
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirhodium tetraacetate binding to a B-DNA double helical dodecamer probed by X-ray crystallography and mass spectrometry†
Gabriella
Tito
 a,
Romualdo
Troisi
a,
Giarita
Ferraro
a,
Andrea
Geri
b,
Lara
Massai
b,
Luigi
Messori
b,
Filomena
Sica
a and
Antonello
Merlino
*a
a,
Romualdo
Troisi
a,
Giarita
Ferraro
a,
Andrea
Geri
b,
Lara
Massai
b,
Luigi
Messori
b,
Filomena
Sica
a and
Antonello
Merlino
*a
aDepartment of Chemical Sciences, University of Naples Federico II, Complesso Universitario di Monte Sant'Angelo via Cintia, I-80126 Napoli, Italy. E-mail: antonello.merlino@unina.it
bDepartment of Chemistry “U. Schiff”, University of Florence, via della Lastruccia 3, 50019 Sesto Fiorentino, Italy
First published on 9th May 2023
Abstract
The reaction of the cytotoxic compound dirhodium tetraacetate with a B-DNA double helical dodecamer was studied by X-ray crystallography and mass spectrometry. The structure of the dirhodium/DNA adduct reveals a dimetallic center binding to an adenine via axial coordination. Complementary information has been gained through ESI MS measurements. Comparison between the present data and those previously obtained for cisplatin indicates that the two metallodrugs react with this DNA dodecamer in a significantly different fashion.
Dirhodium(II) paddlewheel complexes of the general formula [Rh2(O2CR)4]L2 (R = CH3−, CH3CH2−, etc.) contain a Rh(II)–Rh(II) bond, four bridging equatorial O2CR ligands arranged in a lantern-like fashion around the Rh atoms, and two donor ligands (L) coordinated through a dative bond to Rh atoms at the axial coordination site.1 These molecules exert anti-bacterial action against Streptococcus pneumoniae2 and cytostatic activity against L1210 tumours,3,4 P388 leukaemia and sarcoma 180,5 human oral KB6 and Ehrlich-Lettre ascites carcinomas,7 and the MDA-MB-231 breast cancer cell line.8
The exact mechanism at the basis of these properties has not been elucidated yet. Cytotoxic activity could be related to the ability of dirhodium tetracarboxylates to bind nucleic acids,9 to induce damage of nuclear DNA,10 and to inhibit DNA replication and transcription and protein synthesis.11,12 A different mechanism of action involving the inhibition of the ubiquitin–proteasome system has also been proposed.13 Thus, interaction with proteins, RNA and DNA could play a crucial role in these processes. For this reason, it is of fundamental importance to study the dirhodium/protein and dirhodium/nucleic acid recognition mechanism at the molecular level.
X-ray crystallography studies in combination with other biophysical techniques have recently demonstrated that dirhodium tetraacetate ([Rh2(μ-O2CCH3)4])14–17 (Fig. 1) and its derivatives cis-[Rh2(μ-O2CCH3)2(μ-O2CCF3)2]18 and [Rh2(μ-O2CCH3)(μ-O2CCF3)3]19 bind the model proteins hen egg white lysozyme and bovine pancreatic ribonuclease forming a variety of adducts in which the dirhodium core either axially coordinates the N atoms of His side chains or is degraded. In the latter case, monometallic Rh centers bind the side chains of Asp, Lys, Asn and/or the protein C-terminal tail.17
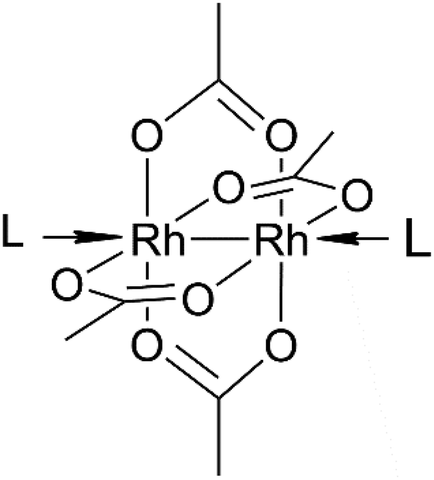 | ||
| Fig. 1 Structure of dirhodium tetraacetate. L, axial ligand. | ||
Several studies on the interaction of [Rh2(μ-O2CCH3)4] and its derivatives with purine bases, nucleotides, and oligonucleotides have also been reported, with contrasting results. It has been demonstrated that the dirhodium unit has a preference for axial binding to purine bases,20 and in particular to adenine (Ade),21,22 but equatorial interaction to purines can also occur.23,24 Axial binding to Ade occurs at N7 or N1 atoms and is stabilized by hydrogen bonds formed by the oxygen atoms of carboxylate ligands and adenine NH2 groups.25 Dirhodium binding to Ade bases can also occur equatorially, in a bridging fashion, through simultaneous binding to N7 and N6 atoms.26,27 Nuclear magnetic resonance (NMR) data demonstrated equatorial binding of dirhodium to Ade and guanine (Gua) bases.28–30 Bidimensional NMR analysis and enzyme digestion studies suggested that the dirhodium center can cross-link the major groove bases cytosine(Cyt)5 and Ade6 of the d(C-T-C-T-C-A-A-C-T-T-C-C)/d(G-G-A-A-G-T-T-G-A-G-A-G) double helix via binding to the N3 atom of Cyt5 and the N7 atom of Ade6.31 In the formed adduct, equatorial binding of a cis-[Rh2(μ-O2CCH3)2(η1-O2CCH3)]+ fragment to the DNA sequence occurs, with the dirhodium center retaining one monodentate and two bridging acetate ligands.31 Matrix-assisted laser desorption ionization and nanoelectrospray coupled to time-of-flight mass spectrometry experiments have shown that dirhodium tetraacetate [Rh2(μ-O2CCH3)4] and its cytotoxic derivatives, [Rh2(μ-O2CCH3)2(CH3CN)6(BF4)2] and [Rh2(μ-O2CCF3)4], are able to bind a DNA model sequence forming adducts in which the Rh–Rh bond and the coordinated equatorial ligands remain intact.32 The reactivity of these molecules in comparison with cisplatin has also been investigated, with cisplatin reacting with DNA tetra- and dodecamers faster than [Rh2(μ-O2CCH3)4] but less quickly than [Rh2(O2CCF3)4].32 Furthermore, it has been demonstrated that GuaGua-containing dodecamers react faster with [Rh2(μ-O2CCF3)4] and [Rh2(μ-O2CCH3)2(CH3CN)6(BF4)2], whereas AdeAde-containing oligonucleotides react faster with [Rh2(μ-O2CCH3)4]. It is possible that in the final dirhodium/DNA adducts the dirhodium center could bind purine bases at the equatorial sites, while the DNA/dirhodium recognition should occur through labile interactions at the axial site.32
To directly compare at the molecular level the interaction of dirhodium compounds and cisplatin with the same DNA double helix, we solved the crystal structure of the adduct formed upon reaction of [Rh2(μ-O2CCH3)4] with the B-DNA dodecamer of the sequence C-G-C-G-A-A-T-T-C-G-C-G, which has been already used to study the reactivity of cisplatin and its derivatives with DNA.33
To evaluate the effect of DNA binding on the electronic properties of the metal compound, UV-vis absorption spectra of [Rh2(μ-O2CCH3)4] in the absence and in the presence of the dodecamer were collected as a function of time in 60 mM sodium cacodylate at pH 6.5, i.e. in the buffer used for crystallization. Under these conditions, the spectrum of [Rh2(μ-O2CCH3)4] shows two peaks at 444 and 587 nm which do not change with time (Fig. S1†). These spectral features are similar to those already reported in sodium citrate (pH 5.1) and ammonium acetate (pH 6.8) solutions.15 Following literature assignment,15 the band at 444 nm was assigned to the Rh2(π*) → Rh–O(σ*) transitions of the tetraacetate ligands, while that at 587 nm was attributed to the Rh2(π*) → Rh2(σ*) transition of the metal–metal single bond. In the presence of DNA (Fig. 2), there is a blue shift of λmax from 587 nm to 582 nm, while the peak at 444 nm remains unaltered. The spectrum experiences a minor change in the absorbance with time. The comparison between the spectra of the compound alone and in the presence of DNA suggests a direct DNA interaction to the dirhodium center. To verify if the binding could alter the structure of the dodecamer, CD spectra of the selected DNA model system with [Rh2(μ-O2CCH3)4] in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 and 1:10 molar ratios were registered. Spectra analysis reveals that the metal compound binding to the DNA does not alter the structure of the dodecamer in solution (Fig. S2†). Similar conclusions can be drawn from thermal shift assays (Table S1†), which reveal that the binding of [Rh2(μ-O2CCH3)4] to the dodecamer does not lead to a change in the DNA melting temperature, contrary to what was previously found with another DNA sequence.31
:2 and 1:10 molar ratios were registered. Spectra analysis reveals that the metal compound binding to the DNA does not alter the structure of the dodecamer in solution (Fig. S2†). Similar conclusions can be drawn from thermal shift assays (Table S1†), which reveal that the binding of [Rh2(μ-O2CCH3)4] to the dodecamer does not lead to a change in the DNA melting temperature, contrary to what was previously found with another DNA sequence.31
 | ||
| Fig. 2 Time course UV-vis spectra of 0.5 mM [Rh2(μ-O2CCH3)4] in 60 mM sodium cacodylate buffer (pH 6.5) in the presence of DNA (DNA to metal molar ratio: 1:2). | ||
Crystals of the DNA adduct with [Rh2(μ-O2CCH3)4] were then obtained by the soaking strategy: crystals of C-G-C-G-A-A-T-T-C-G-C-G were grown, as previously described,33 in 7% v/v MPD, 20 mM MgCl2, 80 mM spermine, and 60 mM sodium cacodylate at pH 6.5 and the adduct with [Rh2(μ-O2CCH3)4] was formed exposing the oligonucleotide crystals to reservoir solutions saturated with the metal compound for 7 days. Crystals of the dirhodium/DNA adduct diffract X-rays at 1.24 Å resolution. The structure contains 487 oligonucleotide atoms, a Mg2+ cation with six water O atoms arranged octahedrally around the metal centre, a chloride ion, 167 water molecules and a dirhodium containing moiety. Mg2+ is often used in oligonucleotide crystallization because of its biological relevance and its ability to stabilize the DNA structure favouring crystal growth. The structure of the dirhodium/DNA adduct refines with a final R-factor value of 19.7% (and a final Rfree value of 22.3%). The overall DNA model in the adduct (Fig. 3) is similar to that of the metallodrug-free DNA (PDB code 3U2N) (Fig. S3†).34 The root mean square deviation (rmsd) between the DNA coordinates is 0.722 Å. The main differences between the two duplexes are located at the 5′ and 3′ ends of the two strands, as evidenced by a detailed comparison of some local base-pair step conformational parameters reported in Fig. S4,† and close to the metal binding site at the level of Ade5. Here, a dimetallic center is bound to the N7 atom via coordination to the axial site (Fig. 4), with low occupancy (0.20). The other axial site is occupied by a water molecule. The other dirhodium ligands are not well defined, but this is not surprising since the Rh–Rh bond allows free rotation of the bridging carboxylate ligands around the Rh–Rh axis and metal ligands are often disordered in the X-ray structures of the adducts with biological macromolecules.35 These results well agree with previous mass spectrometry literature data suggesting that dirhodium moieties with just one or two acetate ligands, or even nude dirhodium moieties, bind the C-G-C-G-A-A-T-T-C-G-C-G and C-C-T-T-C-G-G-T-C-T-C-C sequences.32 Comparison between our results, showing the binding of dirhodium to Ade5, and those obtained with cisplatin (PDB code 5BNA), which binds to Gua4, Gua10 and Gua16 (Fig. 3),33 reveals that the two cytotoxic metallodrugs show a different reactivity with DNA. Notably, comparing the dirhodium tetraacetate binding to proteins14–20 and DNA model systems, it emerges that the metal compound seems to prefer the axial coordination to N atoms rather than the equatorial binding, contrarily to what found in the reactivity of diruthenium compounds with proteins.36–38
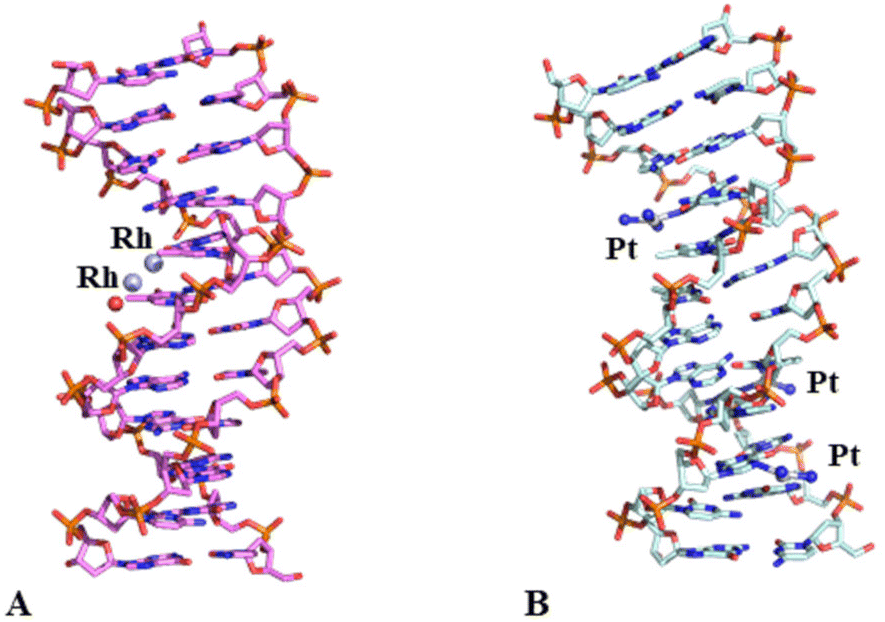 | ||
| Fig. 3 Overall structures of the adducts formed upon reaction of dirhodium tetraacetate (panel A, PDB code 8CE2, here solved) and cisplatin (panel B, PDB code 5BNA)33 with the same DNA oligonucleotide. Metal binding sites are shown. The rmsd between the two structures is 0.727 Å. | ||
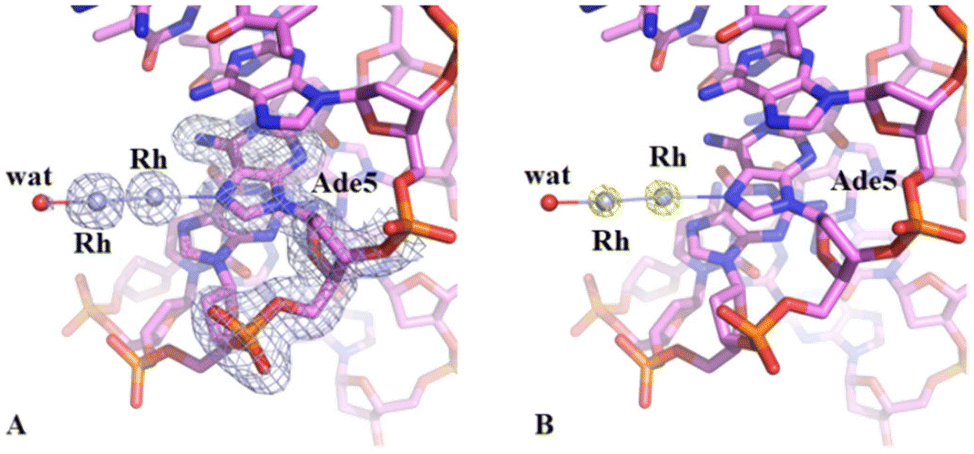 | ||
| Fig. 4 Metal binding site in the adduct formed upon reaction of dirhodium tetraacetate with the C-G-C-G-A-A-T-T-C-G-C-G duplex. 2Fo–Fc electron density map is reported at 0.8 sigma value in panel A. Anomalous difference electron density map is reported at 3.0 sigma value in panel B. | ||
The interactions of dirhodium tetraacetate with the B-DNA Dickerson dodecamer have been further investigated by ESI MS measurements. ESI MS is indeed a very potent tool to analyze the interaction of short DNA oligonucleotides with a variety of ligands as nicely documented by a recent review paper by Gabelica et al.39 The deconvoluted ESI MS spectrum of the untreated DNA duplex in 100 mM ammonium acetate buffer (pH 6.8) is shown in Fig. 5A. Notably, the ESI MS experiment, in line with previous observations,40 causes extensive separation of the two DNA strands so that the spectrum shows intense peaks for both the single strand (SS, 3645.713 Da) and double strand (DS, 7290.466 Da) species. The ratio between the intensities of the two peaks may be finely tuned by adjusting the parameters of the ESI MS experiment. The experiment was then repeated following treatment of the double stranded DNA with [Rh2(μ-O2CCH3)4], in a 1:5 DNA-to-complex ratio, upon 5 hours of incubation at 37 °C. Interestingly, the resulting spectrum shows an additional peak (4086.671 Da) of lower intensity, corresponding to adducts formed between the DNA oligo and the metal complex (Fig. 5B). This adduct is clearly seen exclusively at the level of the SS species; the increase in mass well corresponds to the binding of the dirhodium tetraacetate fragment to the DNA. Notably, upon repeating the experiment after 24 hours of incubation (Fig. 5C), new peaks are observed corresponding to novel adducts of both SS and DS bearing the monorhodium tetraacetate fragment (at 3984.577 Da and 7629.367 Da, respectively). A rather straightforward interpretation of these results may be offered taking into account that the duplex species is very stable in aqueous solution and that strand separation is just the effect of the ESI process. These results suggest that when bound to DNA under the investigated experimental conditions, dirhodium tetraacetate may slowly lose one rhodium atom and may convert to a monorhodium tetraacetate species.
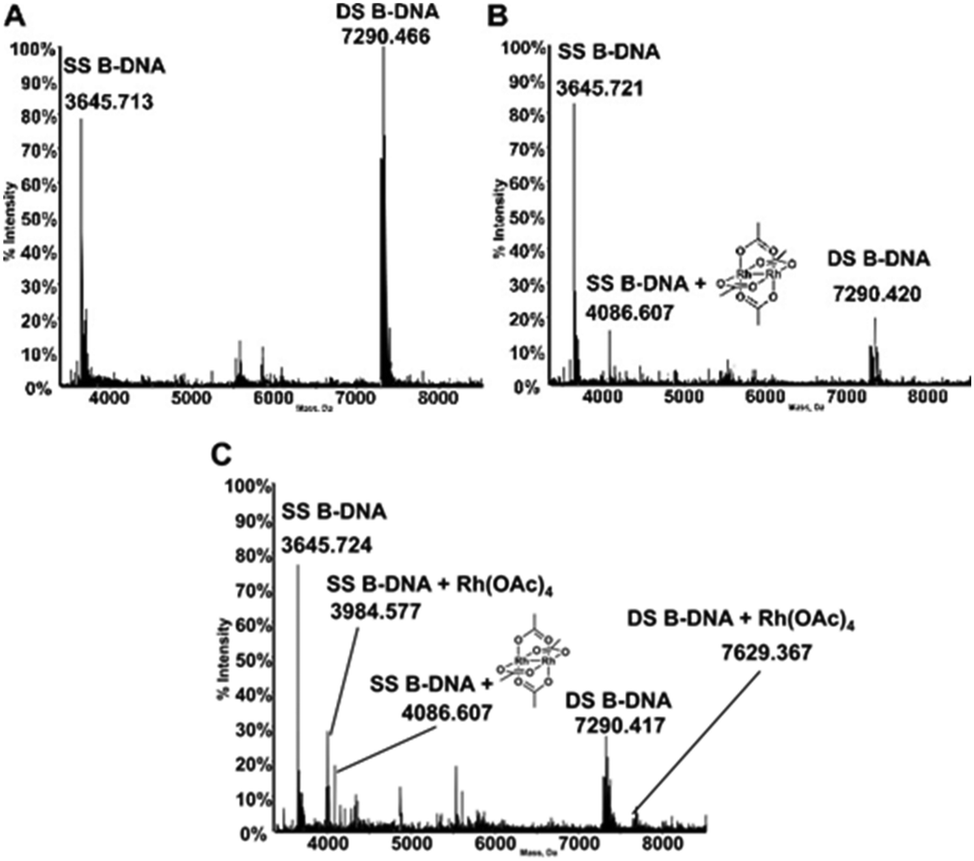 | ||
| Fig. 5 (A) Deconvoluted ESI-MS mass spectrum of B-DNA 5 × 10−6 M in 100 mM NH4OAc at pH 6.8, incubated at 37 °C with dirhodium tetraacetate in a 1:5 DNA-to-complex ratio for (B) 5 h and (C) 24 h; 20% of methanol was added just before injection. | ||
Overall, these ESI MS results are broadly consistent with the crystallographic data and point out that [Rh2(μ-O2CCH3)4] forms adducts with DNA through direct coordination of the rhodium center to DNA nucleobases. Yet, ESI MS results suggest that the Rh–Rh bond may undergo cleavage with time so that the dirhodium tetraacetate fragment is converted to the monorhodium species. Cleavage of the Rh–Rh bond and formation of monometallic Rh-containing fragments were earlier found in the crystal structure of dirhodium/protein adducts.14
Conclusions
In conclusion, we have here reported the first crystal structure of an adduct formed upon reaction of a dirhodium paddlewheel complex, [Rh2(μ-O2CCH3)4], and a DNA duplex model system. A search in the Protein Data Bank reveals that it also represents the first example of a Rh/DNA adduct and a rare example of a Rh compound/DNA complex. The results indicate that [Rh2(μ-O2CCH3)4] interacts with DNA through coordination of the dimetallic center to the N7 atom of an adenine, at the axial position. This view is supported by new ESI MS experiments. The latter point out that at short incubation times, adducts are formed between the DNA and [Rh2(μ-O2CCH3)4] in agreement with crystallographic results. Conversely, for longer incubation times, ESI MS results point out that the dirhodium tetraacetate fragment converts into a monorhodium tetraacetate fragment as the consequence of progressive cleavage of the Rh–Rh bond. In addition, the present data also indicate that dirhodium tetraacetate reacts with DNA differently from cisplatin33 and that its interaction with DNA and proteins14,17,41,42 could occur with a similar mechanism that involves the dirhodium coordination to a N atom at the axial site. Overall, these results support the idea that dirhodium compounds can act through binding to DNA and provide a further step towards the understanding of the molecular basis of the antitumor activity of dirhodium tetracarboxylates. Indeed, our data support the recent observation that vacant axial sites are important for the cytotoxic activity and nuclear accumulation of dirhodium compounds.8Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Elettra synchrotron staff for technical assistance during data collection.References
- R. Hrdina, Eur. J. Inorg. Chem., 2021, 2021, 501–528 CrossRef CAS
.
- X.-Y. Yang, J.-Y. Xu, M. Meng, N. Li, C.-Y. Liu and Q.-Y. He, J. Proteomics, 2019, 194, 160–167 CrossRef CAS PubMed
- R. G. Hughes, J. L. Bear and A. P. Kimball, Cancer Res., 1972, 120 Search PubMed
- R. A. Howard, A. P. Kimball and J. L. Bear, Cancer Res., 1979, 39, 2568–2573 CAS
- J. L. Bear, Precious Metals 1985: Proceedings of the Ninth International Precious Metals Conference, 1986.
- F. Pruchnik and D. Duś, J. Inorg. Biochem., 1996, 61, 55–61 CrossRef CAS
- A. Erck, L. Rainen, J. Whileyman, I.-M. Chang, A. P. Kimball and J. Bear, Exp. Biol. Med., 1974, 145, 1278–1283 CrossRef CAS PubMed
- A. Enriquez Garcia, B. Lai, S. G. Gopinathan, H. H. Harris, C. S. Shemanko and F. Jalilehvand, Chem. Commun., 2019, 55, 8223–8226 RSC
- H. T. Chifotides and K. R. Dunbar, Acc. Chem. Res., 2005, 38, 146–156 CrossRef CAS PubMed
- J. D. Aguirre, A. M. Angeles-Boza, A. Chouai, J.-P. Pellois, C. Turro and K. R. Dunbar, J. Am. Chem. Soc., 2009, 131, 11353–11360 CrossRef CAS PubMed
- Md. M. Rahman, H. Yasuda, K. Takashima and A. Mizuno, Nucleic Acids Symp. Ser., 2007, 51, 89–90 CrossRef PubMed
- K. Sorasaenee, P. K.-L. Fu, A. M. Angeles-Boza, K. R. Dunbar and C. Turro, Inorg. Chem., 2003, 42, 1267–1271 CrossRef CAS PubMed
- F.-M. Siu, I. W.-S. Lin, K. Yan, C.-N. Lok, K.-H. Low, T. Y.-C. Leung, T.-L. Lam and C.-M. Che, Chem. Sci., 2012, 3, 1785 RSC
- D. Loreto and A. Merlino, Coord. Chem. Rev., 2021, 442, 213999 CrossRef CAS
- G. Ferraro, A. Pratesi, L. Messori and A. Merlino, Dalton Trans., 2020, 49, 2412–2416 RSC
- D. Loreto, F. Fasulo, A. B. Muñoz-García, M. Pavone and A. Merlino, Inorg. Chem., 2022, 61, 8402–8405 CrossRef CAS PubMed
- D. Loreto, G. Ferraro and A. Merlino, Int. J. Mater. Sci., 2021, 22, 1496 CAS
- D. Loreto, A. Esposito, N. Demitri, A. Guaragna and A. Merlino, Dalton Trans., 2022, 51, 3695–3705 RSC
- D. Loreto, A. Esposito, N. Demitri, A. Guaragna and A. Merlino, Dalton Trans., 2022, 51, 7294–7304 RSC
- P. Amo-Ochoa, O. Castillo, R. W. Harrington, F. Zamora and A. Houlton, Inorg. Chem., 2013, 52, 2174–2181 CrossRef CAS PubMed
- G. Pneumatikakis and N. Hadjiliadis, J. Chem. Soc., Dalton Trans., 1979, 596–599 RSC
- K. V. Catalan, J. S. Hess, M. M. Maloney, D. J. Mindiola, D. L. Ward and K. R. Dunbar, Inorg. Chem., 1999, 38, 3904–3913 CrossRef CAS
- K. R. Dunbar, J. H. Matonic, V. P. Saharan, C. A. Crawford and G. Christou, J. Am. Chem. Soc., 1994, 116, 2201–2202 CrossRef CAS
- C. A. Crawford, J. H. Matonic, V. P. Saharan, K. Folting, K. R. Dunbar and G. Christou, J. Inorg. Biochem., 1993, 51, 401 CrossRef
- J. R. Rubin, T. P. Haromy and M. Sundaralingam, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 1712–1714 CrossRef PubMed
- H. T. Chifotides and K. R. Dunbar, J. Am. Chem. Soc., 2007, 129, 12480–12490 CrossRef CAS PubMed
- D. V. Deubel, J. Am. Chem. Soc., 2008, 130, 665–675 CrossRef CAS PubMed
- A. M. Palmer, J. D. Knoll and C. Turro, Dalton Trans., 2015, 44, 3640–3646 RSC
- H. T. Chifotides, K. M. Koshlap, L. M. Pérez and K. R. Dunbar, J. Am. Chem. Soc., 2003, 125, 10714–10724 CrossRef CAS PubMed
- S. U. Dunham, H. T. Chifotides, S. Mikulski, A. E. Burr and K. R. Dunbar, Biochemistry, 2005, 44, 996–1003 CrossRef CAS PubMed
- M. Kang, H. T. Chifotides and K. R. Dunbar, Biochemistry, 2008, 47, 2265–2276 CrossRef CAS PubMed
- H. T. Chifotides, J. M. Koomen, M. Kang, S. E. Tichy, K. R. Dunbar and D. H. Russell, Inorg. Chem., 2004, 43, 6177–6187 CrossRef CAS PubMed
- R. M. Wing, P. Pjura, H. R. Drew and R. E. Dickerson, EMBO J., 1984, 3, 1201–1206 CrossRef CAS PubMed
- D. Wei, W. D. Wilson and S. Neidle, J. Am. Chem. Soc., 2013, 135, 1369–1377 CrossRef CAS PubMed
- I. Russo Krauss, G. Ferraro, A. Pica, J. A. Márquez, J. R. Helliwell and A. Merlino, Metallomics, 2017, 9, 1534–1547 CrossRef CAS PubMed
- L. Messori, T. Marzo, R. N. F. Sanches, Hanif-Ur-Rehman, D. de Oliveira Silva and A. Merlino, Angew. Chem., Int. Ed., 2014, 53, 6172–6175 CrossRef CAS PubMed
- I. Tolbatov and A. Marrone, Inorg. Chem., 2022, 61, 16421–16429 CrossRef CAS PubMed
- A. Terán, G. Ferraro, A. E. Sánchez-Peláez, S. Herrero and A. Merlino, Inorg. Chem., 2023, 62(2), 670–674 CrossRef PubMed
- E. Largy, A. König, A. Ghosh, D. Ghosh, S. Benabou, F. Rosu and V. Gabelica, Chem. Rev., 2022, 122, 7720–7839 CrossRef CAS PubMed
- M. Groessl, Y. O. Tsybin, C. G. Hartinger, B. K. Keppler and P. J. Dyson, J. Biol. Inorg. Chem., 2010, 15, 677–688 CrossRef CAS PubMed
- F. Jalilehvand, A. Enriquez Garcia, P. Niksirat, Y. Z. Finfrock and B. S. Gelfand, J. Inorg. Biochem., 2021, 224, 111556 CrossRef CAS PubMed
- I. Tolbatov and A. Marrone, Inorg. Chim. Acta, 2022, 530, 120686 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt00320e |
| This journal is © The Royal Society of Chemistry 2023 |