
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of phenylene-bridged isoxazole and tetrazole-1-ol based energetic materials of low sensitivity†
Sohan
Lal
 a,
Richard J.
Staples
b and
Jean'ne M.
Shreeve
*a
a,
Richard J.
Staples
b and
Jean'ne M.
Shreeve
*a
aDepartment of Chemistry, University of Idaho, Moscow, Idaho 83844-2343, USA. E-mail: jshreeve@uidaho.edu; Fax: (+1) 208-885-5173
bDepartment of Chemistry, Michigan State University, East Lansing, Michigan 48824, USA
First published on 22nd February 2023
Abstract
A variety of phenylene-bridged isoxazole and tetrazole-1-ol based green energetic materials was synthesized, for the first time, in good to excellent yields. The structures of the newly synthesized compounds were confirmed by spectroscopic techniques, elemental analysis, and single-crystal X-ray analysis. The value of the present work is that all newly synthesized compounds have good thermal stabilities ranging between 167–340 °C and acceptable densities between 1.51 g cm−3 to 1.82 g cm−3. Detailed computational insight into the energetic properties of the new compounds shows that they have good energetic properties (propulsive and ballistic) with excellent thermal and mechanical stabilities which makes them promising candidates for solid propulsion systems. Compounds 5, 12 and 14 are the superior candidates as melt-castable energetic materials.
Introduction
Energetic materials (EMs) attract extensive attention from the scientific community because of their wide range of applications as propellants, explosives, and pyrotechnics.1–4 In the last few decades many interesting, energetic materials have been designed and subsequently synthesized including octanitrocubane (ONC),5 2,4,6,8,10,12-hexanitrohexaaza-isowurtizitane (CL-20),6 4,10-dinitro-2,6,8,12-tetra-oxa-4,10-diazatetracyclo[5.5.0.05,9.03,11]-dodecane (TEX),7a formamidinium nitroformate (FANF),7b 1,3,5-trinitroperhydro-1,3,5-triazine (RDX),8 dihydroxylammonium-5,5′-bistetrazolyl-1,1′-diolate (TKX-50),9 and 1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX).10 In addition to these well-known energetic compounds, a few aromatic energetic materials such as 2,4,6-trinitrobenzene-1,3,5-triamine (TATB),11 2,4,6-trinitrotoluene (TNT),11 picric acid (PA),4 and 2,2′,4,4′,6,6′-hexanitrostilbene (HNS)12 also attract the attention of materials chemists due to their high stability and good performance. In general, aromatic compounds with low oxygen balance, especially benzene-containing EMs are known to generate large amounts of soot during their combustion.13 However, this can be improved by introducing appropriate explosophoric groups (–NO2, –ONO2, –N3 and –NHNO2, etc.) on the benzene core. Such EMs possess high positive enthalpies of formation and high density which are governed by their planar structures and high values of their packing coefficients. Some well-known aromatic energetic materials are given with their key performance properties in Fig. 1. | ||
| Fig. 1 Molecular structure and associated energetic properties of commonly used aromatic energetic materials. | ||
Excellent detonation properties of the bridged compounds make them ideal components for space and military applications.14 Some of these energetic materials with their key properties are shown in Fig. 2.9,15 However, the main concern associated with these compounds is their high sensitivity towards impact and friction, which restricts their real-life applications.
 | ||
| Fig. 2 Molecular structure and associated energetic properties of recently reported bistetrazole-1-ol and bis-isoxazole based energetic materials.9–15 | ||
Melt-castable materials are a novel class of energetic materials, having melting temperatures in the range of 70–125 °C which are values significantly different from their decomposition temperatures. TNT- and dinitroanisole- (DNAN) based melt-castable eutectic formulations are well-known in the literature.16 However, environmental issues, low density and poor explosive performance encourage an interest in the scientific community in designing and synthesizing new melt-castable energetic materials that address these concerns. Significant work has been carried out on the design, synthesis, and theoretical calculations of aromatic explosive/propellants. When designing a potential energetic material, performance as well as stability of the desired EM, is important.
A benzene ring with appropriate explosophoric groups is a suitable basic skeleton to provide excellent density, high positive enthalpy of formation, good detonation performance and a π–π stacking interaction which leads to lower sensitivities compared to traditional energetic materials. Now the new compounds have been designed using a benzene ring as a basic skeleton, followed by introduction of azidoxime, tetrazole-1-ol and isoxazole substituted with azidomethyl (–CH2–N3) and nitratomethyl (–CH2–ONO2) groups to improve the nitrogen and oxygen content.
Results and discussion
Synthesis
Terephthaldehyde, 1, is an excellent substrate for the syntheses of phenylene-bridged isoxazoles and tetrazole-N-oxide. Initially, chloro-oxime 3 was prepared via a literature procedure.17 Subsequently, it was converted into bis(azido-oxime) 4 by reacting with sodium azide in dimethylformamide (DMF) at ambient temperature. Then HCl(g) was added to the bis(azido-oxime) 4 which resulted in bis(tetrazole-1-ol) 5 in excellent yield. Further, to improve the energetic content, compound 5 was reacted with selected energetic bases such as hydroxylamine, ammonia, and hydrazine and for comparison with potassium hydroxide to form the potassium salt, 9 (Scheme 1). | ||
| Scheme 1 Synthesis of 5,5′-(1,4-phenylene) bis(1H-tetrazol-1-ol) (5) and its energetic salts (6–9). | ||
In another series, bis (chloro-oxime) 3 was treated with but-2-yne-1,4-diol 10 in the presence of potassium bicarbonate to give isoxazoles with tetramethyl alcohol 11 in excellent yields. Compound 11 was utilized as a key precursor for the synthesis of novel energetic compounds, namely 1,4-phenylenebis (isox-azole-3,4,5-triyl)-tetrakis(methylene)tetra-nitrate 12 (Scheme 2) and 1,4-bis(4,5-bis(azidomethyl)isoxazole-3-yl)-benzene 14 (Scheme 3). Compound 11 was nitrated with 90% HNO3 at 0° C to give compound 12 in excellent yield, 90% (Scheme 2).
 | ||
| Scheme 2 Synthesis of 1,4-bis(4,5-bis(nitratomethyl)isoxazol-3-yl)benzene (12). | ||
 | ||
| Scheme 3 Synthesis of 1,4-bis(4,5-bis(azidomethyl)isoxazol-3-yl)benzene (14). | ||
Compound 11 was treated with SOCl2 in the presence of pyridine to form the corresponding tetrachloro derivative 13, which was subsequently heated with sodium azide at 90 °C to give compound 14 in excellent yield, 84% (Scheme 3).
All the newly synthesized compounds have been fully characterized by 1H, 13C, and 15N NMR, elemental analysis, and IR spectra. The structure of compound 12 was established by single crystal X-ray analysis (Fig. 3).
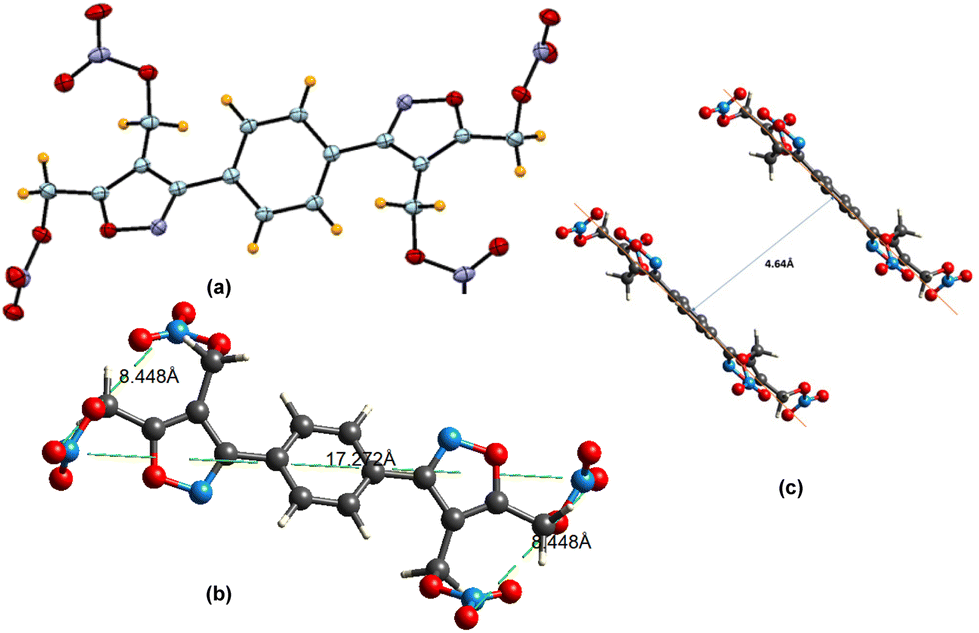 | ||
| Fig. 3 Single crystal structure of compound 12 (CCDC = 2206847) (a) ORTEP diagram. (b) Diagonal length and width of the molecule. (c) Distance between two parallel layers. | ||
Crystal structure
Suitable single yellow plate-shaped crystals of compound 12 were obtained by slow evaporation of methanol.Compound 12 (Fig. 3) crystallizes in the monoclinic space group P21/c with a density of 1.697 g cm−3 at 100 K with a half molecule in the asymmetric unit, which is characterized by the sum formula – in other words Z is 2 and Z′ is 0.5. Compound 12 was found to be packed in a planar structure and the distance between the two parallel layers is about 4.64 Å. The diagonal length and width of the molecule were measured to be 17.27 Å and 8.44 Å, respectively. The packing diagram is shown in Fig. 4. The crystal data collection parameters and other useful crystallographic data are given in the ESI (Tables S4–S11†).
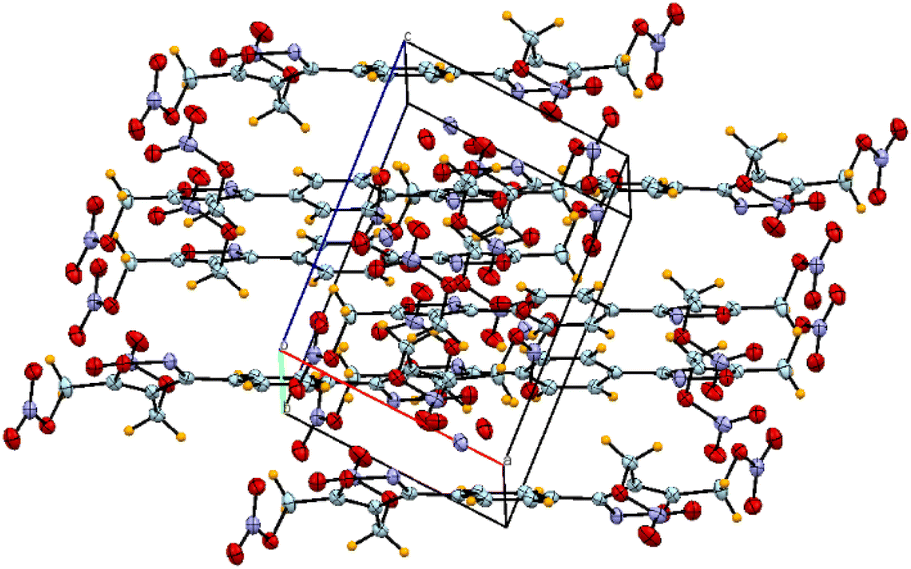 | ||
| Fig. 4 Packing diagram of compound 12. | ||
Computational methods
Ab initio-based computational insight into the title compounds was carried out at B3LYP/6-311++ G (d,p) (atomization method) and MP2/6-311++ G** (isodesmic method, at G2 level18) with the help of the Gaussian 03 suite of programs.19 The gas-phase enthalpies of formation (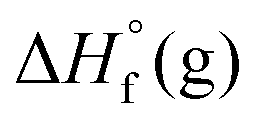 ) of the title compounds were calculated using their isodesmic equations. Then, their solid phase enthalpies of formation (
) of the title compounds were calculated using their isodesmic equations. Then, their solid phase enthalpies of formation ( ) were estimated from their gas phase enthalpy of formation (
) were estimated from their gas phase enthalpy of formation ( ) and enthalpy of sublimation (ΔHsub) using Hess's law (eqn (1)).20
) and enthalpy of sublimation (ΔHsub) using Hess's law (eqn (1)).20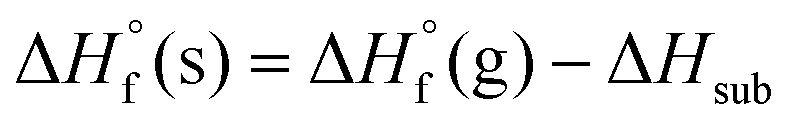 | (1) |
 is the solid-phase enthalpy of formation,
is the solid-phase enthalpy of formation,  is the gas phase enthalpy of formation and ΔHsub is the enthalpy of sublimation.
is the gas phase enthalpy of formation and ΔHsub is the enthalpy of sublimation.
The enthalpy of sublimation was estimated using Trouton's rule according to eqn (2).21
| ΔHsub = 0.188 × T/kJ mol−1 K−1 | (2) |
For salts, the solid-state enthalpies of formation ( ) were obtained using Born–Haber energy cycles.22 The experimental densities of the title compounds were determined at 25 °C using a gas pycnometer. Subsequently, using their solid phase enthalpies of formation (
) were obtained using Born–Haber energy cycles.22 The experimental densities of the title compounds were determined at 25 °C using a gas pycnometer. Subsequently, using their solid phase enthalpies of formation ( ) and experimental densities (Table 1), the related propulsive, ballistic and detonation performances were predicted using EXPLO5 V6.06 software.23
) and experimental densities (Table 1), the related propulsive, ballistic and detonation performances were predicted using EXPLO5 V6.06 software.23
| Compd |
Ω
CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (%) a (%) |
N + Ob [%] | ΔHfc [kJ mol−1] | ρ [g cm−3] | Mp/Tde [°C] |
|---|---|---|---|---|---|
| a Oxygen balance based on CO. b Nitrogen and oxygen contents in percentage. c Calculated solid phase enthalpy of formation. d Measured densities, gas pycnometer at room temperature (25 °C). e Melting points and thermal decomposition temperature (onset) under nitrogen gas (DSC, 5 °C min−1, see ESI†). f Ref. 10 and 25. g Ref. 11. h Ref. 12. | |||||
| 4 | −58.50 | 58.52 | 804.30 | 1.57 | —/167 |
| 5 | −58.50 | 58.52 | 775.02 | 1.63 | 99/209 |
| 6 | −51.20 | 65.36 | 508.69 | 1.62 | —/195 |
| 7 | −68.50 | 61.40 | 381.76 | 1.58 | —/251 |
| 8 | −67.00 | 64.48 | 740.29 | 1.51 | —/228 |
| 9 | −39.70 | 44.69 | −14.25 | 1.82 | —/340 |
| 12 | −122.10 | 60.12 | −57.82 | 1.63 | 123/177 |
| 14 | −74.00 | 52.75 | 1612.71 | 1.60 | 114/207 |
| RDXf | 0.0 | 81.06 | 70.3 | 1.80 | —/204 |
| TNTg | −24.70 | 60.76 | −59.30 | 1.65 | —/300 |
| HNSh | −21.80 | 61.31 | 78.24 | 1.74 | —/316 |
The stabilities of the new compounds were estimated in terms of their electrostatic potentials (ESP), bond dissociation energies (BDEs) (eqn (3)), and energy difference between their HOMOs and LUMOs using B3LYP/6-311++ G (d,p) level of theory with Multiwfn and VMD softwares24 and compared with the experimental stability (IS and FS) values.
| BDE [AB] = E0 [A.] + E0 [B.] − E0 [AB] | (3) |
Densities of all newly synthesized compounds were determined with a gas pycnometer at 25 °C and found to be in the range of 1.51–1.82 g cm−3. The potassium salt 9 has the highest density (1.82 g cm−3) among salts 6–9. It is notable that the relatively higher densities of neutral compounds 5, 6, 12 and 14 (1.63, 1.62, 1.63, and 1.60 g cm−3, respectively), result from π–π stacking interactions and their planar structures. The existence of such extensive interactions among adjacent molecules which are confirmed based on the single crystal X-ray structures of compound 12, resulted in their high densities, and are comparable to the well-known explosive TNT (1.65 g cm−3). Among the salts 6–9, compound 9 has a higher density than that of RDX (ρ = 1.80 g cm−3) as listed in Table 1.
All compounds are solid at room temperature and their decomposition temperatures are found between 167 °C to 340 °C (see ESI, Fig. S6, S13, S19, S26, S31, S38, S43, S49, S55 and S66†). All the title compounds 4–14 have high positive enthalpy of formation, except for compounds 9 and 12. The most positive enthalpy of formation obtained for the azidomethyl-isoxazole 14 (1612.71 kJ mol−1) followed by the azido-oxime 4 (804.30 kJ mol−1), are attributed to the azido groups in the molecules. The lowest  was obtained for the tetranitrate 12 (−57.82 kJ mol−1).
was obtained for the tetranitrate 12 (−57.82 kJ mol−1).
Propulsive properties of the newly synthesized compounds
The suitability of the title compounds as solid propellants was evaluated by using EXPLO5 V6.06 software.23 Their performances were evaluated with three different propellant formulations, viz., (i) monopropellants (neat); (ii) bi-propellants and composite propellants: propellant formulations with AP/HTPB; (iii) composite propellants: propellant formulations with GAP/DOA. As monopropellants, all the new compounds, except compounds 7 and 9, have considerably higher propulsive properties than that of TNT and lower than that of RDX.A similar trend was also observed, when the performance of the new compounds (80%) was studied as composite-propellants with energetic binder GAP (17%) and plasticizer DOA (3%). With exception of the ammonium salt 7, and the potassium salt 9, all compounds show promising propulsion values (Isp = 218–224 s and C* = 1241–1272 m s−1), which are better than those of TNT (∼217 s and C* = 1235 m s−1), which suggest their potential use as solid composite propellants in the rocket industry (Table 2).
| # |
I
sp,vaca [s] |
ρI
sp,vacb [g cm−3 s] |
C*c [m s−1] |
I
sp,vacd [s] |
ρI
sp,vace [g cm−3 s] |
C*f [m s−1] |
I
sp,vacg [s] |
ρI
sp,vach [g cm−3 s] |
I
sp,vaci [s] |
ρI
sp,vacj [g cm−3 s] |
I
spk [s] |
I
sp,vacl [s] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a I sp,vac = vacuum specific impulse at 100% compound (neat). b ρI sp,vac = density vacuum specific impulse at 100% compound (neat). c Characteristic velocity at 100% compound (neat). d I sp,vac = vacuum specific impulse at compound (80%), 17% (GAP) and DOA (3%). e ρI sp,vac = density vacuum specific impulse at compound (80%), 17% (GAP) and DOA (3%). f Characteristic velocity at compound (80%), 17%(GAP) and DOA (3%). g I sp,vac = vacuum specific impulse at 80% AP, 10% binder (HTPB) and 10% compound. h ρI sp,vac = density vacuum specific impulse at 80% AP, 10% binder (HTPB) and 10% compound. i I sp,vac = vacuum specific impulse at 80% compound and 20% HTPB. j ρI sp,vac = density vacuum specific impulse at 80% compound and 20% HTPB. k I sp = specific impulse at compound (20%), and ethanol (80%). l I sp,vac = vacuum specific impulse at compound (20%), and ethanol (80%), for 100% ethanol Isp = 120.09 s and Isp,vac = 129.46 s. | ||||||||||||
| 4 | 232.35 | 364.79 | 1310 | 224.66 | 333.60 | 1272 | 267.04 | 457.09 | 206.52 | 282.21 | 146.51 | 159.35 |
| 5 | 229.33 | 373.82 | 1294 | 222.31 | 339.56 | 1258 | 266.74 | 458.42 | 204.24 | 286.44 | 145.84 | 158.58 |
| 6 | 229.15 | 371.22 | 1308 | 222.88 | 338.87 | 1264 | 267.58 | 459.58 | 206.80 | 288.82 | 146.80 | 159.49 |
| 7 | 198.29 | 313.30 | 1109 | 198.67 | 296.42 | 1109 | 263.32 | 451.04 | 183.04 | 251.24 | 138.49 | 150.03 |
| 8 | 225.37 | 340.31 | 1276 | 220.04 | 317.19 | 1241 | 266.23 | 453.73 | 204.24 | 271.58 | 144.96 | 157.32 |
| 9 | 142.88 | 260.04 | 760 | 160.25 | 265.54 | 853 | 260.72 | 453.06 | 138.22 | 208.86 | 130.96 | 142.05 |
| 12 | 230.59 | 375.86 | 1337 | 223.31 | 341.09 | 1277 | 269.11 | 462.49 | 206.50 | 289.61 | 149.17 | 162.45 |
| 14 | 223.20 | 357.13 | 1245 | 218.05 | 328.43 | 1224 | 265.28 | 455.01 | 200.27 | 277.30 | 144.70 | 157.32 |
| TNT | 222.56 | 368.12 | 1284 | 217.07 | 335.20 | 1235 | 268.36 | 461.90 | 200.55 | 284.10 | 147.37 | 160.38 |
| RDX | 286.98 | 516.56 | 1651 | 259.81 | 427.08 | 1528 | 273.76 | 475.06 | 234.12 | 351.19 | 155.44 | 169.27 |
Additionally, two different composite propellant formulations with ammonium perchlorate (AP) and HTPB (i) 80% AP, 10% HTPB and 10% title compounds (ii) 80% AP and 20% new compounds, have been calculated and the results are listed in Table 2. The specific impulse (Isp) of the title compounds were comparable with that of TNT. However, when the title compounds were used as oxidizers instead of ammonium perchlorate (AP) in the propellent formulations (80% title compound: 20% HTPB), it was revealed that these compounds performed better than TNT but due to lack of oxygen content they had not performed as potential oxidizers (Table 2). Interestingly, in the other formulation with ethanol, the efficiency of ethanol as a fuel can be significantly improved by adding the new compounds (20%) as fuel supplements. The specific impulse (Isp) and vacuum specific impulse (Isp,vac) of the fuel blends were improved by 10–29 s and 13–33 s, respectively as given in Table 2.
The suitability of newly synthesized energetic materials as explosives was evaluated using their calculated  and measured room temperature densities with EXPLO5 V6.06 software (Table 3).
and measured room temperature densities with EXPLO5 V6.06 software (Table 3).
| Compd | DPa [GPa] | Dvb [m s−1] | Q [kJ kg−1] | ISd [J] | FSe [N] |
|---|---|---|---|---|---|
| a Calculated detonation pressure. b Calculated detonation velocity. c Heat of detonation. d Measured impact sensitivity (IS). e Measured friction sensitivity (FS). f Ref. 10. g Ref. 7b. | |||||
| 4 | 17.86 | 6693.04 | 4404.38 | ≥10 | ≥240 |
| 5 | 17.16 | 6885.27 | 4314.17 | ≥40 | ≥360 |
| 6 | 19.98 | 7491.57 | 4228.68 | ≥40 | ≥360 |
| 7 | 16.86 | 7105.19 | 2913.26 | ≥40 | ≥360 |
| 8 | 18.35 | 7371.84 | 3875.11 | ≥40 | ≥360 |
| 9 | 11.49 | 5596.02 | 1822.33 | ≥40 | ≥360 |
| 12 | 18.98 | 6865.72 | 4689.51 | ≥15 | ≥360 |
| 14 | 17.11 | 6621.19 | 4149.31 | ≥15 | ≥360 |
| TNTf | 18.56 | 6839.96 | 4395.70 | ≥15 | ≥353 |
| RDXf | 34.00 | 8867.06 | 5728.99 | ≥7.5 | ≥120 |
| FANFg | 34.13 | 8811.00 | — | ≥100 | ≥360 |
The highest detonation performance values were observed for compound 6 (DP = 19.98 GPa and Dv = 7491 m s−1) and compound 12 (DP = 18.98 GPa and Dv = 6865 m s−1) and are superior to that of TNT. Since all the compounds possess lower detonation performances than RDX, they can be considered safe to use as solid propellants (Table 3).
The new compounds as solid propellants were further ensured to be suitable by measuring their impact sensitivity (IS) and friction sensitivity (FS) with the help of a BAM Fallhammer apparatus and a BAM friction tester,26 respectively. All the new compounds except for 4, 12, and 14 have sensitivities in the range of insensitive energetic materials. However, compounds 4 (IS = 10J, FS = 240N), 12 (IS = 15J, FS = 360N) and 14 (IS = 15J, FS = 360N) can be considered to be more sensitive energetic materials. The new compounds were dissolved in water and held at room temperature for two days. No decomposition was observed.
Furthermore, to understand the structure properties and intermolecular interactions in the crystal packing of compound 12, its Hirshfeld surfaces and two-dimensional fingerprints were calculated by using CrystalExplorer 21.5.27 Red and blue dots on the Hirshfeld surfaces demonstrate the high and low close interactions within the crystal (Fig. 5). The O⋯H (50%) and N⋯H (4%) interactions provide the extra stability to the molecules whereas O⋯N (5%), O⋯C (5%) and N⋯N (3%) are indications of good π–π stacking interactions in the molecules. Similarly, O⋯O (19%) arising from the oxygen atoms of nitrate (–ONO2) groups are contributing to the high density and higher sensitivity of the molecules.
 | ||
| Fig. 5 (a) Hirshfeld surface graph. (b) 2D fingerprint plots in crystal stacking (c) individual atomic contacts percentage in the bar graphs and (d and e) the closest atoms in compound 12. | ||
The O⋯H, N⋯H, O⋯N and O⋯C interactions are dominating over O⋯O interactions which suggests that compound 12 is a well stabilized compound. The kinetic stabilities of the newly synthesized compounds were predicted with the help of electrostatic potential (ESP) measurements as shown in Fig. 6. The global ESP maxima and minima are shown in small gold spheres and small blue spheres, respectively.
 | ||
| Fig. 6 Electrostatic potential (ESP) maps with the representation of surface maxima (small gold spheres) and minima (small blue spheres). | ||
The high stability of compounds 12 and 14 is strongly supported by hydrogen bonds, van der Waals interactions and steric effects as shown in Fig. 7.
 | ||
| Fig. 7 Non-covalent interaction (NCI): reduced density gradient (RDG) and scatter diagram of compounds 12 and 14. | ||
Compounds 4 and 5 have slightly higher ESP maxima (+53.55 and +68.57 kcal mol−1, respectively) and compounds 5, 12 and 14 have significantly lower global ESP minima (−37.52, −23.09, −30.46 kcal mol−1, respectively) compared to RDX (+50.01 and −20.81 kcal mol−1) and TNT (+38.66 and −22.39 kcal mol−1), suggesting that the stability order of the selected compounds is 5 > TNT > 14 > RDX > 12 > 4. The product of σtot2 and balance of charges (ν) suggest that the stability of compounds 4 and 12 would be lower than that of RDX and TNT and that compound 5 will be most stable compound.
Similar trends also are found in their bond dissociation energies and the energy difference between their HOMO and LUMO orbitals as listed in Table 4.
| # | SAtota Å2 | Ratiob (%) | Ratioc (%) | σ 2 tot v (kcal mol−1)2 | BDEe (kJ mol−1) | Ω (eV) |
|---|---|---|---|---|---|---|
| a SAtot = total surface area. b Ratio of positive ESPs. c Ratio of negative ESPs. d Product of total variance and balance of charges. e Bond dissociation energies of trigger bond. f Energies difference between HOMO and LUMO orbitals. | ||||||
| 4 | 263.46 | 42.63 | 57.37 | 13.58 | 23.58 | 4.186 |
| 5 | 248.04 | 65.31 | 34.69 | 61.63 | 510.37 | 4.679 |
| 12 | 480.30 | 59.58 | 40.42 | 20.11 | 126.03 | 4.818 |
| 14 | 448.86 | 57.15 | 42.85 | 28.92 | 231.51 | 5.115 |
| RDX | 209.49 | 55.74 | 44.26 | 27.29 | 130.85 | 5.993 |
| TNT | 221.89 | 57.88 | 42.12 | 22.48 | 238.45 | 4.926 |
Conclusions
Phenylene-bridged isoxazole and tetrazole-1-ol-based green energetic materials were obtained for the first time from readily available terephthalaldehyde in three to five steps in good to excellent yields. The structures of the newly synthesized compounds were established by 1H, 13C and 15N NMR and IR spectra and elemental analysis. Additionally, the X-ray diffraction structure analysis of compound 12 further confirmed its synthesis. Differential scanning calorimetry and thermogravimetric analysis measurements suggest that all the new compounds are thermally and hydrolytically stable and possess good impact and friction sensitivities. They show excellent propulsive and detonation performance. Most of the new compounds are suitable candidates as solid rocket monopropellants and composite propellants for space-related applications. Compounds 6 (DP = 19.98 GPa, Dv = 7492 m s−1) and 12 (DP = 18.98 GPa, Dv = 6866 m s−1) were found to be the best candidates in this series. Compounds 5, 12, and 14 are the best choices as melt-castable energetic materials.Experimental section
General methods
All reagents and solvents were used as received unless otherwise specified (AKSci, Sigma-Aldrich, Acros Organics, VWR). The FT-IR spectra were recorded with KBr plates on a Thermo Nicolet AVATAR 370 spectrometer. Densities were measured at 25 °C with a Micromeritics Accupyc II 1340 gas pycnometer. The melting and decomposition (onset) temperatures were obtained on a differential scanning calorimeter (TA Instruments Company, Model: Q2000) and thermogravimetric analysis (TGA, TA Instruments Company, Model: Q50). The samples were heated from 35 °C to 400 °C at the heating rate of 5 °C min−1. 1H and 13C NMR spectra were recorded on a 300 MHz (Bruker AVANCE 300) nuclear magnetic resonance spectrometer operating at 300.13 and 75.48 MHz, respectively by using DMSO-d6 as the solvent and locking solvent. The 15N NMR spectra were recorded on a 500 MHz (Bruker AVANCE 500) nuclear magnetic resonance spectrometer operating at 50.70 MHz. The chemical shifts are given relative to tetramethylsilane (1H, 13C) and nitromethane (15N) as external standards. Elemental analyses (C, H, N) were performed on a Vario Micro cube Elemental Analyser. The friction sensitivities (FS) and impact sensitivities (IS) were measured with a standard BAM friction tester and BAM drop hammer.The crystals of compound 12 were mounted on a nylon loop with Paratone oil on a XtaLAB Synergy, Dualflex, HyPix diffractometer at 100 K. The structures were solved with the ShelXT28 solution program using dual methods and by using Olex2.29 The model was refined with ShelXL29 using full matrix least squares minimization on F2.
Caution! The new compounds are potentially high-energy materials and could detonate unpredictably under certain conditions. Therefore, it is strongly recommended that during the experiments and their studies proper protective measures, such as Kevlar gloves, face shields, ear protection, and eye protection, should be taken at all times. Only small-scale reactions should be undertaken.
323 (s), 1295 (s), 1213 (m), 980 (s), 858 (s), 821 (m), 531 (m). 1H NMR (DMSO-d6, 300 MHz) δ 7.60 (s, 4H), 8.14 (s, 2H), 11.32 (s, 2H). 13C NMR (DMSO-d6, 75.48 MHz) δ 126.8, 133.9, 147.9. Elemental analysis calcd (%) for C8H8N2O2 (164.16): C 58.53, H 4.91, N 17.06. Found: C 58.53, H 4.97, N 16.69.
:10) to give pure compound 14. Brown solid. 726 mg, 84%, DSC (5 °C min−1): 114 °C (mp), 207 °C (dec.). IR (KBr, cm−1) 3064 (m), 2925 (m), 2112 (br s), 1721 (w), 1626 (m), 1439 (s), 1348 (s), 1281 (s), 1224 (m), 1167 (w), 1124 (w), 1005 (w), 968 (w), 915 (s), 878 (s), 863 (s), 790 (m), 767 (m), 687 (w), 654 (w), 547 (w). 1H NMR (DMSO-d6, 300 MHz) δ 4.65 (s, 4H), 4.94 (s, 4H), 7.93 (s, 4H). 13C NMR (DMSO-d6, 75.48 MHz) δ 41.7, 42.9, 111.2, 128.7, 129.6, 161.3, 166.8. 15N NMR (50.70 MHz, DMSO-d6): δ −5.7, −133.2, −165.6, −166.4, −305.7, −309.6. Elemental analysis calcd (%) for C16H12N14O2·0.5diethylether (469.43): C 46.06, H 3.65, N 41.77. Found: C 46.10, H 3.36, N 41.31.
Author contributions
S. L. conceptualization, investigation, methodology, and manuscript writing-original draft. R. J. S. X-ray data collection and structure solving. S. L. and J. M. S. conceptualization, manuscript writing-review and editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Rigaku Synergy S Diffractometer was purchased with support from the National Science Foundation MRI program (1919565). We are grateful for the support of the Fluorine-19 fund.References
- H. Gao and J. M. Shreeve, Chem. Rev., 2011, 111, 7377–7436 CrossRef CAS PubMed.
- (a) Q. Zhang and J. M. Shreeve, Chem. Rev., 2014, 114, 10527–10574 CrossRef CAS PubMed; (b) H. Gao, Q. Zhang and J. M. Shreeve, J. Mater. Chem. A, 2020, 8, 4193–4216 RSC; (c) S. Lal, R. J. Staples and J. M. Shreeve, Chem. Eng. J., 2023, 452, 139600 CrossRef CAS.
- (a) T. M. Klapötke, Chemistry of High-Energy Materials, Walter de Gruyter GmbH & Co. KG, Berlin/New York, 2011, pp. 179–184 CrossRef; (b) S. Lal, L. Mallick, S. Rajkumar, O. P. Oommen, S. Reshmi, N. Kumbhakarna, A. Chowdhury and I. N. N. Namboothiri, J. Mater. Chem. A, 2015, 3, 22118–22128 RSC.
- J. P. Agrawal and R. D. Hodgson, Organic Chemistry of Explosives, John Wiley & Sons, Ltd., Chichester, 2007, p. 243 Search PubMed.
- (a) P. E. Eaton, R. L. Gilardi and M. X. Zhang, Adv. Mater., 2000, 12, 1143–1148 CrossRef CAS; (b) P. E. Eaton, M.-X. Zhang, R. Gilardi, N. Gelber, S. Iyer and R. Surapaneni, Propellants, Explos., Pyrotech., 2002, 27, 1–6 CrossRef CAS.
- J. V. Viswanath, K. J. Venugopal, N. V. S. Rao and A. Venkataraman, Def. Technol., 2016, 12, 401–418 CrossRef.
- (a) E. C. Koch, Propellants, Explos., Pyrotech., 2015, 40, 374–387 CrossRef CAS; (b) A. F. Baxter, I. Martin, K. O. Christe and R. Haiges, J. Am. Chem. Soc., 2018, 140, 15089–15098 CrossRef CAS PubMed.
- J. Akhavan, The Chemistry of Explosives, RSC Publishing, London, 3rd edn, 2011 Search PubMed.
- (a) N. Fischer, D. Fischer, T. M. KlapÖtke, D. G. Piercey and J. Stierstirfer, J. Mater. Chem., 2012, 22, 20418–20422 RSC; (b) D. Trache, T. M. KlapÖtke, L. Maiz, M. A. Elghany and L. DeLuca, Green Chem., 2017, 19, 4711 RSC.
- T. M. Klapötke, Energetic Materials Encyclopedia, De Gruyter, Berlin/Boston, 2nd edn, 2021 Search PubMed.
- P. W. Cooper, Explosives Engineering, Wiley-VCH, New York, 1996. ISBN 0-471-18636-8 Search PubMed.
- K. G. Shipp, J. Org. Chem., 1964, 29, 2620–2623 CrossRef CAS.
- (a) A. Sankaranarayanan, S. Lal, S. Rashmi, I. N. N. Namboothiri, A. Chowdhury and N. Kumbhakarna, Fuel, 2020, 282, 118816 CrossRef CAS; (b) A. Sankaranarayanan, S. Lal, I. N. N. Namboothiri, S. Rashmi, A. Chowdhury and N. Kumbhakarna, Fuel, 2019, 255, 115836 CrossRef CAS; (c) S. Lal, A. Bhattacharjee, A. Chowdhury, N. Kumbhakarna and I. N. N. Namboothiri, Chem. – Asian J., 2022, 17, e202200489 CrossRef CAS PubMed; (d) S. Lal, A. Chowdhury, N. Kumbhakarna, S. Nandagopal, A. Kumar and I. N. N. Namboothiri, Org. Chem. Front., 2021, 8, 531–548 RSC.
- (a) P. Yin, Q. Zhang and J. M. Shreeve, Acc. Chem. Res., 2016, 49, 4–16 CrossRef CAS PubMed; (b) P. Yin and J. M. Shreeve, Adv. Heterocycl. Chem., 2017, 121, 89–131 CrossRef CAS.
- L. Bauer, M. Benz, T. M. Klapötke, T. Lenz and J. Stierstorfer, J. Org. Chem., 2021, 86, 6371–6380 CrossRef CAS PubMed.
- P. J. Davies and A. S. Provatas, Weapons systems. Characterisation of 2,4-dinitroanisole an ingredient for use in low sensitivity melt cast formulations. Science defence technology organization, Edinburgh, Australia, 2006 Search PubMed.
- R. Banergee, S. Maiti and D. Dhara, Green Chem., 2014, 16, 1365–1373 RSC.
- (a) L. A. Curtiss, K. Raghavachari, G. W. Trucks and J. A. Pople, J. Chem. Phys., 1991, 94, 7221–7232 CrossRef CAS; (b) L. A. Curtiss, K. Raghavachari and J. A. Pople, J. Chem. Phys., 1993, 98, 1293–1298 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. L. Caricato, X. H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Revision D.01 ed, Gaussian, Inc., Wallingford, CT, 2003 Search PubMed.
- P. W. Atkins, Physical Chemistry, Oxford University Press, Oxford, 1982 Search PubMed.
- M. S. Westwell, M. S. Searle, D. J. Wales and D. H. Williams, J. Am. Chem. Soc., 1995, 117, 5013–5015 CrossRef CAS.
- H. Gao, C. Ye, C. M. Piekarski and J. M. Shreeve, J. Phys. Chem. C, 2007, 111, 10718–10731 CrossRef CAS.
- M. Suceska, EXPLO5 6.06, Brodarski Institute, Zagreb Croatia, 2013 Search PubMed.
- T. Lu and F. Chen, Multiwfn, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- (a) S. Lal, H. Gao and J. M. Shreeve, New J. Chem., 2022, 46, 16693–16701 RSC; (b) C. He, Y. Tang, L. A. Mitchell, D. A. Parrish and J. M. Shreeve, J. Mater. Chem. A, 2016, 4, 8969–8973 RSC.
- (a) https://www.bam.de ; (b) A ∼25 mg sample was subjected to a drop-hammer test with a 5 or 10 kg dropping weight. The impact sensitivity was characterized according to the UN recommendations (insensitive, >40 J; less sensitive, 35 J; sensitive, 4 J; very sensitive, 3 J); (c) UN, Committee of Experts on the Transport of Dangerous Goods, Recommendations on the transport of dangerous goods, manual of tests and criteria, United Nations, New York, 5th revised edn, 2009 Search PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2206847. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00166k |
| This journal is © The Royal Society of Chemistry 2023 |