
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The design and synthesis of green emissive iridium(III) complexes guided by calculations of the vibrationally-resolved emission spectra†
Campbell Frank Ross
Mackenzie
 a,
Seung-Yeon
Kwak
b,
Sungmin
Kim
b and
Eli
Zysman-Colman
*a
a,
Seung-Yeon
Kwak
b,
Sungmin
Kim
b and
Eli
Zysman-Colman
*a
aOrganic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, St Andrews, Fife KY16 9ST, UK. E-mail: eli.zysman-colman@st-andrews.ac.uk
bSamsung Advanced Institute of Technology, Samsung Electronics Co. Ltd., Suwon, Gyeonggi-do 16678, Republic of Korea
First published on 28th February 2023
Abstract
A key challenge in developing emissive materials for organic light-emitting diodes is to optimize their colour saturation, which means targeting narrowband emitters. In this combined theoretical and experimental study, we investigate the use of heavy atoms in the form of trimethylsilyl groups as a tool to reduce the intensity of the vibrations in the 2-phenylpyridinato ligands of emissive iridium(III) complexes that contribute to the vibrationally coupled modes that broaden the emission profile. An underutilised computational technique, Frank-Condon vibrationally coupled electronic spectral modelling, was used to identify the key vibrational modes that contribute to the broadening of the emission spectra in known benchmark green-emitting iridium(III) complexes. Based on these results, a family of eight new green-emitting iridium complexes containing trimethylsilyl groups substituted at different positions of the cyclometalating ligands has been prepared to explore the impact that these substituents have on reducing the intensity of the vibrations and the resulting reduction in the contribution of vibrationally coupled emission modes to the shape of the emission spectra. We have demonstrated that locating a trimethylsilyl group at the N4 or N5 position of the 2-phenylpyridine ligand damps the vibrational modes of the iridium complex and provides a modest narrowing of the emission spectrum of 8–9 nm (or 350 cm−1). The strong correlation between experimental and calculated emission spectra highlights the utility of this computational method to understand how the vibrational modes contribute to the profile of the emission spectra in phosphorescent iridium(III) emitters.
Introduction
Organic light-emitting diode (OLED) displays have excellent contrast ratios, high brightness and show very good colour reproduction. Many of these benefits are the result of the high colour purity for each of the red, green, and blue pixels that are used in the display. The colour purity of the emitted light from the device can be improved using both intrinsic and extrinsic solutions. These include modifying the structure of the emitter to produce a narrowing of the emission spectrum,1 incorporating colour filters2 or modifying the device optics with the use of microcavities.3,4 Colour filters reduce the light output and therefore efficiency of the display, while microcavities are optimised for a single emission colour and are difficult to implement in large area displays. The most desirable solution would be to develop narrowband emitters (Fig. 1a) with much smaller full width at half maxima (FWHM), thereby obviating the need for extrinsic colour filters.1 Industry standards for 8k displays and beyond demand even wider colour gamuts and this will require even more saturated colour from each of the red, green and blue emitters. For example, the green colour point in the Rec.2020 colour space for UHDTVs has CIE1931 coordinates of [0.170, 0.797],5 whereas the current HDTV standard (DCI-P3) has CIE coordinates of [0.265, 0.690] (Fig. 1b).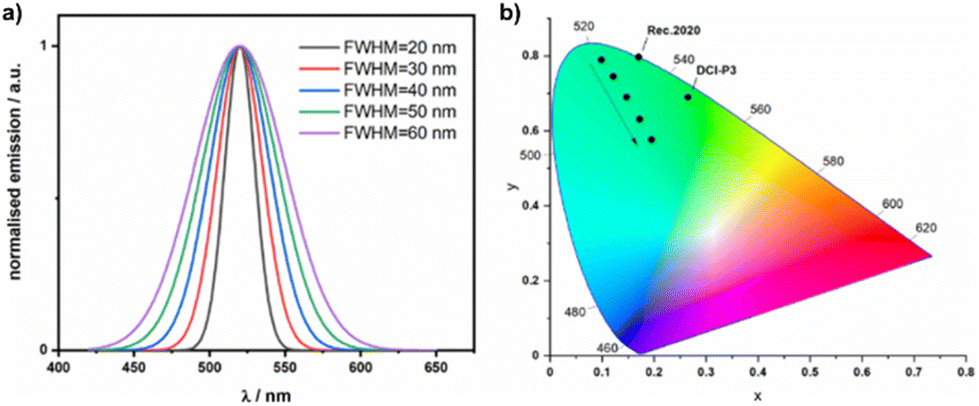 | ||
| Fig. 1 (a) Simulated emission spectra at 520 nm, with Gaussian shaped emission and FWHM ranging from 20 to 60 nm. (b) CIE1931 coordinates of the simulated emission spectra, the arrow shows the shift in coordinates moving from 20 nm to 60 nm FWHM, the CIE coordinates of the green standard for Rec.2020 and DCI-P3 are also marked. | ||
Current OLED displays use phosphorescent iridium complexes as the emitters for the red and green pixels.6,7 The band shape of the emission spectra from emissive iridium complexes is strongly tied to the nature of the emissive excited state. Complexes with mixed metal-to-ligand/ligand-to-ligand charge transfer 3MLCT/3LLCT T1 states show broad, featureless emission, the result of large geometric reorganization in the excited triplet state, while complexes with locally excited 3LC T1 states have narrower and vibronically structured emission profiles.1,8 Unfortunately, emission from 3LC states generally results in longer radiative lifetimes and lower photoluminescence quantum yields (ΦPL) than emission from 3MLCT/3LLCT due to the reduced spin orbit coupling (SOC) as the iridium is not implicated in the former.8,9 To target narrowband phosphorescence while retaining fast, efficient emission from iridium complexes, the goal is to design complexes with mixed 3MLCT/3LLCT/3LC excited states. This is very challenging as both states are sensitive to subtle changes in the electronics of the ligands about the metal centre.8,9 The brute-force way to access emitters with mixed 3MLCT/3LLCT/3LC states is to screen a range of closely related molecules to assess how minor changes in the structure of the ligands impact the nature of the excited state.8,10–12
One of the key requirements for narrowband emission is reducing the vibrational coupling within the molecule that manifests in the presence of additional, lower energy emission bands to the main one.1 There are several design strategies to reduce this vibronic coupling. One is to closely match the geometry of both the ground and excited states, which can be achieved by employing a rigid molecular design.13,14 An alternative strategy is to reduce the amplitude of the vibrations that are strongly coupled to the electronic transitions, which can be achieved by increasing the steric bulk of the ligands on the complex as this dampens high-frequency C–H vibrational modes. There are a large array of different functional groups that can be substituted onto an emissive iridium(III) complex to suppress vibrational coupling without impacting significantly the emission energy. Examples of groups that can suppress vibronic coupling in iridium complexes can be as small as a methyl group,15 or longer alkyl chains,16 aryl groups11,17 or heteroatom-containing functional groups such as silanes.18–24 Previously reported iridium complexes containing trimethylsilyl groups on a ppy ligand are shown in Chart 1 and their photophysical properties are reported in Table 1.
 | ||
| Chart 1 Structures of previously published iridium complexes containing trimethylsilyl groups. | ||
| Complex | λ PL/nm | FWHM/nm | τ PL/μs | Φ PL/% | Ref. |
|---|---|---|---|---|---|
| a In DCM solution. b In toluene solution. c FWHM extracted from the available emission spectrum. | |||||
| fac-Ir(ppyTMS)3 | 521 | 51c | NA | 42 | 18 |
| fac-Ir(mPppyTMS)3 | 527 | 44c | NA | 45 | 18 |
| fac-Ir(dsippy)3 | 519 | 50 | 1.45 | 52 | 19 |
| Ir(F2-m-TMS)2(fptp) | 462 | 49c | 1.8 | 75 | 21 |
| Ir(F2-p-TMS)2(fptp) | 461 | 51c | 2.1 | 76 | 21 |
| GD1 | 520 | 59 | 1.55 | 82 | 22 |
| GD2 | 519 | 58 | 1.74 | 70 | 22 |
| GD3 | 519 | 57 | 1.68 | 71 | 22 |
| GD4 | 519 | 57 | 1.58 | 74 | 22 |
| Ir(PQ)2(TMSppy) | 603 | 78c | NA | 13 | 23 |
| fac-Ir(ppy)3 | 519 | 81c | 1.6 | 90 | 25 |
| IrPic | 505 | 88c | 0.51 | 15 | 26 |
| Ir(ppy)2(acac) | 519 | 70c | 2.35 | 11 | 27 |
One available yet underutilised tool to study vibrational coupling in emissive complexes is to calculate the vibrationally coupled emission spectrum.28–32 Several studies have demonstrated good correlation between experimental and simulated emission spectra of existing iridium- and platinum- containing emitters,29,33–35 but there are very few reports containing vibrational studies that are used as a tool for the design of new complexes.9,36–38
With a view to obtaining improved colour green emitters, here we modelled the vibrationally-coupled emission spectra, of a family of trimethylsilane-substituted green-emissive iridium complexes and then correlated these findings to their photophysical properties.
Results and discussion
Vibrational calculation of iridium complexes
The process to calculate a vibrationally coupled emission spectrum also identifies the main vibrational modes that contribute to the spectral shape. Visualisation of these vibrational modes can be used to identify the parts of the molecule where damping these vibrational modes could have a significant positive impact on the spectral profile. We first modelled the vibrationally coupled emission spectra of three widely studied green-emissive iridium complexes, fac-Ir(ppy)3, Ir(ppy)2(acac) and Ir(ppy)2(pic) (ppy = 2-phenylpyridinato; acac = acetylacetonate; pic = 2-piconilate) using the Adiabatic Hessian (AH) method, which has been demonstrated to work well for phosphorescent iridium complexes.31,32Fig. 2 shows a comparison of the experimental and simulated emission spectra for fac-Ir(ppy)3, IrPic and Ir(ppy)2(acac). The major vibrational modes that contribute to the secondary vibrational peak in the emission spectra at around 580 nm are tabulated in Table S5.† For fac-Ir(ppy)3 and Ir(ppy)2(pic), this emission band results from a single vibrational mode (mode 141 for fac-Ir(ppy)3 and mode 126 for Ir(ppy)2(pic)), while for Ir(ppy)2(acac), there are two almost identical vibrations (modes 125 and 126). Visualisation of the main contributing mode for each molecule provides information on their localization within the molecule (Fig. 3).
 | ||
| Fig. 2 Comparison of simulated and experimental emission spectra (in degassed toluene, λexc = 400 nm) for (a) fac-Ir(ppy)3, (b) Ir(ppy)2(acac) and (c) Ir(ppy)2(pic). (d) An overlay of the simulated emission spectra for these three complexes. | ||
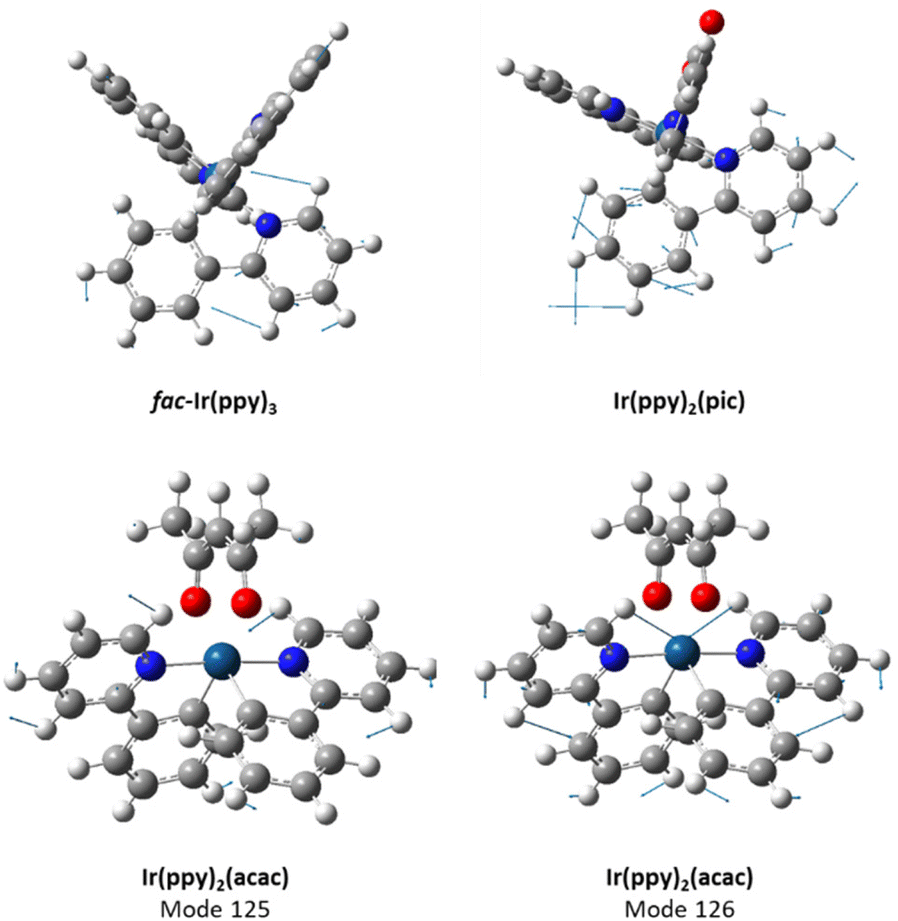 | ||
| Fig. 3 Visualisation of the key vibrational modes for fac-Ir(ppy)3 (mode 141, ν = 1512 cm−1), Ir(ppy)2(pic) (mode 126, ν = 1593 cm−1) and Ir(ppy)2(acac) (mode 125, ν = 1515 cm−1 and mode 126, ν = 1516 cm−1). | ||
The vibration of fac-Ir(ppy)3 can best be described as a ring breathing mode of the pyridyl ring from one of the ppy ligands, with significant contribution from the attached phenyl ring, as well as some minor involvement of the other ligands. The vibration of Ir(ppy)2(acac) can best be described as a ring breathing mode of both pyridyl rings, with contribution from the attached phenyl rings and minor contributions from the acac ligand; in mode 125, the two pyridyl rings are out of phase, while in mode 126 they are in phase. In contrast, the dominant vibration mode in Ir(ppy)2(pic) is best described as a ring breathing mode of the aryl rings in a single ppy ligand.
Based on this analysis, Ir(ppy)2(pic) (Irpic) was chosen as the reference complex for the follow-on study due to the localisation of the key vibrational mode on a single ligand. Irpic and its derivatives have been widely studied, are readily synthesised, have short photoluminescence lifetimes, τPL, and high ΦPL.12
Target molecules
We targeted the use of trimethylsilyl groups as structural dampeners. We designed a family of eight iridium complexes bearing trimethylsilyl substituents on either (or both) of the phenyl and pyridine rings of the cyclometallating ligands (Chart 2). A naming scheme for the complexes was developed based on the substitution pattern of the trimethylsilyl groups, where each complex is identified by the location of the trimethylsilyl group on either the phenyl (C4/C5) and/or pyridine (N4/N5) ring. | ||
| Chart 2 (a) Structure of the parent iridium complex IrPic, (b) atom numbering for scheme for the atoms in the 2-phenylpyridinato ligand, (c) the structures of the eight new trimethylsilyl-substituted complexes synthesised during this project, along with the naming scheme for the complexes. | ||
The introduction of trimethylsilyl groups into emissive iridium complexes has numerous benefits: their synthesis is facile, they increase the vapour pressure and thermal stability of the complex, and they can improve the solubility of the complex.19
Synthesis
The targeted complexes required the synthesis of eight trimethylsilylated 2-phenylpyridine ligands. Of these, three had already been reported in the literature: (2-phenyl-5-trimethylsilylpyridine (N5Sippy), 2-(3-trimethylsilylphenyl)-5-trimethylsilylpyridine (C3N5Si2ppy) and 2-(4-trimethylsilylphenyl)-5-trimethylsilylpyridine (C4N5Si2ppy)),18,19,21,22,39 while the remaining five ligands are new. The ligands were synthesised through the Suzuki–Miyaura coupling of appropriately substituted phenyl boronic acids and 2-bromopyridine (Fig. S1†). Trimethylsilyl-substituted phenyl boronic acids are commercially available, while trimethylsilyl-substituted 2-bromopyridine starting materials were readily obtained from dibromopyridine precursors.40 The target complexes were synthesised using a two-step reaction protocol. Firstly, the ligands were reacted with [Ir(COD)Cl]2 to form a chloro-bridged iridium dimer, which was then reacted with picolinic acid in the presence of an inorganic base to afford the target complexes (Fig. S2 and S3†). Most of the complexes were isolated in good to excellent yields (dimers formed in >80% yield, picolinate complexes in >60% yield). The lower isolated yields for some highly silylated complexes was attributed to their higher solubility, which adversely affected the recovery of material during recrystallisation.The identity of all the ligands and iridium complexes was confirmed by a combination of 1H, 13C and 29Si NMR spectroscopy, as well as high-resolution mass spectrometry of each complex. The purity of the final complexes was verified by high-pressure liquid chromatography (HPLC) and melting point determination.
Electrochemistry
The electrochemical properties of the complexes were studied by cyclic voltammetry and differential pulse voltammetry in degassed acetonitrile. All complexes showed reversible oxidation waves with Eox ranging narrowly between 0.91 and 0.97 V vs. SCE (see Fig. 4 and Table 2). Most complexes exhibit very similar oxidation potentials to the parent IrPic (Eox = 0.96 V vs. SCE). There is a relatively larger ca. 40 mV cathodic shift of the Eox for the three complexes that have trimethylsilyl groups at the 4-position of the phenyl ring: C4, C4N4, and C4N5. While the trimethylsilyl group has minimal electronic influence on an aryl ring (having small Hammet values of σm = −0.04 and σp = −0.07),41 it has previously been shown that the 4-position of the phenyl ring of ppy-type ligands is the position most sensitive to electronic effects of substitution of emissive Ir(ppy)2(L) complexes,42 and suggests that substitution at the other positions on the ppy ligand will have a smaller impact on the state energies of the complex.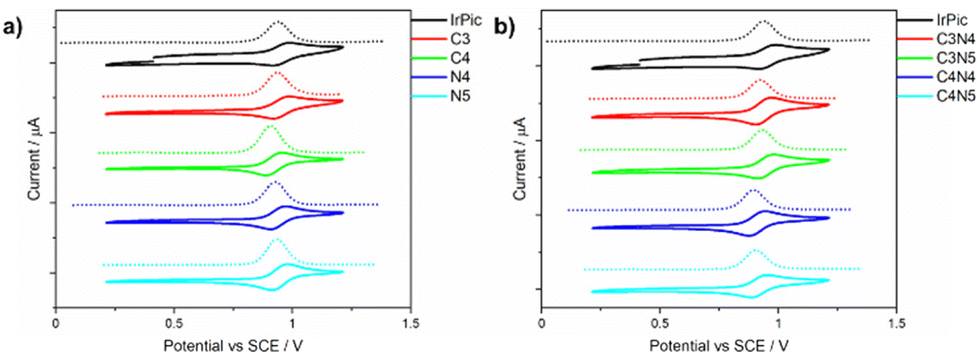 | ||
| Fig. 4 CVs and DPVs of the complexes studied. Measured in degassed acetonitrile with 0.1 M TBAPF6, using Pt disk, Pt wire and Ag/Ag+ working, counter and reference electrodes, respectively. A scan rate of 100 mV s−1 was used, and potentials are reported vs. SCE using Fc/Fc+ as an internal standard (Fc/Fc+ = 0.38 V vs. SCE in acetonitrile).43 | ||
| Complex | CVa | Absorptionb | Emissionc (solution) | Emissiond (doped film) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E ox/V | λ abs/nm | ε/x 103 M−1 cm−1 | λ PL/nm | FWHM/nm | τ PL/μse | CIE | λ PL/nm | FWHM/nm | τ PL/μse | CIE | Φ PL/%f |
k
r × 105 s−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) h h |
k
nr × 105 s−1i |
|
| a Measured in degassed acetonitrile with 0.1M TBAPF6, using Pt disk, Pt wire and Ag/Ag+ working, counter and reference electrodes respectively. Scan rate = 100 mV s−1, and potentials are reported vs. SCE using Fc/Fc+ as an internal standard (E1/2 (Fc/Fc+) vs. SCE = 0.38 V)43. b Measured in dilute toluene solution. c Measured in dilute degassed toluene solution (λexc = 400 nm). d Measured as 10 wt% doped films in mCBP, spin-coated on a quartz substrate from chlorobenzene (λexc = 340 nm). e λ exc = 378 nm. f Determined using an integrating sphere (λexc = 340 nm), the estimated uncertainty is ±5%. g Absorption measurements for IrPic were recorded in DCM solution due to the limited solubility in toluene. h k r = ΦPL/τaverage. i k nr = (1-ΦPL)/τaverage. | ||||||||||||||
| IrPic | 0.96 | 398, 446g | 5.39, 3.68g | 507 | 81 | 0.75 | [0.27, 0.58] | 514 | 80 | 0.79 (23) 1.75 (77) | [0.30,0.61] | 53 | 4.6 | 4.1 |
| C3 | 0.96 | 401, 453 | 3.82, 2.39 | 506 | 74 | 1.22 | [0.28,0.61] | 510 | 74 | 1.37 (73) 6.44 (27) | [0.28,0.59] | 19 | 0.4 | 1.8 |
| C4 | 0.92 | 412, 452 | 6.02, 4.68 | 524 | 76 | 1.78 | [0.34,0.62] | 524 | 77 | 1.19 (79) 4.13 (21) | [0.34,0.60] | 30 | 1.2 | 2.7 |
| N4 | 0.94 | 407, 460 | 2.89, 1.78 | 518 | 73 | 0.35 | [0.30,0.63] | 526 | 75 | 0.78 (58) 1.88 (42) | [0.32,0.61] | 50 | 3.4 | 3.4 |
| N5 | 0.97 | 409, 455 | 4.58, 3.61 | 515 | 72 | 0.86 | [0.30,0.63] | 519 | 77 | 0.57 (22) 1.61 (78) | [0.32,0.61] | 51 | 3.4 | 3.2 |
| C3N4 | 0.94 | 407, 458 | 5.58, 3.42 | 516 | 72 | 0.63 | [0.30,0.63] | 516 | 74 | 0.87 (65) 4.74 (35) | [0.31.0.61] | 43 | 1.1 | 1.5 |
| C3N5 | 0.95 | 409, 455 | 5.03, 3.99 | 513 | 72 | 0.90 | [0.30,0.63] | 515 | 75 | 0.51 (24) 1.40 (76) | [0.31,0.60] | 32 | 2.4 | 5.2 |
| C4N4 | 0.91 | 416, 466 | 4.39, 2.99 | 533 | 75 | 0.12 | [0.36,0.61] | 532 | 76 | 1.55 (67) 7.31 (33) | [0.36,0.60] | 53 | 0.9 | 0.8 |
| C4N5 | 0.92 | 419, 467 | 5.74, 5.03 | 532 | 77 | 0.24 | [0.36,0.61] | 532 | 77 | 1.03 (69) 2.53 (31) | [0.36,0.60] | 60 | 3.3 | 2.2 |
Photophysics
The absorption spectra of the complexes were measured in toluene solution (Fig. S56†). All possess two low-energy, low intensity absorption bands at ca. 400 and 450 nm, with molar extinction coefficients, ε, between ca. 2000 and 6000 M−1 cm−1 (Table 2). The two low energy bands in most of the complexes are assigned to S0 → S2 and S0 → S4 transitions that have larger oscillator strengths (f > 0.02) than the S0 → S1 and S0 → S3 transitions (f < 0.01). The S2 excited state comes from a HOMO → LUMO + 1 transition, while the S4 excited state comes from a HOMO → LUMO + 3 transition, both of these excited states possess mixed 1MLCT/LLCT character. However, the three complexes with a trimethylsilyl group at the C4 position (C4, C4N4 and C4N5) show distinct absorption profiles as the lowest energy absorption bands for these complexes are assigned to the S0 → S1 transition. High energy transitions below 300 nm result from local π–π* excitation of the ppy ligands, as previously reported for ppy-containing iridium complexes.25The emission properties of the nine complexes in degassed dilute toluene are shown in Fig. 5. Each of the complexes showed bright green emission with λPL between 506 and 533 nm (Table 2). There is a modest reduction in the FWHM of the emission spectra of the trimethylsilyl-substituted complexes, with FWHM ranging between 72 and 77 nm (2510–2680 cm−1), compared with the FWHM of 81 nm (2940 cm−1) for IrPic. CIE 1931 colour coordinates are reported in Table 2 and the coordinates plotted in Fig. 6. Complexes with substitution at the 4-position of the phenyl ring (C4, C4N4 and C4N5) possess a red-shifted emission (λPL = 524 to 533 nm) that is consistent with the destabilized HOMO observed in the electrochemistry. The most saturated green emission is present in N4 and N5.
 | ||
| Fig. 5 PL spectra in degassed toluene solution (λexc = 400 nm). | ||
 | ||
| Fig. 6 CIE diagram for the PL in degassed toluene solution (λexc = 400 nm). | ||
The PL lifetimes of the complexes in toluene varied widely between 0.12 and 1.78 μs, with all complexes except C3 and C4 possessing fast emission lifetimes of less than 0.90 μs. This is consistent with other moderately emissive IrPic derivatives26 that show moderate solution-state ΦPL and short τPL.
We next determined the PL properties of the complexes as 10 wt% doped films in mCBP (Fig. S57†), This doping concentration and host were chosen as these are representative of the emissive layer in high performance OLEDs.19,44 Like the solution-state study, all complexes emitted in the green, with λPL between 510 and 532 nm (Table 2). As in solution, there is a modest reduction in the FWHM of the emission of the trimethylsilyl-substituted complexes (FWHM ranging from 74–77 nm or 2540–2700 cm−1) in comparison to IrPic (FWHM = 80 nm or 2830 cm−1). The CIE coordinates of the doped mCBP files largely mirror those in solution (Fig. S58†).
All complexes showed biexponential emission decay in the mCBP doped films. The short lifetime component varied from 0.51 to 1.55 μs, while the longer component varied from 1.40 to 7.31 μs. The contribution of the shorter lifetime component ranges from 22 to 79% with no trend discernible in terms of the relative contributions. The weighted average lifetimes range from 1.3 to 5.6 μs. The ΦPL ranged from 50–60% for complexes N4, N5, C4N4 and C4N5, which are very similar to that of Irpic (53%). These are slightly lower than the doped film ΦPL values observed for FIrpic (78% in CBP),45 but consistent with those observed for a series of Firpic derivates in doped film (40–84%).46 In contrast, C3, C4, C3N4 and C3N5 showed lower ΦPL values between 19 and 43%, which is attributed to the significant reduction in kr from 4.6 × 105 in IrPic to as low as 4.1 × 104 in C3 (Table 2).
Based on the results, the complexes showing the most saturated green emission are N4 and N5. These two complexes have slightly narrower emission (FWHM decreased by 8–9 nm), the same short τPL (<1.0 μs) and similar ΦPL (ca. 50%). This is consistent with the previously reported narrowing (FWHM decreased by 6 nm) of the emission spectra for complexes BG1-BG4 that have a trimethylsilyl group in the N5 position of the ppy ligands.
Computational studies
The electronic properties of the eight new silylated complexes and the parent complex IrPic were investigated using density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations. All complexes showed extremely similar electron density distributions (see Table S1†), with the addition of the trimethylsilyl group having little impact on the calculated electronic properties of most of the complexes. The HOMO of all the complexes is distributed across the iridium center and the phenylpyridine ligands, while the LUMO is located on the ancillary picolinate ligand, the energy levels are tabulated in Table 3. Complexes that contain trimethylsilyl groups at C4, (C4, C4N4 and C4N5), possess LUMOs that are distributed across both the phenylpyridine and picolinate ligands. The LUMO energy levels remain essentially the same across the series of complexes, while there is a moderate destabilisation of up to 90 mV of the HOMO compared to Irpic.| Complex | HOMO/eV | LUMO/eV | S 1 (f)/eV | T 1/eV | Calculated emissiona/nm |
|---|---|---|---|---|---|
| a Emission wavelength calculated as a vertical transition from the optimised T1 structure. | |||||
| IrPic | −5.57 | −1.97 | 2.89 (0.01) | 2.65 | 544 |
| C3 | −5.54 | −1.97 | 2.87 (0.01) | 2.66 | 544 |
| C4 | −5.54 | −1.96 | 2.86 (0.11) | 2.59 | 554 |
| N4 | −5.54 | −1.95 | 2.88 (0.01) | 2.65 | 549 |
| N5 | −5.54 | −1.97 | 2.86 (0.03) | 2.63 | 551 |
| C3N4 | −5.51 | −1.95 | 2.86 (0.01) | 2.66 | 547 |
| C3N5 | −5.50 | −1.97 | 2.84 (0.02) | 2.64 | 545 |
| C4N4 | −5.52 | −1.96 | 2.84 (0.12) | 2.58 | 568 |
| C4N5 | −5.48 | −1.97 | 2.80 (0.12) | 2.56 | 568 |
The emissive T1 state is of 3MLCT/LLCT character. The T1 state of complexes C4, C4N4 and C4N5 have almost equal contributions of HOMO–LUMO and HOMO–LUMO + 1 transitions, while the remaining complexes have a T1 state dominated by the HOMO–LUMO + 1 transition (see Table S2 for tabulated excited state energies and transitions for all complexes†). Compared to Irpic, the largest though modest stabilization of the excited state energies is seen for C4, C4N4 and C4N5, consistent with the red-shift observed in the emission spectra of these complexes.
We next calculated the vibronically resolved emission spectra of the complexes to discern what effect if any trimethylsilyl substitution would have on the emission profile, see Table 4. The AH method used to evaluate the parent IrPic molecule at the beginning of this study wasn't suitable for molecules containing trimethylsilyl groups due to the change in geometry of the complex between the optimised geometries of the excited and ground states due to rotation of the trimethylsilyl groups (see Fig. S65 for a diagram showing this rotation†). With this change in geometry, the Franck–Condon integrals between vibrational modes cannot be calculated.28 Instead, we attempted to evaluate the vibrationally resolved emission spectra using the vertical hessian (VH) method, in which the geometry at the initial state (excited state for emission) is used to calculate the vibrational frequencies of the molecule in both the initial and final states. Fig. 7 shows the calculated vibrationally resolved emission spectra using this method. The calculated spectra are a poor simulation of the experimental spectra, as all the complexes have the same spectral shape and FWHM of 93–96 nm. In this case, by not accounting for the slight geometrical changes around the iridium atom between the excited and ground states during the emission spectra, the VH spectra do a poor job of replicating the experimental spectra, although the method has been shown to work for some phosphorescent platinum complexes.29
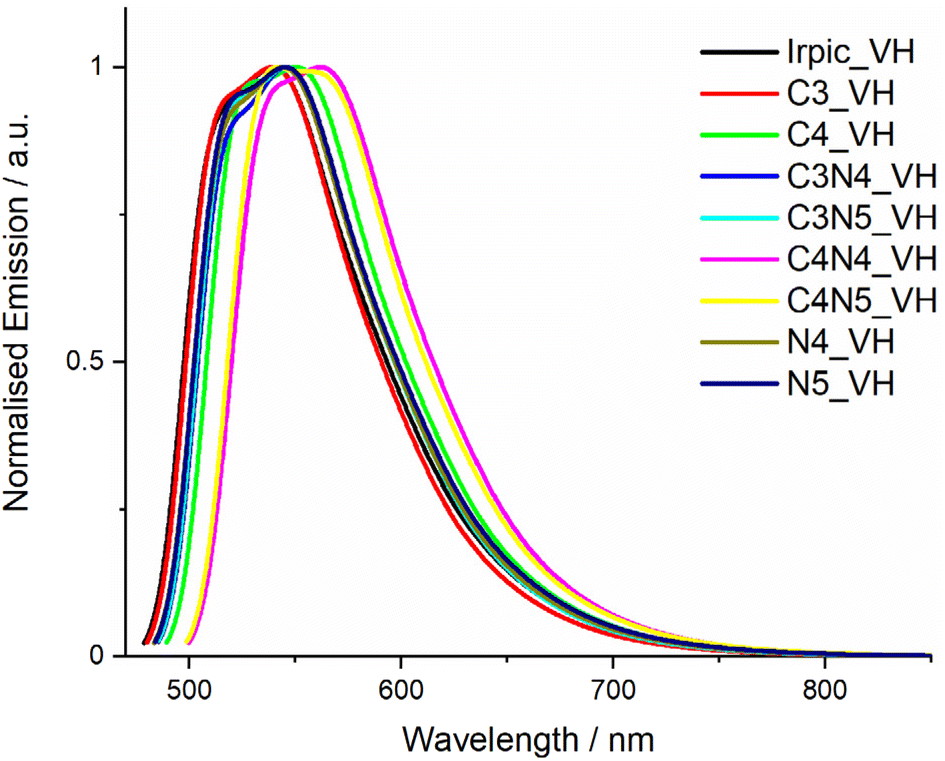 | ||
| Fig. 7 Plot of calculated vibrational emission spectra for all complexes in this study using the VH method. | ||
| Complex | Experimental | VH | AHa | ||||||
|---|---|---|---|---|---|---|---|---|---|
| λ max/nm | FWHM/nm | FWHM/cm−1 | λ max/nm | FWHM/nm | FWHM/cm−1 | λ max/nm | FWHM/nm | FWHM/cm−1 | |
| a Calculated for model complexes with SiMe3 groups replaced with SiH3 groups. | |||||||||
| IrPic | 507 | 81 | 2940 | 539 | 95.9 | 3250 | 544 | 106.1 | 3190 |
| C3 | 506 | 74 | 2680 | 539 | 92.3 | 3140 | 540 | 112.4 | 3380 |
| C4 | 524 | 76 | 2600 | 550 | 94.2 | 3070 | 560 | 138.9 | 3770 |
| N4 | 518 | 73 | 2570 | 544 | 94.8 | 3160 | 561 | 96.5 | 2780 |
| N5 | 515 | 72 | 2580 | 545 | 95.9 | 3200 | 558 | 93.6 | 2740 |
| C3N4 | 516 | 72 | 2550 | 546 | 93.7 | 3110 | 555 | 103.7 | 3010 |
| C3N5 | 513 | 72 | 2550 | 546 | 93.5 | 3110 | 551 | 95.8 | 2860 |
| C4N4 | 533 | 75 | 2510 | 562 | 95.3 | 2980 | 577 | 115.4 | 3080 |
| C4N5 | 532 | 77 | 2540 | 541 | 93.5 | 2960 | 572 | 107 | 2930 |
Where rotation of pendant groups on a molecule prevents use of the AH method to calculate vibrational emission spectra, one method to allow these calculations is to replace the pendant group with a smaller group or atom that doesn't undergo the same geometric change.29 In this example, replacing the trimethylsilyl groups with a silane (SiH3) group will retain the heavy atom to damp the vibrational modes of the aryl rings, but remove the methyl groups that rotate to prevent use of the AH model.
The spectra calculated using the AH method on model complexes with silane groups (SiH3) more accurately reproduce the relative heights of the main emission peak and the principal vibronic emission peak, while the trends in the FWHM correlate well with the experimental values. Comparison plots of the experimental spectrum, and the VH and AH calculated spectra for each complex are shown in Fig. S68–S76.†
There are two key features in the calculated AH emission spectra (Fig. 8) that determine the broadness of the emission. Firstly, there is the relative height of the main vibrational band (at ca. 600 nm), which for Irpic, is 95% of the 0–0 transition band, while for N4 and N5, it is only 85% of the intensity of the 0–0 transition band. Secondly, there is the size of the low energy shoulder, which is smallest for N4 and N5 and increases moving to Irpic and then the remaining complexes.
 | ||
| Fig. 8 Plot of calculated emission spectra for all complexes in this study using the AH method on complexes with trimethylsilyl groups replace with silane (SiH3) groups. | ||
Comparison of the calculated vibronic transitions for the trimethylsilyl-substituted complexes shows that there are fewer coupled vibrational modes contributing to the emission spectra compared to IrPic (Table S6†). In addition, visualisation of the vibrational modes of the new complexes shows a damping of the ring breathing modes that contributed strongly to the vibrational modes of Irpic. The vibrational modes for the complexes are tabulated in Table S7,† while selected vibrational modes are shown in Fig. 9.
 | ||
| Fig. 9 Representation of selected vibrational modes in the iridium complexes. (A) Irpic, mode 126, (b) C3, mode 138, (c) N4, mode 138, (d) C3N4, mode 154. | ||
Conclusions
This study has documented the synthesis and characterisation of a family of trimethylsilyl-substituted iridium complexes and their photophysical properties. The addition of the trimethylsilyl groups to the pyridyl ring of the phenylpyridine ligands dampens the vibrational modes that contribute to broadness of the emission spectra, narrowing their emission profile. The complexes with substitution of trimethylsilyl groups at the 4- or 5- positions of the pyridine ring showed a modest reduction in the FWHM of 3 to 4 nm, while retaining short emission lifetimes and moderately high ΦPL. In contrast, substitution at the 4- position of the phenyl ring led to a destabilisation of the HOMO and a reordering of the molecular orbitals, resulting in a red-shifting and broadening of the emission spectra as the geometry of the frontier molecular orbitals changed.The computed vibrationally resolved emission spectrum of the parent iridium complex IrPic allowed the identification of an aryl breathing mode of one of the phenylpyridine ligands as being the most significant contributor to the broadness of the emission. Vibrational emission spectra calculated for the trimethylsilyl-substituted complexes show a decrease in the coupling between the vibrational modes and the emissive electronic transition of the complexes.
Despite the modest impact that trimethyl substitution has on the overall emission spectrum, this study does demonstrate how calculation of the vibrationally resolved emission spectra of iridium complexes can be used to rationalize spectral shape, thus serving as a potentially valuable tool for designing complexes targeted at showing narrowband emission.
Experimental
Synthesis of materials
Full synthetic procedures are provided in the ESI.† The identity of all heteroleptic complexes was determined through NMR spectroscopy and mass spectrometry, while the purity of the final complexes was verified by HPLC.DFT calculations
DFT calculations were performed using the Gaussian16 (Rev. C.01) suite of software.47 All calculations were performed using the B3LYP functional48 and the 6-31G(d,p) basis set49 for non-metal atoms and the SBKJC VDZ ECP basis set50 for Ir atoms, using the conductor-like polarizable continuum model (CPCM) for acetonitrile solution.51–53 Singlet ground state geometry optimizations were carried out using the crystallographic structure of Ir(ppy)2(pic),12 with trimethylsilyl groups added in Gaussview v6.0,54 as the starting point. Geometry optimizations of the triplet excited state were performed using the optimized singlet structures as starting points. Vibrational frequency calculations were performed on all optimised structures to ensure that the optimized geometries represented the local minima. Excited state energies were calculated using the TD-DFT formalism.55,56 The energy of the singlet state at the optimised triplet structure was calculated and used to estimate the vertical energy gap between these two states and estimate the phosphorescent emission energy of the complexes. Calculated structures and Kohn–Sham orbitals were visualized with Gaussview v6.0.54Vibrational calculations
Vibrational calculations were performed using the Gaussian16 (Rev. C.01) suite of software.47 Vibrational emission spectra were calculated using either the Adiabatic Hessian (AH)57 or Vertical Hessian (VH)29 implementation of the Franck–Condon principle. The calculations were performed using the B3LYP functional48 and the 6-31G(d,p) basis set49 for non-metal atoms and the SBKJC VDZ ECP basis set50 for Ir atoms, this level of theory has shown to provide a good match between calculated and experimental emission spectra.31,57 The excited state (T1) vibrational frequencies were calculated at the TD-DFT optimised structure, while vibrational frequencies were calculated in the ground state (S0) at the same structure (VH method) or the optimised ground state geometry (AH). Where imaginary (negative) frequencies were found for the T1 or S0 structures, the low frequency components of the vibrational spectra were removed. FC calculations were performed with a unitary transition dipole, considering 10 classes, with a maximum of 1010 integrals considered and a Gaussian HWHM of 500 cm−1 used to simulate the emission spectra.Author contributions
C. F. R. M. performed the experimental work, data analysis, computational chemistry and prepared the original manuscript. E. Z.-C. was responsible for project administration and supervision of work. All authors contributed to the design of the study and the writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Samsung for funding this research.References
- J. M. Ha, S. H. Hur, A. Pathak, J.-E. Jeong and H. Y. Woo, NPG Asia Mater., 2021, 13, 53 CrossRef CAS.
- Y. Huang, E. L. Hsiang, M. Y. Deng and S. T. Wu, Light: Sci. Appl., 2020, 9, 105 CrossRef CAS PubMed.
- J. G. Kim, Y. Hwang, H. Hwang, J. H. Choi, Y. W. Park and B. K. Ju, Sci. Rep., 2020, 10, 5631 CrossRef CAS PubMed.
- M. Wang, J. Lin, Y. C. Hsiao, X. Liu and B. Hu, Nat. Commun., 2019, 10, 1614 CrossRef PubMed.
- ITU-R BT.2020-2, Internation Telecommunications Union, Geneva, 2015, https://www.itu.int/rec/R-REC-BT.2020-2-201510-I/en.
- A. Monkman, ACS Appl. Mater. Interfaces, 2022, 14, 20463–20467 CrossRef CAS PubMed.
- M. Frobel, F. Fries, T. Schwab, S. Lenk, K. Leo, M. C. Gather and S. Reineke, Sci. Rep., 2018, 8, 9684 CrossRef PubMed.
- S. DiLuzio, V. Mdluli, T. U. Connell, J. Lewis, V. Van Benschoten and S. Bernhard, J. Am. Chem. Soc., 2021, 143, 1179–1194 CrossRef CAS PubMed.
- P. A. Scattergood, A. M. Ranieri, L. Charalambou, A. Comia, D. A. W. Ross, C. R. Rice, S. J. O. Hardman, J. L. Heully, I. M. Dixon, M. Massi, F. Alary and P. I. P. Elliott, Inorg. Chem., 2020, 59, 1785–1803 CrossRef CAS PubMed.
- M. S. Lowry, W. R. Hudson, R. A. Pascal and S. Bernhard, J. Am. Chem. Soc., 2004, 126, 14129–14135 CrossRef CAS PubMed.
- A. Hohenleutner, S. Schmidbauer, R. Vasold, D. Joosten, P. Stoessel, H. Buchholz and B. König, Adv. Funct. Mater., 2012, 22, 3406–3413 CrossRef CAS.
- E. Baranoff, I. Jung, R. Scopelliti, E. Solari, M. Gratzel and M. K. Nazeeruddin, Dalton Trans., 2011, 40, 6860–6867 RSC.
- X.-J. Liao, J.-J. Zhu, L. Yuan, Z.-P. Yan, Z.-L. Tu, M.-X. Mao, J.-J. Lu, W.-W. Zhang and Y.-X. Zheng, Mater. Chem. Front., 2021, 5, 6951–6959 RSC.
- X.-J. Liao, J.-J. Zhu, L. Yuan, Z.-P. Yan, X.-F. Luo, Y.-P. Zhang, J.-J. Lu and Y.-X. Zheng, J. Mater. Chem. C, 2021, 9, 8226–8232 RSC.
- K. P. S. Zanoni, A. Ito and N. Y. Murakami Iha, New J. Chem., 2015, 39, 6367–6376 RSC.
- S. Y. Baek, S. Y. Kwak, S. T. Kim, K. Y. Hwang, H. Koo, W. J. Son, B. Choi, S. Kim, H. Choi and M. H. Baik, Nat. Commun., 2020, 11, 2292 CrossRef CAS PubMed.
- M.-J. Kim, S.-J. Yoo, J. Hwang, S.-J. Park, J.-W. Kang, Y.-H. Kim, J.-J. Kim and S.-K. Kwon, J. Mater. Chem. C, 2017, 5, 3107–3111 RSC.
- H. U. Kim, H. J. Park, J.-H. Jang, W. Song, I. H. Jung, J. Y. Lee and D.-H. Hwang, Dyes Pigm., 2018, 156, 395–402 CrossRef CAS.
- S. O. Jung, Q. Zhao, J.-W. Park, S. O. Kim, Y.-H. Kim, H.-Y. Oh, J. Kim, S.-K. Kwon and Y. Kang, Org. Electron., 2009, 10, 1066–1073 CrossRef CAS.
- Y. Bi, J. Wei, S. Chen, H. Zhao and X. Zhang, J. Phys. Chem. C, 2021, 125, 24671–24684 CrossRef CAS.
- C. Y. Kim, D.-G. Ha, H. H. Kang, H.-J. Yun, S.-K. Kwon, J.-J. Kim and Y.-H. Kim, J. Mater. Chem., 2012, 22, 22721 RSC.
- J. M. Kim, K. Y. Hwang, S. Kim, J. Lim, B. Kang, K. H. Lee, B. Choi, S. Y. Kwak and J. Y. Lee, Adv. Sci., 2022, 9, e2203903 CrossRef PubMed.
- H. U. Kim, J.-H. Jang, H. J. Park, J. Y. Lee and D.-H. Hwang, J. Nanosci. Nanotechnol., 2017, 17, 5587–5592 CrossRef CAS.
- X. Xu, X. Yang, Y. Wu, G. Zhou, C. Wu and W. Y. Wong, Chem. - Asian J., 2015, 10, 252–262 CrossRef CAS PubMed.
- T. Hofbeck and H. Yersin, Inorg. Chem., 2010, 49, 9290–9299 CrossRef CAS PubMed.
- R. Davidson, Y.-T. Hsu, C. Bhagani, D. Yufit and A. Beeby, Organometallics, 2017, 36, 2727–2735 CrossRef CAS.
- Y. Zhou, H. Gao, X. Wang and H. Qi, Inorg. Chem., 2015, 54, 1446–1453 CrossRef CAS PubMed.
- F. Santoro, R. Improta, A. Lami, J. Bloino and V. Barone, J. Chem. Phys., 2007, 126, 084509 CrossRef PubMed.
- F. Vazart, C. Latouche, J. Bloino and V. Barone, Inorg. Chem., 2015, 54, 5588–5595 CrossRef CAS.
- J. Bloino, A. Baiardi and M. Biczysko, Int. J. Quantum Chem., 2016, 116, 1543–1574 CrossRef CAS.
- F. Egidi, M. Fuse, A. Baiardi, J. Bloino, X. Li and V. Barone, Chirality, 2018, 30, 850–865 CrossRef CAS PubMed.
- F. Santoro and D. Jacquemin, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 460–486 CAS.
- C. Latouche, D. Skouteris, F. Palazzetti and V. Barone, J. Chem. Theory Comput., 2015, 11, 3281–3289 CrossRef CAS PubMed.
- R. Schira and C. Latouche, Dalton Trans., 2021, 50, 746–753 RSC.
- A. Stoliaroff, J. Rio and C. Latouche, New J. Chem., 2019, 43, 11903–11911 RSC.
- M. Fecková, S. Kahlal, T. Roisnel, J. Y. Saillard, J. Boixel, M. Hruzd, P. Poul, S. Gauthier, F. Robin-le Guen, F. Bureš and S. Achelle, Eur. J. Inorg. Chem., 2021, 1592–1600 CrossRef.
- M. Hruzd, N. le Poul, M. Cordier, S. Kahlal, J. Y. Saillard, S. Achelle, S. Gauthier and F. Robin-le Guen, Dalton Trans., 2022, 51, 5546–5560 RSC.
- W. Xu, L. Lystrom, Y. Pan, X. Sun, S. A. Thomas, S. V. Kilina, Z. Yang, H. Wang, E. K. Hobbie and W. Sun, Inorg. Chem., 2021, 60, 15278–15290 CrossRef CAS PubMed.
- M. Waki, N. Mizoshita, T. Tani and S. Inagaki, Angew. Chem., Int. Ed., 2011, 50, 11667–11671 CrossRef CAS PubMed.
- M. Alessi, A. L. Larkin, K. A. Ogilvie, L. A. Green, S. Lai, S. Lopez and V. Snieckus, J. Org. Chem., 2007, 72, 1588–1594 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- J. Frey, B. F. E. Curchod, R. Scopelliti, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and E. Baranoff, Dalton Trans., 2014, 43, 5667–5679 RSC.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- L. S. Cui, J. U. Kim, H. Nomura, H. Nakanotani and C. Adachi, Angew. Chem., Int. Ed., 2016, 55, 6864–6868 CrossRef CAS PubMed.
- Y. Kawamura, K. Goushi, J. Brooks, J. J. Brown, H. Sasabe and C. Adachi, Appl. Phys. Lett., 2005, 86, 071104 CrossRef.
- K. S. Bejoymohandas, A. Kumar, S. Varughese, E. Varathan, V. Subramanian and M. L. P. Reddy, J. Mater. Chem. C, 2015, 3, 7405–7420 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- J. S. Binkley, J. A. Pople and W. J. Hehre, J. Am. Chem. Soc., 1980, 102, 939–947 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi and V. Barone, J. Chem. Phys., 2001, 115, 4708–4717 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- R. Dennington, T. Keith and J. Millam, Gaussview, Version 6, Semichem Inc., Shawnee Mission KS, 2019 Search PubMed.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- R. C. E. Sia, R. A. Arellano-Reyes, T. E. Keyes and J. Guthmuller, Phys. Chem. Chem. Phys., 2021, 23, 26324–26335 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: A document containing synthetic details, NMR spectra, HPLC traces, mass spectra, additional photophysics data and tabulated DFT results is available online. Coordinates for all DFT optimised geometries of the complexes are available in an xyz file. See DOI: https://doi.org/10.1039/d3dt00304c. The research data supporting this publication can be accessed at https://doi.org/10.17630/723407b5-e795-437d-ab9f-6ee27fde870d |
| This journal is © The Royal Society of Chemistry 2023 |