallic cooperativity has witnessed a rapid resurgence in the last decades, in great part due to the prospects it offers for bond activation and catalysis.1 The same can be stated about heterogeneous2 and enzymatic catalysis,3 where bi- and multimetallic synergisms are at the heart of many successful chemical transformations. Beyond catalytic applications, molecular dinuclear structures offer a range of possibilities that have led to the observation of remarkable electronic, magnetic or photophysical properties, exotic bonding schemes and highly unusual reactivity, including an outstanding capacity to trap otherwise fleeting species.1 For instance, our group recently unlocked a new mode of ligand non-innocent behavior discernible only through a bimetallic approach. More precisely, we described the direct involvement of the widespread pentamethyl cyclopentadienyl ([C5Me5]−, Cp*) ligand, whose popularity largely relies on its assumed robust spectator character, in the migration of a hydride from one of its methyl groups in compound [(η5-C5Me5)Rh(PMe3)2] (1a) to the rhodium centre (Fig. 1),4 a process which was so far restricted to early transition metals.5 As introduced above, we were capable of characterizing this reversible event leveraging a bimetallic approach, more precisely by introducing the highly electrophilic and sterically shielded [(PCyp2ArXyl2)Au]+ fragment6 (Cyp = cyclopentyl; ArXyl2 = C6H3-2,6-(C6H3-2′,6′-Me2)2). | ||
| Fig. 1 Cp* non-innocence by reversible hydride migration in a Rh(I)/Au(I) system and systematic investigation carried out in this work on the effects of tuning the Rh(I) fragment. | ||
We could conclude that hydride migration from Cp* occurs in a reversible manner, suggesting that there is potential to design proton-coupled-electron-transfer (PCET) catalysis based on this approach.7 In fact, this has already been demonstrated for hydrogen evolution and dinitrogen reduction by means of a related reversible proton migration between late transition metals and the internal carbon atoms of Cp ligands.8 However, before embarking ourselves into similar endeavors based on the methyl functionalities of Cp*, it is necessary to better understand the factors influencing the described non-innocence, as well as the precise role associated to the gold moiety. Besides, in terms of intermolecular reactivity, the aforesaid Rh(I)/Au(I) pairs render potential for bimetallic bond activation processes. Thus, as highly constrained metallic fragments of opposed Lewis character, they may behave as bimetallic frustrated Lewis pairs (FLPs),9 a nascent topic to which we have contributed in recent years.10 Nonetheless, the Cp* non-innocence described herein advises for the exploration of alternative aromatic capping ligands in our continuous search for bimetallic FLPs.
In this context, we have decided to modify the Lewis basic Rh(I) precursor [(η5-C5Me5)Rh(PMe3)2] (1a) to investigate the effects on ligand non-innocence and bimetallic Rh(I)/Au(I) reactivity. To do so we have tuned the stereoelectronic and chelating properties of the phosphine ligands and the outcomes resulting from substituting the Cp* fragment by the also prevalent indenyl ([C9H7]−) ligand, which lacks C(sp3)–H groups (Fig. 1). We provide a thorough computational investigation to ascertain the mechanism by which the hydride reversibly exchanges positions between Cp* and rhodium and for the cooperative bimetallic activation of polar N–H bonds in ammonia.
Results and discussion
We first decided to examine the effects of modifying the simple trimethyl phosphine ligands in 1a. To circumvent the possibility of ligand dissociation during Cp* and small molecule activation, we selected the chelating bisphoshpines 1,2-bis(dimethylphosphino)ethane (dmpe) and 1,2-bis(diphenylphosphino)ethane (dppe), which led to compounds 1b and 1c, respectively (see Scheme 1). These compounds were prepared following the same procedure employed to access 1a, that is, by reducing dimer [(η5-C5Me5)RhCl2]2 with sodium amalgam in the presence of the corresponding phosphine (see ESI† for details). In terms of the electrophilic gold fragment, we focused on two precursors of equal formula, [(PR2ArXyl2)Au(NTf2)] (2Me, R = Me; 2Cyp, R = Cyp; NTf2 = [N(SO2CF3)2]−), stabilized by bulky terphenyl phosphine ligands. Although these phosphines are comparable in terms of donating properties,11 their steric profile is markedly different. Thus, PCyp2ArXyl2 is considerably bulkier than PMe2ArXyl2, as evidenced by their percentage buried volumes, recently reported for their corresponding dicoordinate gold–ethylene complexes (PMe2ArXyl2, 38.2% vs. PMe2ArCyp2, 53.5%).12 | ||
| Scheme 1 Contrasting bimetallic reactivity between Lewis basic Rh(I) compounds 1b and 1c and Lewis acidic Au(I) compounds 2Me and 2Cyp. | ||
As we anticipated, the combination of the rhodium precursor 1b with gold compounds 2Me and 2Cyp led to analogous reactivity to that observed for the Rh(I) species 1a (Scheme 1a),4 owing to their similar stereoelectronic properties. Therefore, the smaller [(PMe2ArXyl)Au]+ fragment leads to quantitative formation of the corresponding Rh(I) → Au(I) metal-only Lewis pair (MOLP)13 (3bMe). In contrast, the more sterically congested [(PCyp2ArXyl)Au]+ unit triggers the immediate migration of a hydride towards the rhodium centre with formation of a new Au–CH2 bond in the Cp*-functionalized compound 4bCyp, without any trace of bimetallic dative bonding. The formation of these bimetallic species is easily inferred from multinuclear NMR spectroscopy. A distinctive AB2 pattern demonstrating the formation of a bimetallic adduct arises in the 31P{1H} NMR spectrum of 3bMe, with an apparent double triplet at 14.6 ppm (2JPRh = 12, 3JPP = 9 Hz) and a double doublet at 45.5 ppm (1JPRh = 154, 3JPP = 9 Hz), due to PMe2ArXyl2 and dmpe ligands, respectively (cf.3aMe, 13.9 ppm (3JPP = 12, 2JPRh = 10 Hz), −3.1 ppm ( 1JPRh = 155, 3JPP = 12 Hz)).4 In turn, the 1H NMR spectrum of 4bCyp reveals the formation of a new hydride ligand resonating at −13.60 ppm (td, 2JHP = 34, 1JHRh = 26 Hz; cf. 4aCyp, −13.34 ppm, dt, 2JHP = 36, 1JHRh = 25 Hz) and the distinctive asymmetry of the Cp* ligand, now transformed into the {C5Me4CH2AuP} moiety, leading to three resonances at 1.74, 1.69 and 1.21 ppm (d, 3JHP = 7.7 Hz) in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1 ratio (cf.4aCyp, 1.84, 1.73 and 1.05 ppm (3JHP = 9.6 Hz)). The molecular structure of 4bCyp was authenticated by X-ray diffraction studies from single crystals grown by slow diffusion of pentane into a saturated benzene solution (Fig. 2). Although the overall quality of the crystals was poor, the connectivity was rather clear and certified that the rhodium centre adopts a piano-stool conformation to accommodate the newly form hydride ligand, while the [(PCyp2ArXyl2)Au]+ unit displays an orthogonal arrangement relative to the Cp* plane defined by the central five-membered ring (85.22°).
:3:1 ratio (cf.4aCyp, 1.84, 1.73 and 1.05 ppm (3JHP = 9.6 Hz)). The molecular structure of 4bCyp was authenticated by X-ray diffraction studies from single crystals grown by slow diffusion of pentane into a saturated benzene solution (Fig. 2). Although the overall quality of the crystals was poor, the connectivity was rather clear and certified that the rhodium centre adopts a piano-stool conformation to accommodate the newly form hydride ligand, while the [(PCyp2ArXyl2)Au]+ unit displays an orthogonal arrangement relative to the Cp* plane defined by the central five-membered ring (85.22°).
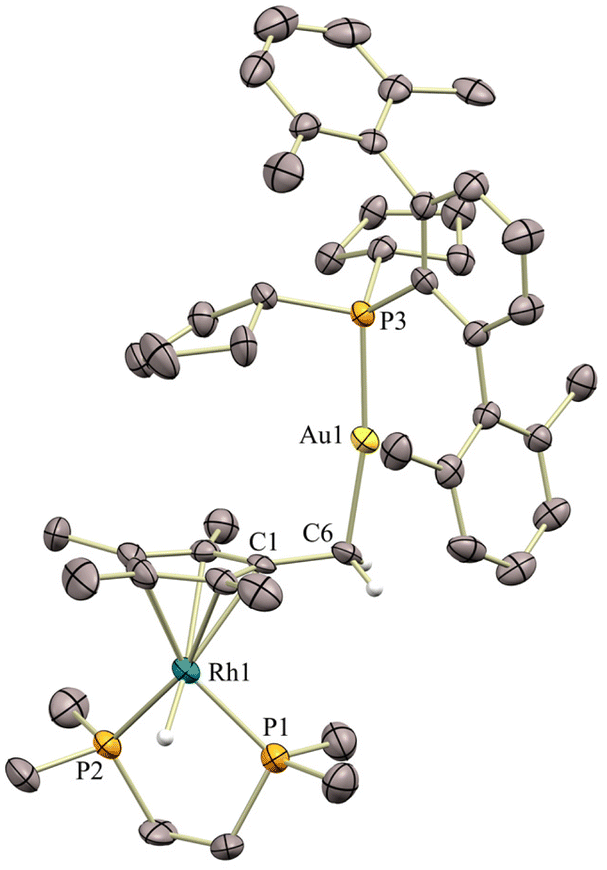 | ||
| Fig. 2 ORTEP diagram of compound 4bCyp; for the sake of clarity most hydrogen atoms, as well as solvent molecules and triflimide counteranions are excluded. Thermal ellipsoids are set at 50% probability. | ||
We envisioned that the bulkier and less basic dppe ligand would induce differences in the bimetallic reactivity of precursor 1c due to steric and electronic effects. In this case, reaction with the smaller PMe2ArXyl2-based gold complex 2Me not only produced the predicted bimetallic Lewis adduct 3cMe, but also small amounts of the Cp*-functionalized product 4cMe in around 30% spectroscopic yield (Scheme 1b). Nonetheless, the latter species rapidly evolves to the thermodynamic product 3cMe, though its formation suggest that the higher steric pressure exerted by dppe compared to dmpe partially hampers the approximation of the two metal sites and facilitates the detection of 4cMe, not discernible for neither 1a nor 1b for the smaller PMe2ArXyl2 gold system. This finding also supports the reversibility of the hydride migration from Cp* to rhodium. Although the isolation of the minor species 4cMe was not possible, its existence is corroborated by multinuclear NMR analysis. Thus, distinctive resonances in the 1H NMR are found at −12.30 ppm for the hydride ligand and at 1.99, 1.91 and 0.37 (d, 3JHP = 9 Hz) for the functionalized Cp* ring. 31P{1H} NMR resonances recorded at 73.4 (1JPRh = 139 Hz) and 40.8 (2JPRh = 9 Hz) ppm due to dppe and PMe2ArXyl2 ligands, respectively, are also in agreement with the analogous signals associated to 4aCyp and 4bCyp.
In stark contrast, when we carried out the same study with the bulkier gold complex 2Cyp a different scenario ensues: there is virtually no reaction at low temperature (−60 °C). However, upon warming to 25 °C we observe the formation of both the Cp*-activated product 4cCyp and the bimetallic Lewis adduct 3cCyp, but only as minor species (ca. 30% overall), while the monometallic precursors 1c and 2Cyp remain as the major components for several hours (Scheme 1b). It is important to remark that despite being the more congested Rh(I) compound, the formation of the bimetallic adduct 3cCyp is now preferred over 4cCyp, which differs from Rh(I) precursors 1a and 1b. We attribute this difference to a shift from kinetic control favouring compounds 4 in the prior systems, to thermodynamic control leading to adducts 3 in the present case, in line with our computational investigations (vide infra, Fig. 5). Unexpectedly, the activation product 4cCyp is initially formed but it disappears after several hours in favour of a mixture of 1c, 2Cyp and 3cCyp, evincing the reversibility of the formation of the Au–C bond and concomitant hydride migration, as occurs for the PMe2ArXyl2 system. Although we are yet unsure about the reasons on why 4cCyp is only discernible initially, it is also true that its overall concentration is variable among different experiments and its initial amount minimal when directly conducting the experiment at 25 °C. In any case, it seems rather clear that 3cCyp and 4cCyp form from independent reactions from compounds 1c and 2Cyp, in agreement with our computational investigations and also with our prior isotopic labelling experiments carried out during ammonia activation studies.
Compounds 1c, 2Cyp and 3cCyp are in dynamic equilibrium, in line with the reduced basicity of 1c, which allowed us to spectroscopically investigate the thermodynamics of such a process. A solution of complexes 1c and 2cCyp in benzene-d6 was monitored for 24 hours to ensure the complete disappearance of 4cCyp, then a van't Hoff analysis over a 60 K range yielded thermal parameters for the equilibrium of ΔH = 2.0 ± 0.5 kcal mol−1 and ΔS = 8 ± 2 cal K−1 mol−1, corresponding to ΔG298 = −0.4 ± 1.0 kcal mol−1 for the formation of the bimetallic adduct 3cCyp. The very small but positive enthalpic value, along with the only moderately positive entropic component, enable the persistence of variable amounts of the monometallic fragments in solution, a prerequisite for exhibiting bimetallic FLP reactivity.10
Therefore, it seems that the higher steric profile of dppe compared to PMe3 and dmpe, along with the reduced basicity of the rhodium centre, partly impedes the formation of an inactive bimetallic adduct. We tested whether this modulation in bimetallic reactivity has a direct effect on the activation of small molecules, for which we selected the activation of the N–H bond in ammonia. Our choice substantiates on the still challenging character of this activation for transition metal complexes14 and on our prior finding that 4aCyp has already shown success.4 Compounds 4aCyp and 4bCyp mediate the heterolytic cleavage of ammonia under mild conditions (1 bar, 25 °C; Scheme 2a), with the former being more rapid (t1/2 ≈ 30 min), to yield [(η5-C5Me5)Rh(L2)H]+ (5a, L = PMe3; 5b, L2 = dmpe) and [(PCyp2ArXyl2)Au(NH2)].4 Contrarily, the equilibrium mixture comprised of 1c, 2Cyp and 3cCyp fails to cleave the N–H bond under similar conditions (Scheme 2b). Instead, only 2Cyp readily converts into the corresponding ammonia adduct [(PCyp2ArXyl2)Au(NH3)](NTf2). Subsequently, the mixture slowly evolves to a solution containing the latter adduct and Rh(I) precursor 1c in detriment of the bimetallic pair 3cCyp. Therefore, the reduced basicity of dppe compared to dmpe or PMe3 hampers the use of the corresponding Lewis basic Rh(I) precursor as a metallic FLP component under these conditions.
 | ||
| Scheme 2 (a) FLP-type N–H bond activation in ammonia by Rh(I)/Au(I) bimetallic pairs vs. (b) no cooperative activation and ammonia adduct formation. | ||
Next, we examined the activation of ammonia by computational means. To do so, we decided to focus on the related gold fragment 2Tripp based on the terphenyl ligand PMe2ArTripp2 (ArTripp2 = C6H3-2,6-(C6H2-2′,4′,6′-iPr3)2). We made this choice of ligand to limit the computational difficulties associated to the fluxionality of the cyclopentadienyl groups in PCyp2ArXyl2, while at the same time offering similar experimental results to the latter in terms of bimetallic Cp* and ammonia activation.4 Both metal centers in complex 4aTripp are saturated, but the evolution of 4aTripp towards the Lewis adduct 3aTripp clearly indicates that the independent fragments are accessible in solution (+4.6 kcal mol−1 in Fig. 3, 4aTripp as reference). Then, the weakly coordinating triflimide in 2Tripp is readily displaced by ammonia, a reaction that is largely exergonic (9.5 kcal mol−1). Binding to the electrophilic gold(I) center lowers the barrier for deprotonation by the basic Rh center to 15.3 kcal mol−1,15 in a way that is resemblant of conventional FLPs.16 However, this reaction is thermoneutral relative to the fragments, but overall endergonic relative to 4aTripp. We also computed these fragments independently to ensure translational entropy was not responsible for the endergonicity (Fig. 4). We therefore evaluated that, under the reaction conditions, the generated gold amido complex and unreacted 2Tripp could rapidly evolve to form a bridged amido, cationic digold complex featuring an aurophilic interaction,4 an event that is more than sufficiently exergonic to drive the reaction forward.
 | ||
| Fig. 3 Free energy profile for the cooperative activation of NH3, where the zero has been assigned to the Cp*-activated bimetallic compound 4aTripp. | ||
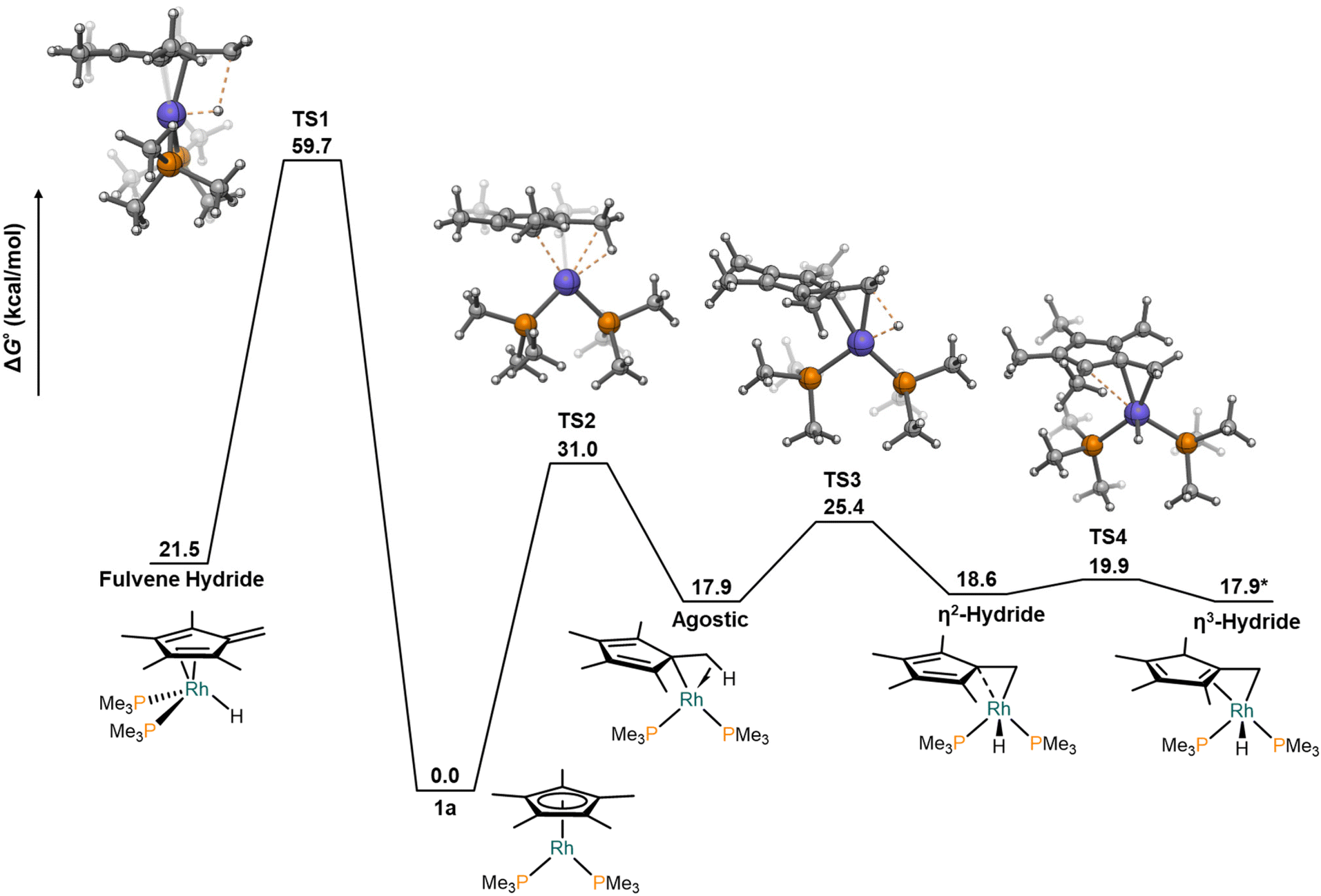 | ||
| Fig. 4 Free energy profile for the intramolecular conversion of 1a into a fulvene hydride complex. *SCF energy relative to TS4. | ||
Beyond the mechanism by which cooperative N–H bond activation occurs, we wondered about the precise pathway that accounts for the migration of a hydride from the Cp* ligand to the Rh centre. In our original communication4 we suggested that an intramolecular migration from a methyl group of the Cp* towards the metal in 1a would produce a hydride fulvene structure in a process being redox neutral at rhodium, as previously proposed for early transition metals.5 This highly reactive species could then be trapped by the electrophilic complexes 2. However, our computational studies indicate that the concerted transition state for the direct transfer of the hydride presented an exceedingly large barrier (TS1 in Fig. 4, 59.7 kcal mol−1) to yield the aforementioned fulvene hydride complex at 21.5 kcal mol−1. Interestingly, Cp* slippage to form an agostic complex presented a much lower barrier (TS2, 31.0 kcal mol−1), from which C–H bond breaking to form the hydride is much more facile (TS3, 25.4 kcal mol−1). Subsequent partial restoration of the Cp* hapticity does not seem rate limiting either (TS4). Nonetheless, these barriers are overly large to be consistent with experimental reaction conditions,4 which rules out an unassisted intramolecular equilibrium of 1a to form a fulvene hydride complex. We then sought whether gold complexes 2 could facilitate any of these or other reaction pathways.
Both the Xyl and Tripp systems were computationally studied to gain insight into the role of steric hindrance in the reaction outcome (Fig. 5). Experimentally, we previously observed4 that while the combination of 1a and 2Xyl readily leads to the bimetallic adduct 3aXyl, the use of 2Tripp produces a mixture of 4aTripp and 3aTripp that eventually evolves towards the later. Thus, the formation of species 4aTripp seems to be under kinetic control, as the thermodynamic product is the Lewis adduct. Our computational studies are in agreement with these experimental findings. The Xyl system presents a low barrier (Fig. 5, TS5-Xyl, 11.4 kcal mol−1) for the formation of the Lewis adduct, as expected due to the lower steric demand of the phosphine. In turn, for the Tripp system the transition state lies at 25.3 kcal mol−1, which enables the binding of the gold center to a carbon atom of the anionic Cp* ring (TS6-Tripp, 19.7 kcal mol−1), forming a Rh-diene complex at 5.0 kcal mol−1 (Cp-Au). This species cannot be accessed by the Xyl system, as the barrier for the formation of 3aXyl is much lower.
 | ||
| Fig. 5 Free energy profile encompassing Lewis adduct formation, binding of Au(I) to the Cp* and thermodynamics of formation of 4aXyl (orange) and 4aTripp (black). | ||
Next, we sought to investigate whether the binding of the electrophilic gold center to the Cp* was indeed key to form 4aTripp, that is, to promote hydride transfer to the Rh center concomitant with the formation of the Au–CH2 bond. Using Cp-AuXyl as a reference, it was clear that despite the pyramidalization that accompanies the formation of the Au–C bond pushing a methyl group towards rhodium, the concerted hydride transfer and gold migration to the nascent CH2 moiety remains inaccessible (TS7 in Fig. S46,† 49.4 kcal mol−1). In turn, the barrier to form an agostic complex via slippage drops to 22.5 kcal mol−1 (TS8-Xyl, cf. TS2 at 31.0 kcal mol−1), from which hydride formation (TS9-Xyl) lies at 25.0 kcal mol−1, comparable to the unassisted pathway (TS3 in Fig. 4).
We then moved to study the bulkier Tripp system, to ascertain whether these effects were not countered by increased steric demands, as Cp* activation is not observed for the Xyl system. The formation of the corresponding agostic complex for the Tripp system presents a barrier of 25.7 kcal mol−1 (TS8-Tripp in Fig. 6), competitive with that of the formation of the Lewis adduct (TS5-Tripp in Fig. 5, 25.3 kcal mol−1), and thus consistent with the aforesaid generation of a mixture of 3aTripp and 4aTripp. Subsequent steps for the formation of the Rh hydride and for the migration of the gold center towards the CH2 moiety present similar barriers of 25.7 and 25.4 kcal mol−1, respectively (TS9-Tripp and TS10), affordable under experimental conditions. Although no saddle points could be found, relaxed potential energy scans indicated that the restoration of the Cp* aromaticity to yield 4aTripp from AuCH2slipped was accessible. To complete our computational screening, two alternative pathways, involving hydride abstraction by the Lewis acid and concerted Au–C bond formation concomitant with HNTf2 release were found to be higher energy and are briefly discussed in the ESI (Fig. S47 and S48†).
 | ||
| Fig. 6 Free energy profile for the conversion of Cp-AuTripp into 4aTripp. | ||
Therefore, the mechanism for the unusual Cp* non-innocent behavior described herein encompasses the initial binding of the Lewis acidic gold fragment to the Cp* inner ring, which facilitates the migration of the hydride to the rhodium centre by a series of conformational rearrangements that involve the direct participation of both Cp* and gold. These results may have implications in a variety of transformations that imply the use of Cp*-based complexes in the presence of electrophiles. The simplest electrophile would naturally be a proton, for which the already discussed protonation of the inner Cp* ring prevails.8 However, the use of more elaborated electrophiles as additives or co-catalysts in a range of catalytic transformations mediated by Cp*-based complexes is in continuous development.17 It would not be surprising that the same kind of Cp* non-innocence triggered by the electrophile might be operating in some of these systems, but it could have been overlooked due to difficulties for its identification associated to reversibility and low catalyst loadings. In addition, our studies highlight the importance of controlling the bulkiness of the electrophile (i.e. gold fragment), whose subtle tuning dictates selectivity between Lewis adduct formation vs. Cp* activation.
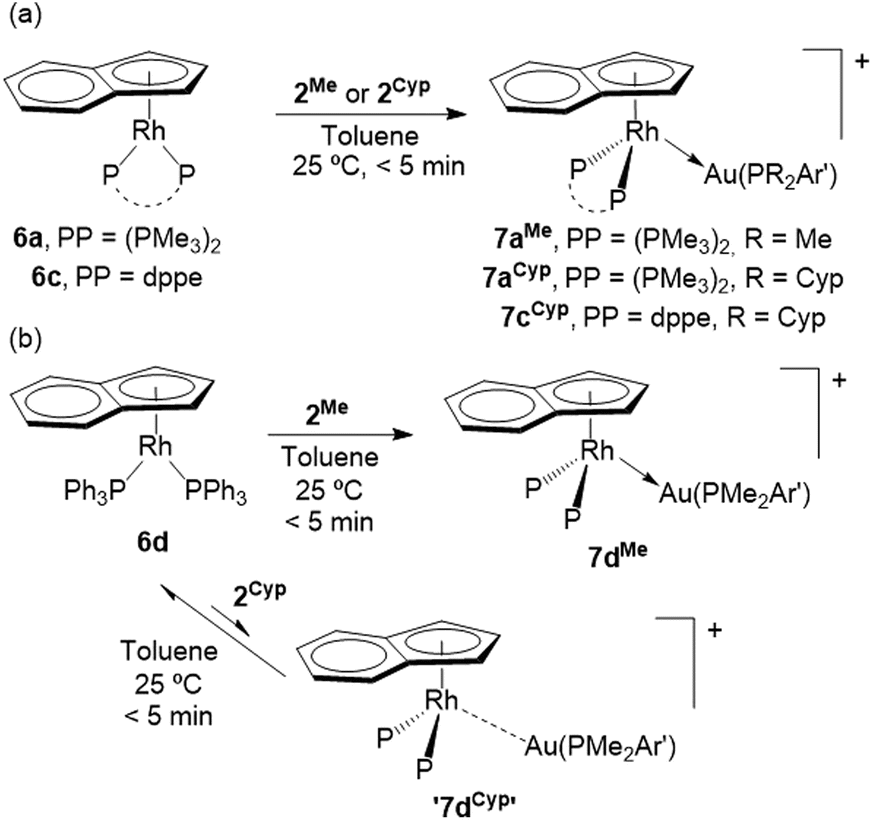
With the aim of circumventing the discussed non-innocence character of Cp* to access a broader family of Rh(I)/Au(I) bimetallic FLPs we decided to study analogous Rh(I) precursors based on the well-known indenyl ligand ([C9H7]−).18 Besides, we postulated that the greater capacity of the latter ligand to navigate through variable hapticities (from η1 to η5) might offer a richer reactivity after FLP-type bimetallic bond activation. Initially, we prepared the previously reported compound [(η5-C9H7)Rh(PMe3)2]19 (6a) as our benchmark indenyl-based species. To carry out comparative studies we synthesized analogous Rh(I) species bearing both dppe (6c) and the non-chelating and more sterically demanding PPh3 (6d) (see ESI† for synthetic protocols, spectroscopic characterization and X-ray diffraction studies of 6c), though we omitted a related dmpe version, anticipating identical behavior to 6a in line to the previously discussed results.
Not surprisingly, the equimolar reaction of 6a and the smaller gold precursor 2Me led to the corresponding bimetallic adduct 7aMe (Scheme 3a). In this case, the reaction with the bulkier 2Cyp similarly yielded the metal–metal bonded adduct 7aCyp, a behavior attributed to the reduced steric pressure exerted by the indenyl ligand compared to Cp*. The two new adducts exhibit a similar pattern in their 31P{1H} NMR spectra, with two doublets due to PMe3 (7aMe, δ −3.9, 1JPRh = 158 Hz; 7aCyp, δ −6.2, 1JPRh = 159 Hz) and the terphenyl phosphine (7aMe, δ 4.6, 2JPRh = 18 Hz; 7aCyp, δ 43.4, 2JPRh = 19 Hz). At variance with bimetallic adduct 3aMe, there is no observable scalar coupling between the two distinct phosphines, a feature attributable to the slightly different geometric environment around rhodium, as evidenced by X-ray diffraction studies (Fig. 7). For instance, the Rh–Au–P angles in the indenyl systems are wider (7aMe, 171.61(7); 7aCyp, 168.21(10)°) than for the Cp* complex 3aTripp (157.90(11)°), while the greater coordination flexibility of indenyl reflects into wider offset angles between the rhodium centre and the centroid of the η5-coordinated ring (3aTripp, 1.85°; 7aMe, 10.88°; 7aCyp, 12.35°). Nonetheless, the three structures exhibit comparable Rh–Au bond distances of 2.593(1) (3aTripp), 2.5541(8) (7aMe) and 2.5970(12) (7aCyp) Å, in all cases slightly lower than the sum of their covalent radii (2.78 Å).20 Analogously, the calculated formal shortness ratio (FSR),21 namely the ratio between the M–M bond distance and the sum of their metallic radii, are almost identical and accounts for 1.00 (3aTripp), 0.99 (7aMe) and 1.00 (7aCyp).
 | ||
| Scheme 3 Bimetallic reactivity between Lewis basic indenyl-Rh(I) compounds 6a, 6c and 6d with Lewis acidic Au(I) compounds 2Me and 2Cyp. | ||
 | ||
| Fig. 7 ORTEP diagrams of indenyl compounds 7aMe, 7aCyp and 7dMe; for the sake of clarity most hydrogen atoms, as well as solvent molecules and triflimide counteranions are excluded. Some fragments are represented in wireframe format and thermal ellipsoids are set at 50% probability. | ||
We then examined the combination of the more congested Rh(I) precursor 6c with the bulkier Au(I) complex 2Cyp. Despite increasing the steric pressure, this mixture cleanly evolves to the corresponding metallic Lewis pair 7cCyp (Scheme 3a), as inferred from two doublets in the 31P{1H} NMR spectrum at 74.7 and 47.0 ppm associated to dppe and PCyp2ArXyl2, respectively. Crystals suitable for X-ray diffraction studies were grown by slow diffusion of pentane into a saturated benzene solution of 7cCyp. Interestingly, its ORTEP diagram (Fig. S44†) reveals an elongated Rh–Au bond length of 2.6314(10) Å, though still within the sum of the corresponding covalent radii and with an FSR value of 1.02. Moving towards more congested systems, we examined the analogous reaction with the PPh3-containing rhodium complex 6d, in which case the formation of the bimetallic adduct does not take place (Scheme 3b). Instead, we observe broad 31P{1H} NMR resonances associated to the corresponding monometallic precursors, indicating dynamic behaviour between these species and the bimetallic adduct. When the reaction is monitored at −20 °C in the NMR probe the resonances narrow, as previously observed for other bimetallic FLPs investigated in our group.10 The reluctance to form a stable Rh → Au dative bond in this case is most likely due to steric reasons. In fact, treating compound 6d with the smaller gold complex 2Me does yield the corresponding bimetallic pair 7dMe, which was fully characterized by spectroscopic means (see ESI† for details). Besides, its molecular formulation was authenticated by X-ray diffraction studies (Fig. 7), demonstrating the presence of a dative Rh → Au bond characterized by a length of 2.5828(3) Å.
We have previously demonstrated the importance of accessing monometallic fragments in highly polarized and unsupported bimetallic pairs to enhance their cooperative reactivity.10,22 As such, we promptly explored the reactivity of the non-bonded pair 6d:2Cyp towards a variety of small molecules containing both non-polar or weakly polarized (H2, C2H2, C2H4) and polar bonds (NH3, H2O, MeOH). However, instead of the foreseen FLP-type bond activation, we rapidly detected the formation of the heteroleptic gold compound [Au(PCyp2ArXyl2)(PPh3)] (8) (Scheme 4), defined by two distinctive doublets (2JPP = 309 Hz) at 59.4 and 44.3 ppm by 31P{1H} NMR spectroscopy. To assure its formulation this compound was independently prepared and its full characterization is included in the ESI.† Compound 8 is the major and, in some cases, the only discernible gold species obtained during our reactivity studies. Interestingly, the lability of PPh3 is only evidenced upon addition of small molecules, but it also requires the presence of the gold precursor, since no phosphine dissociation was observed in the absence of 2Cyp. Further proof of phosphine lability was obtained after attempts to activate ethylene, which led among other species to the presumable formation of [(η5-C9H7)(PPh3)(C2H4)Rh → Au(PCyp2ArXyl2)](NTf2), inferred from mass spectrometry analysis (see ESI†). Herein, we postulate that substitution of PPh3 by the smaller ethylene ligand likely facilitates the formation of the dative Rh → Au bond. Likewise, initial efforts to activate isonitriles in an FLP-like manner (i.e. 1,1- or 1,2-addition) led to similar results. As an example, addition of 2,6-dimethylphenylisocyanide to a solution of 6d:2Cyp led to a complex mixture from which a small crop of crystals of [(η5-C9H7(PPh3)(XylNC)Rh → Au(PCyp2ArXyl2)](NTf2) could be grown and studied by X-ray diffraction (Fig. S44†), once more revealing the lability of PPh3 from Rh(I) and thus limiting its applicability at least in Au(I)-based bimetallic FLPs.
 | ||
| Scheme 4 Attempts to carry out bimetallic bond activation with a range of substrates (H2, C2H2, C2H4, NH3, H2O, MeOH and 2,6-dimethylphenylisocyanide) leading to formation of [Au(PCyp2ArXyl2)(PPh3)]. | ||
We wondered whether the superior kinetic stability of the chelating dppe ligand would circumvent this drawback while still facilitating the prevalence of the monometallic fragments (bearing in mind the aforesaid elongated Rh → Au bond). However, analogous experiments based on precursor 6b led to identical results, since the major gold-containing species in all our studies was digold compound [{(PCyp2ArXyl2)Au}2(μ-dppe)](NTf2)2, characterized by 31P{1H} NMR resonances at 74.9 and 46.5 ppm (2JPP = 164 Hz) and whose X-ray diffraction structure is provided as well in the ESI (Fig. S44†). We attribute, at least in part, the unexpected facility by which phosphines dissociate in the presence of gold to the weakening of the Rh–P bonds upon the approach of electrophiles. We have computed in recent studies that the strength of the dative bonding between Rh in compound 1a and a range of metallic electrophiles correlates with greater contributions of σ(Rh–P) orbitals, thus weakening Rh–P bonds.23
Conclusions
In summary, we provide a systematic study on a series of Rh(I)/Au(I) bimetallic pairs under sterically congested environments. This study evidences the potential for bimetallic cooperation and contrasting reactivity of two metals together compared to their individual mononuclear species. The nature of phosphine ligands bound to either Rh(I) or Au(I) precursors is crucial to control selectivity, where the dominance of either kinetic or thermodynamic products can be precisely tuned. Thus, in the case of [(η5-C5Me5)Rh(PR3)2] compounds, a highly unusual Cp* non-innocent behavior has been described upon addition of the bulkier gold species [(PCyp2ArXyl2)Au(NTf2)], while the less congested [(PMe2ArXyl2)Au(NTf2)] system leads to the formation of Rh → Au bimetallic adducts. Nonetheless, the basicity of the phosphines bound to Rh also plays an important role, since for the more congested but less basic dppe ligand the Cp*-activated complexes only appear as transient species. In addition, the stereoelectronic properties of the phosphines dictate the equilibria between bimetallic adducts and individual monometallic compounds for the [(η5-C9H7)Rh(PR3)2] precursors. These equilibria are directly associated to the ability of bimetallic pairs to exhibit frustrated Lewis pair (FLP) behavior, which was observed only for the bulkier Cp*-based Rh system. In contrast, the indenyl analogues were unable to mediate FLP-type bond activation due to the lability of the Rh–P bonds in the presence of gold electrophiles.Our theoretical studies provide insights about the factors that influence the divergent bimetallic pathways described in this work and the cooperative N–H bond activation of ammonia. Importantly, we now rule out the previously suggested involvement of a fulvene structure trapped by electrophilic gold to account for the reported Cp* non-innocence, as invoked for mononuclear systems based on early transition metals. At variance, we favor a genuine bimetallic pathway that implies the initial binding of gold to the inner Cp* ring, which facilitates proton migration to the basic Rh site and exemplify the potential of bimetallic synergisms. It is likely that this unusual cooperative ligand non-innocence operates in catalytic systems that combine Cp*-based complexes with bulky electrophiles, but its reversible nature and low Cp*-catalyst loadings may have prevented its identification in the past.
Author contributions
M. G. A. synthesized and characterized all compounds. J. J. M. carried out computational studies. M. G. A. and C. M. carried out XRD studies. M. G. A., J. J. M. and J. C. wrote the original draft. J. C. supervised the overall project. All authors contributed to review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the European Research Council (ERC Starting Grant, CoopCat, 756575). We also thank Grant PID2019-110856GA-I00 funded by MCIN/AEI/10.13039/501100011033, Junta de Andalucía (P18-FR-4688) and US/JUNTA/FEDER, UE (US-1380849). J. J. M. thanks Junta de Andalucía for the postdoctoral program “Personal Investigador Doctor” (ref. DOC_00153). The authors gratefully acknowledge the use of CESGA computational facilities.References
- L. H. Gade, Angew. Chem., Int. Ed., 2000, 39, 2658 CrossRef CAS PubMed; J. Park and S. Hong, Chem. Soc. Rev., 2012, 41, 6931 RSC; B. G. Cooper, J. W. Napoline and C. M. Thomas, Catal. Rev., 2012, 54, 1 CrossRef; J. P. Krogman and C. M. Thomas, Chem. Commun., 2014, 50, 5115 RSC; P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28 CrossRef PubMed; J. F. Berry and C. M. Thomas, Dalton Trans., 2017, 46, 5472 RSC; J. F. Berry and C. C. Lu, Inorg. Chem., 2017, 56, 7577 CrossRef PubMed; D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705 RSC; I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936 CrossRef; R. C. Cammarota, L. J. Clouston and C. C. Lu, Coord. Chem. Rev., 2017, 334, 100 CrossRef; N. P. Mankad, Chem. Commun., 2018, 54, 1291 RSC; C. M. Farley and C. Uyeda, Trends Chem., 2019, 1, 497 CrossRef; J. Campos, Nat. Rev. Chem., 2020, 4, 696 CrossRef; B. Chatterjee, W.-C. Chang, S. Jena and C. Werlé, ACS Catal., 2020, 10, 14024 CrossRef; K. Koessler and B. Butschke, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2021, DOI:10.1002/9781119951438.eibc2782; A. M. Borys and E. Hevia, Trends Chem., 2021, 3, 803 CrossRef; G. Sciortino and F. Maseras, Top. Catal., 2022, 65, 105 CrossRef; M. Navarro, J. J. Moreno, M. Pérez-Jiménez and J. Campos, Chem. Commun., 2022, 58, 11220 RSC.
- See for example: S. Zhang, L. Nguyen, J.-X. Liang, J. Shan, J. Liu, A. I. Frenkel, A. Patlolla, W. Huang, J. Li and F. Tao, Nat. Commun., 2015, 6, 7938 CrossRef CAS PubMed; T. Chen and V. O. Rodionov, ACS Catal., 2016, 6, 4025 CrossRef; M. Filez, Angew. Chem., Int. Ed., 2019, 58, 13220 CrossRef PubMed; A. K. Goulas, S. Sreekumar, Y. Song, P. Kharidehal, G. Gunbas, P. J. Dietrich, G. R. Johnson, Y. C. Wang, A. M. Grippo, L. C. Grabow, A. A. Gokhale and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 6805 CrossRef PubMed.
- P. A. Lindahl, J. Inorg. Biochem., 2012, 106, 172 CrossRef CAS PubMed; J. Clausen and W. Junge, Nature, 2004, 430, 480 CrossRef PubMed; S. J. Lee, M. S. McCormick, S. J. Lippard and U.-S. Cho, Nature, 2013, 494, 380 CrossRef PubMed.
- M. G. Alférez, N. Hidalgo, J. J. Moreno and J. Campos, Angew. Chem., Int. Ed., 2020, 59, 20863 CrossRef PubMed.
- J. E. Bercaw, J. Am. Chem. Soc., 1974, 96, 5087 CrossRef CAS; C. McDade, J. C. Green and J. E. Bercaw, Organometallics, 1982, 1, 1629 CrossRef; A. R. Bulls, W. P. Schaefer, M. Serfas and J. E. Bercaw, Organometallics, 1987, 6, 1219 CrossRef; F. G. N. Cloke, J. P. Day, J. C. Green, C. P. Morley and A. C. Swain, J. Chem. Soc., Dalton Trans., 1991, 789 RSC.
- N. Hidalgo, J. J. Moreno, M. Pérez-Jiménez, C. Maya, J. López-Serrano and J. Campos, Chem. – Eur. J., 2020, 26, 5982 CrossRef CAS PubMed.
- T. Yatabe, T. Kishima, H. Nagano, T. Matsumoto, M. Yamasaki, K.-S. Yoon and S. Ogo, Chem. Lett., 2017, 46, 74 CrossRef CAS.
- L. M. A. Quintana, S. I. Johnson, S. L. Corona, W. Villatoro, W. A. Goddard III, M. K. Takase, D. G. VanderVelde, J. R. Winkler, H. B. Gray and J. D. Blakemore, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 6409 CrossRef CAS PubMed; C. L. Pitman, O. N. L. Finster and A. J. M. Miller, Chem. Commun., 2016, 52, 9105 RSC; M. J. Chalkley, T. J. Del Castillo, B. D. Matson, J. P. Roddy and J. C. Peters, ACS Cent. Sci., 2017, 3, 217 CrossRef PubMed; S. Pal, S. Kusumoto and K. Nozaki, Organometallics, 2018, 37, 906 CrossRef; M. J. Chalkley, T. J. Del Castillo, B. D. Matson and J. C. Peters, J. Am. Chem. Soc., 2018, 140, 6122 CrossRef PubMed; M. J. Chalkley, P. H. Oyala and J. C. Peters, J. Am. Chem. Soc., 2019, 141, 4721 CrossRef PubMed.
- M. G. Alférez, N. Hidalgo and J. Campos, in Frustrated Lewis Pairs, ed. C. Slootweg and A. Jupp, Springer, 2020 Search PubMed; M. Navarro and J. Campos, in Adv. Organomet. Chem, ed. P. J. Pérez, Elsevier, Amsterdam, 2021, ch. 3, vol. 75 Search PubMed.
- J. Campos, J. Am. Chem. Soc., 2017, 139, 2944 CrossRef CAS PubMed; N. Hidalgo, J. J. Moreno, M. Pérez-Jiménez, C. Maya, J. López-Serrano and J. Campos, Chem. – Eur. J., 2020, 26, 5982 CrossRef PubMed; N. Hidalgo, J. J. Moreno, M. Pérez-Jiménez, C. Maya, J. López-Serrano and J. Campos, Organometallics, 2020, 39, 2534 CrossRef PubMed; H. Corona, M. Pérez-Jiménez, F. de la Cruz-Martínez, I. Fernández and J. Campos, Angew. Chem., Int. Ed., 2022, 61, e202207581 CrossRef PubMed.
- J. J. Moreno, M. F. Espada, J. Campos, J. López-Serrano, S. A. Macgregor and E. Carmona, J. Am. Chem. Soc., 2019, 141, 2205 CrossRef CAS PubMed.
- M. Navarro, M. G. Alférez, M. de Sousa, J. Miranda-Pizarro and J. Campos, ACS Catal., 2022, 12, 4227 CrossRef CAS PubMed.
- J. Bauer, H. Braunschweig and R. D. Dewhurst, Chem. Rev., 2012, 112, 4329 CrossRef CAS PubMed.
- Selected examples of ammonia activation by transition metal complexes: (a) J. Zhao, A. S. Goldman and J. F. Hartwig, Science, 2005, 307, 1080 CrossRef CAS PubMed; (b) C. M. Fafard, D. Adhikari, B. M. Foxman, D. J. Mindiola and O. V. Ozerov, J. Am. Chem. Soc., 2007, 129, 10318 CrossRef CAS PubMed; (c) E. Morgan, D. F. MacLean, R. McDonald and L. Turculet, J. Am. Chem. Soc., 2009, 131(40), 14234 CrossRef CAS PubMed; (d) J. Abbenseth, M. Kinauer, F. W. Heinemann, C. Wertele, B. de Bruin, S. Schneider and M. G. Scheibel, Inorg. Chem., 2015, 54, 9290 CrossRef PubMed; (e) C. C. Almquist, N. Removski, T. Rajeshkumar, B. S. Gelfand, L. Maron and W. E. Piers, Angew. Chem., Int. Ed., 2022, 61, e202203576 CrossRef CAS PubMed.
- M. J. Bezdek, S. Guo and P. J. Chirik, Science, 2016, 354, 730 CrossRef CAS PubMed.
- Selected examples of FLPs involving initial binding of the substrate to the acidic site: M. A. Dureen, C. C. Brown and D. W. Stephan, Organometallics, 2010, 29, 6594 CrossRef CAS; R. Liedtke, R. Fröhlich, G. Kehr and G. Erker, Organometallics, 2011, 30, 5222 CrossRef; O. Ekkerta, G. G. Mieraa, T. Wiegand, H. Eckert, B. Schirmer, J. L. Petersen, C. G. Daniliuc, R. Fröhlich, S. Grimme, G. Kehr and G. Erker, Chem. Sci., 2013, 4, 2657 RSC; M. M. Rahman, M. D. Smith and D. V. Peryshkov, Inorg. Chem., 2016, 55, 5101 CrossRef PubMed; N. Aders, L. Keweloh, D. Pleschka, A. Hepp, M. Layh, F. Rogel and W. Uhl, Organometallics, 2019, 38, 2839 CrossRef; I. Bhattacharjee, S. Bhunya and A. Paul, Inorg. Chem., 2020, 59, 1046 CrossRef PubMed; A. Ullah and G. Q. Chen, Org. Chem. Front., 2022, 9, 4421 RSC.
- See for example: P. A. Deck, C. L. Beswick and T. J. Marks, J. Am. Chem. Soc., 1998, 120(8), 1772 CrossRef CAS; S. E. Smith, J. M. Sasaki, R. G. Bergman, J. E. Mondloch and R. G. Finke, J. Am. Chem. Soc., 2008, 130(6), 1839 CrossRef PubMed; S. M. Rummelt and A. Fürstner, Angew. Chem., Int. Ed., 2014, 53, 3626 CrossRef PubMed; Q. Lu, S. Vásquez-Céspedes, T. Gensch and F. Glorius, ACS Catal., 2016, 6, 2352 CrossRef; J. H. Kim, S. Greßies and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 5577 CrossRef PubMed; D.-A. Roşca, K. Radkowski, L. M. Wolf, M. Wagh, R. Goddard, W. Thiel and A. Fürstner, J. Am. Chem. Soc., 2017, 139, 2443 CrossRef PubMed; F. Zhao, B. Xu, D. Ren, L. Han, Z. Yu and T. Liu, Organometallics, 2018, 37, 1026 CrossRef; R. Yamada, N. Iwasawa and J. Takaya, Angew. Chem., Int. Ed., 2019, 58, 17251 CrossRef PubMed; J. Liu, H. Song, T. Wang, J. Jia, Q.-X. Tong, C.-H. Tung and W. Wang, J. Am. Chem. Soc., 2021, 143, 409 CrossRef PubMed; R. Tanaka, Y. Hirata, M. Kojima, T. Yoshino and S. Matsunaga, Chem. Commun., 2022, 58, 76 RSC.
- V. B. Kharitonov, D. V. Muratov and D. A. Loginov, Coord. Chem. Rev., 2019, 399, 213027 CrossRef CAS.
- D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492 CrossRef CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- L. Pauling, J. Am. Chem. Soc., 1947, 69, 542 CrossRef CAS.
- N. Hidalgo, F. de la Cruz-Martínez, M. T. Martín, M. C. Nicasio and J. Campos, Chem. Commun., 2022, 58, 9144 RSC; N. Hidalgo, C. Romero-Pérez, C. Maya, I. Fernández and J. Campos, Organometallics, 2021, 40, 1113 CrossRef CAS PubMed; N. Hidalgo, C. Maya and J. Campos, Chem. Commun., 2019, 55, 8812 RSC.
- S. Bajo, M. G. Alférez, M. M. Alcaide, J. López-Serrano and J. Campos, Chem. – Eur. J., 2020, 26, 16833 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR studies and computational details. CCDC 2223979–2223987. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00410d |
| This journal is © The Royal Society of Chemistry 2023 |