DOI:
10.1039/D3DT00089C
(Paper)
Dalton Trans., 2023,
52, 5882-5894
Group 13 salphen compounds (In, Ga and Al): a comparison of their structural features and activities as catalysts for cyclic carbonate synthesis†
Received
10th January 2023
, Accepted 21st February 2023
First published on 22nd February 2023
Abstract
Many complexes based on group 13 elements have been successfully applied as catalysts for the synthesis of cyclic carbonates from epoxides and CO2 and to date these have provided some of the most active catalysts developed. It is notable that most reports have focused on the use of aluminium-based compounds likely because of the well-established Lewis acidity of this element and its cost. In comparison, relatively little attention has been paid to the development of catalysts based on the heavier group 13 elements, despite their known Lewis acidic properties. This study describes the synthesis of aluminium, gallium and indium compounds supported by a readily prepared salphen ligand and explores both their comparative structures and also their potential as catalysts for the synthesis of cyclic carbonates. In addition, the halide ligand which forms a key part of the compound has been systematically varied and the effect of this change on the structure and catalytic activity is also discussed. It is demonstrated that the indium compounds are actually, and unexpectedly, the most active for cyclic carbonate synthesis, despite their lower Lewis acidity when compared to their aluminium congeners. The experimental observations from this work are fully supported by a Density Functional Theory (DFT) study, which provides important insights into the reasons as to why the indium catalyst with bromide, [InBr(salphen)], is most active.
Introduction
The synthesis of cyclic carbonates through the atom efficient coupling of epoxides to carbon dioxide (CO2) presents an attractive application of CO2 as a C1 reagent (Scheme 1a). Cyclic carbonates have found use as solvents in Li-ion batteries, as polar aprotic solvents, as intermediates in fine chemical synthesis and as monomers in polymer synthesis, amongst many other applications.1 As a result of these diverse applications, the demand for these commodity chemicals is continually increasing. However, the high oxidation state of the C in CO2 and the linearity of the molecule make it highly stable and as such, harsh reaction conditions are generally required for its activation and utilization. The use of catalysts is a well-established approach to reduce the energy needed for reactions to proceed and it is therefore no surprise that a large number of catalysts have been developed to overcome the inherent inertness of the CO2 molecule.2 Two notable approaches in the context of cyclic carbonate synthesis are: (i) use of Lewis acids3 or (ii) use of organocatalysts (typically as hydrogen-bond donors).4 In general, the use of Lewis acids has provided the most success, with a number of highly active catalysts reported to date. When developing novel catalysts, careful selection of both metal and supporting ligand selection are key to a successful outcome. Ligands which are easily prepared are frequently applied and, in this context, the salphen ligand which is readily obtained from the condensation of an ortho-phenylenediamine with two equivalents of a salicylaldehyde has found diverse applications.5 In the context of cyclic carbonate synthesis, as long ago as 2010, Kleij and co-workers demonstrated that easily prepared [Zn(salphen)] complexes were efficient catalysts for the coupling of epoxides and CO2 under mild reaction conditions.6 Since this report, other [M(salphen)] complexes (M = metal) have also been successfully applied as catalysts for this reaction.7 In these reports, Kleij and co-worker observed some catalytic activity with an [AlCl(salphen)] complex (Scheme 1b (i)),6b whilst North and co-worker demonstrated that a bimetallic [AlCl(salphen)]2 complex was highly active under mild reaction conditions and low catalyst loadings.7a This latter report also indicated that the corresponding monometallic [AlCl(salphen)] complex was significantly less active. It should be noted that this increased activity for the dimeric species arises from the bridging O-atom in [AlCl(salphen)]2 acting as a Lewis base activator for CO2.
 |
| | Scheme 1 (a) Reaction of epoxides and CO2 to form cyclic carbonates. (b) (i) Previously reported [AlCl(salphen)] compound successfully applied as catalyst, and (ii) a recently reported Ga catalyst found to be more active than its Al congener (indicative results obtained from ref. 6b, 7a and 19). (c) Overview of the compounds studied in this work. | |
Meanwhile, Ga and In complexes bearing Schiff-base ligands have previously been applied as catalysts for the ring-opening polymerization (ROP) of lactide, highlighting their potential in Lewis acid catalysis.8–14 Specifically related to the work in this study, in 2018, Williams and co-workers described the application of [InCl(salphen)] complexes and derivatives for the co-polymerization of epoxides and CO2 to efficiently form polycarbonates.15 More recently, Mehrkhodavandi and co-workers have also developed active In-based catalysts for this conversion.16 In addition to this, recently, we have reported that a Ga(aminotrisphenolate) compound is more active than its Al congener (Scheme 1b(ii))17 for the synthesis of cyclic carbonates. Whilst also recently and highly relevant, Mehrkhodavandi and co-workers have demonstrated that InBr3 is more active than other group 13 trihalides.18 With these precedents in mind, we decided to study and compare the potential of Al, Ga and In salphen complexes (Scheme 1c) as catalysts for the synthesis of cyclic carbonates to see if it is possible to improve on the relatively poor observed catalytic activity of the [AlCl(salphen)] reported by both Kleij and North. Herein, we report these findings, not only changing the group 13 metal but also the halide in the [MX(salphen)] complexes (M = metal, X = halide) in order to fully optimise the activity of the compounds for catalysis of the cycloaddition of epoxides and CO2. The comparative structural features of the compounds are also discussed, and the experimental work is supported by important insights from an in-depth computational study in order to shed further light on these relatively simple group 13 compounds.
Results and discussion
Synthesis of group 13 salphen complexes
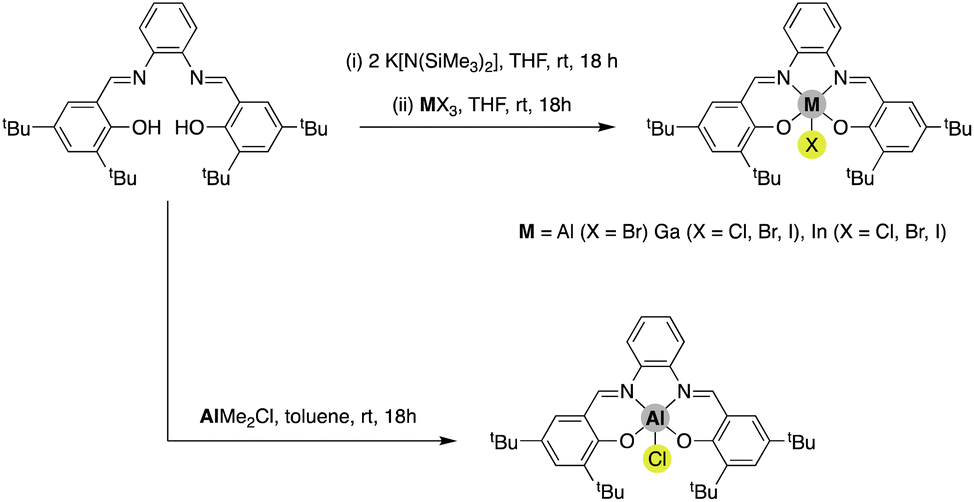
Initially, the Al, Ga and In salphen complexes were synthesised. The products were obtained via two distinct approaches (Scheme 2); the direct reaction of the salphen ligand with AlMe2Cl was selected for the synthesis of [AlCl(salphen)], a standard route for the preparation of this compound. This compound is well described in the literature and has found a variety of applications.19 The lack of ready availability of similar M-alkyl reagents for Ga and In led us to search for other routes for the synthesis of [GaCl(salphen)] and [InCl(salphen)]. A salt-metathesis using K[N(SiMe3)2] reported by Williams and co-workers15 for synthesis of their [InCl(salphen)] complex could be readily transferred and furnished good yields for preparation of these salphen complexes. This synthetic methodology could also be used to successfully prepare the other group-13 salphen complexes; [GaBr(salphen)], [GaI(salphen)], [InBr(salphen)] and [InI(salphen)]. Of these compounds, [AlCl(salphen)], [AlBr(salphen)], [GaCl(salphen)] and [InBr(salphen)] have been previously reported, albeit in several cases they have been prepared by more complicated routes.19–22
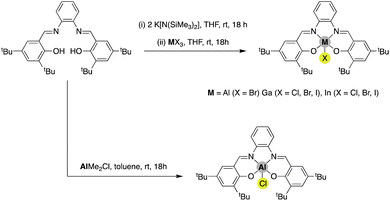 |
| | Scheme 2 Synthesis of Al, Ga and In salphen complexes [MX(salphen)] with different halides used in this study. Note: despite several attempts and modifications, it was not possible to prepare [AlI(salphen)]. | |
We found that [AlBr(salphen)] was more challenging to prepare and this was reflected in the isolated yield which was significantly lower than that of the other compounds. Interestingly, it appears that in the solid state, this compound proceeds to decompose over time, indicating poor structural stability. Finally, despite exhaustive attempts to prepare [AlI(salphen)] we have been unable to obtain this compound. Indeed, to the best of our knowledge it has not been previously reported in the literature. The difficulty in preparing this compound is likely related to an extension of the poor stability of the aforementioned [AlBr(salphen)] compound, whereby exchange for a larger iodide ligand may be expected to impart even lower stability. We are continuing to study other potential methods of synthesis to confirm this, but to date we have not been successful.
Single crystal X-ray structures
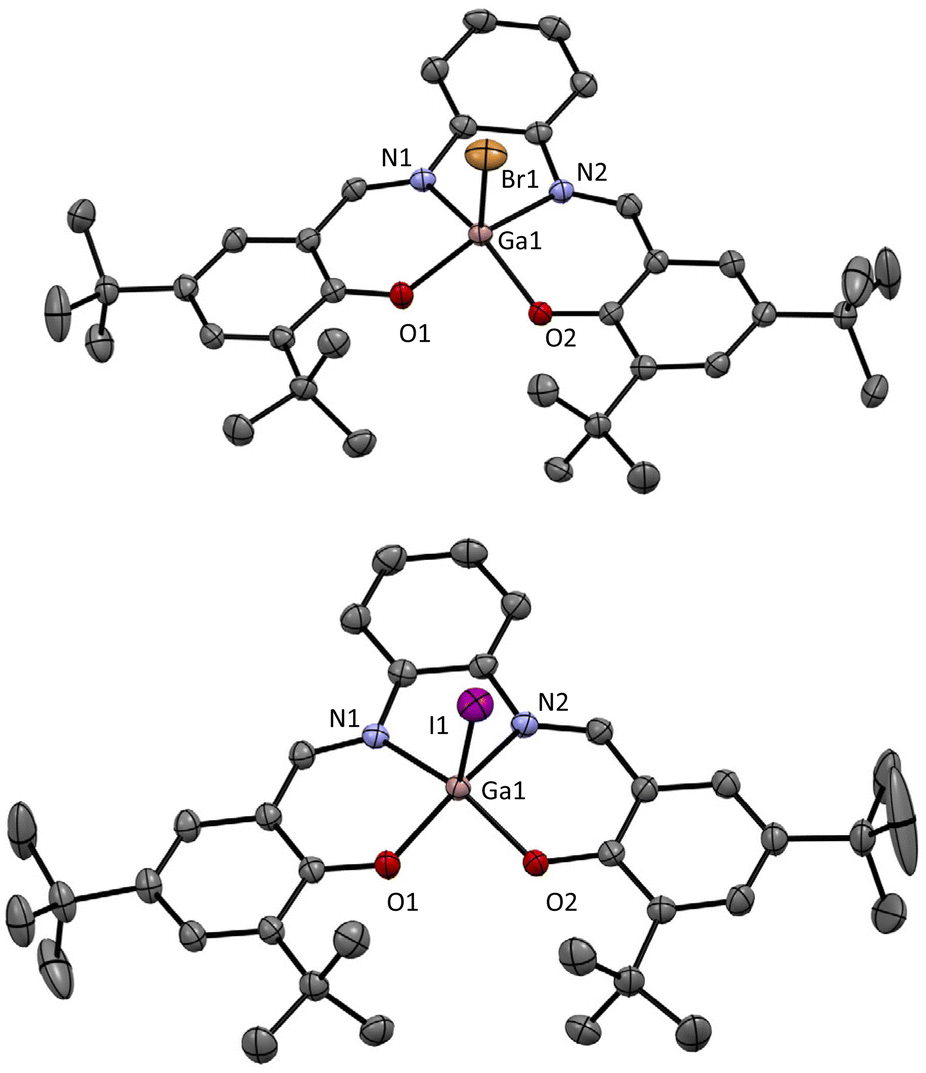
During these studies, it was possible to obtain crystals of both [GaBr(salphen)] and [GaI(salphen)] which were suitable for single-crystal X-ray diffraction studies. The obtained solid state structure for [GaBr(salphen)] (Fig. 1) exhibits a geometry around the Ga which is a little distorted from square pyramidal geometry. This distortion can be quantified using the τ5 value.23
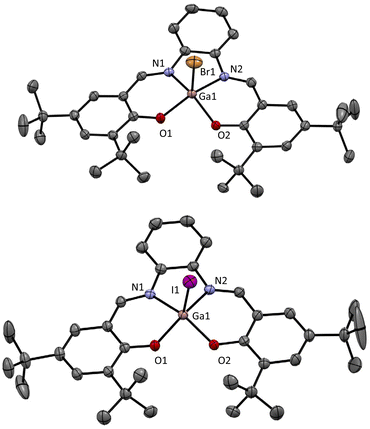 |
| | Fig. 1 X-ray crystal structures obtained for (top) [GaBr(salphen)] and (bottom) [GaI(salphen)] (hydrogen atoms omitted for clarity). τ5 = 0.14 and 0.18, respectively (both slightly distorted square pyramidal geometry). Full structural information can be found in the ESI and CCDC 2173026 or CCDC 2177923.† | |
Comparison of the obtained τ5 value from the solid state structure of [GaBr(salphen)] (Table 2, entry 4; 0.14), with the solid state structure τ5 values for [AlCl(salphen)],19a[AlBr(salphen)]22b and[GaCl(salphen)]20 provides interesting insights (Table 1, entries 1–3; either directly reported or calculated from the archived crystal structure data available from the CCDC). In the [AlX(salphen)] compounds (X = Cl and Br), changing Cl for Br leads to a significant increase in the τ5 value in the solid state structure and hence the distortion (Table 1, entry 1 vs. entry 2). In the case of [AlBr(salphen)], inspection of the solid state structure reveals the presence of strong bifurcated hydrogen bonding interactions between the Br bonded to the Al and both the imine proton (3.874 Å, 154.04°) and the closest proton of the adjacent aromatic ring (3.893 Å, 156.02°). These interactions could be responsible for this distortion. Interestingly, in the [AlCl(salphen)] complex, there are also hydrogen bonding interactions, however, they are weaker and only involve the halide and the proton of the adjacent aromatic ring (3.871 Å, 171.68°). As the distortion is significantly increased in [AlBr(salphen)] and given the rigid and planar nature of the salphen ligand, the compound likely becomes less stable. Indeed, as mentioned above, after prolonged storage on the bench, whilst all the other synthesised compounds are clearly stable, the [AlBr(salphen)] appears to decompose in the solid state (see ESI for comparative 1H NMR spectra†). It would therefore be predicted that [AlI(salphen)] may have an even higher τ5 value, leading to even poorer stability of this compound, making it very difficult to obtain, as we have observed. Importantly, in comparison, the change in the τ5 value in the solid state structure from [GaCl(salphen)] to [GaBr(salphen)] is very small (Table 1, entry 3 vs. entry 4) which is probably a result of the increased size of the Ga atom compared to the Al atom. Furthermore, in contrast to the structures of the Al complexes, in the case of Ga structures, the halide ligand does not establish any hydrogen bonding interactions with adjacent ligands. The reduced distortion in the Ga complexes allows for the synthesis of [GaI(salphen)] as a bench-stable compound which itself displays a relatively small distortion in the solid state (τ5 = 0.19), a value which is much lower than that observed for [AlBr(salphen)]. Although no structural solid state data is available for the [InX(salphen)] compounds, it may be expected that the further increased size of the In atom results in smaller distortions and more stable compounds with all three halides.
Table 1 Comparison of experimental (obtained from X-ray diffraction structures) bond lengths (Å) for previously reported [MX(salphen)] compounds with [GaBr(salphen)] and [GaI(salphen)] from this work for comparisona
[AlCl(salphen)]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a,b (ref. 19a) a,b (ref. 19a) |
[AlBr(salphen)]
(ref. 22b) |
|
Two independent molecules found in the unit cell.
Structural data obtained from CCDC 2046323.
Structural data obtained from CCDC 276667.
Structural data obtained from CCDC 1221216.
This work; structural data available from CCDC 2173026.†
This work; structural data available from CCDC 2177923.†
|
| Al–Cl |
2.182(2)/2.183(2) |
Al–Br |
2.3508(12) |
| Al–N(1) |
2.000(4)/2.009(4) |
Al–N(1) |
1.998(4) |
| Al–N(2) |
2.002(3)/1.998(3) |
Al–N(2) |
1.992(4) |
| Al–O(1) |
1.792(4)/1.783(2) |
Al–O(1) |
1.776(3) |
| Al–O(2) |
1.786(2)/1.792(3) |
Al–O(2) |
1.783(3) |
|
[GaCl(salphen)]
(ref. 20) |
[GaBr(salphen)]
(this work)
|
| Ga–Cl |
2.199(3) |
Ga–Br |
2.3419(4) |
| Ga–N(1) |
2.045(7) |
Ga–N(1) |
2.034(2) |
| Ga-N(2) |
2.016(7) |
Ga–N(2) |
2.048(2) |
| Ga–O(1) |
1.862(6) |
Ga–O(1) |
1.8745(17) |
| Ga–O(2) |
1.881(6) |
Ga–O(2) |
1.8726(17) |
|
[GaI(salphen)]
(this work)
|
| Ga–I |
2.5499(3) |
|
|
| Ga–N(1) |
2.0250(18) |
|
|
| Ga–N(2) |
2.0500(17) |
|
|
| Ga–O(1) |
1.8791(15) |
|
|
| Ga–O(2) |
1.8701(16) |
|
|
Table 2 Comparison of experimental (solid state) and calculated (gaseous state) τ5 values for the [MX(salphen)] compoundsa
|

|
| Entry |
Compound |
τ
5
|
Ref. |
|
τ
5 Values calculated using (β − α)/60°, where β > α and which correspond to the two greatest valence angles of the coordination centre. When τ5 is close to 0 the geometry is similar to square pyramidal, while if τ5 is close to 1 the geometry is similar to trigonal bipyramidal.
Data obtained either directly from the published article or calculated using data obtained from the CCDC.
Two independent molecules found in the unit cell.
Structural data obtained from CCDC 2046323.
Structural data obtained from CCDC 276667.
Structural data obtained from CCDC 1221216.
Structural data available from CCDC 2173026.†
Structural data available from CCDC 2177923.†
|
| 1c,d |
[AlCl(salphen)]
|
0.10/0.12 |
19a
|
| 2e |
[AlBr(salphen)]
|
0.32 |
22b
|
| 3 |
[AlI(salphen)]
|
–– |
–– |
| 4f |
[GaCl(salphen)]
|
0.11 |
20
|
| 5g |
[GaBr(salphen)]
|
0.14 |
This work |
| 6h |
[GaI(salphen)]
|
0.19 |
This work |
| 7 |
[InCl(salphen)]
|
–– |
–– |
| 8 |
[InBr(salphen)]
|
–– |
–– |
| 9 |
[InI(salphen)]
|
–– |
–– |
Calculated structures
With only a limited number of solid state structures available in the published literature, we decided to perform geometry optimization on all of the compounds. The obtained structures are shown in Fig. 2 and are relevant to solution studies. This approach has allowed more insight to be gained into the effects of changing the metal and the halide. These structures were calculated using RI-B97-D3/def2-SVP and the resulting τ5 values are included in Fig. 2. Interestingly there are significant differences between the experimental solid state structural data and the calculated structures. In the case of the [AlX(salphen)] complexes, the calculated structures present a very similar τ5 value for all the halide ligands. Meanwhile, the solid state structure of [AlCl(salphen)] displays a lower τ5 value than the calculated value (Table 2, entry 1; 0.10/0.12 vs. calculated: 0.18). In contrast, the experimental solid state structure of [AlBr(salphen)] displays a significantly higher τ5 value than the calculated value (Table 2, entry 2; 0.18 vs. calculated: 0.32), which could be influenced by the presence of a strong hydrogen bonding interaction. The τ5 values for the calculated [GaX(salphen)] complexes are more in line with the reduced distortions of the solid state structures. Finally, for the [InX(salphen)] compounds no solid state structural data is available, however, it can be seen from the calculated structures that the distortions from square pyramidal geometry are significantly reduced compared to their Al and Ga congeners (Fig. 2). Indeed, the value for [InI(salphen)] of τ5 = 0.07 presents an almost square pyramidal geometry. These low τ5 values are most likely a result of the increased size of the In atom compared to Ga and Al. Charges for the metal centre within each catalyst system are given in the ESI (Table S6†).
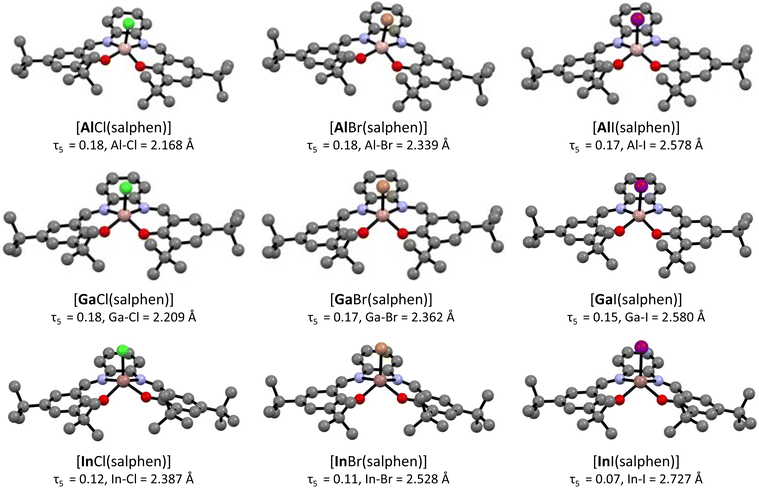 |
| | Fig. 2 Calculated structures (RI-B97-D3/def2-SVP) of the [MX(salphen)] compounds (M = Al, Ga and In; X = Cl, Br and I) with τ5 values and M–X bond lengths. Hydrogen atoms omitted for clarity. | |
Comparative evaluation as catalysts for the synthesis of cyclic carbonates from CO2 and epoxides
With the eight complexes in hand, and their structures studied and compared, attempts were made to evaluate their potential as catalysts for the conversion of styrene oxide and CO2 to styrene carbonate (Table 3). The availability of Al, Ga and In compounds with the corresponding Cl, Br or I ligand in this work, provides an opportunity for a systematic study into the effect of changing the metal and halide. Initial trials were performed using a binary catalyst system of 0.25 mol% catalyst, 1.0 mol% co-catalyst (TBAI; tetra-butylammonium iodide) at 50 °C under solvent-free conditions. These initial studies resulted in two important observations. Firstly, changing from Al to Ga and then In, led to an increase in the obtained yield from 47% to 67% and then 98% (Table 3, entries 1, 3 and 6). This was unexpected as it would be proposed that the Al complex is the most Lewis acidic and therefore should be the best catalyst for the Lewis acid-catalyzed reaction. This is however in agreement with our previous work where we established that the strength of the Lewis acid is not the only important factor in Lewis acid-catalyzed cyclic carbonate synthesis from epoxides and CO2.17 It should be noted that when using the [AlCl(salphen)] compound, on occasion a precipitate was observed at the end of the reaction, an observation previously made by Kleij and co-workers for this compound.7b Secondly, it can be seen that in the case of both [AlX(salphen)] and [GaX(salphen)] compounds, changing from Cl to Br and then (where available) I, the catalytic activity increases (Table 3, entries 1–5). In order to perform the same study on the [InX(salphen)] compounds it was necessary to reduce the binary catalyst system loading to observe differences in reactivity as [InCl(salphen)] provided almost quantitative conversion under the reaction conditions used for the [AlX(salphen)] and [GaX(salphen)] comparison. As such, the binary catalyst system loading was reduced to half (0.125 mol% [MX(salphen)] and 0.5 mol% TBAI). Under these conditions, the [InX(salphen)] compounds provided yields of 63, 70 and 62%, for X = Cl, Br and I, respectively (Table 3, entries 7–9). With these results, the [InBr(salphen)] was selected as the most active catalyst for further optimisation. At this point, to further optimise, two approaches were taken. Firstly, increasing the binary catalyst system to 0.20 mol% [InBr(salphen)] and 0.8 mol% TBAI, provided a yield of 98% (Table 3, entry 10). Meanwhile, increasing the temperature to 60 °C using 0.125 mol% [InBr(salphen)] and 0.5 mol% TBAI resulted in a yield of 95% (Table 3, entry 11), a significant improvement on the reaction at 50 °C (70%; Table 3, entry 8). A combination of a slight increase in the binary catalyst system loading (0.15 mol% catalyst and 0.6 mol%) provided quantitative yield of cyclic carbonate product (Table 3, entry 12). Importantly, very low yield (4%) was obtained in the absence of catalyst and no conversion was observed in the absence of TBAI indicating that the halide ligands are unable to act as co-catalyst nucleophiles under these conditions (Table 3, entries 13 and 14). Attempts to study the Lewis acidity of the compounds using the Gutmann-Beckett method failed, producing precipitates which could not be analysed by NMR spectroscopy.24 However, it is notable that a general trend between the chemical shift of the imine proton of the compounds and the activity of the catalyst is observed, which is likely a result of the distinct Lewis acidities of the three different metals. This is particularly evident with the [MCl(salphen)] compounds (Fig. 3); [AlCl(salphen)] (47%; 8.95 ppm), [GaCl(salphen)] (67%; 8.93 ppm) and [InCl(salphen)] (98%; 8.79 ppm). These are the values obtained in CDCl3, however, a similar trend is observed in non-polar C6D6.
 |
| | Fig. 3 Comparison of 1H NMR spectra (CDCl3, 298 K) of the imine and aromatic region for (yields at 0.25 mol% catalyst, 1.0 mol% TBAI and 50 °C, along with chemical shift of the imine proton): (a) [AlCl(salphen)] (47%, 8.95 ppm); (b) [GaCl(salphen)] (67%, 8.93 ppm); (c) [InCl(salphen)] (98%, 8.79 ppm). *Indicates the imine proton. | |
Table 3 Comparison of the catalytic activity of the [MX(salphen)] compounds and reaction condition optimization using styrene oxidea
|

|
| Entry |
Compound ([MX(salphen)]) |
M loading (mol%) |
TBAI (mol%) |
T (°C) |
Yieldb,c (%) |
|
General reaction conditions: styrene oxide (10 mmol), [MX(salphen)], TBAI, 8 bar CO2, T, 18 h, sealed high-pressure reactor.
Yield determined by inspection of the 1H NMR spectra of the crude reaction mixture using mesitylene as the internal standard.
Selectivity towards the cyclic carbonate product >99% in all cases. TBAI = tetra-butylammonium iodide. rt = room temperature.
|
| 1 |
[AlCl(salphen)]
|
0.25 |
1.0 |
50 |
47 |
| 2 |
[AlBr(salphen)]
|
0.25 |
1.0 |
50 |
65 |
| 3 |
[GaCl(salphen)]
|
0.25 |
1.0 |
50 |
67 |
| 4 |
[GaBr(salphen)]
|
0.25 |
1.0 |
50 |
86 |
| 5 |
[GaI(salphen)]
|
0.25 |
1.0 |
50 |
95 |
| 6 |
[InCl(salphen)]
|
0.25 |
1.0 |
50 |
98 |
| 7 |
[InCl(salphen)]
|
0.125 |
0.5 |
50 |
63 |
| 8 |
[InBr(salphen)]
|
0.125 |
0.5 |
50 |
70 |
| 9 |
[InI(salphen)]
|
0.125 |
0.5 |
50 |
62 |
| 10 |
[InBr(salphen)]
|
0.20 |
0.8 |
50 |
98 |
| 11 |
[InBr(salphen)]
|
0.125 |
0.5 |
60 |
95 |
|
12
|
[InBr(salphen)]
|
0.15
|
0.6
|
60
|
>99
|
| 13 |
–– |
–– |
0.6 |
60 |
4 |
| 14 |
[InBr(salphen)]
|
0.15 |
–– |
60 |
0 |
|
Room temperature study:
|
| 15 |
[InBr(salphen)]
|
1.0 |
4.0 |
Rt |
>99 |
| 16 |
[InBr(salphen)]
|
–– |
4.0 |
Rt |
4 |
|
17
|
[InBr(salphen)]
|
0.75
|
3.0
|
Rt
|
>99
|
| 18 |
[InBr(salphen)]
|
0.5 |
2.0 |
Rt |
92 |
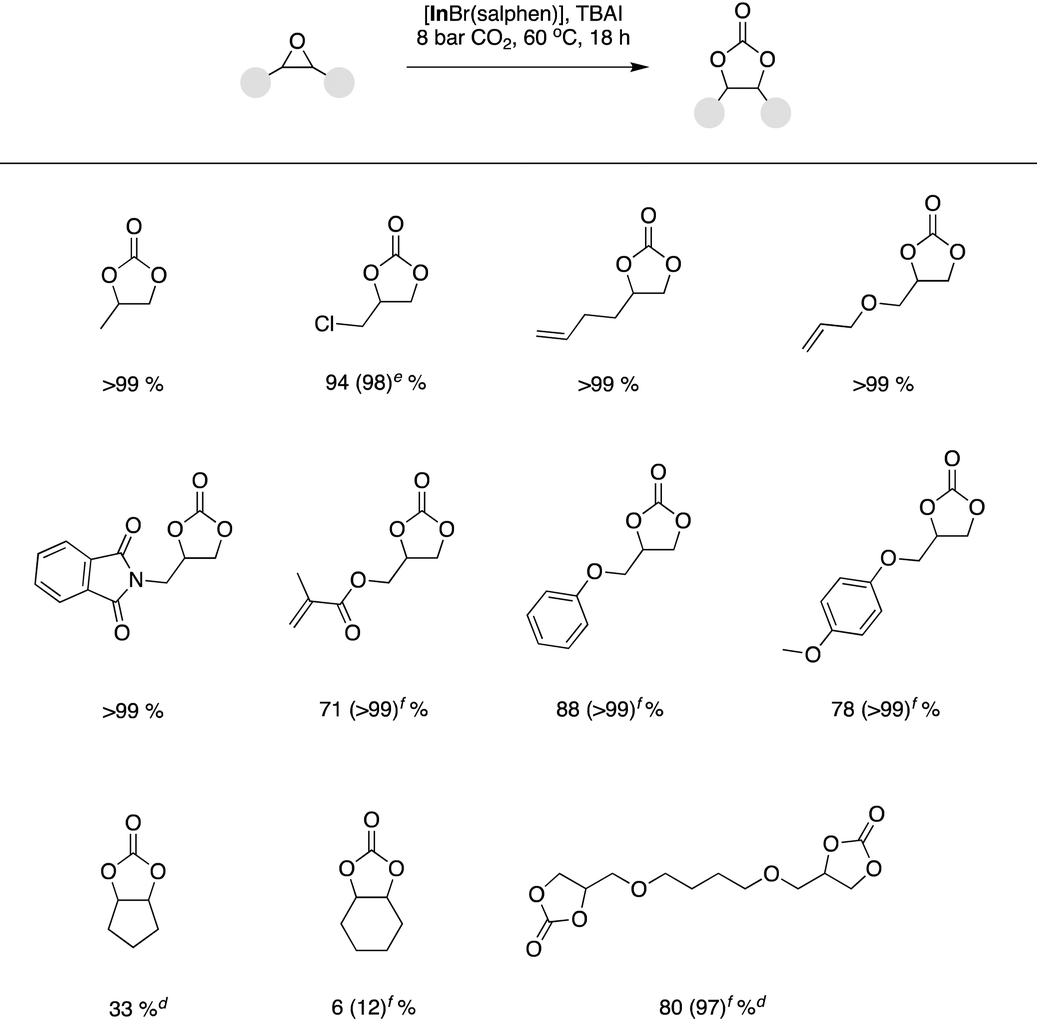
The optimized binary catalyst system based on [InBr(salphen)] and TBAI was also transferrable to a range of substrates beyond styrene oxide (Table 4). In general, terminal epoxides with a variety of functionalities could be readily converted in good to quantitative yields. However, in some cases, to obtain desirable quantitative yields, it was necessary to increase the binary catalyst system loading to 0.225 mol% [InBr(salphen)] and 0.9 mol% TBAI or 0.30 mol% [InBr(salphen)] and 0.12 mol% TBAI. Even under increased binary catalyst system loadings it was not possible to obtain significant yields starting from internal epoxides (cyclopentene oxide and cyclohexene oxide).
Table 4 Substrate scope using [InBr(salphen)] under the optimized reaction conditions,a,b,c and substrate optimized conditionsd,e,f
|
General reaction conditions: rt experiments; substrate (10 mmol), [InBr(salphen)] (0.75 mol%), TBAI (3.0 mol%), 8 bar CO2, rt, 18 h, sealed high-pressure reactor or 60 °C experiments; substrate (10 mmol), [InBr(salphen)] (0.15 mol%), TBAI (0.6 mol%), 8 bar CO2, 60 °C, 18 h, sealed high-pressure reactor.
Yield determined by inspection of the 1H NMR spectra of the crude reaction mixture.
Selectivity towards the cyclic carbonate product >99% in all cases.
5.0 mmol scale.
Value in parenthesis relates to result obtained using 0.225 mol% [InBr(salphen)] and 0.9 mol% TBAI.
Value in parenthesis relates to result obtained using 0.30 mol% [InBr(salphen)] and 1.2 mol% TBAI.
|
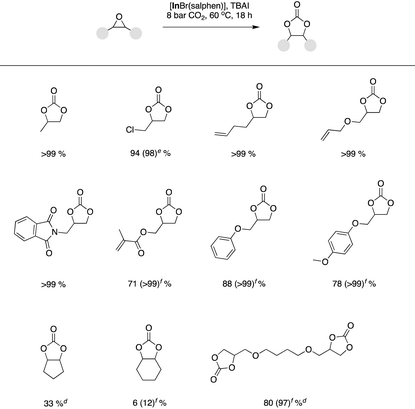
|
With a further desire to develop a catalyst system which is applicable at room temperature, we proceeded to further optimize (Table 3, entries 15–18). An initial experiment applying a binary catalyst system loading of 1.0 mol% [InBr(salphen)] and 4.0 mol% TBAI gratifyingly resulted in a quantitative yield of the cyclic carbonate product (Table 3, entry 15). In the absence of [InBr(salphen)], the same reaction yielded only 4% of the styrene carbonate product (Table 3, entry 16). Reduction of the binarycatalyst system loading to 0.75 mol% [InBr(salphen)] and 3.0 mol% TBAI also resulted in a >99% product yield (Table 3, entry 17). However, further reduction to 0.50 mol% [InBr(salphen)] and 2.0 mol% TBAI did not allow for complete substrate conversion and as such the optimised binary catalyst system loading for room temperature conversion was selected as 0.75 mol% [InBr(salphen)] and 3.0 mol% TBAI. As with the elevated temperature study, the substrate scope was studied, where it was found that a range of terminal epoxides could be converted in high to excellent yields (Table 4).
Further to the study at room temperature, optimisation for the conversion of internal epoxides, which failed to provide suitable yields at 60 °C was pursued (Table 5). This study was performed using cyclopentene oxide as substrate at 80 °C with the optimal binary catalyst system loading for terminal epoxides at 60 °C, 48% yield was obtained (Table 5, entry 1). Increase of the binary catalyst system loading allowed for a 91% yield (Table 5, entry 3; 1.2 mol% [InBr(salphen)] and 4.8 mol% TBAI). Whilst a final increase in the reaction temperature to 90 °C provided a 94% yield. Importantly, in the absence of [InBr(salphen)], but still using 4.8 mol% TBAI, at this temperature only 2% yield was observed (Table 5, entry 5). These final reaction conditions were then transferred towards cyclohexene oxide (Table 6; 60% yield) and both the cis and trans-isomers of 2,3-epoxybutane (Table 6, 97 and 84% yields, respectively). In this latter case, the product was observed to be a mixture of the cis and trans-cyclic carbonates.
Table 5 Comparison of the catalytic activity of the [MX(salphen)] catalysts and reaction condition optimization using styrene oxidea
|

|
| Entry |
Catalyst loading (mol%) |
TBAI (mol%) |
T (°C) |
Yieldb,c (%) |
|
General reaction conditions: cyclopentene oxide (10 mmol), [MX(salphen)], TBAI, 8 bar CO2, T, 18 h, sealed high-pressure reactor.
Yield determined by inspection of the 1H NMR spectra of the crude reaction mixture using mesitylene as the internal standard.
Selectivity towards the cyclic carbonate product >99% in all cases. TBAI = tetra-butylammonium iodide.
|
| 1 |
0.15 |
0.6 |
80 |
48 |
| 2 |
0.30 |
1.2 |
80 |
56 |
| 3 |
1.2 |
4.8 |
80 |
91 |
| 4 |
1.2 |
4.8 |
90 |
94 |
| 5 |
–– |
4.8 |
90 |
2 |
Table 6 Substrate scope using [InBr(salphen)] catalyst under the optimized reaction conditionsa,b,c,d
|
General reaction conditions: substrate (10 mmol), [InBr(salphen)] (1.2 mol%), TBAI (4.8 mol%), 8 bar CO2, 90 °C, 18 h, sealed high-pressure reactor.
Yield determined by inspection of the 1H NMR spectra of the crude reaction mixture.
Selectivity towards the cyclic carbonate product >99% in all cases.
5.0 mmol scale.
|

|
DFT study into the mechanism and differences in the catalytic reactivities of the group-13 compounds
With the binary catalyst system optimized and the substrate scope studied, the operative mechanism was elucidated using Density Functional Theory (DFT) methods. These initial studies involve the ligand with o-tert-butyl groups to reduce computational costs (note that the effect of these substituents has also been studied in this work and found to be rather small; these results are provided in the ESI†). Fig. 4 shows the concentration corrected (see computational method in ESI†) calculated solvated free energy surface (ΔG298 K) for the three group 13 [MCl(salphen)] pathways (M = Al, Ga and In; in blue, red and black, respectively). All three [MCl(salphen)] compounds follow the generally accepted mechanism for the Lewis acid catalysed synthesis of carbonates from CO2 and epoxides. Initial endergonic coupling of the epoxide substrate and catalyst affords the IC adduct. From IC, halide assisted epoxide ring opening occurs (TS1) leading to the alkoxide species Int1α. The barrier for this step ranges from 14.51 kcal mol−1 to 20.09 kcal mol−1 for these [MCl(salphen)] catalysts and is similar to previously reported barriers. At this point, to allow for insertion of CO2 into the metal alkoxide bond, Int1α must dissociate the trans halide ion, affording Int1β. For each of the binary catalyst systems the relative energies for halide abstraction by either TBA+ (tetrabutylammonium cation) or another [MCl(salphen)] catalyst have been calculated (Table 7). As can been seen in Table 7, for all metals formation of a dihalide species, [MCl2(salphen)]−, is energetically more favourable than abstraction by TBA+. In addition, it is interesting to note that the [AlCl2(salphen)]−, and subsequently Int1β, are significantly more stable compared to the energies of the corresponding Ga or In congeners. From Int1β, insertion of CO2, viaTS2, is the rate determining step for the reaction. The overall barriers for the Al and In catalysts are similar; approximately 29 kcal mol−1, whilst Ga is 36.15 kcal mol−1. This is consistent with the experimental observation of [GaCl(salphen)] being less active compared to [InCl(salphen)]. As mentioned earlier, and in previous literature,7b the Al-based compounds show solubility issues, which are presumed to be related to the halide dissociation, however further investigation is beyond the scope of this computational study. From Int2β several divergent pathways are available, all of which afford the observed product. Re-association of the halide, leading to Int2α, is energetically favourable for [GaCl(salphen)] and [InCl(salphen)], yet unfavourable for [AlCl(salphen)], highlighting the greater stability of the [AlCl2(salphen)]− complex and thus reduced catalytic performance. Ring closure, with subsequent loss of I−, from Int2α can occur via one of two pathways, R1 or R2. R1 presents a direct ring closure from Int2α whereas R2 presents an isomerisation prior to the ring closing, forming the final complex (FC). Transitions states for both pathways, within each metal system, are energetically very similar and accessible under the experimental conditions used. Of note, for [InCl(salphen)] it was observed that the isomerisation step preferentially occurs prior to re-association of the halide ion.To the best of our knowledge, the loss of the halide ion to facilitate the CO2 insertion step, is the first mechanistic proposal of this type for group 13 Lewis acid catalysed epoxide and CO2 coupling reactions. This new insight combines aspects of both Zn(II) salphen25 and transition metal salen26 mechanisms previously reported.
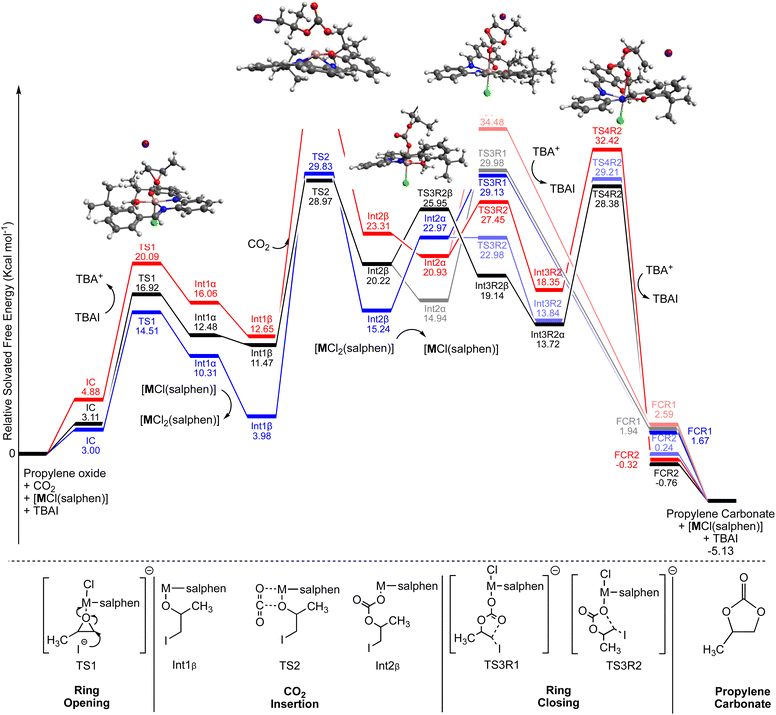 |
| | Fig. 4 Calculated free energy surface (ΔG298 K), RIJCOSX-ωB97M-D3BJ/def2-TZVPP, for cyclic carbonate synthesis using propylene oxide as substrate with [MCl(salphen)] (without p-tert-butyl substituents) compounds as catalyst. M = Al (blue), Ga (red) and In (black). All energies are concentration corrected to model the experiment conditions. | |
Table 7 Comparison of calculated relative energy (ΔG298 K at RIJCOSX-ωB97M-D3BJ/def2-TZVPP) for the halide abstraction routes for each [MCl(salphen)] compound
| Abstraction route |
Relative stability (kcal mol−1) |
| Y = alkoxide arising from ring-opening of the epoxide with I−. |
|
[MYCl(salphen)]− + [MCl(salphen)] → [MY(salphen)] + [MCl2(salphen)]− |
|
[AlCl(salphen)]
|
−6.33 |
|
[GaCl(salphen)]
|
−3.40 |
|
[InCl(salphen)]
|
−1.00 |
|
[MYCl(salphen)]− + TBA+ → [MY(salphen)] + TBACl |
| TBA+ ([AlCl(salphen)] Pathway) |
−2.11 |
| TBA+ ([GaCl(salphen)] Pathway) |
−1.35 |
| TBA+ ([InCl(salphen)] Pathway) |
+8.21 |
With the underlying mechanism in-hand, we turned our attention to the effect of variation of the halide on the reactivity for the [InX(salphen)] compounds. In this case as there was an optimised catalyst system, the ligand with tert-butyl substituents was included. Fig. 4 shows the free energy surface (ΔG298 K) for the [InX(salphen)] catalysed reaction between CO2 and epoxeide to cyclic carbonate, where X = Cl, Br and I. As expected, the reaction follows similar mechanistic pathways with th different halides, with only small energy differences separating the catalysts systems. In agreement with the slightly improved experimental results, the [InBr(salphen)] catalyst shows lower barriers for the epoxide ring opening and the CO2 insertions steps, as well as a more energetically favourable catalyst-carbonate adduct (FCR1) formation. Concurrently, the [InI(salphen)] catalyst shows higher barriers and the least favourable FCR1 intermediate, again confirming the experimental observations (Fig. 5).
 |
| | Fig. 5 Calculated free energy surface (ΔG298 K), RIJCOSX-ωB97M-D3BJ/def2-TZVPP, for cyclic carbonate synthesis using propylene oxide as substrate [InX(salphen)] compounds as catalyst. X = Cl (blue), Br (black) and I (red). All tert-butyl substituents included in the calculations. All energies are concentration corrected to model the experiment conditions. | |
Lewis acidity is often evoked as an important catalyst design descriptor for this type of reaction. In an attempt to further explore the Lewis acidity of these catalysts we performed Fluoride Ion Association (FIA) calculations, based on the TMS-anchored isodesmic reaction scheme, originally proposed by Krossing27 and recently updated by Greb.28 The absolute FIA values, shown in Table 8, suggest that the [AlX(salphen)] compounds are most Lewis acidic, followed by the [InX(salphen)] compounds and then finally, [GaX(salphen)] compounds. The [GaX(salphen)] compounds being least Lewis acidic agrees with the higher calculated energy barriers shown in Fig. 3. The significantly higher FIA values for the [AlX(salphen)] compounds also confirms the reason for the increased stability of the [AlCl2(salphen)]− complex, leading to the observed relative energy differences for the halide abstraction and re-association processes (Fig. 3; Int1α to Int1β and Int2β to Int2α). Interestingly, the FIA values for the [InX(salphen)] compounds are invariant, suggesting very little effect from changing the halide. Although [InBr(salphen)] has been shown to be most active, the differences between the different halides using In were found to be less pronounced than with the [AlX(salphen)] and [GaX(salphen)] compounds. Actually, the greater Lewis acidity expressed by the [AlX(salphen)] compounds do not directly result in greater catalytic activity. In agreement to some previous works16,29 clearly there is more to understanding these reactions than Lewis acidity alone.
Table 8 Comparison of calculated isodesmic absolute Fluoride Ion Association (FIA) values for each catalytic species. Calculated at RIJSCOSX-PW6B95-D3BJ/def2-qzvpp
| Catalyst |
Absolute FIA (kJ mol−1) |
|
[AlCl(salphen)]
|
317.87 |
|
[AlBr(salphen)]
|
331.31 |
|
[GaCl(salphen)]
|
273.19 |
|
[GaBr(salphen)]
|
280.37 |
|
[GaI(salphen)]
|
289.90 |
|
[InCl(salphen)]
|
289.88 |
|
[InBr(salphen)]
|
289.57 |
|
[InI(salphen)]
|
288.80 |
Conclusions
In conclusion, we have prepared a range of Al, Ga and In compounds with systematic variation of the halide ligand (Cl, Br and I). The features of the solid state structures obtained in this work have been compared with the available literature structures of [AlCl(salphen)], [AlBr(salphen)] and [GaCl(salphen)]. It has been observed that strong distortions from ideal square pyramidal geometry are observed in the solid state for [AlBr(salphen)], with this structure displaying strong bifurcated hydrogen bonding interactions. These interactions are not observed in the case of the Ga congeners. Meanwhile, the calculated structures of all the compounds have also been obtained and compared. All the synthesised compounds have been studied for their potential as catalysts for the synthesis of cyclic carbonates from epoxides and CO2. The general trend that [InX(salphen)] > [GaX(salphen)] > [AlX(salphen)] has been found, where [InBr(salphen)] was identified as the most active catalyst for the conversion. This catalyst allowed access to quantitative yields of product at relatively low catalyst and co-catalyst loadings (0.15 mol%/0.6 mol%, respectively) at 60 °C. The optimal binary catalyst system has been found to be transferrable to other terminal epoxides beyond styrene oxide, although with slight increases in binary catalyst system loading required in some cases. Further to this, the catalyst system has also been successfully applied at room temperature after an increase in catalyst system loading. Meanwhile, at elevated temperature/catalyst system loading conversion of challenging internal epoxides is also possible. A DFT study has provided information as to why the [InX(salphen)] compounds are the most active catalysts despite their apparent lower Lewis acidity compared with the [AlX(salphen)] congeners. As a final comment, overall, it is clear that this specific catalyst system displays a demanding CO2 insertion step, and that more Lewis acidic metals produce less reactive metal-alkoxide species after initial ring-opening of the epoxide substrate. Therefore, this study highlights that not in all cases a more Lewis acidic metal centre provides a stronger catalyst. Indeed, there is a clear interplay between Lewis acidity and ligand coordination fluxionality. As a result, this work contributes as a further example to the cases described in the recent review by Kleij and co-worker concerning the mechanistic details of cyclic carbonate synthesis from epoxides and CO2.30 In summary, this work should inspire other researchers to investigate the potential of catalysts based on the heavier group 13 elements for use as catalysts for cyclic carbonate synthesis, a field which is currently dominated by Al. Meanwhile we suggest that it may also be possible to transfer the [InBr(salphen)] compound as an improvement to [AlCl(salphen)] for the host of applications that this well-reported compound has previously found.19
Experimental part
All solvents and reagents were purchased from Fisher Scientific or Cymit Quimica and were used without further purification. 1H, 13C{1H} and COSY NMR spectra were recorded on a Bruker AV-400 spectrometer in CDCl3 and referenced to the residual solvent peak at 7.26 ppm (1H) or 77.16 ppm (13C). Elemental analysis was performed by the Centro de Espectometría de Masas y Análisis Elemental (CEMAETA) at the Universidad de Alcalá. The salphen ligand (6,6′-((1E,1′E)-(1,2-phenylenebis(azaneylylidene))bis(methaneylylidene))bis(2,4-di-tert-butylphenol)) was prepared according to a reported literature procedure.31 All syntheses of the [MX(salphen)] compounds were carried out using a Schlenk line and with dry solvents in order to exclude air or moisture. All catalytic reactions were performed in Berghof High-pressure reactors (BR-40, PTFE liner, 70 mL volume) using high-purity carbon dioxide (>99.995%) purchased from Linde (no further purification), with an initial starting pressure of 8.0 bar.
General synthetic procedures for [MX(salphen)] preparation
General method [A]: Ligand (H2salphen) was dissolved in toluene in a Schlenk tube under argon and cooled to −78 °C. To the solution was slowly added 1.0 equiv. of dimethylaluminum chloride (AlMe2Cl) solution (1.0 M in hexane). The reaction was allowed to warm to room temperature and stirred overnight. The next day, the solvent was removed under reduced pressure and the crude solid was washed with hexane (3 times) to remove residual unreacted ligand. The resulting solid was further dried under vacuum to provide analytically pure [AlCl(salphen)] as a yellow powder. General method [B]:32 Ligand (H2salphen) was dissolved in THF in a Schlenk tube under an argon atmosphere. To the solution was slowly added 2.0 equiv. of K[N(SiMe3)2] as a solid. The reaction was sealed and left to stir overnight. The next day, the solution was filtered into a solution of MX3 in THF using a filter cannula. The reaction mixture was stirred for a further 24 hours, during which time a white precipitate formed. The solution was filtered, and the solvent removed under reduced pressure. The crude residue was washed with hexane (3 times) to remove residual unreacted ligand. Finally, the solid was taken up into dichloromethane, filtered and the solvent removed from the filtrate under reduced pressure. The resulting solid was further dried under vacuum to provide analytically pure [MX(salphen)] as a yellow powder.
Synthesis of [AlCl(salphen)]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) 19h
19h
Prepared using general method A; yellow powder (82%). 1H NMR (400 MHz, 298 K, CDCl3) δ 8.95 (s, 2H), 7.76–7.72 (m, 2H), 7.69 (d, 4JHH = 2.6 Hz, 2H), 7.40–7.35 (m, 2H), 7.24 (d, 4JHH = 2.6 Hz, 2H), 1.62 (s, 18H), 1.36 (s, 18H) ppm. 13C{1H} NMR (101 MHz, 298 K, CDCl3) δ 164.4, 162.5, 141.6, 139.7, 137.8, 133.1, 128.3, 128.2, 118.5, 115.4, 35.7, 34.2, 31.3, 29.9 ppm. IR (ATR): 2946.5, 2903.6, 2868.2, 1613.9, 1584.1, 1537.5, 1386.6, 1358.6, 1200.2, 1183.4, 846.1, 592.6 cm−1.
Synthesis of [AlBr(salphen)]22b
Prepared using general method B; yellow powder (31%). 1H NMR (400 MHz, 298 K, CDCl3) δ 8.98 (s, 2H), 7.79–7.75 (m, 2H), 7.69 (d, 4JHH = 2.6 Hz, 2H), 7.44–7.41 (m, 2H), 7.25 (d, 4JHH = 2.6 Hz, 2H), 1.60 (s, 18H), 1.36 (s, 18H) ppm. 13C{1H} NMR (101 MHz, 298 K, CDCl3) δ 164.2, 162.5, 141.7, 139.9, 137.5, 133.2, 128.3, 128.3, 118.5, 115.5, 35.7, 34.2, 31.3, 29.9 ppm. IR (ATR): 2959.5, 2868.2, 1613.9, 1580.4, 1537.5, 1384.7, 1360.5, 1202.1, 1183.4, 848.0, 758.5, 568.4 cm−1.
Synthesis of [GaCl(salphen)]20
Prepared using general method B; yellow powder (82%). 1H NMR (400 MHz, 298 K, CDCl3) δ 8.93 (s, 2H), 7.77–7.73 (m, 2H), 7.64 (d, 4JHH = 2.6 Hz, 2H), 7.43–7.38 (m, 2H), 7.16 (d, 4JHH = 2.6 Hz, 2H), 1.59 (s, 18H), 1.35 (s, 18H). 13C{1H} NMR (101 MHz, 298 K, CDCl3) δ 167.6, 162.2, 142.4, 139.2, 135.8, 132.8, 128.6, 128.2, 117.0, 115.2, 35.8, 34.1, 31.2, 29.8. IR (ATR): 2946.5, 2868.2, 1604.6, 1584.1, 1533.8, 1386.6, 1358.6, 1198.3, 1183.4, 743.6, 542.3 cm−1.
Synthesis of [GaBr(salphen)]
Prepared using general method B; yellow powder (91%). 1H NMR (400 MHz, 298 K, CDCl3) δ 8.90 (s, 2H), 7.76–7.71 (m, 2H), 7.65 (d, 4JHH = 2.6 Hz, 2H), 7.40–7.36 (m, 2H), 7.16 (d, 4JHH = 2.6 Hz, 2H), 1.61 (s, 18H), 1.35 (s, 18H) ppm. 13C{1H} NMR (101 MHz, 298 K, CDCl3) δ 167.4, 162.1, 142.4, 139.3, 135.5, 132.9, 128.7, 128.2, 117.0, 115.3, 35.8, 34.1, 31.3, 29.8 ppm. IR (ATR): 2952.1, 2905.5, 2868.2, 1602.8, 1582.3, 1531.9, 1388.4, 1358.6, 1196.5, 1179.7, 747.3, 538.6 cm−1. Elemental analysis for C36H46GaBrN2O2. Calc. C: 62.81%, H: 6.74%, N: 4.65%. Found: C: 62.64%, H: 6.68%, N: 4.40%.
Synthesis of [GaI(salphen)]
Prepared using general method B; yellow powder (88%). 1H NMR (400 MHz, 298 K,CDCl3) δ 8.89 (s, 2H), 7.78–7.74 (m, 2H), 7.65 (d, 4JHH = 2.6 Hz, 2H), 7.45–7.40 (m, 2H), 7.16 (d, 4JHH = 2.6 Hz, 2H), 1.59 (s, 18H), 1.35 (s, 18H) ppm. 13C{1H} NMR (101 MHz, 298 K, CDCl3) δ 167.1, 161.8, 142.5, 139.3, 135.2, 133.0, 128.7, 128.2, 117.0, 115.4, 35.8, 34.1, 31.2, 29.9 ppm. IR (ATR): 2953.9, 2903.6, 2866.3, 1600.9, 1582.3, 1531.9, 1358.6, 1248.7, 1196.5, 1179.7, 747.3, 538.6 cm−1. Elemental analysis for C36H46GaIN2O2. Calc. C: 58.80%, H: 6.31%, N: 3.81%. Found: C: 58.84%, H: 6.13%, N: 4.08%.
Synthesis of [InCl(salphen)]21b
Prepared using general method B; yellow powder (72%). 1H NMR (400 MHz, 298 K, CDCl3) δ 8.79 (s, 2H), 7.72–7.67 (m, 2H), 7.59 (d, 4JHH = 2.6 Hz, 2H), 7.44–7.40 (m, 2H), 7.09 (d, 4JHH = 2.6 Hz, 2H), 1.53 (s, 18H), 1.33 (s, 18H) ppm. 13C{1H} NMR (101 MHz, 298 K, CDCl3) δ 169.6, 165.4, 143.0, 138.6, 137.1, 132.3, 129.8, 128.4, 117.8, 116.3, 35.7, 34.1, 31.3, 29.6 ppm. IR (ATR): 2953.9, 2905.5, 2866.3, 1600.9, 1580.4, 1528.2, 1429.4, 1388.4, 1172.2, 749.2, 533.0 cm−1.
Synthesis of [InBr(salphen)]21b
Prepared using general method B; yellow powder (93%). 1H NMR (400 MHz, 298 K, CDCl3) δ 8.78 (s, 2H), 7.71–7.67 (m, 2H), 7.59 (d, 4JHH = 2.6 Hz, 2H), 7.43–7.39 (m, 2H), 7.10 (d, 4JHH = 2.6 Hz, 2H), 1.54 (s, 18H), 1.34 (s, 18H) ppm. 13C{1H} NMR (101 MHz, 298 K, CDCl3) δ 169.5, 165.2, 143.1, 138.5, 137.0, 132.3, 129.8, 128.4, 117.8, 116.4, 35.7, 34.1, 31.3, 29.6 ppm. IR (ATR): 2953.9, 2905.5, 2868.2, 1599.0, 1578.5, 1528.2, 1429.4, 1388.4, 1250.5, 1172.2, 751.1, 533.0 cm−1.
Synthesis of [InI(salphen)]
Prepared using general method B; yellow powder (88%). 1H NMR (400 MHz, 298 K, CDCl3) δ 8.76 (s, 2H), 7.72–7.68 (m, 2H), 7.58 (d, 4JHH = 2.6 Hz, 2H), 7.45–7.41 (m, 2H), 7.09 (d, 4JHH = 2.6 Hz, 2H), 1.53 (s, 18H), 1.33 (s, 18H) ppm. 13C{1H} NMR (101 MHz, 298 K, CDCl3) δ 169.2, 164.8, 143.1, 138.4, 137.0, 132.2, 129.7, 128.4, 117.9, 116.4, 35.7, 34.1, 31.3, 29.7 ppm. IR (ATR): 2952.1, 2903.6, 2868.2, 1597.2, 1578.5, 1526.3, 1425.7, 1386.6, 1250.5, 1172.2, 747.3, 531.1 cm−1. Elemental analysis for C36H46InIN2O2. Calc. C: 55.40%, H: 5.94%, N: 3.59%. Found: C: 55.18%, H: 5.89%, N: 3.82%.
General procedure for the catalytic conversion of epoxides and CO2 to cyclic carbonates
A high-pressure reactor, equipped with a stirrer bar, was charged with catalyst, TBAI and epoxide. The reactor was then filled with CO2 to 2 bar and partially vented, a procedure that was repeated 3 times, before being finally filled with CO2 to a pressure of 8 bar. The reactor was left stirring for the required time at the required temperature. At the end of the reaction the reactor was cooled and slowly vented before a known amount of mesitylene was added. An aliquot was then removed and dissolved in CDCl3. The percentage conversion and NMR yield of the reaction was obtained from the 1H NMR spectrum of this crude sample using the mesitylene as internal standard and comparison to published spectra.17
General comments for computational study
All DFT calculations undertaken using the ORCA 4.2.1 computational software.33 Optimizations and analytical frequency calculations were performed at the RI-B97-D3/def2-SVP level of theory34–36 and single-point energies and solvation corrections calculated at RIJCOSX-ωB97M-D3BJ/def2-TZVPP.36–38 Solvation correction was implemented with the CPCM model39 with a dielectric constant value of 16 to represent the epoxide solvent and 69 for cyclic carbonate environments. Analytical frequencies were calculated for inclusion of the Zero Point Energy (ZPE) correction and entropic contributions to the free energy term (ΔG298 K), as well as confirming all intermediate were true with no imaginary modes and all transition states had the correct critical frequency of decomposition (imaginary mode). All thermodynamic parameters were calculated in the standard state (298.15 K and 1 atm). Numerical precision integration grids were increase beyond the default settings, to Grid4 for the SCF step and Grid5 for the final energy evaluation. Concentration correction for the individual species was applied as a free energy correction based on RT ln(ci/catm)40 where ci is the experimental concentration of the relevant species. This is particularly important in systems such as this, given the dual role of the epoxide, as both reagent and solvent. Graphical visualization and structural analysis performed from the DFT calculations using Avogadro 1.2.0.41 Quantum Theory of Atoms in Molecules (QTAIM) analysis was conducted with the Multiwfn software package.42 Comparison of the metal-to-ligand bonding descriptors are given in the ESI (Table S5†).
Fluoride ion affinity calculations
FIA calculations using the method developed by Krossing27 and updated by Greb43 have been calculated on the full ligand system for all experimentally studied catalysts. The TMS-anchored isodesmic reaction scheme method is shown in the ESI (Fig. S36†) and the “level of choice” for our calculations was RIJSCOSX-PW6B95/def2-qzvpp,44 based on the previously optimised structures. This method is the recommended DFT method for these calculations.43
X-ray crystal structure analyses
Diffraction data were collected using an Oxford Diffraction Supernova diffractometer, equipped with an Atlas CCD area detector and a four-circle kappa goniometer. For the data collection, Mo source with multilayer optics was used. Data integration, scaling, and empirical absorption correction were carried out using the CrysAlis Program package.45 The structures were solved using direct methods and refined by Full-Matrix-Least-Squares against F2 with SHELX46 under OLEX2.47 The non-hydrogen atoms were refined anisotropically, and hydrogen atoms were placed at idealized positions and refined using the riding model. Graphics were made with OLEX2 and MERCURY.48
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
CW would like to thank the Comunidad de Madrid (Spain) for funding (Programa de Atracción de Talento 2019: Modalidad 1; Award number 2019-T1/AMB-13037, and CM/JIN/2021-018). All authors would like to acknowledge funding from the Spanish Government (RTI2018-094840-BC31 and PID2020-113046RA-I00/AEI/10.13039/501100011033) and the Universidad de Alcalá (UAH-AE-2017-2). DJC thanks the Comunidad de Madrid (Spain) and European Union for funding a contract under the Programa INVESTIGO (47-UAH-INV). AH and RL would like to thank Sheffield Hallam University and the Biomolecular Sciences Research Centre for funding and computational resource access.
References
-
(a) P. Rollin, L. K. Soares, A. M. Barcellos, D. R. Araujo, E. J. Lenardão, R. G. Jacob and G. Perin, Appl. Sci., 2021, 11, 5024 CrossRef CAS;
(b) W. Guo, J. E. Gómez, À. Cristòfol, J. Xie and A. W. Kleij, Angew. Chem., Int. Ed., 2018, 57, 13735–13747 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- For general overviews, see:
(a) P. P. Pescarmona, Curr. Opin. Green Sustainable Chem., 2021, 29, 100457 CrossRef CAS;
(b) H. Büttner, L. Longwitz, J. Steinbauer, C. Wulf and T. Werner, Top. Curr. Chem., 2017, 375, 50 CrossRef PubMed;
(c) C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- For specific reviews, see:
(a) L. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC;
(b) M. Liu, X. Wang, Y. Jiang, J. Sun and M. Arai, Catal. Rev. - Sci. Eng., 2019, 61, 214–269 CrossRef CAS;
(c) M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651–2684 RSC.
- C. J. Whiteoak, G. Salassa and A. W. Kleij, Chem. Soc. Rev., 2012, 41, 622–631 RSC.
-
(a) A. Decortes, M. Martínez Belmonte, J. Benet-Buchholz and A. W. Kleij, Chem. Commun., 2010, 46, 4580–4582 RSC;
(b) A. Decortes and A. W. Kleij, ChemCatChem, 2011, 3, 831–834 CrossRef CAS.
- For selected examples, see:
(a) X. Wu and M. North, ChemSusChem, 2017, 10, 74–78 CrossRef CAS PubMed;
(b) J. A. Castro-Osma, M. North and X. Wu, Chem. – Eur. J., 2016, 22, 2100–2107 CrossRef CAS PubMed;
(c) J. A. Castro-Osma, K. J. Lamb and M. North, ACS Catal., 2016, 6, 5012–5025 CrossRef CAS;
(d) Z. Hošťalek, R. Mundil, I. Císařová, O. Trhlikova, E. Grau, F. Peruch, H. Cramail and J. Merna, Polymer, 2015, 63, 52–61 CrossRef;
(e) Y. Ren, Y. Shi, J. Chen, S. Yang, C. Qia and H. Jiang, RSC Adv., 2013, 3, 2167–2170 RSC;
(f) Y. Wang, Y. Qin, X. Wang and F. Wang, Catal. Sci. Technol., 2014, 4, 3964–3972 RSC.
- S. Ghosh, E. Glöckler, C. Wölper, A. Tjaberings, A. H. Gröschel and S. Schulz, Z. Anorg. Allg. Chem., 2021, 647, 1594–1601 CrossRef CAS.
- A. B. Kremer, R. J. Andrews, M. J. Milner, X. R. Zhang, T. Ebrahimi, B. O. Patrick, P. L. Diaconescu and P. Mehrkhodavandi, Inorg. Chem., 2017, 56, 1375–1385 CrossRef CAS PubMed.
- K. M. Osten and P. Mehrkhodavandi, Acc. Chem. Res., 2017, 50, 2861–2869 CrossRef CAS PubMed.
- C. Goonesinghe, H. Roshandel, C. Diaz, H.-J. Jung, K. Nyamayaro, M. Ezhova and P. Mehrkhodavandi, Chem. Sci., 2020, 11, 6485–6491 RSC.
- C. Diaz, J. Fu, S. Soobrattee, L. Cao, K. Nyamayaro, C. Goonesinghe, B. O. Patrick and P. Mehrkhodavandi, Inorg. Chem., 2022, 61, 3763–3773 CrossRef CAS PubMed.
- D. Specklin, C. Fliedel, F. Hild, S. Mameri, L. Karmazin, C. Bailly and S. Dagorne, Dalton Trans., 2017, 46, 12824–12834 RSC.
- For an overview of other In and Ga complexes bearing other ligands for ring-opening catalysis, see:
(a) S. Dagorne, M. Normand, E. Kirillov and J.-F. Carpentier, Coord. Chem. Rev., 2013, 257, 1869–1886 CrossRef CAS.
- A. Thevenon, A. Cyriac, D. Myers, A. J. P. White, C. B. Durr and C. K. Williams, J. Am. Chem. Soc., 2018, 140, 6893–6903 CrossRef CAS PubMed.
- H. A. Baalbaki, K. Nyamayaro, J. Shu, C. Goonesinghe, H.-J. Jung and P. Mehrkhodavandi, Inorg. Chem., 2021, 60, 19304–19314 CrossRef CAS PubMed.
- L. Álvarez-Miguel, J. Damián Burgoa, M. E. G. Mosquera, A. Hamilton and C. J. Whiteoak, ChemCatChem, 2021, 13, 4099–4110 CrossRef.
- H. A. Baalbaki, H. Roshandel, J. E. Hein and P. Mehrkhodavandi, Catal. Sci. Technol., 2021, 11, 2119–2129 RSC.
- For selected examples, see:
(a) X.-L. Chen, B. Wang, L. Pan and Y.-S. Li, Macromolecules, 2022, 55, 3502–3512 CrossRef CAS;
(b) J.-C. Yang, J. Yang, W.-B. Li, X.-B. Lu and Y. Liu, Angew. Chem., Int. Ed., 2022, 61, e202116208 CAS;
(c) C. A. L. Lidston, B. A. Abel and G. W. Coates, J. Am. Chem. Soc., 2020, 142, 20161–20169 CrossRef CAS PubMed;
(d) A. Gualandi, M. Marchini, L. Mengozzi, H. T. Kidanu, A. Franc, P. Ceroni and P. G. Cozzi, Eur. J. Org. Chem., 2020, 10, 1486–1490 CrossRef;
(e) B. A. Abel, C. A. L. Lidston and G. W. Coates, J. Am. Chem. Soc., 2019, 141, 12760–12769 CrossRef CAS PubMed;
(f) D. J. Saxon, M. Nasiri, M. Mandal, S. Maduskar, P. J. Dauenhauer, C. J. Cramer, A. M. LaPointe and T. M. Reineke, J. Am. Chem. Soc., 2019, 141, 5107–5111 CrossRef CAS PubMed;
(g) M. E. Fieser, M. Mandal, M. J. Sanford, N. J. V. Zee, L. A. Mitchell, C. R. Dunbar, D. M. Urness, C. J. Cramer, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2017, 139, 15222–15231 CrossRef CAS PubMed;
(h) Y. Zhou, R. Duan, X. Li, X. Pang, X. Wang and X. Chen, Chem. – Asian J., 2017, 12, 3135–3140 CrossRef CAS PubMed;
(i) M. J. Sanford, L. Peña Carrodeguas, N. J. V. Zee, A. W. Kleij and G. W. Coates, Macromolecules, 2016, 49, 6394–6400 CrossRef CAS;
(j) B. T. Whiting and G. W. Coates, J. Am. Chem. Soc., 2013, 135, 10974–10977 CrossRef CAS PubMed;
(k) E. H. Nejad, A. Paoniasari, C. G. W. van Melis, C. E. Koning and R. Duchateau, Macromolecules, 2013, 46, 631–637 CrossRef CAS;
(l) D. J. Darensbourg, P. Ganguly and D. Billodeaux, Macromolecules, 2005, 38, 5406–5410 CrossRef CAS.
- M. S. Hill and D. A. Atwood, Eur. J. Inorg. Chem., 1998, 1, 67–72 CrossRef.
-
(a) C. Diaz, T. Tomković, C. Goonesinghe, S. G. Hatzikiriakos and P. Mehrkhodavandi, Macromolecules, 2020, 53, 8819–8828 CrossRef CAS;
(b) M. S. Hill and D, A. Atwood, Main Group Chem., 1998, 2, 191–202 CrossRef CAS.
-
(a) R. R. Butala, S. Parkin, J. H. Walrod and D. A. Atwood, Inorg. Chem., 2021, 60, 4456–4462 CrossRef CAS PubMed;
(b) A. Mitra, L. J. DePue, S. Parkin and D. A. Atwood, J. Am. Chem. Soc., 2006, 128, 1147–1153 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- We propose that the following reaction takes place: 2[MX(salphen)] + Et3PO → [M(salphen)]2O + [Et3PX]X and even with excess of Et3PO we were unable to observe the signals required for calculation of the Acceptor Number (AN).
- F. Castro-Gómez, G. Salassa, A. W. Kleij and C. Bo, Chem. – Eur. J., 2013, 19, 6289–6298 CrossRef PubMed.
- T. T. Wang, Y. Xie and W. Q. Deng, J. Phys. Chem. A, 2014, 118, 9239–9243 CrossRef CAS PubMed.
- H. Böhrer, N. Trapp, D. Himmel, M. Schleep and I. Krossing, Dalton Trans., 2015, 44, 7489–7499 RSC.
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, ChemPhysChem, 2020, 21, 987–994 CrossRef CAS PubMed.
- H. Plommer, L. Stein, J. N. Murphy, N. Ikpo, N. Mora-Diez and F. M. Kerton, Dalton Trans., 2020, 49, 6884–6895 RSC.
- F. D. Monica and A. W. Kleji, Catal. Sci. Technol., 2020, 10, 3483–3501 RSC.
- Y. Wang, Y. Qin, X. Wang and F. Wang, Catal. Sci. Technol., 2014, 4, 3964–3972 RSC.
- This method is based on the procedure reported by Williams and co-workers for the synthesis of [InCl(salphen)]: see ref. 14.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- F. Neese, J. Comput. Chem., 2003, 24, 1740–1747 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
-
(a) F. Weingrad and R. Aldrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC;
(b) A. Schaefer, H. Horn and R. Aldrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef CAS.
- F. Neese, F. Wennmohs, A. Henson and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- N. Mardirossian and M. Head-Gordon, J. Chem. Phys., 2016, 144, 214110 CrossRef PubMed.
- S. Sinnecker, A. Rafendran, A. Klamt, M. Diedenhofen and F. Neese, J. Phys. Chem. A, 2006, 110, 2235–2245 CrossRef CAS PubMed.
- J.N. Harvey, F. Himo, F. Maseras and L. Perrin, ACS Catal., 2019, 9, 6803–6813 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, ChemPhysChem, 2020, 21, 987–994 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2005, 109, 5656–5667 CrossRef CAS PubMed.
-
CrysAlisPro: Data Collection and Integration Software, version 1.171.37.35, Agilent Technologies UK Ltd., Oxford, U.K., 2011 Search PubMed.
- G. M. Sheldrick, Crystallogr, 2008, A64, 112–122 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. J. Puschmann, Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- MERCURY:
(a) I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, B58, 389–397 CrossRef CAS PubMed;
(b) C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. J. van de Streek, Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, including all synthesis and characterization data. CCDC 2173026 and 2177923. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00089c |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Lucía
Álvarez-Miguel
a,
Lucía
Álvarez-Miguel